

Abstract
RATIONALE:
Metabolomics analyses using gas chromatography mass spectrometry (GC/MS) - based metabolomics are heavily impeded by the lack of high-resolution mass spectrometers and limited spectral libraries to complement the excellent chromatography that GC platforms offer, a challenge that is being addressed with the implementation of high resolution (HR) platforms such as 1D-GC/Orbitrap-MS.
METHODS:
We used serum samples from a non-human primate (NHP), a baboon (Papio hamadryas), with suitable quality controls to quantify the chemical space using an advanced HR MS platform for confident metabolite identification and robust quantification to assess the suitability of the platform for routine clinical metabolomics research. In a complementary approach, we also analyzed the same serum samples using a two-dimensional gas chromatography time-of-flight mass-spectrometer (2D-GC/ToF-MS) for metabolite identification and quantification following established standard protocols.
RESULTS:
Overall, the 2D-GC/ToF-MS (~5000 peaks per sample) and 1D-GC/Orbitrap-MS (~500 peaks per sample) analyses enabled identification and quantification of a total of 555 annotated metabolites from the NHP serum with a spectral similarity score Rsim ≥ 900 and S/N ratio of > 25. A common set of 30 metabolites with HMDB and KEGG IDs was quantified in the serum samples by both platforms where 2D-GC/ToF-MS enabled quantification of a total 384 metabolites (118 HMDB IDs) and 1D-GC/Orbitrap-MS analysis quantification of a total 200 metabolites (47 HMDB IDs). Thus, roughly 30–70% of the peaks remain unidentified or un-annotated across both platforms.
CONCLUSIONS:
Our study provides insights into the benefits and limitations of the use of a higher mass resolution and mass accuracy instrument for untargeted GC/MS-based metabolomics with multi-dimensional chromatography in future studies addressing clinical conditions or exposome studies.
Keywords: metabolomics, Orbitrap, GC/MS, serum, non-human primate, high resolution
1 |. INTRODUCTION
Metabolomics, the study of systematic quantification of small molecules in the molecular weight range of 50–2000 Daltons in biological systems (cells, tissues, organs, biofluids, or whole organisms), has proven to be an indispensable platform for systems biology and precision medicine.1 Instrumentation such as gas and liquid chromatography with mass-spectrometry (GC/MS, LC-MS)2,3 alongside spectroscopy platforms (NMR, IR)4,5 has aided the capture of the biochemical space closer to the phenotype6 as a functional genomics tool. Gas chromatography, amenable to polar primary metabolites7and fatty acids, provides advantages over other chromatographic systems by virtue of its excellent chromatographic resolution, and it can be used effectively for routine quantitative metabolomic applications. With the advent of modern instruments, such as Orbitrap technology-based mass spectrometers, that are capable of sub-ppm mass accuracy at high mass resolutions (i.e., > 50,000), calculation of predicted molecular formulae based on the mass defect of a detected ion is now possible.8 Orbitrap-based high resolution accurate mass instrumentation offers the ability to obtain mass spectral data at high resolving power with mass accuracies <1 ppm. Since the implementation of GC/Orbitrap technology9–11, the technology has only been used in a few studies on biofilms12 and pesticide analysis.13 However, no metabolomics studies have used this unique platform for metabolomic analysis of primate biofluids or tissues. With the recent realization of the importance of the exposome (defined as the entirety of physicochemical and biological exposure endured by an organism from birth till death) in influencing human health14 and disease risk,15 the blood exposome can be of paramount interest in uncovering causes of a wide range of diseases.16 In this research area, the use of gas chromatography (GC)/high resolution accurate mass (HRAM) mass spectrometer (MS) for improved detection and quantitation of metabolites may be of particular interest and benefit.
Two-dimensional gas chromatography time-of-flight mass-spectrometry (2D-GC/ToF-MS), developed in the 1990s, has gained growing popularity as a powerful analytical tool for volatile, semi-volatile and derivatization-amenable polar compounds, as the instrumentation provides unprecedented selectivity (three separation dimensions, related to volatility, polarity, and mass), high sensitivity (through band compression), enhanced separation power, and increased speed.17 Using human serum samples, Winnike et al 18 compared 1D-GC/ToF-MS and 2D-GC/ToF-MS platforms and showed that a 2D system detected about three times as many peaks (for instance, at S/N=50 505 and 1517 peaks were quantified using 1D-GC/ToF-MS and 2D-GC/ToF-MS platforms, respectively). The 2D-GC/ToF-MS platform provides better mass spectral quality and improved sensitivity owing to enhanced resolution, alongside improved peak capacity, resulting in a combined power of chromatographic and mass spectral deconvolution resolution known as ‘analytical purity’.19 In contrast, a typical 1D-GC-TOF-MS performed for serum and urine samples yields 200–700 metabolite peaks.20 Thus, there is a clear need to expand the chemical space of biofluids, more specifically, the blood-based biomarker metabolites,16 and for robust and sensitive quantification of metabolites. To our knowledge, our report is the first attempt to identify and quantify serum metabolites, and to evaluate the power of separation achieved via dimensions (2D-GC/ToF-MS) and mass resolution (1D-GC/Orbitrap-MS) in a comparative basis.
For over 50 years, baboons (Papio hamadryas) have served as an experimental non-human primate (NHP) model for a wide range of human cardiometabolic disorders owing to their impeccable similarities to humans in genome sequence, pathophysiology (in cardiovascular diseases, obesity, type 2 diabetes), and development.21 Given the importance of baboons as a NHP model system, we embarked on the investigation described here to capture and analyze serum-specific metabolites. The principal aims were to assess the capabilities of the two instrument platforms (1D-GC/Orbitrap-MS and 2D-GC/ToF-MS) for the identification of metabolites in baboon serum and the robustness of quantification, and to evaluate the performance of increased chromatographic dimensions to high mass resolution for expanding the biochemical space of serum metabolites.
2 |. EXPERIMENTAL
2.1 |. Chemicals
Acetonitrile, isopropanol, and pyridine were of HPLC grade, and methoxyamine hydrochloride (MeOX), 1% TMCS in N-methyl-N-trimethylsilyl-trifluoroacetamide (MSTFA), adonitol, and d4-succinic acid were obtained from Sigma-Aldrich, St. Louis, MO, USA.
2.2 |. Baboon serum sample
For this study, we utilized a healthy adult male olive baboon (Papio hamadryas) maintained as part of the baboon colony at the Southwest National Primate Research Center, located on the campus of the Texas Biomedical Research Institute, San Antonio, Texas, USA. The male baboon used in this study was 18 yrs. old. The baboon had been raised and maintained on a standard monkey chow diet (high complex carbohydrates; low fat) prior to the fasting blood collection. All procedures involving animals were reviewed and approved by the Texas Biomedical Research Institute’s Institutional Animal Care and Use Committee (IACUC). Freshly collected serum samples were stored in aliquots at −80 °C until analysis.
2.3 |. Serum sample extraction and derivatization for GC/MS analysis on both platforms
Aliquots of serum (30 µL) samples were subjected to sequential solvent extraction once each with 1 mL of acetonitrile: isopropanol: water (3:3:2, v/v) ratio and 500 µL of acetonitrile: water (1:1, v/v) ratio mixtures at 4 °C.22 Adonitol and d4-succinic acid (both 5 µL from 10 mg/mL stock) were added to each aliquots as two internal standards prior to the extraction. The pooled extracts (~ 1500 µL) from the two steps were dried under vacuum at 4 °C prior to chemical derivatization. Dummy extractions performed on blank tubes served as extraction blanks to account for background (extraction solvent, derivatization reagents) noise and other sources of contamination (septa, liner, column, vials, handling etc.). Blanks were only used to see that no carry overs occurred during randomized run orders and to manually filter out contaminating chemicals from the combined list of features. Six (S1, S2, S3, S6, S7, S8) samples were then sequentially derivatized with methoxyamine hydrochloride (MeOX) and 1% TMCS in N-methyl-N-trimethylsilyl-trifluoroacetamide (MSTFA) as described elsewhere.23, 24 The steps involved the addition of 10 μL of MeOX (20 mg/mL) in pyridine incubated under shaking at 55 °C for 60 min followed by trimethylsilylation at 60 °C for 60 min after adding 90 μL MSTFA. We summarize the entire workflow for the two platforms used in this study in Figure 1.
FIGURE 1.

A flow chart demonstrating the two workflows used for sample preparation, data acquisition and analysis, and interpretation for 2D-GC/ToF-MS and 1D-GC/Orbitrap-MS.
2.4 |. 1D-GC/Orbitrap-MS instrument parameters
The acquisition sequence started with blank solvent (pyridine) injections, followed by randomized lists of extraction blanks (B), reagent blanks (R) and samples (S). QC samples were injected at scheduled intervals for tentative identification and monitoring shifts in retention indices (RI) as well as serving as quality control (QC) checks. Derivatized samples (S6, S7, S8; and three technical replicates from each, i.e., nine runs), blank solvent and reagents, and QC samples were injected in randomized order across the batch, for a total of nine injection runs for the three independent samples. A robotic arm TriPlus™ RSH autosampler (Thermo Scientific™, Bremen, Germany) injected 1 µL of derivatized sample into a split/splitless (SSL) injector at 250 °C using a 1:100 split flow on TRACE™ 1310 gas chromatography (Thermo Scientific™, Austin, TX, USA). Helium carrier gas at a flow rate of 1 mL/min was used for separation on a Thermo Scientific™ TraceGOLD™ TG-5SILMS column (30 m length × 0.25 mm i.d. × 0.25 µm film thickness). The initial oven temperature was held at 70 °C for 4 min, followed by an initial gradient of 20 °C/min ramp rate. The final temperature was 320 °C and this was held for 8 min. Eluting peaks were transferred through an auxiliary transfer temperature of 250 °C into a Q Exactive™-GC mass spectrometer (Thermo Scientific™, Bremen, Germany). The mass spectrometer has an ExtractaBrite Ion Source technology, patented RF lens, with resolving power (RP) 120,000 FWHM @ m/z 200 with EI, chemical ionization (CI), full-scan, timed-SIM and MS/MS capabilities. From the ion source, an AQT quadrupole is used for precursor ion isolation, which leads to the Orbitrap Mass analyzer or HCD cell for MS/MS fragmentation. Electron ionisation (EI) at 70 eV energy, emission current of 50 µA with an ion source temperature of 230 °C was used in all experiments. A filament delay of 5.3 min was selected to prevent excess reagents from being ionized. High-resolution EI spectra were acquired using 60,000 resolution (FWHM at m/z 200) with a mass range of m/z 50–650.
2.5 |. 1D-GC/Orbitrap-MS data processing
The acquired data were processed using Thermo Scientific™ TraceFinder™ 4.1 software for untargeted analysis. Initial analysis of collected spectra included baseline correction, peak filtering, quantification, assignment of a unique mass and retention index, signal-to-noise calculation and compound identification based on the mass spectral pattern compared with EI spectral libraries such as NIST Mass Spectral Reference Library (NIST14/2014; National Institute of Standards and Technology, Gaithersburg, MD, USA), the MSRI spectral libraries from Golm Metabolome Database25 available from Max-Planck-Institute for Plant Physiology, Golm, Germany (http://csbdb.mpimp-golm.mpg.de/csbdb/gmd/gmd.html), MassBank,26 MoNA (Mass Bank of North America, (http://mona.fiehnlab.ucdavis.edu/) and a supplied high resolution (HR)-MS mass spectral library for the GC/MS dataset using proprietary TraceFinder™ software.
2.6 |. Metabolomic analysis by 2D-GC/ToF- MS
Gas chromatography-mass spectrometry was performed as described by Lisec et al.23 Derivatized samples (S1, S2, S3; and three technical replicates from each, i.e., nine runs) were injected in splitless mode using an autosampler (VCTS, Gerstel™, Linthicum, MD, USA) consisting of an Agilent© 7890 B gas chromatography (Agilent Technologies, Palo Alto, CA, USA) in line with a Pegasus ® 4D ToF-MS instrument (LECO Corp., St. Joseph, MI, USA) equipped with an electron ionization (EI) source. The injection temperature was set at 250 °C (front inlet) and the helium (carrier gas) flow rate was set to 1 mL min−1. Separation by gas chromatography was achieved using two columns, a primary Rxi®−5Sil MS capillary column (Cat. No. 13623–6850, Restek, Bellefonte, PA, USA) (30 m × 0.25 mm × 0.25 μm) in line with a secondary Rxi®−17Sil capillary column (Cat. No. 40201–6850, Restek) (2 m × 0.15 mm × 0.15 μm). The columns were connected using a press-fit connector before the modulator, with modulation occurring on the secondary column. The temperature program for the primary column started isothermal at 70 °C for 1 min followed by a 6 °C min−1 ramp to 310 °C and a final 11 min hold at 310 °C. The secondary oven temperature was programmed with an offset of 5 °C whereas the modulator temperature (i.e., the temperature at which eluates from the primary (first) column are injected into the secondary (second) column, regulated by a dual-stage Quad-jet thermal modulation that uses a liquid nitrogen stream as a coolant and heating from the secondary oven itself) offset was 15° C relative to the first oven temperature. The modulation period (i.e., second-dimension separation time) was 4 s divided into hot and cold pulse times of 0.6 s and 1.4 s, respectively between the two stages. The transfer line temperature was maintained at 250 °C and the ion source temperature was 250 °C. All samples were run with an offset time of 470 seconds to allow the solvents and derivatizing reagents to pass through without reaching the detector. The system was then temperature-equilibrated at 70 °C for 5 min before the next sample. The acquisition sequence started with blank solvent (pyridine) injections, followed by a randomized list of extraction blanks (B), reagent blanks (R) and samples (S). Furthermore, pooled QC samples were injected at scheduled intervals and at the start for tentative identification and monitoring shifts in retention time. Mass spectra were collected at 200 scans/s with a range of m/z 40–600.
2.7 |. 2D-GC/ToF-MS data processing
The 2D-GC/ToF-MS data sets were processed (clean-up, aligned, and annotated) using ChromaToF version 4.71.0.0 (LECO Corp.) as described by Winnike et al 18 with settings such as S/N: 25; peak width: 0.15 s, base line offset: 1; m/z range: 50–800, library matching score cut off at > 500 as cut-off). The consulted EI spectral libraries were same as described for the Orbitrap platform. The filtered raw GC/MS data included data from three technical replicates and ~600 manually curated analytes. Base peak areas (BPCs) of the fragment ions (m/z) were normalized using median normalization and log2 transformation. The peak areas were further normalized by dividing each peak area value by the area of the internal standard for a specific sample.
For both platforms the metabolite annotation and assignment followed the metabolomics standards initiative (MSI) guidelines for metabolite identification, i.e., Level 2: identification was based on spectral database (match factor >80%) and Level 3: only compound groups were known, e.g. specific ions and RT regions of metabolites.
The raw datasets and the metadata associated with both the platforms have now been deposited at the Metabolomics Workbench (Study ID: ST000972) and are available at https://bit.ly/2IH4MxB.
2.8 |. Statistical analysis
Statistical processing of both GC/MS data sets was performed using statistical software R (Version 3.4.3).27, 28 Imputted, outlier removed, and scaled peak area representative of relative metabolite amounts obtained from using DeviumWeb29 are presented.
2.9 |. Univariate and multivariate analysis
Hierarchical clustering analysis (HCA) was performed on Pearson distances using PermutMatrix30. The raw metabolite abundance values were Z-score normalized, and the color scale represents +2 (high) to −2 (low) abundance in the heat map. The correlations reported are Spearman rank correlations. Furthermore, Pearson correlations were visualized as heat maps, based on Z-score normalized data ranging from +1 (positive, red), 0 (no correlation, black), and −1 (negative, green) correlation of metabolite abundance across nine technical replicates. Principal components analysis (PCA) and partial least squared discriminant analyses (PLSDA) were performed using DeviumWeb,29 where the output displayed score plots to visualize the sample groups. The data were scaled with unit variance without any transformation.
2.10 |. Pathway enrichment and clustering analysis
Pathway enrichment was performed using MetaboAnalyst (www.Metaboanalyst.ca).31 For ID conversions, the Chemical Translation Service (CTS: http://cts.fiehnlab.ucdavis.edu/conversion/batch) was used to convert the common chemical names into their KEGG, HMDB, Metlin, PubChem CID, and ChEBI identifiers.
3 |. RESULTS AND DISCUSSION
3.1 |. Platform dependent identification of metabolites in baboon serum
Using the 1D-GC/Orbitrap-MS platform, we identified a total of 200 metabolites (47 HMDB IDs) out of 1565 metabolite peaks detected at least in 50% of the runs using a similar criterion36, 37 obtained from the studied sample sets (Table S1, supporting information). Furthermore, of the 74 polar metabolites identified in the global GC/MS approach in human serum, only 33 could be clearly quantified38. We focused our analysis on known compounds (i.e., those that could be annotated after a significant match score and superior S/N) to be able to interpret the biological relevance of the compounds, which is not feasible with unknown metabolites in a prototype study. These metabolites represent 50 different KEGG-based metabolic pathways such as protein biosynthesis, glucose-alanine cycle, malate-aspartate shuttle, glutathione metabolism, galactose metabolism, ammonia recycling, alanine metabolism, urea cycle, arginine and proline metabolism, alpha-linolenic and linoleic acid metabolism, aspartate metabolism, beta-alanine metabolism, starch and sucrose metabolism among others (Table S2, supporting information). Using the 2D-GC/ToF-MS platform, we identified 384 metabolites (118 HMDB IDs) out of 3154 metabolite peaks detected at least in 50% of the runs, obtained from the studied sample sets (Table S3, supporting information). These belonged to 60 different KEGG-based pathways such as protein biosynthesis, urea cycle, galactose metabolism, alanine metabolism, ammonia recycling, alpha linolenic acid and linoleic acid metabolism, arginine and proline metabolism, aspartate metabolism, glucose-alanine cycle, beta-alanine metabolism among others (Table S4, supporting information).
Altogether, the two platforms allowed detection of both endogenous and exogenous compounds. For instance, cholesterol, amino acids, sugars, sugar alcohols, organic acids, fatty acids and lipid classes, polyamines, nitrogenous compounds, phosphate intermediates were observed, which are typically quantified in human serum samples7. Other physiologically interesting compounds (adrenaline, O-acetylcarnitine, xanthine, taurine, sialyllactose, NAD, tocopherol), metabolites that were associated with KEGG and HMDB identifiers and with biological relevance and constitute the endogenous compounds.
Compounds identified such cholesterol, gamma-tocopherol, CA cycle intermediates (citric acid, oxalic acid), sugars (sucrose, glucose, mannose, isomaltose), amino acids (glutamic acid, alanine, proline, aspartic acid, cysteine, pyroglutamic acid, arginine, valine, tryptophan, leucine/isoleucine), sugar alcohols (my-inositol), phosphates (glycerol-2-phosphate, myo-inositol-phosphate), nitrogenous compounds (hypoxanthine, xanthine), fatty acids (heptadecanoic acid, stearic) and organic acids (lactic acid, hippuric acid) are well established components of the human serum38. Elaidic acid, (trans-9 C18:1, pure fatty acid with esterified oleic acid in triglyceride) is a component of hydrogenated vegetable oils as a part of the monkey chow diet fed to the baboon. Palatinose (i.e., isomaltulose) present in sugar crops, could of baboon dietary origin. Phenethylamine is a monoaminergic neuromodulator/ neurotransmitter in the human central nervous system that occurs naturally in the body. Hippuric acid, is a component tin biofluids, and is a result of consumption of phenolic compounds, possibly in the plant-based chow diet of the baboon.
For the baseline correction, mass spectral deconvolution, peak picking, integration, library search, and signal-to-noise filtering, we used proprietary software supplied by the vendors, i.e., ChromaToF™ (LECO Corp.) and TraceFinder™ (Thermo Scientific). Typical total ion current (TIC) chromatograms for a 2D-GC/ToF-MS and 1D-GC/Orbitrap-MS are shown in Figures 2 A and 2B, respectively. The median signal intensities for identified metabolites were 5.3E+7 and 5.6E+4, for 1D-GC/Orbitrap-MS and 2D-GC/ToF-MS, respectively. The average TIC intensities were 2.092E+11 and 1.443E+8 for 1D-GC/Orbitrap-MS and 2D-GC/ToF-MS, respectively. Earlier studies have shown that 5 ppm mass accuracy of the Orbitrap mass analyzer is reached with >95% probability at a dynamic range of more than 5000, which is at least an order of magnitude higher than typical values for ToF instruments.32 These higher magnitudes of intensity ranges for the high-resolution platform aids in better and more robust quantification than ToF and QQQ instruments with GC chromatographic systems.
FIGURE 2.
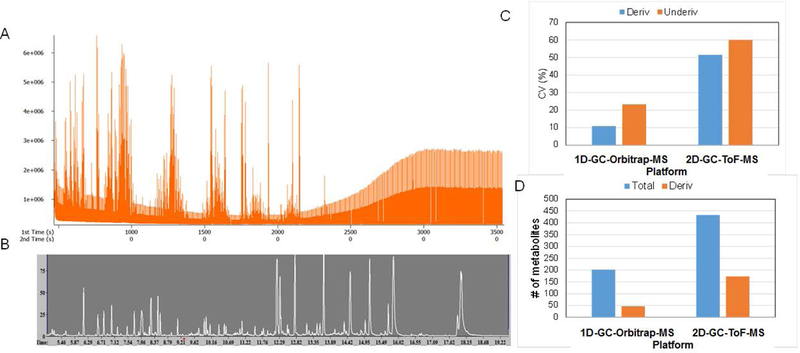
(A) The total ion chromatogram (TIC) for 2D-GC/ToF-MS, and (B) 1D-GC/Orbitrap-MS runs, (C) CV expressed in % for derivatized and underivatized compounds in 1D-GC-Orbitrap-MS and 2D-GC/ToF-MS platforms, and (D) number of confidently quantified metabolites in 1D-GC/Orbitrap-MS and 2D-GC/ToF-MS platforms.
As an example, we demonstrate the EI-spectra of the TMS derivatives of four amino acids (aspartic acid, 3TMS; glutamic acid, 3TMS; valine, 2TMS, and tryptophan, 3TMS) obtained from the high resolution 1D GC-Orbitrap-MS platform (Figure 3 A-D).
FIGURE 3.

EI-spectra of four amino acids (A) aspartic acid, 3TMS; (B) glutamic acid, 3TMS; (C) valine, 2TMS, and (D) tryptophan, 3TMS) obtained from the high-resolution 1D GC/Orbitrap-MS platform.
In addition to the above well-studied metabolites of biological relevance, 2D-GC-ToF-MS and 1D-GC-Orbitrap-MS yielded an additional 192 and 112 compounds, respectively, without any biological pathway identifiers (i.e., HMDB and KEGG IDs), suggesting a possible exposomic/exogenous origin or those that were not captured by existing stringent noise filtering criteria. These exogenous compounds range from drugs and pharmaceuticals (norlidocaine, ketamine, 9-OH-risperidone, cetirizine N-oxide) possibly administered to the baboon during its life course to dietary constituents (trans-zeatin, coumarin, kinetin-9-riboside, aesculetin) as well as environmental compounds or exposures through their human handlers. Furthermore, identification of the exposomic or exogenous origin compounds remains challenging as the biological (spectral) DBs do not include them, which can further complement the high resolution data generated by advanced platforms. In an illustration, we provide the EI-MS spectra examples two metabolites, glycerol-3TMS and urea-2TMS obtained for 1D-GC-Orbitrap-MS and 2D-GC-ToF-MS, respectively (Table S1, supporting information).
Overall, the metabolites identified in our analysis outnumber the list of identified metabolites in previously reported primate studies in rhesus macaques and chimpanzees, 33 where 177 of the 399 metabolites extracted from liver were associated with an identifier and had an associated name. Clearly, the compounds reported in our study, identified with stringent criteria in place (S/N> 25, R(sim)> 900, and intensity counts of at least 1E+3), will need to be validated using other orthogonal techniques or platforms. Nonetheless, the results provide a potential larger foundation for exposomic studies in NHPs.
Previous studies using 2D-GC/ToF-MS using spleen extracts have led to the detection of around 1200 compounds compared with only 500 in the 1D-GC/ToF-MS of the same samples.19 Very recently, the power of 2D LC/HRMS was exploited for the analysis of rice metabolome.34 Three-dimensional (3D) gas chromatography with time-of-flight mass spectrometric detection (GC3/ToFMS) allowed the analytical advantages of employing three varied chemical selectivities in the 3D separation coupled with ToF-MS for the resolution of complex mixtures in equivalent modulation period conditions.35
3.2 |. Robust quantification with the high mass resolution platform
In addition to our assessment of the qualitative differences, we evaluated the metabolite abundance data for their quantitative variations. Technical variability can significantly impede the statistical inferences in a given metabolomics datasets, increasing the risk for spurious and misinterpreted results and limited reproducibility. To this end, we evaluated the CV, a measure of variance of quantified metabolites [CV%= standard deviation/ mean) *100] for the metabolites identified using both platforms across the biological replicates, averaging out the technical replicates. The results indicate that the median CVs for derivatized compounds were 10.9% and 51.5% for 1D-GC/Orbitrap-MS and 2D-GC/ToF-MS, respectively (Figure 2C). For underivatized metabolites, the median CV values were 23.3% and 59.9% for 1D-GC/Orbitrap-MS and 2D-GC/ToF-MS, respectively. These measures are useful in providing a decent measure of instrument performance for metabolomics applications for a given platform, and not their comparability. Furthermore, algorithms that account for the variation of measurement error with intensity were found to provide the most accurate estimates of differential abundance in mass-spectrometry.39 Use of the total MS signal is one of the better methods for the non-targeted metabolomic and proteomic analysis of the biological fluid samples, and is a very common data processing method toward normalization.40 However, the total number of metabolites identified were higher for the 2D-GC/ToF-MS platform. Using high resolution 1D-GC/Orbitrap-MS only on EI capabilities we identified fewer metabolites (Figure 2D). Of the total of 200 quantified metabolites, 46 were derivatized (trimethylsilyated) for the1D-GC/Orbitrap-MS platform, while for the 2D-GC/ToF-MS platform, 117 of the 384 metabolites were derivatized.
In addition to the CV% calculations, we explored multivariate statistical analyses to assess whether either of the platforms performed better for clustering the technical replicates and the samples among each other. With a multitude of multivariate analysis, such as the unsupervised PCA analyses, we observed that the first two PCs (PC1, PC2) could explain 40.5% of the variance for the metabolites quantified using the 2D-GC/ToF-MS platform, whereas for the metabolites quantified using the 1D-GC/Orbitrap-MS platform, the first two PCs could explain 64.8% of the total variance (Figures 4A and4B). Using unsupervised PLS-DA analyses, we observed that the first two PCS (PC1, PC2) could explain 34.7% of the variance for the 2D-GC/ToF-MS platform, whereas for the 1D-GC/Orbitrap-MS platform, the first two PCs could explain 64.8% of the total variance (Figures 4C and4D). For the 2D-GC/ToF-MS platform, the PLS-DA models were validated using R2 and Q2 based on leave-one-out cross-validation (LOOCV) where a five-component model was selected as optimized model with R2=0.92, Q2=0.22. For the 1D-Orbitrap-MS platform, the PLS-DA models, a five-component model was optimal with R2=0.98, Q2=0.26. Furthermore, a metabolite-metabolite (Pearson’s) correlation based on the relative metabolite abundance data revealed that the metabolites form tighter/ stronger clusters (i.e., modules) for GC/Orbitrap-based metabolite abundance data than those for 2D-GC/ToF-MS-based metabolite data (Figure 5).
FIGURE 4.
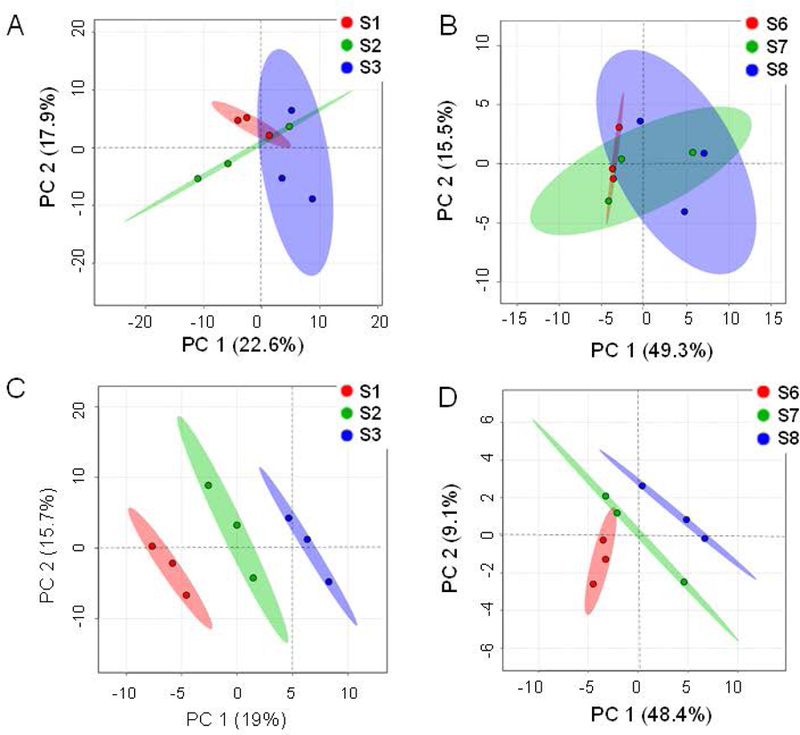
Multivariate analysis of the metabolite abundance data. PCA score-plots for (A) 2D-GC/ToF-MS; first two PCs (PC1, PC2) could explain 40.5% of the variance for the metabolites quantified using the 2D-GC/ToF-MS platform and (B) 1D-GC/Orbitrap-MS datasets where the first two PCs could explain 64.8% of the total variance. The PLS-DA score plots for (C) 2D-GC-ToF-MS demonstrate that the first two PCS (PC1, PC2) could explain 34.7% of the variance and for (D) 1D-GC/Orbitrap-MS datasets, the first two PCs could explain 64.8% of the total variance. S1, S2, S3 were serum samples run by 2D-GC/ToF-MS and S6, S7, S8 were serum samples run by 1D-GC/Orbitrap-MS.
FIGURE 5.
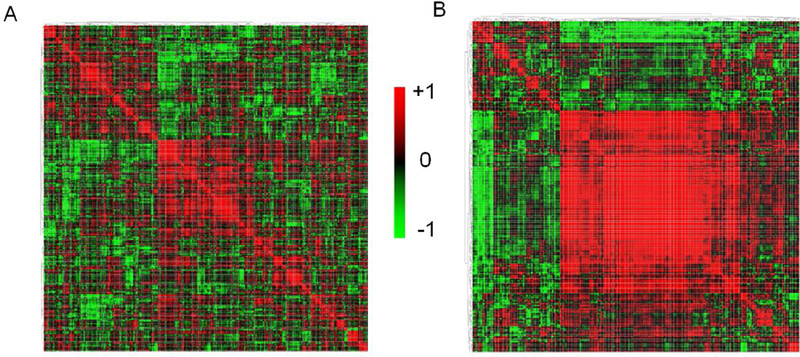
Metabolite-metabolite (Pearson’s) correlation for (A) 2D-GC/ToF-MS quantified and (B) 1D-GC/Orbitrap-MS quantified metabolite abundance data. The diffused small modules (clusters) in A and the larger central module in B indicate tighter clustering based on quantitative data for the 1D-GC/Orbitrap-MS platform.
3.3 |. Platform specificity of the metabolites quantified
The two platforms used here, 2D-GC/ToF-MS and the 1D-GC/Orbitrap-MS, offer different capabilities and advantages. First, the combination of both low polar (Rtx-5, cross bond diphenyl dimethyl polysiloxane fused silica) and mid-polar (Rxi-17Sil, cross bond phase) columns helped in capturing a wide range of metabolites ranging from volatiles and semi-volatiles (underivatized) to polar metabolites (upon derivatization) for the 2D-GC/ToF-MS platform. This advantage was obviously not achieved in the case of the 1D-GC/Orbitrap-MS platform that utilized only one low polar column. Second, the 2D-GC/ToF-MS instrument with its ToF capabilities provides only unit resolution, albeit at a faster scan rate and increased peak capacity allowing identification of a larger number of compounds. Third, the 2D instrument provided another additional dimension owing to the thermal modulation capabilities from the secondary oven. Finally, the high-resolution 1D-GC/Orbitrap-MS platform provides higher mass accuracy (< 5 ppm), in addition to a higher dynamic range for compound quantification. Thus, the list of metabolites generated from the two platforms have significant overlap, although system-specific sets of metabolites are also discovered.
Hierarchical clustering analysis (HCA) for the three samples (i.e., nine technical replicates altogether) revealed the grouping of the top 25 metabolites for both platforms (Figures 6A and6B). A closer look at the metabolite IDs showed differences in discrimination power for the 3 samples across the two platforms. The metabolites quantified using 2D-GC/ToF-MS showed two clusters (top and bottom) where the first cluster consisted of sugars (fructose, glucose), organic acids (citric acid, ribonic acid) and amino acids (threonine, aspartate, phenylalanine, arginine), and the second cluster consisted of several fatty acid metabolites (octadecanoic acid, decanal, hexanoic acid, heptanoic acid, nonanal) among others, which helped define the differences between sample S1, and S2 and S3 (Figure 6A). For the 1D-GC/Orbitrap-MS platform, the upper cluster, which was the major cluster, consisted of amino acids (leucine, arginine), fatty acid metabolites (cholesterol, elaidic acid, undecanone, 9-eicosene, decanal, 3-methyl-2-oxovaleric acid) among others, thus discriminating samples S8 from S6 and S7 (Figure 5B). We do not envisage that the samples will be quantitatively or qualitatively very different from each other given the consistency in internal standards observed for the nine technical replicates across each platform, but the chemical specificities and differences in their CVs allowed discrimination of the samples.
FIGURE 6.

Heat map displaying hierarchical clustering analysis (HCA) analysis for relative abundance data for (A) 2D-GC/ToF-MS and (B) 1D-GC/Orbitrap-MS quantified metabolites. Both platforms were capable of clustering the independent technical-runs/replicates together while retaining the biological replicates separate. Furthermore, the discriminatory metabolites (amino acids, and fatty acids) are common to both platforms in addition to unique metabolites captured by each of the platforms.
30 metabolites (with biological identifiers) were confidently identified (with similarity/ matching score > 900, S/N > 25) by both platforms. 2D-GC/ToF-MS allowed the identification and quantification of an additional 118 unique metabolites, probably by virtue of two different column chemistries in series, and additional thermal modulation, and the HR-GC/MS platform allowed identification and quantification of 46 unique metabolites that were not captured using 2D-GC/ToF-MS (Figure 7A). These identified metabolites converted into 49 shared pathways, and 11 and 1 unique pathways for 2D-GC/ToF-MS and 1D-GC/Orbitrap-MS, respectively (Figure 7B). The higher coverage of pathways that are biologically relevant would be useful in discovery metabolomics, while a more confident and robust quantification in HR-MS would be useful for targeted approaches. Given that the majority of the identified compounds do not pertain to KEGG-based and known biological pathways, these identified metabolites could be exogenous, and may constitute an example of the NHP exposome.
FIGURE 7.

Two-way Venn diagrams showing the unique and shared (A) identified metabolites and (B) KEGG-based metabolic pathways for the 2D-GC/ToF-MS and 1D-GC/Orbitrap-MS platforms.
Our initial assessment clearly has several limitations. First, we did not use the chemical ionization (CI)-mass spectrometry capabilities available with1D-GC/Orbitrap-MS in this preliminary investigation, nor the MS/MS capabilities available for the unknown screening using chemical structure predictions with chemical databases and cheminformatics approaches. Clearly, the analytical chemistry and metabolomics research community lacks an experimentally derived chemical-ionization (CI)-database or one with GC/MS/MS information, which would help circumvent some of the challenges associated with high resolution EI and CI data. This probably limited the number of metabolites confidently identified using 1D-GC/Orbitrap-MS. Moreover, instead of specialized applications or biomarker discovery adopting a targeted approach, we focused on the two platforms for their routine use in laboratories or core facilities for untargeted data acquisition from biological samples. In such a setting, the power of two dimensions presented by two column chemistries in series belonging to two different polarities clearly surpasses the unidimensional chromatography capabilities of the Orbitrap platform, at least in the standard application shown here. Future studies using more complex biological samples (such as tissue extracts) and samples derived from a wider range of animals or patients will help assess whether the demonstrated higher reproducibility of the high-resolution instrument will be advantageous, especially with further optimization of run protocols. Finally, the unknown peaks in any given mass chromatograms are challenging to uncover without <1 ppm mass accuracy, which would be achievable using the capabilities available for the 1D-GC/Orbitrap-MS platform in future studies.
4 |. CONCLUSIONS
The current investigation was designed as a proof-of-concept for metabolomics data from NHP serum acquired in high-resolution mode using 1D-GC/Orbitrap-MS, and demonstrated the capabilities of resolving a plethora of quantified compounds with high confidence. We show that the 2D-GC/ToF-MS platform, by virtue of the two column chemistries in just one single run from a given sample, can identify almost twice as many metabolites. For the first time, we show the capabilities of 1D-GC/Orbitrap-MS platform in obtaining robust quantification of a decently sized representative chemical space from NHP serum and in discovery of low abundance metabolites such as exogenous compounds that are part of the exposome. Clearly, depending on applications, the metabolomics research community can opt for a high-resolution instrument for precision in quantification and/or added dimension for better resolution and identification of more metabolites in a given complex biological sample. These findings should help investigators and researchers choose between the two platforms, depending on the applications and objectives for future clinical studies and complex samples, for confidence in measurement as well as expanding the metabolite space while performing untargeted GC/MS-based metabolomics experiments.
Supplementary Material
ACKNOWLEDGEMENTS
BBM designed the research; BBM and EB performed the experiments; MO provided essential reagents and materials, BBM and EB analyzed the data, BBM, ACB, DK, LAC, MO wrote the manuscript, and BBM interpreted the data and has the primary responsibility for the final content and edits. All authors read and approved the final manuscript. The authors would like to thank Jason Cole, Thermo Fisher Scientific for critical reading and insights for the manuscript.
REFERENCES
- 1.Wishart DS. Emerging applications of metabolomics in drug discovery and precision medicine. Nat Rev Drug Discov 2016;15:473–484. [DOI] [PubMed] [Google Scholar]
- 2.Lu W, Bennett BD, Rabinowitz JD. Analytical strategies for LC–MS-based targeted metabolomics. J Chromatogr B 2008;871:236–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trygg J, Gullberg J, Johansson AI, et al. Extraction and GC/MS analysis of the human blood plasma metabolome. Anal Chem 2005;77:8086–8094. [DOI] [PubMed] [Google Scholar]
- 4.Markley JL, Brüschweiler R, Edison AS, et al. Wishart DS The future of NMR-based metabolomics. Curr Opin Biotechnol 2017;43:34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ellis DI, Goodacre R. Metabolic fingerprinting in disease diagnosis: biomedical applications of infrared and Raman spectroscopy. Analyst 2006;131:875–885. [DOI] [PubMed] [Google Scholar]
- 6.Fiehn O Metabolomics—the link between genotypes and phenotypes. In Functional Genomics Springer; Netherlands, 2002; pp. 155–171. [PubMed] [Google Scholar]
- 7.Fiehn O Metabolomics by gas chromatography–mass spectrometry: Combined targeted and untargeted profiling. Curr Protoc Mol Biol 2016;30–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kind T, Tsugawa H, Cajka T, et al. Identification of small molecules using accurate mass MS/MS search. Mass Spectrom Rev 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peterson AC, Balloon AJ, Westphall MS, Coon JJ. Development of a GC/Quadrupole-Orbitrap mass spectrometer, part II: new approaches for discovery metabolomics. Anal Chem 2014;86:10044–10051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peterson AC, Hauschild JP, Quarmby ST, et al. Development of a GC/quadrupole-Orbitrap mass spectrometer, part I: design and characterization. Anal Chem 2014;86:10036–10043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peterson AC, McAlister GC, Quarmby ST, Griep-Raming J, Coon JJ. Development and characterization of a GC-enabled QLT-Orbitrap for high-resolution and high-mass accuracy GC/MS. Anal Chem 2010;82:8618–8628. [DOI] [PubMed] [Google Scholar]
- 12.Weidt S, Haggarty J, Kean R, et al. A novel targeted/untargeted GC-Orbitrap metabolomics methodology applied to Candida albicans and Staphylococcus aureus biofilms. Metabolomics 2016;12:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uclés S, Uclés A, Lozano A, Bueno MM, Fernández-Alba AR. Shifting the paradigm in gas chromatography mass spectrometry pesticide analysis using high resolution accurate mass spectrometry. J. Chromatogr A 2017;1501:107–116. [DOI] [PubMed] [Google Scholar]
- 14.Rappaport SM. Implications of the exposome for exposure science. J Expo Sci Environ Epidemiol 2011;21:5–9. [DOI] [PubMed] [Google Scholar]
- 15.Rappaport SM, Smith MT. Environment and disease risks. Science 2010;330:460–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rappaport SM, Barupal DK, Wishart D, Vineis P, Scalbert A. The blood exposome and its role in discovering causes of disease. Environ Health Perspect 2014;122:769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mondello L, Tranchida PQ, Dugo P, Dugo G Comprehensive two‐dimensional gas chromatography‐mass spectrometry: A review. Mass Spectrom Rev 2008;27:101–124. [DOI] [PubMed] [Google Scholar]
- 18.Winnike JH, Wei X, Knagge KJ, et al. Comparison of GC/MS and GC× GC/MS in the analysis of human serum samples for biomarker discovery. J Proteome Res 2005;14:1810–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Welthagen W, Shellie RA, Spranger J, et al. Comprehensive two-dimensional gas chromatography–time-of-flight mass spectrometry (GC× GC-TOF) for high resolution metabolomics: biomarker discovery on spleen tissue extracts of obese NZO compared to lean C57BL/6 mice. Metabolomics 2005;1:65–73. [Google Scholar]
- 20.Dunn WB, Broadhurst D, Ellis DI, et al. A GC-TOF-MS study of the stability of serum and urine metabolomes during the UK Biobank sample collection and preparation protocols. Int J Epidemiol 2008;37:i23–i30. [DOI] [PubMed] [Google Scholar]
- 21.Cox LA, Olivier M, Spradling-Reeves K, et al. Nonhuman primates and translational research—cardiovascular disease. ILAR J 2017;58:235–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fiehn O, Wohlgemuth G, Scholz M, et al. Quality control for plant metabolomics: reporting MSI‐compliant studies. Plant J 2008;53:691–704. [DOI] [PubMed] [Google Scholar]
- 23.Lisec J, Schauer N, Kopka J, Willmitzer L, Fernie AR. Gas chromatography mass spectrometry-based metabolite profiling in plants. Nat Protoc 2006;1:387–396. [DOI] [PubMed] [Google Scholar]
- 24.Wachsmuth CJ, Vogl FC, Oefner PJ, Dettmer K. Gas chromatographic techniques in metabolomics. Chromatographic methods in metabolomics; Hyotylainen T, Wiedmer S, Eds, 2013; pp.87–105. [Google Scholar]
- 25.Kopka J, Schauer N, Krueger S, et al. GMD@ CSB. DB: the Golm metabolome database. Bioinformatics 2005;21:1635–1638. [DOI] [PubMed] [Google Scholar]
- 26.Horai H, Arita M, Kanaya S, et al. MassBank: a public repository for sharing mass spectral data for life sciences. J Mass Spectrom 2010;45:703–714. [DOI] [PubMed] [Google Scholar]
- 27.Team RC. R: A language and environment for statistical computing [Internet] Vienna, Austria: R Foundation for Statistical Computing; 2015. [Google Scholar]
- 28.Sokal RR, Rohlf FJB. The principles and practice of statistics in biological research Freeman, WH; 1981. [Google Scholar]
- 29.Grapov D DeviumWeb: version 0.3.2. ZENODO 2014. doi: 10.5281/zenodo.12879, https://github.com/dgrapov/DeviumWeb [DOI] [Google Scholar]
- 30.Caraux G, Pinloche S. PermutMatrix: a graphical environment to arrange gene expression profiles in optimal linear order. Bioinformatics 2005;21:1280–1281. [DOI] [PubMed] [Google Scholar]
- 31.Xia J, Sinelnikov IV, Han B, Wishart DS. MetaboAnalyst 3.0—making metabolomics more meaningful. Nucleic Acids Res 2015;43:W251–W257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Makarov A, Denisov E, Lange O, Horning S. Dynamic range of mass accuracy in LTQ Orbitrap hybrid mass spectrometer. J Am Soc Mass Spectrom 2006;17:977–982. [DOI] [PubMed] [Google Scholar]
- 33.Blekhman R, Perry GH, Shahbaz S, et al. Comparative metabolomics in primates reveals the effects of diet and gene regulatory variation on metabolic divergence. Sci Rep 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Navarro-Reig M, Jaumot J, Baglai A, et al. Untargeted comprehensive two-dimensional liquid chromatography coupled with high-resolution mass spectrometry analysis of rice metabolome using multivariate curve resolution. Anal Chem 2017;89:7675–7683. [DOI] [PubMed] [Google Scholar]
- 35.Watson NE, Bahaghighat HD, Cui K, Synovec RE, Comprehensive three-dimensional gas chromatography with time-of-flight mass spectrometry. Anal Chem 2017;89:1793–1800. [DOI] [PubMed] [Google Scholar]
- 36.Dunn WB, Wilson ID, Nicholls AW, et al. The importance of experimental design and QC samples in large-scale and MS-driven untargeted metabolomic studies of humans. Bioanalysis 2012;4:2249–2264. [DOI] [PubMed] [Google Scholar]
- 37.Dettmer K, Almstetter MF, Appel IJ, et al. Comparison of serum versus plasma collection in gas chromatography-Mass spectrometry based metabolomics. Electrophoresis 2010; 31:2365–2373. [DOI] [PubMed] [Google Scholar]
- 38.Psychogios N, Hau DD, Peng J, et al. The human serum metabolome. PLoS One 2011; 6: 16957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carrillo B, Yanofsky C, Laboissiere S, et al. Methods for combining peptide intensities to estimate relative protein abundance. Bioinformatics 2009;26:98–103. [DOI] [PubMed] [Google Scholar]
- 40.Mizuno H, Ueda K, Kobayashi Y, et al. The great importance of normalization of LC-MS data for highly accurate non-targeted metabolomics. Biomedical Chromatography 2017;31: [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.