

Abstract
κ opioid receptor (KOR) antagonists are potential pharmacotherapies for the treatment of migraine and stress-related mood disorders including depression, anxiety and drug abuse, thus the development of novel KOR antagonists with an improved potency/selectivity profile and medication-like duration of action has attracted the interest of the medicinal chemistry community. In this paper, we describe the discovery of 1-(6-ethyl-8-fluoro-4-methyl-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)-N-(tetrahydro-2H-pyran-4-yl)piperidin-4 amine (CYM-53093, BTRX-335140) as a potent and selective KOR antagonist, endowed with favorable in vitro ADMET and in vivo pharmacokinetic profiles and medication-like duration of action in rat pharmacodynamic experiments. Orally administered CYM-53093 showed robust efficacy in antagonizing KOR agonist-induced prolactin secretion and in tail-flick analgesia in mice. CYM-53093 exhibited a broad selectivity over a panel of off-target proteins. This compound is in Phase 1 clinical trials for the treatment of neuropsychiatric disorders wherein dynorphin is thought to contribute to the underlying pathophysiology.
Graphical Abstract
INTRODUCTION
The opioid receptors belong to the superfamily of G-protein coupled receptors and are generally classified into four subtypes: μ opioid receptor (MOR), δ opioid receptor (DOR), κ opioid receptor (KOR) and the nociceptin/orphanin FQ (N/OFQ) receptor. The opioid receptors show a high degree of sequence homology, however activation of these receptors by selective endogenous and exogenous ligands has been shown to produce striking differences in pharmacological and physiological effects.1,2 The KOR is a Gαi/o-coupled receptor primarily activated by endogenous dynorphin opioid peptides.3–4 The KOR is distributed throughout the spinal cord, brain stem and human brain.5 In the brain, KORs are particularly expressed in the anterior cingulate cortex, amygdala, insula, putamen, neocortical region, caudate, thalamus, globus pallidus, pons, substantia nigra and hippocampus.5–9 Numerous lines of evidence from preclinical and clinical studies have suggested the KOR as a central player in a variety of neuropsychiatric and neurological disorders such as depression, epilepsy, Alzheimer’s disease, substance and alcohol abuse and schizophrenia.10–19 Studies suggest that the KOR may play a role in post-traumatic stress disorder (consistent with the modulatory effects of dynorphin on reward, mood, and stress) and in migraine prophylaxis.20–22 As a consequence of these findings, the development of selective KOR antagonists has stimulated great interest in both academia and the pharmaceutical industry. Archetypical KOR antagonists nor-BNI (1), GNTI (2) and non-morphinan JDTic (3) (Figure 1) exhibit a delay in the onset of action of hours or days, and their antagonism effects are measurable even for several weeks at minimally-effective doses; in contrast, these compounds show a rapid decline in plasma levels.23 Concerns about the feasibility of developing medications with archetypical KOR-antagonists have centered on their abnormal long duration of action. These concerns have led to the development of KOR antagonists with medication-like duration of action from which JNJ-67953964 (4) (formerly known as LY-2456302 and CERC-501) and PF-04455242 (5) have been evaluated in clinical trials (Figure 1).24 5 showed single digit nanomolar activity at the KOR and good selectivity against the DOR, but poor selectivity against the MOR.25 Phase 1 clinical trials of 5 were terminated due to toxicology findings in animals exposed to the compound for three months.26 4 displayed in vitro sub-nanomolar KOR antagonism with a selectivity of approximately 21-fold over the MOR and 135-fold over the DOR, and efficacy in animal models of substance abuse and depression.24, 27, 28, 29 4 was until recently the only KOR antagonist undergoing clinical development as monotherapy and has been shown to be safe in humans with mild to moderate side effects at daily doses of 10 mg (p.o.).30,31 Remarkably, 4 has shown clinically meaningful results in a phase II proof of concept study for treating patients with treatment-resistant depression32 but dose escalation may be limited as effects of MOR antagonism begin to emerge.31,33
Figure 1.

Prototypical and Clinical Stage KOR Antagonists
The compound ALKS-5461 is a combination of buprenorphine (6) (partial MOR-agonist and KOR-antagonist) and samidorphan (7) (MOR-antagonist) that has been postulated to act primarily as a KOR-antagonist.34 ALKS-5461 is well tolerated in humans with limited concerns for abuse potential. The compound has been studied as adjunctive therapy for patients with treatment-resistant depression and showed clinically meaningful antidepressant effects in a few randomized double-blind placebo-controlled trials.35 As a result of this work a new drug application (NDA) has recently been filed for ALKS-5461 for the adjunctive treatment of major depressive disorder (MDD) in patients with inadequate response to standard antidepressant therapy.36
RESULTS AND DISCUSSION
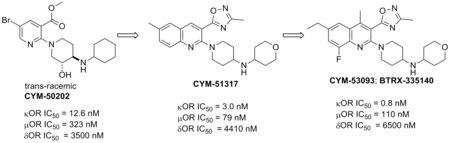
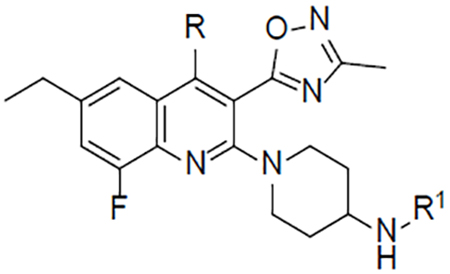
In an effort to identify novel selective KOR antagonists with desirable pharmacokinetic and pharmacodynamic properties, a high-throughput screening (HTS) campaign of the Molecular Libraries-Small Molecule Repository (MLSMR) was carried out by our laboratories and identified the hit 3-ethyl-5-methyl-2-(4-((2-hydroxycyclohexyl)amino)piperidin-1-yl)-6-methylpyridine-3,5-dicarboxylate 8 (Figure 2) as a weak KOR antagonist (IC50 = 410 nM) with poor selectivity against the MOR (11-fold).37 The initial structure–activity relationship (SAR) investigations of the hit were focused on increasing the potency while trying to improve the selectivity against the MOR. Iterative SAR led to the discovery of the pyridine CYM-50202 (9) as a potent KOR antagonist (IC50 = 12.6 nM) with good and moderate selectivity against the DOR and MOR (278- and 27-fold), respectively (Figure 2). 9 displayed high solubility, an encouraging pharmacokinetic (PK) profile in rat and excellent CNS-penetration. Perhaps more importantly, 9 did not display long lasting pharmacodynamic effects observed with prototypical KOR antagonists.37 From a panel of 52 pharmacologically relevant off-target proteins, 9 exhibited >50% inhibition at only the sodium channel site 2 (IC50 = 1.8 μM) and ORL-1 (IC50 = 3 μM).37 Unfortunately, we identified a number of issues with this molecule. First, 9 showed significant inhibition of CYP2D6 at 10μM raising concerns of potential drug-drug interaction liabilities. In addition, the activity at sodium channel site 2 raised concerns of potential cardiovascular liabilities. Moreover, the methyl ester and bromide group were identified as undesirable structural motifs of this chemotype due to the likely ester cleavage in vivo and a structural alert in the case of the bromide. Consequently, additional efforts to develop KOR antagonists with improved potency (single-digit nanomolar), selectivity (>100 fold against MOR) and safety profile were undertaken and the results of the SAR studies are reported in this manuscript. For the purpose of exploring the SAR of 9, the molecule was divided into three fragments: the pyridine head group A, the piperidine linker B and the amine tail C.
Figure 2.

Early KOR Antagonist HTS Hit 8 and 9
We started our iterative SAR studies by replacing the ester group with an oxadiazole isostere and by changing the bromine for small alkyl groups while exploring single diastereomers in the piperidine-region B. These efforts culminated in the discovery of single digit nanomolar KOR antagonist 16 (IC50 = 1.3 nM) with modest and high selectivity against the MOR and DOR (24 and >100-fold), respectively (Table 1). All synthesized compounds were evaluated for antagonist activity at the MOR, DOR, and KOR using our established assays.37 16 is endowed with physicochemical properties that are consistent with CNS-penetrant compounds38 (cLogP = 2.2, tPSA = 81.8, MW = 371) and devoid of the structural liabilities of 9. Unfortunately selectivity over the MOR remained suboptimal (optimal >100 fold). The synthesis of 16 is depicted in Scheme 1. Amine 12 was synthesized through reductive-amination of diastereomerically-pure amine 10 with cyclohexanone followed by Cbz-hydrogenolysis. The oxadiazole intermediate 15 was synthesized from the carboxylic acid 13 via coupling-cyclization with acetamidoxime 14 in the presence of EDCI and HOBt. The final compound 16 was obtained via microwave-assisted nucleophilic aromatic substitution (SNAr) of chloride 15 with amine 12 (Scheme 1).
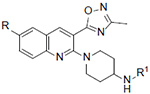
Table 1.
KOR, MOR, DOR Antagonist Activity of Compounds 16, 27–39
 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | R | R1 | IC50 (nM)a | Selectivity | Calculated Physicochemical Properties50, 51 | ||||
| KORb | MOR | DOR | MOR/KOR | cLogP | tPSA | cpKa | |||
| 16 | 1.3 | 31.1 | 2410 | 24 | 2.2 | 82 | 10.2 | ||
| 27 | Me | 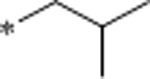 |
1.3 | 176 | >10000 | 135 | 3.4 | 62 | 10.7 |
| 28 | Me | 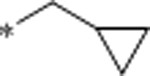 |
2.2 | 829 | >10000 | 377 | 2.9 | 62 | 10.6 |
| 29 (CYM-51317) | Me | 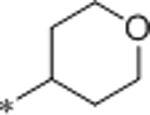 |
3.0 | 79 | 4410 | 26 | 1.6 | 71 | 10.5 |
| 30 | Et | 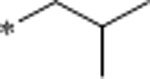 |
1.3 | 4390 | >10000 | 3377 | 3.9 | 62 | 10.7 |
| 31 | Et | 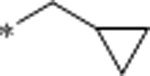 |
1.7 | 2930 | >10000 | 1723 | 3.4 | 62 | 10.6 |
| 32 | Et | 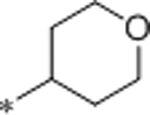 |
7 | 183 | >10000 | 26 | 2.1 | 71 | 10.5 |
| 33 | Et | 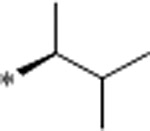 |
0.76 | 3330 | >10000 | 4382 | 4.2 | 62 | 10.8 |
| 34 | Et | 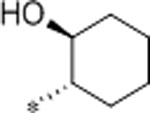 |
0.59 | 583 | >10000 | 988 | 3.4 | 82 | 10.2 |
| 35 | Et | 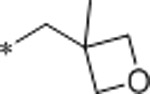 |
4.5 | >10000 | >10000 | >2222 | 2.1 | 71 | 10.3 |
| 36 | Et | 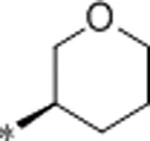 |
0.77 | 1460 | >10000 | 1896 | 3.0 | 71 | 9.9 |
| 37 | Et | 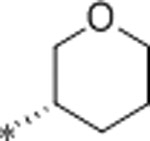 |
1.5 | 1500 | >10000 | 1000 | 3.0 | 71 | 9.9 |
| 38 | Et | 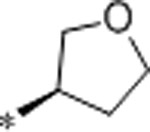 |
9.5 | >10000 | >10000 | >1053 | 2.6 | 71 | 9.8 |
| 39 | Et | 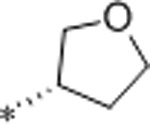 |
10.3 | >10000 | >10000 | >971 | 2.6 | 71 | 9.8 |
Values are reported as mean of n = 3 determinations.
This assay uses Tango OPRK1-bla U2OS cells which express KOR linked to a GAL4-VP16 transcription factor via a TEV protease site. Stimulation of the KOR by U-50488 causes migration of the β-arrestin fusion protein to the GPCR, and through proteolysis liberates GAL4-VP16 from the receptor. Assay protocols are described in ref 37.
Scheme 1.

Synthesis of 16
Encouraged by these data we sought to improve the selectivity of 16 by undertaking a multiparameter-SAR optimization of the three molecular regions of 16 (A, B and C). For the optimization process we outlined specific tactics for each molecular region: 1) in region A to replace the pyridine for a quinoline ring while keeping the oxadiazole fixed in position 3 (Scheme 2); 2) in region B to simplify the piperidine ring by removing the 3-hydroxy group to rapidly prepare a variety of analogs in the quinoline ring and tail-amine in a parallel-chemistry format; 3) in region C to explore a variety of structurally-diverse amines with the goal of optimizing the physicochemical properties (pKa, tPSA, MW, cLogP and hydrogen bond donors) for good oral bioavailability and brain penetration.38
Scheme 2.

Synthesis of 27–39
The synthesis of these compounds is depicted in Scheme 2 and their biological activities are listed in Table 1. Oxadiazole intermediates 19 and 20 were synthesized from the corresponding commercially available carboxylic acids 17 and 18 via acyl chloride formation followed by coupling-cyclization with acetamidoxime 14. Ketones 24 and 25 were synthesized via microwave-assisted SNAr of 19 and 20 with amine 21 followed by ketal deprotection. Finally, reductive amination of ketones 24 and 25 with a variety of amines 26 furnished the final compounds 27–39.
Based on iterative SAR optimization cycles, the 6-position of the quinoline ring was found to be an essential structural feature to enhance selectivity against the MOR and DOR. For example, the 6-ethyl analogs 30 and 31 showed similar potency at the KOR and enhanced selectivity against the DOR and MOR (>1700-fold) as compared to the 6-methyl counterparts 27 and 28. This SAR analysis also revealed that the location of a hydrogen bond acceptor in the amine tail was important for selectivity. For instance, both 6-methylquinoline 4-THP derivative 29 (CYM-51317) and 6-ethylquinoline 4-THP analog 32 showed single-digit nanomolar activity at the KOR but modest selectivity against the MOR (26-fold). Further modifications at the tail region were carried out while keeping the ethyl group at the 6-position. The enantiomeric pair of 3-THP-analogs 36 and 37 showed similar activity at KOR but (R)-3-THP 36 was more selective against the MOR. The 5-membered ring analogs (R)-3-THF 38 and (S)-3-THP 39 were ~12 and ~17-fold less potent than their corresponding 6-membered ring enantiomers 36 and 37, and showed good selectivity against the MOR. Interestingly, the bulkier and lipophilic analog 33 as well as the polar aminoalcohol 34 showed sub-nanomolar potency at the KOR and exquisite selectivity against the MOR (4382- and 988-fold, respectively). Furthermore, the oxetane 35 showed nanomolar potency at the KOR and remarkable selectivity against the MOR. This diverse set of molecules highlighted the potential of the amine tail to tune-in the desired pharmacokinetic properties while maintaining potency and selectivity.
A set of selected compounds 27, 29, 33, 34, 35, 36 were assayed for stability in rat, mouse and human liver microsomes (Table 2). The tested compounds showed relatively high intrinsic clearance in human hepatic microsomes and, in general, compounds with lower cLogP values (e.g. 29, 35) displayed lower intrinsic clearance in hepatic microsomes than compounds with higher cLogP values (e.g. 27, 33). Representative compounds 29, 34, 35, 36 were further profiled in additional pharmacokinetic and off-target assays (Table 3). The compounds were moderately bound by plasma proteins (64.0–98.1% bound) across species and a correlation was observed between cLogP values and plasma protein binding. Remarkably, many compounds (at 10μM concentration) showed both low cytochrome P450 inhibition and hERG inhibition profile. Specifically, 29 was tested in a panel of four relevant cardiac ion channels in a patch clamp assay and found devoid of activity at 10 μM (<50% inhibition). Additionally, 29 showed high membrane permeability (A-B) in a Caco-2 cell line with low efflux (Table 3). Based on these results we decided to examine the in vivo pharmacokinetic profile of 29 in rats (Table 4). 29 distributed well into tissues (Vss = 13 L/kg) and had a high clearance (CL) after 1 mg kg−1 intravenous dosing (i.v.) and exhibited plasma exposures out to 6 hours (110.16 ng mL−1 at Cmax and 1705 min·ng mL−1 AUC0−∞), after a dose of 2 mg kg−1 p.o. The absolute oral bioavailability (F) was 20% (Table 4). Additionally, 29 displayed a brain to plasma ratio of 2.9 in mice at 30 minutes and of 3.8 at 120 minutes post intraperitoneal (i.p.) administration of a 10 mg kg−1 dose (Table 4). Although 29 was only moderately selective against the MOR, it showed promising overall pharmacokinetic properties and off-target profile thus it was selected as a tool compound for further in vivo studies.
Table 2.
Intrinsic Clearance (Clint) in Hepatic Microsomes of Compounds 27, 29, 33, 34, 35 and 36
| Compound | Calculated Physicochemical properties | Microsomal Clint (mL/min/kg)a | ||||
|---|---|---|---|---|---|---|
| MW | tPSA | cLogP | human | rat | mouse | |
| 27 | 379 | 62 | 3.4 | 49.5 | 49.5 | ND |
| 29 | 407 | 71 | 1.6 | 15.1 | 7.1 | 14.1 |
| 33 | 407 | 62 | 4.2 | 46.5 | ND | 58.7 |
| 34 | 435 | 82 | 3.4 | 36.1 | ND | 34.8 |
| 35 | 421 | 71 | 2.1 | 23.1 | 35.4 | 17.2 |
| 36 | 421 | 71 | 3.0 | 40.3 | 51.3 | 58.2 |
ND = No data
Test compounds (1 μM) were incubated with 0.2 mg mL−1 pooled human, rat and mouse hepatic microsomes at 37°C with continuous shaking. Further details for the assays can be found in the experimental section and reference 37.
Table 3.
ADMET Properties of 29, 34, 35, 36
| Compound | 29 | 34 | 35 | 36 | |
|---|---|---|---|---|---|
| Protein plasma binding (% bound) at 1 μMa | human | 71 | 98.1 | 91.7 | 96.5 |
| mouse | 64 | 95.5 | 89 | 91.4 | |
| rat | 78 | 97 | 92.4 | 96 | |
| Human cytochrome P450 inhibition at 10μM (%)a | CYP3A4 | 4 | −28 | −13 | −22 |
| CYP2D6 | 20 | 9 | −40 | 1 | |
| CYP2C9 | −6 | −13 | −11 | −18 | |
| CYP1A2 | 5 | 13 | 10 | 20 | |
| Ion channel % Inhibition at 10 μMa,b |
Voltage-gated sodium (Nav1.5) | 32 | 72 | ||
| hERG | 35 | 77 | 19 | 6 | |
| Voltage-gated calcium (Cav1.2) | <25 | 28 | ND | ND | |
| Inward-rectifying voltage-gated potassium (Kir2.1) | <25 | <25 | ND | ND | |
| Hyperpolarization-activated cyclic nucleotide-gated potassium (HCN4) | <25 | <25 | ND | ND | |
| Caco-2 cells | A-B (10−6 cm/s) | 26.2 | ND | ND | ND |
| Efflux ratio | 1.0 | ND | ND | ND |
ND = No data. A-B = movement of the compound from the apical to the basal side of the membrane; a surrogate for permeability
Further details for the assays can be found in the experimental section and reference 37.
Compounds were profiled in the Eurofin cardiac profiler panel. Further details can be found in the supporting information.
Table 4.
Plasma Pharmacokinetic Profile of Compound 29 in Rodentsa
| Rat (n = 3, mean values) | |||||||
|---|---|---|---|---|---|---|---|
| i.v. (1 mg·kg−1) | p.o. (2 mg kg−1) | ||||||
| CL (mL·min−1·kg−1) | T ½ (h) | AUC∞ min.ng/mL | Vss (L·kg−1) | T ½ (h) | Cmax (ng/mL) | AUC0−∞ min.ng/mL | F (%) |
| 83 | 1.98 | 12,740 | 12.95 | 1.56 | 110.16 | 1,705 | 20 |
| Brain:plasma ratio in mice (i.p. administration, 10 mg kg−1, mean values) | 30 min | 2.9 | |||||
| 120 min | 3.8 | ||||||
29 was dosed as the monotartrate salt.
In a mouse tail-flick assay 29 did not show a long duration of action in blocking the antinociceptive effect of the KOR-agonist, U69,593.22 Perhaps most importantly, in an injury-free rat model of cephalic pain with features of migraine and medication overuse headache, oral administration of 29 showed preventive benefits on blocking stress induced allodynia thus suggesting that KOR antagonists could provide a new therapeutic strategy in preventing migraine.22 In parallel, the potential mutagenic activity of this compound class was tested on selected compounds 33, 34, 35 and 36 in a bacterial reverse mutation assay (Ames assay). Four Salmonella typhimurium strains TA100, TA1535, TA1537, TA98 as well as the E. coli strain WP2uvrA were used in the presence and absence of an exogenous metabolic activation system (S9). Both 34 and 35 exhibited mutagenicity in the TA1537 strain in the presence and absence of S9; 33 and 36 also showed a mutagenic response in the TA1537 strain but only in the absence of S9. Positive mutagenic activity in the TA1537 strain often reflects intercalation of the tested molecules with DNA, and this intercalation is commonly associated with the planarity, π-stacking and hydrogen bonding interaction capabilities of molecules.39 Based on the hypothesis that the quinoline A was the portion of the molecule involved in the intercalation with the DNA, two strategies were evolved to address the genotoxicity liability: 1) reduce the basicity of the quinoline-nitrogen to disrupt hydrogen-bond interactions between DNA and the quinoline ring and 2) disrupt the planarity of the quinoline-oxadiazole axis to hinder π-stacking interactions. To implement these strategies, a fluorine atom was introduced at the 8-position of the quinoline-ring to substantially reduce the pKa of the quinoline nitrogen (up to 2.5 pKa units). In addition, a methyl group was installed in the 4-position of the quinoline ring in an attempt to disrupt the planarity.
For the construction of poly-substituted quinoline rings we decided to use the Friedländer methodology since it is one of the most robust strategies for the modification and manipulation of quinoline rings.40 The synthesis of these compounds is described in Scheme 3 (without a 4-methyl group) and Scheme 4 (with a 4-methyl group) and their biological activity reported in Table 5. The aldehyde 41 was synthesized from carboxylic acid 40 via reduction with BH3-THF complex followed by oxidation with manganese dioxide in dichloromethane. The quinoline intermediate 43 was synthesized by condensation of acetic acid 42 with aldehyde 41 in phosphorus oxychloride. A SNAr reaction of 43 with amine 21, followed by Suzuki-Miyaura coupling with Et3B and ketal deprotection afforded the key ketone-intermediate 45. Finally, reductive amination of ketone 45 with a series of amines 26 furnished the final compounds 46–50 (Scheme 3). Preparation of ketone 54 was accomplished via iodination of aniline 51 followed by Heck coupling with ethylene glycol vinyl ether 53.41 Condensation of acetic acid 42 with ketone 54 in phosphorus oxychloride afforded the poly-substituted quinoline 55. A SNAr reaction of amine 21 with 55 followed by ketal deprotection afforded the ketone intermediate 57. The final compounds 58-62 were synthesized via reductive amination of 57 with amines 26 (Scheme 4).
Scheme 3.

Synthesis of 46–50
Scheme 4.

Synthesis of 58–62
Table 5.
KOR, MOR, DOR Antagonist Activity of Compounds 29, 46–50, 58–62
| Compound | R | R1 | IC50(nM)a | Selectivity | Calculated Physicochemical Properties50, 51 | ||||
|---|---|---|---|---|---|---|---|---|---|
| KORb | MOR | DOR | MOR/KOR | cLogP | tPSA | cpKa | |||
| 29 | 3.0 | 79 | 4410 | 26 | 1.6 | 71 | 10.5 | ||
| 46 | H | 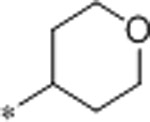 |
3.6 | 1200 | >10000 | 333 | 2.2 | 71 | 10.5 |
| 47 | H | 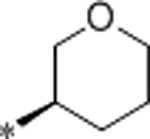 |
1.1 | 1400 | >10000 | 1273 | 3.1 | 71 | 9.9 |
| 48 | H | 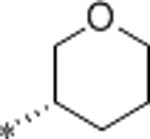 |
2.1 | 6200 | >10000 | 2952 | 3.1 | 71 | 9.9 |
| 49 | H | 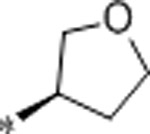 |
2.3 | 6400 | >10000 | 2783 | 2.7 | 71 | 9.8 |
| 50 | H | 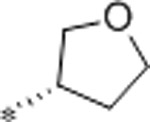 |
2.4 | 6500 | >10000 | 2708 | 2.7 | 71 | 9.8 |
| 58 | Me | 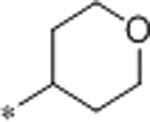 |
0.8 | 110 | 6500 | 138 | 2.4 | 71 | 10.5 |
| 59 | Me | 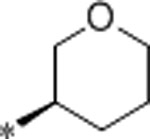 |
0.26 | 100 | 4500 | 384 | 3.3 | 71 | 9.9 |
| 60 | Me | 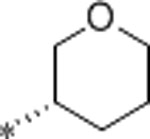 |
0.53 | 400 | >10000 | 754 | 3.3 | 71 | 9.9 |
| 61 | Me | 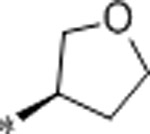 |
0.78 | 1100 | >10000 | 1410 | 2.9 | 71 | 9.8 |
| 62 | Me |  |
0.88 | 1400 | >10000 | 1591 | 2.9 | 71 | 9.8 |
Values are reported as mean of n = 3 determinations.
This assay uses Tango OPRK1-bla U2OS cells which express KOR linked to a GAL4-VP16 transcription factor via a TEV protease site. Stimulation of the KOR by U-50488 causes migration of the β-arrestin fusion protein to the GPCR, and through proteolysis liberates GAL4-VP16 from the receptor. Assay protocols are described in ref 37.
For the selection of tail-amines we sought to modulate the physicochemical properties in a range compatible with oral bioavailability and CNS penetration utilizing the SARs gleaned from the previous series. The THP analogs 47 (CYM-53052), 48 and 46 were approximately equipotent to the non-fluorinated analogs 36, 37 and 32, with higher selectivity against the MOR in the case of 46 and 48 and lower in the case of 47. Furthermore, the 3-amino-THF enantiomers 49 and 50 were ~4-fold more potent than the non-fluorinated analogs 38 and 39, and both enantiomers (49 and 50) retained excellent selectivity against the MOR. The introduction of a methyl group in the 4-position of the quinoline ring generally increased potency at the KOR while decreasing the selectivity against the MOR. For example, the 4-amino-THP 58 (CYM-53093, BTRX-335140) and the 3-amino-THP enantiomers 59 and 60 were ~4-fold more potent than the demethylated analogs 46-48 but less selective against the MOR. The 5-membered ring analogs 61 and 62 were ~3-fold more potent than 49 and 50 at the KOR and moderately less selective against the MOR. From these analogs, compounds 58 and 47 were selected as representative molecules of the 4-methyl- and non-methylated series for Ames assays. Neither analog showed mutagenicity in the Ames assays, with or without the addition of S9, highlighting the success of the aforementioned stereoelectronic design principles.
In a variety of in vitro pharmacokinetic assays, 47 and 58 exhibited low intrinsic clearance in human hepatic microsomes and hepatocytes and moderate to high clearance in other species (Table 6). In vivo pharmacokinetic studies in rats (Table 7) indicated that 58 distributed well into tissues (high apparent Vss of 13.8 L kg−1) and had a high plasma clearance rate (105 mL min −1 kg−1) after a 1 mg kg−1 i.v. dose. After a 5 mg kg−1 p.o. dose, 58 exhibited plasma exposure levels out to 8 hours (39.1 ng mL−1 at Cmax and 232 h·ng mL−1 AUC0-t). The absolute oral bioavailability was 30%. The pharmacokinetic parameters observed in mice were similar. In rats, 58 distributed well into the CNS with a high brain to plasma ratio of 2.2:1 and adequate CSF exposure (~4 fold its IC50) 1 hour after an oral administration of 5 mg kg−1 (Table 8).
Table 6.
Intrinsic Clearance (Clint) in Hepatic Microsomes and Hepatocytes for 47, 49, 58, 59, 61 and 62
| Compd | Physicochemical Properties | Microsomal Clint (mL/min/kg) | Hepatocytes Clint (mL/min/kg) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MW | tPSA | cLogP | human | rat | mouse | dog | human | rat | mouse | dog | |
| 47 | 439 | 71 | 3.1 | 12.0 | 22.7 | ND | 54.4 | 14.8 | 209 | ND | ND |
| 49 | 425 | 71 | 2.7 | 6.69 | 83.0 | 112.0 | ND | ND | ND | ND | ND |
| 58 | 453 | 71 | 2.4 | 1.95 | 17.5 | 49.1 | 55.2 | 8.3 | 118 | ND | 84.7 |
| 59 | 453 | 71 | 3.3 | 36.2 | 57.5 | ND | ND | ND | ND | ND | ND |
| 61 | 439 | 71 | 2.9 | 21.0 | 54.6 | ND | ND | ND | ND | ND | ND |
| 62 | 439 | 71 | 2.9 | 22.0 | 89.5 | ND | 39.5 | ND | ND | ND | ND |
ND = No data.
Details for the assays can be found in the supporting information. Compounds were profiled by Agilux.
Table 7.
Pharmacokinetic Profile of Compound 58 in Rodentsa
| i.v. (rat, 1 mg·kg−1; mice, 3 mg·kg−1) | p.o. (rat, 5 mg kg−1; mice, 10 mg·kg−1) | |||||||
|---|---|---|---|---|---|---|---|---|
| CL (mL·min−1·kg−1) | T ½ (h) | AUC0-t (h·ng mL−1) | Vss (L·kg−1) | T ½ (h) | Cmax (ng mL−1) | AUC0-t (h·ng mL−1) | F (%) | |
| Mice | 66.5 | 1.91 | 725 | 7.72 | 2.57 | 80.9 | 265 | 12 |
| Rats (fed) | 105 | 1.81 | 153 | 13.8 | 6.19 | 39.1 | 232 | 30.2 |
58 was dosed as the monotartrate salt.
Table 8:
CNS Exposure of 58 in Male Rats (n = 2, mean values)a
|
58 (fasted) |
p.o., 5 mg kg−1, 1h | 0.276 | 0.54 | 0.0031 | 2.2 |
58 was dosed as the monotartrate salt.
Having a few compounds in the series with promising in vitro and in vivo attributes in hand, we sought to assess the potential for our KOR antagonists to elicit an on-target pharmacodynamic response in an in vivo model. Prolactin (PRL) is a hormone involved in many biological processes that is mainly secreted by lactotroph cells of the anterior pituitary gland. Prolactin is primarily regulated via tonic inhibition by dopamine released from the tuberoinfundibular dopaminergic (TIDA) neurons.42,43 Activation of the opioid system, particularly of the KOR believed to be localized on dopaminergic neurons within TIDA and in the mesolimbic-mesocortical system modulates dopamine release.44,45,46 PRL overproduction by KOR agonists has been investigated in clinical and preclinical models and its modulation has been postulated as a KOR-mediated translatable biomarker,44,46–48 thus reduction of KOR agonist-induced plasma PRL in rats by KOR antagonists may be used as an estimation of KOR antagonist target engagement. We selected this preclinical pharmacodynamic model as a screen to guide the decision-making process for the further advancement of our KOR antagonists. For practical reasons the rat PRL model was adapted to mice and leveraged to rapidly screen KOR antagonist compounds. U69,593 was chosen as a reference KOR agonist and administered via intraperitoneal injection. KOR antagonist compounds were initially assessed via intraperitoneal (i.p.) dosing at a single concentration, typically at 10 or 30 mg kg−1, and those demonstrating inhibition of PRL release were further evaluated at lower concentrations. Compounds that showed robust i.p. PRL inhibition were tested orally (p.o.). Each KOR antagonist was injected 1h prior to U69,593 in the i.p. experiments, while in the p.o. experiments the KOR antagonists were administered by oral gavage 2h before U69,593. From these p.o. studies we found that at 30 and 10 mg kg−1 47 and 49 were effective in reducing plasma prolactin to basal levels but were ineffective at lower doses; at 3 mg kg−1, 58 and 62 were effective in reducing U69,593-stimulated plasma prolactin secretion to basal levels (Figure 3 and 4). Furthermore, 58 showed effective PRL reduction at 0.1 mg kg−1 after oral administration (Figure 4).
Figure 3.

Inhibition of KOR Agonist Stimulated Prolactin Via p.o. Administration of 47, 49 and 62a,b
a KOR antagonists were dosed as the monotartrate salts.
b 3 mice per dose group where 2 samples were collected per mouse.
Figure 4.

Inhibition of KOR Agonist Stimulated Prolactin Via p.o. Administration of 58a, b
a 58 was dosed as the monotartrate salt.
b 4 mice per dose group where 2 samples were collected per mouse.
From this set, 47, 49, 58 and 62 were tested in further pharmacokinetic and off-target assays (Table 9). 58 was 93.4% bound to human plasma proteins and 98.4% bound to rat brain homogenate. The other tested compounds were protein bound to a similar extent in human plasma and in plasma across other species. Moderate solubility was observed for this series of compounds (70–85 μM) and permeability was moderate to high for 58 with low efflux ratio (7.1 and 20.9 10−6 cm/s in Caco-2 cells and MDCK-MDR1 cells, respectively; ER <2.3). Furthermore, 58 showed minimal inhibition of CYP450 enzymes 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 3A4 with IC50 values of >30 μM and there was no change in the potency of 58 upon pre-incubation for any of the CYP enzymes. 58 also was profiled in a very comprehensive panel of over 500 off-targets including ion channels, kinases, GPCRs and other receptors and transporters and there were no hits identified with IC50 values <1μM. Few off-target hits were identified with IC50 values between 10 and 1 μM; in a patch clamp assay conducted a physiological temperature, the compound inhibited the hERG channel with an IC50 of 1 μM.
Table 9.
Additional ADMET Properties of 47, 49, 58, and 62a
| 47 | 49 | 58 | 62 | ||
|---|---|---|---|---|---|
| Plasma protein binding (% bound) |
human | 92.4 | 94.8 | 93.4 | 97.3 |
| rat | ND | 94.3 | 91.5 | ND | |
| dog | ND | 96.3 | 91.5 | ND | |
| Binding in rat brain homogenate (2μM) (% bound) | ND | ND | 98.4 | ND | |
| MDCK-MDR1 cell line (P-gp) | A-B (10−6 cm/s) | 8.76 | 13.5 | 7.1 | 4.6 |
| Efflux ratio | 1.5 | 0.896 | 2.3 | 5.2 | |
| Caco-2 cell line | A-B (10−6 cm/s) | 19.2 | ND | 20.9 | ND |
| Efflux ratio | 0.493 | ND | 0.38 | ND | |
| Solubility (μM) at pH 7.4 | 75 | 84 | 74 | 70 |
KOR-antagonists were used as monotartrate salts. ND = No data. A-B = movement of the compound from the apical to the basal side of the membrane; a surrogate for permeability.
In a radioligand binding assay using [3H]diprenorphine, 58 exhibited a Ki of 1.5 nM at the human KOR and 300-fold selectivity over the human MOR (Ki = 450 nM).
Based on these results, we decided to evaluate in vivo the duration of action of 58 in a hot water (50°C) tail-flick test in male mice by assessing its ability to antagonize the antinociceptive effect of U-50488. 58 or vehicle were administered to mice at 1 and 24 h prior to the administration of U-50488 (15 mg kg−1, i.p.) and latencies for tail-flick were measured at baseline, 30 and 60 minutes after agonist injection (Figure 5). Baseline tail-flick latency of naïve mice was 5.76 ± 0.16 s. Injection of U-50488 produced antinociception indicated by a significant increase in tail-flick latency, which peaked at 30 min post-dose. Unlike nor-BNI22, 58 (1 mg kg−1, i.p.) blocked the U-50488-induced antinociception at 1 h, but not at 24 h pretreatment time, indicating a medication-like duration of action in blocking the KOR in vivo (Figure 5A-D). There was no observable change in the rat tail-flick latency response induced by i.p. 58 (1 mg kg−1) alone at 1 h post-dose (Figure 5E, F). Remarkably, oral administration of 58 (3, 10, and 30 mg kg−1) also blocked U-50488-induced antinociception in a dose-dependent manner (Figure 5G, H).
Figure 5.

KOR blockade by 58 in the mouse tail-flick assay. A, B. Systemic injection of U50488 (15 mg kg−1, i.p.) induced robust antinociception within 30 min in the mouse tail-flick test (50°C water bath), which was blocked by 58 (1 mg kg−1, i.p.) given at 1 h prior to U50488. C, D. 58 (1 mg kg−1, i.p.) given at 24 h prior to U50488 (15 mg kg−1, i.p.) failed to block U50488-induced antinociception assessed at 30 min post-U50,488. E, F. 58 (1 mg kg−1, i.p.) alone did not alter tail flick latency at 60 min post-dose. G, H. Oral 58 (3, 10, 30 mg kg−1, p.o.) given at 1 h prior to U50488 (15 mg kg−1, i.p.) dose-dependently blocked U50488-induced antinociception assessed at 30 min post-U50488. Data represent mean ± SEM. %Maximum Possible Effect (MPE) = (Post dose latency – Pre dose latency)/(Cutoff – Pre dose latency) × 100. Cutoff = 15 sec to prevent tissue damage. Statistics performed using two-way ANOVA followed by Sidak’s multiple comparisons post-hoc test (A-F) or linear regression (G, H). *P<0.05 vs. baseline (BL) of the corresponding treatment group. Group sizes are shown in the figure. 58 was dosed as the monotartrate salt.
CONCLUSIONS
In summary, we have developed a novel chemical class of potent and selective KOR antagonists via multiparametric optimization approach originating from a sub-micromolar HTS-hit 8. 29 showed favorable pharmacokinetic properties including good CNS penetration. Remarkably, 29 was tested in a novel injury-free rat model of migraine and was found to ameliorate stress-induced allodynia, thus providing support for the hypothesis that KOR antagonists could have a therapeutic role in the preventive treatment of migraine. 29 and 58 displayed reversibility of analgesic effects induced by a KOR-agonist within 24h in a tail-flick assay in mice indicating medication-like duration of action of their KOR antagonist activity in vivo. The pharmacokinetic attributes of 58, including its CNS exposure provide confidence in the drug-like properties and developability of the compound. A low oral dose of 58 to mice effectively reduced the elevation of U69,593-induced plasma PRL. 58 did not exhibit significant inhibition of the major cytochrome P450 isoforms and was highly selective for KOR against a broad off-target panel. 58 was advanced into Phase 1 clinical trials49 based on its overall in vitro and in vivo properties across nonclinical species as well as its safety profile.
Figure 5. Blockade of Antinociceptive Response by 58 in the Mouse Tail-Flick Assay
EXPERIMENTAL SECTION
Methods and Materials
Plasma protein binding
Plasma protein binding of compounds 29, 34, 35 and 36 to human, mouse, and rat plasma was evaluated using equilibrium dialysis (Pierce RED system). Compound (1.0 μM) was added to the plasma compartment and after eight hours the concentration of drug was measured in the plasma and buffer compartments by LC-MS/MS. The concentration in the buffer compartment is considered to be the free fraction of compound.
Plasma protein binding of compounds 47, 49, 58 and 62 were performed by Agilux Laboratories (now Charles River Laboratories) (Worcester, MA) (Assay protocols are described in supporting information).
Hepatic microsome stability
Test compounds (27, 29, 33, 34, 35 and 36) (1 μM) were incubated (separately) with 0.2 mg mL−1 pooled human, mouse and rat hepatic microsomes at 37 °C with continuous shaking to determine the rate of metabolism. Samples were collected at 0, 5, 10, 20, 40, and 60 min and acetonitrile was added to quench the reactions and precipitate the proteins. Samples were then centrifuged and the concentration of test compound was determined using HPLC coupled to a triple quadrupole mass spectrometer (LC/MS-MS).
Microsomal hepatic stability of compounds 47, 49, 58, 59, 61 and 62 and hepatic stability of 47 and 58 were performed by Agilux Laboratories (now Charles River Laboratories) (Worcester, MA) (Assay protocols are described in supporting information).
Cytochrome P450 inhibition
The metabolism by CYP1A2 (phenaceten demethylated to acetaminophen), CYP2C9 (tolbutamide hydroxylated to hydroxytolbutamide), CYP2D6 (bufuralol hydroxylated to 4’-hydroxybufuralol), and CYP3A4 (midazolam hydroxylated to 1’-hydroxymidazolam) in the presence or absence of 10 μM test compound (29, 34, 35 and 36) were evaluated. The concentration of each marker substrate is approximately its Km. Specific inhibitors for each isoform were used to validate the system. Compound concentrations were determined by LC-MS/MS. CYP inhibition of 58 was performed by Cyprotex (Assay protocols are described in supporting information)
Bidirectional permeability assays.
The MDCK-MDR1 permeability assays of 47, 49, 58 and 62 were performed by AMRI and Agilux Laboratories (Detailed methods in the supporting information).
The Caco-2 permeability assays of 47 and 58 were performed by Agilux Laboratories (now Charles River Laboratories) (Worcester, MA) (Assay protocols are described in supporting information).
Solubility assay.
The solubility assays were performed by AMRI (Assay protocols are described in supporting information).
Brain homogenate assay.
The purpose of this assay is to asses brain tissue binding. The rat brain homogenate assay was performed by Agilux Laboratories (Assay protocols are described in supporting information).
Electrophysiological assays
The electrophysiological assays for 29, 34, 35 and 36 were performed by Eurofins Pharma Bioanalytics Services US Inc. The electrophysiological assays profiled the activity of these compounds at 10μM on the ion channel targets described in Table 3 using the IonWorks Quattro electrophysiological platform (Assay protocols are described in supporting information).
Procedure for Pharmacokinetic Studies.
Pharmacokinetics of 29 monotartrate salt was assessed in Sprague-Dawley male rats (350–400 grams). The compound was prepared as a solution in 10:10:80 dimethyl sulfoxide/Tween 80/water (v/v/v) at a concentration of 1 mg ml−1 for i.v. and p.o. administration of 1 and 2 mg kg−1, respectively. Compound concentrations were determined using peripheral whole blood. Blood was obtained at 5, 15 and 30 min and 1, 2, 4, 6 and 8 hours postdose for both i.v. and p.o. experiments. All procedures and handling were according to standard operating procedures approved by the Institutional Animal Care and Use Committee (IACUC).
Brain levels of 29. 29 monotartrate salt was dosed i.p. at 10 mg kg−1 in a 1 mg ml−1 solution containing 1 part DMSO, 1 part Tween 80, and 8 parts water into C57Bl6 mice (n = 6). Blood and brain were taken at 30 minutes and 120 minutes. Blood was collected into EDTA-containing tubes and plasma was generated using standard centrifugation techniques. Brain was homogenized and proteins were precipitated with acetonitrile and compound concentrations were determined by LC-MS/MS. Data were fit by WinNonLin using a noncompartmental model and compound concentration in plasma and brain is calculated.
The pharmacokinetic studies and CNS exposure of 58 monotartrate salt were conducted at BioDuro Inc. (Shanghai, China).
Pharmacokinetics of 58 monotartrate salt was assessed in Sprague-Dawley male rats (241–287 g). The compound was prepared as a solution in PBS at a concentration of 0.25 mg ml−1 for i.v. and p.o. administration of 1 and 5 mg kg−1, respectively. Compound concentrations were determined using peripheral whole blood. For i.v. route aliquots of 250 μL of blood were taken via a jugular vein cannula (n = 3) at 5, 15 and 30 min and 1, 2, 4, 8, and 24 hours postdose. For oral route aliquots of 250 μL of blood were taken via a jugular vein cannula (n = 3) at 15 and 30 min and 1, 2, 4, 8, and 24 hours postdose. LC−MS/MS was used as the analytical method for the establishment of compound concentration in blood. Summary statistics were calculated using Microsoft Excel 2013, and graphs were prepared with GraphPad Prism Version 7.
Pharmacokinetics of 58 monotartrate salt was assessed in CD-1 male mice (26–27 g). The compound was prepared as a solution in 0.5% MC at a concentration of 1 mg ml−1.
All procedures and handling were according to standard operating procedures approved by the Institutional Animal Care and Use Committee (IACUC).
Prolactin assay
The assay to measure KOR antagonist inhibition of KOR agonist stimulated prolactin release was conducted at The Scripps Research Institute (TSRI; La Jolla, California), and was approved through the TSRI Institutional Animal Use and Care Committee. Eight to ten week old C57BL/6J male mice from the colony maintained by the Department of Animal Resources were used in this study (n = 3–4/group). Animals were maintained on a 12-hr light/dark cycle and had access to food and water ad lib.
For oral administration, KOR antagonist monotartrate salts were solubilized in water at a concentration of 0.5 mg ml−1 and administer by gavage. The KOR agonist, U69,593 (Sigma) was solubilized in 45% 2-hydroxypropyl-cyclodextrin at a concentration of 10 mg ml−1 and was administered (0.3mg kg−1) subcutaneously in the nape of the neck. The KOR antagonists were administered 2 h before U69,593 administration. Sample collection and prolactin determination: Thirty minutes post agonist treatment mice were euthanized with carbon dioxide, and cardiac puncture was performed. Blood was collected and placed into a microtainer coated with K2EDTA and kept cold. Blood was spun at 4°C, 1,000 × g for 10 minutes. Plasma was removed and prolactin levels determined using the Milliplex Mouse Pituitary Magnetic Bead Panel (MPTMAG-49K) on a Milliplex Analyzer Luminex 200 and pg/mL of prolactin was quantitatively determined using Milliplex Analyst 5.1 software.
Tail flick-test
Tail flick test of 58 monotartrate salt was conducted in the University of Arizona. Adult male ICR mice (weight 25–30 g) were purchased from Envigo (Indianapolis, IN, USA). All procedures were performed in accordance with the guidelines of the Committee for Research and Ethical Issues of IASP under protocols approved by the University of Arizona Institutional Animal Care and Use Committee. Animals were housed three per cage on a 12 h light-dark cycle with food and water ad libitum.
Mice were held gently and 1/2 to 2/3 of the tail was dipped into 50°C hot water. The latency to a rapid flick of the tail was recorded by an investigator blinded to the treatment groups. The cut-off time was set at 15 s to prevent tissue damage. U50488 (KOR agonist, Tocris, Bristol, BS, UK) was dissolved in saline. The dose volume was 10 ml/kg for i.p. injections in mice.
Bacterial reverse mutation assay (Ames assay)
The potential mutagenic activity of compounds 33, 34, 35 and 36 was conducted by BioReliance Corporation in Rockville Maryland. The monotartaric salts of 33, 34, 35 and 36 were evaluated for their ability to induce reverse mutations at the histidine locus of strains of Salmonella typhimurium (TA98, TA100, TA1535, TA1537) and Escherichia coli (WP2 uvrA) in the presence and absence of rat liver S9. Positive controls were employed for each strain and metabolic activation system. The compounds were tested at 8 concentrations ranging from 1.5 to 5000 μg/plate.
The Ames assay of 47 and 58 were conducted by Charles River Laboratories Montreal ULC. The monotartaric salts of 47 and 58 were tested at concentrations between 0.075 and 250 μg/well in the micro Ames duplicate with tester strains TA98, TA100, TA1535, TA97a and WP2 uvrA. Both monotartaric salts were also tested at concentrations between 1.58 and 5000 μg/plate in the standard Ames in triplicate with tester strain TA1537 (Assay protocols are described in supporting information).
Chemistry. General Methods.
Unless otherwise stated, commercially available reagents and solvents were used without purification. Solvents for extraction: ACS grade. Solvents for reaction: reagent grade. Reagents: unless otherwise noted, from Alfa Aesar, Fisher and Aldrich highest quality available. TLC: silica gel 60 F254 aluminum plates, (Whatman, type Al Sil G/UV, 250 μm layer); visualization by UV absorption. Flash chromatography was performed on silica gel 60 (0.40–0.63 mm, 230–440 mesh, EM Science). Biotage Flash+ systems were used for medium-pressure column chromatography. NMR: 1H and 13C spectra were obtained using a Bruker DRX-500 MHz spectrometer. 1H, and 13C NMR data are reported with chemical shifts (δ) in parts-per-million (ppm) relative to the residual signal of the deuterated solvent as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, qn = quintet, m = multiplet, and br = broad), coupling constant in Hz. Reactions were monitored by LC/MS using Shimadzu LC/MS2020. Purity was determined by LCMS using a luna C-18 column (5 μm, 4.6 mm × 50 mm, Phenomenex) and detection was performed with a UV DAD at 254 and 230 nm wavelength. Elution was carried out with a 10−95% gradient over 5 min of CH3CN in water containing 0.1% AcOH at a flow rate of 1.0 mL/min at 25 °C. The purity of all test compounds is higher than 95%. High resolution mass spectra were obtained on an Agilent 6230 TOF LC/MS system using electrospray ionization (ESI) in positive mode.
Synthesis of (3S,4S)-4-(cyclohexylamino)piperidin-3-ol (12).
A mixture of amine 10 (500 mg, 2 mmol), cyclohexanone 11 (207μL, 2 mmol), NaBH(OAc)3 (635 mg, 3 mmol) and AcOH (171μL, 3 mmol) in 1,2-dichloroethane (5 mL) was stirred at room temperature (rt) for 24h. The mixture was filtered through Celite, concentrated under reduced pressure and purified by column chromatography using CH2Cl2/MeOH (0 to 12 % MeOH) to give (3S,4S)-benzyl 4-(cyclohexylamino)-3-hydroxypiperidine-1-carboxylate as a white solid in 76% yield (510 mg, 1.53 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 7.37–7.30 (m, 5H), 6.58 (br s, 2H), 5.17–5.04 (m, 2H), 4.46–4.12 (m, 2H), 3.66–3.57 (m, 1H), 2.90–2.59 (m, 4H), 2.05–1.96 (m, 3H), 1.82–1.74 (m, 2H), 1.66–1.59 (m, 2H), 1.44–1.36 (m, 1H), 1.31–1.13 (m, 4H). 13C NMR (125 MHz, CDCl3): δ (ppm) 155.1, 136.5, 128.6, 128.2, 128.1, 67.6, 59.3, 54.6, 48.9, 42.7, 31.9, 30.3, 28.0, 25.4, 24.9, 24.6, 24.1.
LCMS: (M+1) m/z = 333.
A solution of (3S,4S)-benzyl 4-(cyclohexylamino)-3-hydroxypiperidine-1-carboxylate (500 mg, 1.5 mmol) in 12 mL of EtOH was added Pd/C (10 %wt, 5 mg). The reaction mixture was purged three times with hydrogen and stirred under hydrogen atmosphere for 2h. The reaction mixture was filtered through Celite and condensed to give 97% yield (290 mg, 1.46 mmol) of 12 as a pale brown solid. 1H NMR (500 MHz, CDCl3): δ (ppm) 3.29–3.22 (m, 1H), 3.18–3.14 (m, 1H), 3.05–2.98 (m, 1H), 2.61–2.52 (m, 2H), 2.46–2.37 (m, 2H), 2.06–1.99 (m, 1H), 1.93–1.86 (m, 1H), 1.76–1.66 (m, 3H), 1.62–1.54 (m, 1H), 1.31–1.08 (m, 6H), 1.02–0.94 (m, 1H). 13C NMR (125 MHz, CDCl3): δ (ppm) 72.3, 59.3, 53.2, 51.9, 45.9, 35.3, 33.6, 32.6, 26.2, 25.3, 24.8.
LCMS: (M+1) m/z = 199.
Synthesis of 5-(2-chloro-5-methylpyridin-3-yl)-3-methyl-1,2,4-oxadiazole (15).
To a solution of carboxylic acid 13 (600 mg, 3.49 mmol) in 10mL DMF were added EDCI (870 mg, 4.54 mmol) and HOBt (694 mg, 4.54 mmol) and the mixture was stirred at rt for 10 min. Acetamidoxime 14 (284 mg, 3.84 mmol) was added and the mixture was stirred 30 min at rt. The solution was heated at 110 °C under microwave irradiation (mw) for 1h. The mixture was quenched with brine (100 mL) and extracted with EtOAc (3×60mL). The combined organic phases were washed with brine (3 × 100 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The 15 was purified by column chromatography using Hexanes/EtOAc (0 to 30 % EtOAc) to give the product as a pale brown solid in 71% yield (519mg, 2.46 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.40 (d, J=2.4 Hz, 1H), 8.18 (d, J=2.4 Hz, 1H), 2.51 (s, 3H), 2.42 (s, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 172.8, 167.9, 152.9, 147.0, 140.9, 132.8, 120.2, 17.7, 11.8.
LCMS: (M+1) m/z = 210.
Synthesis of (3S,4S)-4-(cyclohexylamino)-1-(5-methyl-3-(3-methyl-1,2,4-oxadiazol-5-yl)pyridin-2-yl)piperidin-3-ol (16).
A mixture of chloropyridine 15 (20 mg, 0.095 mmol), 12 (38 mg, 0.19 mmol) and DIPEA (33 μL, 0.19 mmol) in EtOH (250 μL) was heated at 135 °C under microwave irradiation for 4h. After cooling to room temperature, the mixture was concentrated under reduced pressure and purified by column chromatography using CH2Cl2/MeOH (0 to 10 % MeOH) to give pyridine 16 as a pale yellow solid in 62% yield (22 mg, 0.059 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.19 (d, J=2.3 Hz, 1H), 8.00 (d, J=2.3 Hz, 1H), 3.70–3.66 (m, 1H), 3.58–3.53 (m, 1H), (td, J= 8.2, 4.0 Hz, 1H), 2.97–2.91 (m, 2H), 2.62–2.55 (m, 2H), 2.46 (s, 3H), 2.28 (s, 3H), 2.09–2.04 (m, 1H), 1.94–187 (m, 1H), 1.78–1.67 (m, 3H), 1.62–1.56 (m, 1H), 1.48–1.40 (m, 1H), 1.31–1.10 (m, 5H), 1.04–0.96 (m, 1H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.0, 167.6, 158.3, 151.6, 141.0, 125.5, 109.1, 70.5, 57.8, 54.4, 53.6, 49.0, 35.1, 33.7, 29.7, 26.2, 25.3, 24.9, 17.3, 11.8.
HRMS (ESI-TOF) calcd for C20H29N5O2 [M + H]+ 372.2394; found 372.2399.
Synthesis of 5-(2-chloro-6-methylquinolin-3-yl)-3-methyl-1,2,4-oxadiazole (19).
A mixture of acid 17 (2.0 g, 9.0 mmol), SOCl2 (2.0 mL, 27.0 mmol) and DMF (cat.) in CH2Cl2 was heated at 60 °C for 2h. After cooling to rt, the mixture was concentrated under reduced pressure. The solid residue was dissolved in 1,4-dioxane (12 mL) and N-hydroxyacetamidine 14 (866 mg, 11.7 mmol) and DIPEA (3.14 mL, 18 mmol) were added. The resulting mixture was stirred at rt for 30 min and 3h at 110 °C. After cooling to rt, the mixture was concentrated under reduced pressure. The crude was dissolved in EtOAc (200 ml) and washed with brine (3×100mL). The organic phase was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography using hexanes/EtOAc (0 to 25 % EtOAc) to give quinoline 19 as a yellow solid in 42% yield (981 mg, 3.77 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.79 (s, 1H), 7.98 (d, J=8.4 Hz, 1H), 7.70 (d, J=8.4 Hz, 1H), 7.69 (s, 1H), 2.57 (s, 3H), 2.55 (s, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 173.1, 168.0, 147.2, 146.1, 141.3, 138.7, 135.6, 128.4, 127.3, 126.0, 118.2, 21.8, 11.9.
LCMS: (M+1) m/z = 260.
Synthesis 5-(2-chloro-6-ethylquinolin-3-yl)-3-methyl-1,2,4-oxadiazole (20).
A mixture of acid 18 (1.76 g, 7.5 mmol), SOCl2 (1.6 mL, 27.0 mmol) and DMF (cat.) in CH2Cl2 (10 mL) was heated at 60 °C for 2h. After cooling to room temperature, the mixture was concentrated under reduced pressure. The solid residue was dissolved in 1,4-dioxane (12 mL) and N-hydroxyacetamidine 14 (719 mg, 9.7 mmol) and DIPEA (2.6 mL, 14.93 mmol) were added. The resulting mixture was stirred at rt for 30 min and 3h at 110 °C. After cooling to rt, the mixture was concentrated under reduced pressure. The crude was dissolved in EtOAc (200 ml) and washed with brine (3×100 mL). The organic phase was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography using hexanes/EtOAc (0 to 25 % EtOAc) to give quinoline 20 as a yellow solid in 39% yield (800 mg, 2.92 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.81 (s, 1H), 8.00 (d, J=8.6 Hz, 1H), 7.73 (d, J=8.6 Hz, 1H), 7.70 (s, 1H), 2.87 (q, J=7.6 Hz, 2H), 2.55 (s, 3H), 1.35 (t, J=7.6, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 173.1, 168.0, 147.5, 146.2, 144.8, 141.5, 134.6, 128.6, 126.2, 126.0, 118.2, 29.0, 15.2, 11.9.
LCMS: (M+1) m/z = 274.
Synthesis of 8-(6-methyl-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)-1,4-dioxa-8-azaspiro[4.5]decane (22).
A mixture of 19 (1.0 g, 3.8 mmol), 21 (1.1 g, 7.7 mmol) and DIPEA (1.34 mL, 7.7 mmol) in EtOH (15 mL) was heated at 130 °C under microwave irradiation for 3h. The mixture was concentrated under reduce pressure and purified by column chromatography using hexanes/EtOAc (0 to 30 % EtOAc) to give quinoline 22 as a yellow solid in 86% yield (1.205 g, 3.28 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.52 (s, 1H), 7.71 (d, J=8.5 Hz, 1H), 7.50 (d, J=8.5 Hz, 1H), 7.49 (s, 1H), 3.99 (s, 4H), 3.45–3.40 (m, 4H), 2.51 (s, 3H), 2.48 (s, 3H), 1.91–186 (m, 4H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.5, 167.9, 156.7, 147.2, 141.7, 134.4, 134.3, 127.4, 127.2, 123.5, 111.5, 107.6, 64.5, 48.2, 34.8, 21.4, 11.9.
LCMS: (M+1) m/z = 367.
Synthesis of 8-(6-ethyl-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)-1,4-dioxa-8-azaspiro[4.5]decane (23).
A mixture of 20 (500 mg, 1.82 mmol), 21 (523 mg, 3.65 mmol) and DIPEA (636 μL, 3.65 mmol) in EtOH (8 mL) was heated at 130 °C under microwave irradiation for 3h. The mixture was concentrated under reduce pressure and purified by column chromatography using hexanes/EtOAc (0 to 30 % EtOAc) to give quinoline 23 as a yellow solid in 82% yield (569 mg, 1.49 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.54 (s, 1H), 7.74 (d, J=8.6 Hz, 1H), 7.54 (dd, J=8.6, 2.0 Hz, 1H), 7.50 (s, 1H), 3.98 (s, 4H), 3.45–3.41 (m, 4H), 2.77 (q, J=7.6 Hz, 2H), 2.50 (s, 3H), 1.91–1.86 (m, 4H), 1.30 (t, J=7.6 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.5, 167.9, 156.7, 147.4, 141.8, 140.7, 133.3, 127.5, 125.9, 123.5, 111.4, 107.6, 64.5, 48.2, 34.8, 28.7, 15.5, 11.9.
LCMS: (M+1) m/z = 381.
Synthesis of 1-(6-methyl-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)piperidin-4-one (24).
To a solution of 22 (1.5 g, 4.09 mmol) in THF (7 mL) was added 10% aq. H2SO4 (120 mL) at rt. The mixture was stirred at rt for 3h. The mixture was neutralized with sat. aq. NaOH and the product extracted with EtOAc (×3). The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography using hexanes/EtOAc (0 to 30% EtOAc) to give quinoline 24 as a yellow solid in 91% yield (1.20 g, 3.72 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.63 (s, 1H), 7.75 (d, J=8.4 Hz, 1H), 7.55 (d, J=8.4 Hz, 1H), 7.54 (s, 1H), 3.66 (t, J=6.0 Hz, 4 H), 2.66 (t, J=6.0 Hz, 4H), 2.52 (s, 3H), 2.50 (s, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 208.8, 175.1, 168.0, 156.0, 146.9, 142.0, 135.1, 134.7, 127.5, 127.3, 123.9, 111.3, 49.8, 41.4, 21.5, 11.9.
LCMS: (M+1) m/z = 323.
Synthesis of 1-(6-ethyl-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)piperidin-4-one (25).
To a solution of 23 (580 mg, 1.52 mmol) in THF (4 mL) was added 10% aq. H2SO4 (50 mL) at rt. The mixture was stirred at rt for 3h. The mixture was neutralized with sat. aq. NaOH and the product extracted with EtOAc (×3). The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography using hexanes/EtOAc (0 to 30% EtOAc) to give quinoline 25 as a yellow solid in 87% yield (447 mg, 1.32 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.67 (s, 1H), 7.77 (d, J=8.6 Hz, 1H), 7.59 (dd, J=8.6, 2.0 Hz, 1H), 7.56 (s, 1H), 3.66 (t, J=6.0 Hz, 4 H), 2.80 (q, J=7.6 Hz, 2H), 2.66 (t, J=6.0 Hz, 4H), 2.52 (s, 3H), 1.32 (t, J=7.6 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 208.8, 175.1, 168.0, 156.0, 147.2, 142.2, 141.5, 133.6, 127.6, 126.0, 123.9, 111.3, 49.8, 41.4, 28.8, 15.5, 11.9.
LCMS: (M+1) m/z = 337.
General procedure for the synthesis of final compounds.
Procedure 1.
A mixture of ketone (1.0 equiv.), amine (1.1–2.0 equiv.), NaBH(OAc)3 (2.0 equiv.) and AcOH (2.0 equiv.) in 1,2-dichloroethane was stirred at rt overnight. After filtration through Celite, the filtrate was concentrated under reduced pressure and purified.
Procedure 2.
A mixture of ketone (1.0 equiv.), amine-HCl salt (1.1–2.0 equiv.), DIPEA (1.1–2.0 equiv.), NaBH(OAc)3 (2.0 equiv.) and AcOH (2.0 equiv.) in 1,2-dichloroethane was stirred at rt overnight. After filtration through Celite, the filtrate was concentrated under reduced pressure and purified.
Synthesis of N-isobutyl-1-(6-methyl-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)piperidin-4-amine (27).
Prepared according to procedure 1 using 24 (30 mg, 0.093 mmol) and isobutylamine (18.6 μL, 0.186 mmol) with purification by prep-HPLC (10–95% ACN in H2O with 0.1% TFA for 12 min) to give 27 as a pale yellow solid in 81% yield (28.5 mg, 0.075 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.50 (s, 1H), 7.71 (d, J=8.6 Hz, 1H), 7.50 (dd, J=8.6, 1.9 Hz, 1H), 7.47 (s, 1H), 3.70–3.64 (m, 2H), 2.99–2.92 (m, 2H), 2.66–2.59 (m, 1H), 2.51 (s, 3H), 2.47 (s, 3H), 2.46 (d, J=6.7 Hz, 2H), 1.97–1.90 (m, 2H), 1.76–1.68 (m, 1H), 1.63–1.54 (m, 2H), 1.44 (br s, 1H), 0.91 (d, J=6.7 Hz, 6H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.7, 167.8, 157.2, 147.2, 141.6, 134.3, 134.2, 127.3, 127.2, 123.5, 111.7, 55.3, 55.0, 49.3, 32.6, 28.8, 21.4, 20.9, 11.9.
HRMS (ESI-TOF) calcd for C22H29N5O [M + H]+ 380.2445; found 380.2449.
N-(cyclopropylmethyl)-1-(6-methyl-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)piperidin-4-amine (28).
Prepared according to the general procedure 1 using 24 (40mg, 0.119 mmol) and cyclopropylmethylamine (21 μL, 0.248 mmol) with purification by prep-HPLC (10–95% ACN in H2O for 12 min) to give 28 as a yellow solid in 88% yield (41 mg, 0.108 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.50 (s, 1H), 7.70 (d, J=8.5 Hz, 1H), 7.49 (dd, J=8.5, 2.0 Hz, 1H), 7.47 (s, 1H), 3.70–3.63 (m, 2H), 2.94 (t, J=13.6 Hz, 2H), 2.70–2.64 (m, 1H), 2.51 (d, J=6.9 Hz, 2H), 2.50 (s, 3H), 2.46 (s, 3H), 1.96–1.90 (m, 2H), 1.63–1.55 (m, 2H), 1.00–0.92 (m, 1H), 0.50–0.46 (m, 2H), 0.13–0.09 (m, 2H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.6, 167.8, 157.1, 147.1, 141.6, 134.3, 134.2, 127.3, 127.2, 123.5, 111.7, 55.0, 52.0, 49.3, 32.5, 21.4, 11.9, 11.6, 3.6.
HRMS (ESI-TOF) calcd for C22H27N5O [M + H]+ 378.2288; found 378.2293.
1-(6-methyl-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)-N-(tetrahydro-2H-pyran-4-yl)piperidin-4-amine (29).
Prepared according to the general procedure1 using 24 (1.0 g, 3.1 mmol) and 4-aminotetrahydropyran (352 μL, 3.41 mmol) with purification on silica gel column chromatography using CH2Cl2/MeOH (0 to 10 % MeOH) to give 29 as a yellow solid in 89% yield (1.125 g, 2.76 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.50 (s, 1H), 7.70 (d, J=8.6 Hz, 1H), 7.49 (dd, J=8.6, 2.0 Hz, 1H), 7.47 (s, 1H), 4.00–3.94 (m, 2H), 3.70–3.64 (m, 2H), 3.39 (td, J=11.7, 2.0 Hz, 2H), 2.98–2.91 (m, 2H), 2.89–2.80 (m, 2H), 2.50 (s, 3H), 2.46 (s, 3H), 1.95–1.89 (m, 2H), 1.83–1.77 (m, 2H), 1.61–1.53 (m, 2H), 1.44–1.35 (m, 2H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.7, 167.8, 157.1, 147.1, 141.7, 134.4, 134.3, 127.3, 127.2, 123.5, 111.7, 67.0, 51.0, 50.3, 49.3, 34.4, 33.0, 21.4, 11.9.
HRMS (ESI-TOF) calcd for C23H29N5O2 [M + H]+ 408.2394; found 408.2399.
Synthesis of 1-(6-ethyl-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)-N-isobutylpiperidin-4-amine (30).
Prepared according to the general procedure 1 using 25 (30 mg, 0.089 mmol) and isobutylamine (17.8 μL, 0.178 mmol) with purification on silica gel column chromatography using CH2Cl2/MeOH (0 to 10 % MeOH) to give 30 as a yellow solid in 80% yield (28 mg, 0.071 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.53 (s, 1H), 7.73 (d, J=8.6 Hz, 1H), 7.53 (dd, J=8.6, 2.0 Hz, 1H), 7.49 (s, 1H), 3.71–3.64 (m, 2H), 3.00–2.92 (m, 2H), 2.77 (q, J=7.6 Hz, 2H), 2.66–2.60 (m, 1H), 2.51 (s, 3H), 2.46 (d, J=6.8 Hz, 2H), 1.97–1.92 (m, 2H),1.76–1.68 (m, 1H), 1.62–1.54 (m, 2H), 1.39 (br s, 1H), 1.30 (t, J=7.6 Hz, 3H), 0.91 (d, J=6.7 Hz, 6H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.7, 167.8, 157.2, 147.4, 141.8, 140.6, 133.2, 127.4, 126.0, 123.5, 111.6, 53.3, 55.0, 49.3, 32.6, 28.8, 28.7, 20.9, 15.5, 11.9.
HRMS (ESI-TOF) calcd for C23H31N5O [M + H]+ 394.2601; found 394.2607.
Synthesis of N-(cyclopropylmethyl)-1-(6-ethyl-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)piperidin-4-amine (31).
Prepared according to the general procedure 1 using 25 (40 mg, 0.119 mmol) and cyclopropylmethylamine (20 μL, 0.238 mmol) with purification on silica gel column chromatography using CH2Cl2/MeOH (0 to 10% MeOH) to give 31 as a yellow solid in 90% yield (42.1 mg, 0.107 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.52 (s, 1H), 7.73 (d, J=8.6 Hz, 1H), 7.53 (dd, J=8.6, 2.0 Hz, 1H), 7.49 (s, 1H), 3.70–3.64 (m, 2H), 2.98–2.90 (m, 2H), 2.77 (q, J=7.6 Hz, 2H), 2.70–2.63 (m, 1H),2.51 (d, J=6.8 Hz, 2H), 2.50 (s, 3H), 1.96–1.91 (m, 2H), 1.63–1.55 (m, 2H), 1.29 (t, J=7.6 Hz, 3H),0.98–0.91 (m, 1H), 0.50–0.46 (m, 2H), 0.13–0.09 (m, 2H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.7, 167.8, 157.2, 147.4, 141.8, 140.6, 133.2, 127.4, 125.9, 123.5, 111.6, 55.1, 52.1, 49.3, 32.6, 28.7, 15.5, 11.9, 11.7, 3.5.
HRMS (ESI-TOF) calcd for C23H29N5O [M + H]+ 392.2445; found 392.2450.
Synthesis of 1-(6-ethyl-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)-N-(tetrahydro-2H-pyran-4-yl)piperidin-4-amine (32).
Prepared according to the general procedure 1 using 25 (40 mg, 0.119 mmol) and 4-aminotetrahydropyran (24.6 μL, 0.238 mmol) with purification on silica gel column chromatography using CH2Cl2/MeOH (0 to 10 % MeOH) to give 32 as a yellow solid in 87% yield (43.8 mg, 0.104 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.53 (s, 1H), 7.73 (d, J=8.6 Hz, 1H), 7.53 (dd, J=8.6, 2.0 Hz, 1H), 7.49 (s, 1H), 3.99–3.93 (m, 2H), 3.70–3.64 (m, 2H), 3.39 (td, J=11.7, 2.3 Hz, 2H), 2.98–2.92 (m, 2H), 2.89–2.80 (m, 2H), 2.76 (q, J=7.6 Hz, 2H), 2.50 (s, 3H), 1.95–1.89 (m, 2H), 1.83–1.77 (m, 2H), 1.62–1.53 (m, 2H), 1.44–1.35 (m, 2H), 1.29 (t, J=7.6 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.6, 167.8, 157.1, 147.3, 141.8, 140.7, 133.2, 127.4, 125.9, 123.5, 111.6, 67.0, 51.0, 50.3, 49.3, 34.4, 33.0, 28.7, 15.5, 11.9.
HRMS (ESI-TOF) calcd for C24H31N5O2 [M + H]+ 422.255; found 422.2556.
Synthesis of (S)-1-(6-ethyl-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)-N-(3-methylbutan-2-yl)piperidin-4-amine (33).
Prepared according to the general procedure 1 using 25 (200 mg, 0.59 mmol) and (S)-2-amino-3-methylbutane (91 μL, 0.77 mmol) with purification on silica gel column chromatography using CH2Cl2/MeOH (0 to 10 % MeOH) to give 33 as a yellow solid in 92% yield (222 mg, 0.545 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.52 (s, 1H), 7.73 (d, J=8.6 Hz, 1H), 7.53 (dd, J=8.6, 2.0 Hz, 1H), 7.48 (s, 1H), 3.70–3.63 (m, 2H), 3.00–2.90 (m, 2H), 2.79–2.69 (m, 3H), 2.64–2.59 (m, 1H), 2.50 (s, 3H), 1.96–1.87 (m, 2H), 1.68–1.46 (m, 3H), 1.29 (t, J=7.6 Hz, 3H), 1.23 (br s, 1H), 0.96 (d, J=7.6 Hz, 3H), 0.89 (d, J=6.8 Hz, 3H), 0.86 (d, J=6.8 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.7, 167.8, 157.2, 147.4, 141.8, 140.6, 133.2, 127.4, 125.9, 123.4, 111.6, 54.5, 52.1, 49.4, 49.3, 33.4, 32.7, 28.7, 19.5, 17.4, 16.9, 15.5, 11.9.
HRMS (ESI-TOF) calcd for C24H33N5O [M + H]+ 408.2758; found 408.2763.
Synthesis of (1S,2S)-2-((1-(6-ethyl-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)piperidin-4-yl)amino)cyclohexanol (34).
Prepared according to the general procedure 2 using 25 (1.16 g, 3.46 mmol) and (1S,2S)-2-aminocyclohexanol HCl (530mg, 3.46 mmol) with purification on silica gel column chromatography using CH2Cl2/MeOH (0 to 10% MeOH) to give 34 as a yellow gum in 78% yield (1200 mg, 2.75 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.53 (s, 1H), 7.74 (d, J=8.6 Hz, 1H), 7.54 (dd, J=8.6, 2.0 Hz, 1H), 7.49 (s, 1H), 3.68–3.60 (m, 2H), 3.11–3.06 (m, 1H), 3.03–2.91 (m, 2H), 2.85–2.73 (m, 3H), 2.50 (s, 3H), 2.33–2.28 (m, 1H), 2.10–2.02 (m, 2H), 1.98–1.92 (m, 1H), 1.87–1.79 (m, 1H), 1.74–1.61 (m, 3H), 1.56–1.48 (m, 1H), 1.30–1.21 (m, 7H), 0.98–0.89 (m, 1H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.7, 167.8, 157.1, 147.4, 141.8, 140.7, 133.2, 127.4, 126.0, 123.5, 111.6, 74.2, 60.6, 51.9, 49.0, 48.9, 34.0, 33.1, 32.5, 31.8, 28.7, 25.5, 24.4, 15.5, 11.9.
HRMS (ESI-TOF) calcd for C25H33N5O2 [M + H]+ 436.2707; found 436.2713.
Synthesis of 1-(6-ethyl-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)-N-((3-methyloxetan-3-yl)methyl)piperidin-4-amine (35).
Prepared according to the general procedure 1 using 25 (1.59 g, 4.73 mmol) and (3-methyloxetan-3-yl)methanamine (0.65 mL, 6.15 mmol) with purification on silica gel column chromatography using CH2Cl2/MeOH (0 to 10% MeOH) to give 35 as a yellow solid in 91% yield (1.81 g, 4.29 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.54 (s, 1H), 7.74 (d, J=8.6 Hz, 1H), 7.54 (d, J=8.6 Hz, 1H), 7.50 (s, 1H), 4.45 (d, J=5.7 Hz, 2H), 4.37 (d, J=5.7 Hz, 2H), 3.70–3.65 (m, 2H), 3.01–2.94 (m, 2H), 2.86 (s, 2H), 2.78 (q, J=7.6 Hz, 2H), 2.69–2.62 (m, 1H), 2.51 (s, 3H), 2.00–1.94 (m, 2H), 1.64–1.55 (m, 2H), 1.32–1.28 (m, 6H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.7, 167.8, 157.2, 147.4, 141.9, 140.7, 133.2, 127.4, 126.0, 123.5, 111.6, 81.4, 55.6, 54.4, 49.2, 39.8, 32.6, 28.7, 22.2, 15.5, 11.9.
HRMS (ESI-TOF) calcd for C24H31N5O2 [M + H]+ 422.255; found 422.2556.
Synthesis of (R)-1-(6-ethyl-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)-N-(tetrahydro-2H-pyran-3-yl)piperidin-4-amine (36).
Prepared according to the general procedure 2 using 25 (150 mg, 0.446 mmol) (R)-3-aminotetrahydropyran HCl (73.6 mg, 0.535 mmol) with purification on silica gel column chromatography using CH2Cl2/MeOH (0 to 10% MeOH) to give 36 as a yellow solid in 87% yield (164 mg, 0.389 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.52 (s, 1H), 7.73 (d, J=8.6 Hz, 1H), 7.53 (dd, J=8.6, 2.0 Hz, 1H), 7.48 (s, 1H), 3.90–3.86 (m, 1H), 3.80–3.76 (m, 1H), 3.69–3.63 (m, 2H), 3.42–3.36 (m, 1H), 3.14 (dd, J=11.0, 8.5 Hz, 1H), 3.00–2.91 (m, 2H), 2.82–2.72 (m, 4H), 2.50 (s, 3H), 1.96–1.84 (m, 3H), 1.70–1.52 (m, 4H), 1.38–1.31 (m, 2H), 1.29 (t, J=7.6 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.6, 167.8, 157.1, 147.3, 141.8, 140.6, 133.2, 127.4, 125.9, 123.5, 111.6, 73.1, 68.3, 52.0, 50.1, 49.20, 49.18, 33.1, 33.0, 31.0, 28.6, 24.8, 15.5, 11.9.
HRMS (ESI-TOF) calcd for C24H31N5O2 [M + H]+ 422.255; found 422.2560.
Synthesis of (S)-1-(6-ethyl-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)-N-(tetrahydro-2H-pyran-3-yl)piperidin-4-amine (37).
Prepared according to procedure 2 using 25 (30 mg, 0.089 mmol) and (S)-3-aminotetrahydropyran HCl (24.5 mg, 0.178 mmol) with purification on silica gel column chromatography using CH2Cl2/MeOH (0 to 10 % MeOH) to give 37 as a yellow solid in 75% yield (28 mg, 0.066 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.52 (s, 1H), 7.73 (d, J=8.6 Hz, 1H), 7.53 (dd, J=8.6, 2.0 Hz, 1H), 7.49 (s, 1H), 3.90–3.86 (m, 1H), 3.80–3.76 (m, 1H), 3.69–3.64 (m, 2H), 3.42–3.37 (m, 1H), 3.15 (dd, J=11.0, 8.5 Hz, 1H), 2.98–2.92 (m, 2H), 2.82–2.72 (m, 4H), 2.50 (s, 3H), 1.96–1.84 (m, 3H), 1.70–1.52 (m, 4H), 1.38–1.31 (m, 2H), 1.29 (t, J=7.6 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.7, 167.8, 157.1, 147.4, 141.8, 140.7, 133.2, 127.4, 125.9, 123.5, 111.6, 73.1, 68.3, 52.1, 50.1, 49.21, 49.20, 33.1, 33.0, 31.0, 28.7, 24.8, 15.5, 11.9.
HRMS (ESI-TOF) calcd for C24H31N5O2 [M + H]+ 422.255; found 422.2556.
Synthesis of (R)-1-(6-ethyl-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)-N-(tetrahydrofuran-3-yl)piperidin-4-amine (38).
Prepared according to the general procedure 2 using 25 (30 mg, 0.089 mmol) and (R)-3-aminotetrahydrofuran HCl (22 mg, 0.178 mmol) with purification on silica gel column chromatography using CH2Cl2/MeOH (0 to 10% MeOH) to give 38 as a yellow solid in 91% yield (33 mg, 0.0809 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.54 (s, 1H), 7.73 (d, J=8.6 Hz, 1H), 7.54 (dd, J=8.6, 2.0 Hz, 1H), 7.49 (s, 1H), 3.95–3.90 (m, 1H), 3.85 (dd, J=8.4, 5.5 Hz, 1H), 3.80–3.75 (m, 1H), 3.71–3.65 (m, 2H), 3.59–3.51 (m, 2H), 3.00–2.92 (m, 2H), 2.77 (q, J=7.6 Hz, 2H), 2.72–2.66 (m, 1H), 2.50 (s, 3H), 2.16–2.09 (m, 1H), 1.97–1.89 (m, 2H), 1.73–1.67 (m, 1H), 1.64–1.55 (m, 2H), 1.48 (br s, 1H), 1.30 (t, J=7.6 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.6, 167.8, 157.1, 147.4, 141.8, 140.7, 133.2, 127.4, 126.0, 123.5, 111.6, 74.0, 67.3, 55.4, 53.6, 49.2, 33.8, 32.83, 32.81, 28.7, 15.5, 11.9.
HRMS (ESI-TOF) calcd for C23H29N5O2 [M + H]+ 408.2394; found 408.2401.
Synthesis of (S)-1-(6-ethyl-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)-N-(tetrahydrofuran-3-yl)piperidin-4-amine (39).
Prepared according to the general procedure 2 using 25 (30 mg, 0.089 mmol) and (S)-3-aminotetrahydrofuran p-toluenesulfonate (46 mg, 0.178 mmol) with purification on silica gel column chromatography using CH2Cl2/MeOH (0 to 10% MeOH) to give 39 as a yellow solid in 85% yield (31 mg, 0.076 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.54 (s, 1H), 7.73 (d, J=8.6 Hz, 1H), 7.54 (dd, J=8.6, 2.0 Hz, 1H), 7.49 (s, 1H), 3.95–3.90 (m, 1H), 3.85 (dd, J=8.4, 5.5 Hz, 1H), 3.80–3.75 (m, 1H), 3.70–3.65 (m, 2H), 3.58–3.51 (m, 2H), 2.99–2.92 (m, 2H), 2.77 (q, J=7.6 Hz, 2H), 2.72–2.65 (m, 1H), 2.50 (s, 3H), 2.16–2.09 (m, 1H), 1.97–1.89 (m, 2H), 1.73–1.67 (m, 1H), 1.64–1.55 (m, 2H), 1.30 (t, J=7.6 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.6, 167.8, 157.1, 147.4, 141.8, 140.7, 133.2, 127.4, 126.0, 123.5, 111.6, 74.0, 67.3, 55.4, 53.6, 49.2, 33.8, 32.83, 32.81, 28.7, 15.5, 11.9.
HRMS (ESI-TOF) calcd for C23H29N5O2 [M + H]+ 408.2394; found 408.2401.
Synthesis of 2-amino-5-bromo-3-fluorobenzaldehyde (41).
To a solution of 40 (8.0 g, 34 mmol) in anhydrous THF (50 mL) was added a 1M solution of BH3.THF complex in THF (102.5 mL, 102.5 mmol) dropwise at 0 °C. The mixture was then stirred at rt for 18h. The reaction was quenched by slow addition of ~20 mL of MeOH at 0 °C. The mixture was concentrated under reduced pressure and the residue was partitioned between EtOAc and brine. The organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude (2-amino-5-bromo-3-fluorophenyl)methanol was obtained as a brown solid in 76% yield (5.65 g, 25.68 mmol) and was used in the next reaction without further purification. 1H NMR (500 MHz, CD3OD): δ (ppm) 7.10 (s, 1H), 7.09 (dd, J=11.7, 2.2 Hz, 1H), 4.86 (s, 1H), 4.56 (s, 2H). 13C NMR (125 MHz, CD3OD): δ (ppm) 152.7 (J=240.1 Hz), 134.6 (J=13.1 Hz), 130.8 (J=4.2 Hz), 127.3 (J=2.9 Hz), 118.1 (J=22.6 Hz), 108.1 (J=9.6 Hz), 62.2.
LCMS: (M+1) m/z = 419, 421.
A mixture of crude (2-amino-5-bromo-3-fluorophenyl)methanol (5.6 g, 25.45 mmol) and activated MnO2 (13.27 g, 152.7 mmol) in anhydrous CH2Cl2 (120 mL) was stirred at rt for 8h. The filtrate was concentrated after filtration through celite. The residue was purified with a short silica gel path using hexanes/EtOAc (0 to 30 % EtOAc) to give 41 as a pale yellow solid in quantitative yield (5.55 g, 25.4 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 9.81 (s, 1H), 7.43 (s, 1H), 7.28 (d, J=8.7 Hz), 6.19 (br s, 2H). 13C NMR (125 MHz, CDCl3): δ (ppm) 192.3, 150.8 (J=245.1 Hz), 138.1 (J=13.5 Hz), 132.6 (J=3.4 Hz), 122.8 (J=21.1 Hz), 121.1 (J=4.6 Hz), 105.4 (J=8.7 Hz).
Synthesis of 5-(6-bromo-2-chloro-8-fluoroquinolin-3-yl)-3-methyl-1,2,4-oxadiazole (43).
A mixture of aldehyde 41 (1.50 g, 6.88 mmol), oxadiazole acid 42 (1.173 g, 8.25 mmol) and POCl3 (8 mL) was stirred at 80 °C for 1 h. After cooling to rt, excess POCl3 was removed under reduced pressure. The crude was quenched with ice/H2O and the mixture was stirred at 0 °C for 10 min. The precipitate was filtered, washed with H2O and dried under reduced pressure. The product was purified by column chromatography using hexanes/EtOAc (0 to 30% EtOAc) to give 43 as a yellow solid in 14% yield (330 mg, 0.963 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.82 (d, J=1.4 Hz, 1H), 7.92 (s, 1H), 7.70 (dd, J=9.2, 2.0 Hz, 1H), 2.56 (s, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 172.1, 168.2, 156.7 (J=264.2 Hz), 148.2, 140.5 (J=2.8 Hz), 137.5 (J=11.9 Hz), 128.1 (J=2.2 Hz), 126.3 (J=5.0 Hz), 121.4 (J=21.6 Hz), 121.4, 120.6. 11.9.
LCMS: (M+1) m/z = 341, 343.
Synthesis of 8-(6-bromo-8-fluoro-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)-1,4-dioxa-8-azaspiro[4.5]decane (44).
A suspension of 43 (300 mg, 0.875 mmol), 21 (168 μL, 1.31 mmol) and DIPEA (229 μL, 1.31 mmol) in EtOH (70 mL) was heated at 130 °C under microwave irradiation for 2h. After cooling to rt, the mixture was concentrated under reduced pressure and purified by column chromatography using hexanes/EtOAc (0 to 50% EtOAc) to give quinoline 44 in 45% yield as a yellow solid (200 mg, 0.445 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.46 (s, 1H), 7.65 (s, 1H), 7.48 (dd, J=9.7, 1.6 Hz, 1H), 3.98 (s, 4H), 3.51–3.46 (m, 4H), 2.51 (s, 3H), 1.89–1.84 (m, 4H). 13C NMR (125 MHz, CDCl3): δ (ppm) 174.7, 168.1, 156.7, 156.5 (J=259.1 Hz), 141.0 (J=2.2 Hz), 137.6 (J=11.5 Hz), 126.0 (J=4.7 Hz), 125.4 (J=3.2 Hz), 120.1 (J=21.9 Hz), 115.6 (J=8.7 Hz), 112.9, 107.3, 64.5, 47.8, 34.7, 11.9.
LCMS: (M+1) m/z = 449, 451.
Synthesis of 1-(6-ethyl-8-fluoro-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)piperidin-4-one (45).
To a mixture of 44 (110 mg, 0.24 mmol), Cs2CO3 (239 mg, 0.73 mmol) and Pd(dppf)Cl2.CH2Cl2 (19.6 mg, 0.024 mmol) in THF (4 mL) was added 1M solution of Et3B in THF (730 μL, 0.73 mmol) at rt. The mixture was heated at 70 °C for 1h. After cooling to rt, the mixture was partitioned between brine (30 mL) and EtOAc (30 mL). The aqueous layer was extracted with EtOAc (2×30 mL) and the combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by column chromatography using hexanes/EtOAc (0 to 70 % EtOAc) to give 8-(6-ethyl-8-fluoro-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)-1,4-dioxa-8-azaspiro[4.5]decane in 74% yield as a yellow solid (71 mg, 0.178 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.52 (d, J=1.5 Hz, 1H), 7.29 (s, 1H), 7.25 (dd, J=11.5, 2.0 Hz, 1H), 3.98 (s, 4H), 3.49–3.43 (m, 4H), 2.75 (q, J=7.6 Hz, 2H), 2.50 (s, 3H), 1.90–1.85 (m, 4H), 1.29 (t, J=7.6 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.2, 167.9, 156.6 (J=253.7 Hz), 156.5, 141.6 (J=3.1 Hz), 140.7 (J=6.4 Hz), 137.1 (J=11.9 Hz), 124.9 (J=2.9 Hz), 121.5 (J=4.1 Hz), 117.1 (J=18.1 Hz), 112.1, 107.5, 64.5, 48.0, 34.7, 28.7, 15.3, 11.9.
LCMS: (M+1) m/z = 399.
To a solution of 8-(6-ethyl-8-fluoro-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)-1,4-dioxa-8-azaspiro[4.5]decane (60 mg, 0.15 mmol) in THF (1 mL) was added 10% aq. H2SO4 (5 mL) and the mixture was stirred at 45 °C for 2h. After cooling to rt, the mixture was neutralized with sat. aq. NaOH and extracted with EtOAc (3×20 mL). The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The ketone 45 was obtained as a yellow solid in 94% yield (50 mg, 0.144 mmol) and was used for the next reaction without further purification. 1H NMR (500 MHz, CDCl3): δ (ppm) 8.65 (d, J=1.6 Hz, 1H), 7.36 (s, 1H), 7.30 (dd, J=11.5, 1.7 Hz, 1H), 3.70 (t, J=6.0 Hz, 4H), 2.78 (q, J=7.6 Hz, 2H), 2.66 (t, J=6.1 Hz, 4H), 2.52 (s, 3H), 1.31 (t, J=7.6 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 208.5, 174.7, 168.1, 156.6 (J=254.4 Hz), 155.9, 142.0 (J=3.1 Hz), 141.6 (J=6.3 Hz), 136.9 (J=11.9 Hz), 125.3 (J=2.8 Hz), 121.6 (J=4.1 Hz), 117.5 (J=18.0 Hz), 112.1, 49.5, 41.2, 28.8, 15.3, 11.9.
LCMS: (M+1) m/z = 355.
Synthesis of 1-(6-ethyl-8-fluoro-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)-N-(tetrahydro-2H-pyran-4-yl)piperidin-4-amine (46).
Prepared according to the general procedure 1 using 45 (53 mg, 0.149 mmol) and 4-aminotetrahydropyran (23 μL, 0.224 mmol) with purification on silica gel column chromatography using CH2Cl2/MeOH (0 to 10% MeOH) to give 46 as a yellow solid in 82% yield (54 mg, 123 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.52 (d, J=1.5 Hz, 1H), 7.29 (s, 1H), 7.25 (dd, J=11.6, 1.8 Hz, 1H), 4.00–3.94 (m, 2H), 3.75–3.68 (m, 2H), 3.40 (td, J=11.8, 2.1 Hz, 2H), 3.03–2.96 (m, 2H), 2.90–2.82 (m, 2H), 2.75 (q, J=7.6 Hz, 2H), 2.51 (s, 3H), 1.97–1.89 (m, 2H), 1.84–1.77 (m, 2H), 1.62–1.52 (m, 2H), 1.45–1.35 (m, 3H), 1.29 (t, J=7.6 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 174.4, 167.9, 156.9, 156.6 (J=253.3 Hz), 141.7 (J=3.0 Hz), 140.7 (J=6.4 Hz), 137.1 (J=11.8 Hz), 124.9 (J=2.9 Hz), 121.5 (J=4.0 Hz), 117.1 (J=18.1 Hz), 112.4, 67.1, 51.0, 50.4, 49.1, 34.5, 32.9, 28.7, 15.3, 11.9.
HRMS (ESI-TOF) calcd for C24H30FN5O2 [M + H]+ 440.2456; found 440.2464.
Synthesis of (R)-1-(6-ethyl-8-fluoro-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)-N-(tetrahydro-2H-pyran-3-yl)piperidin-4-amine (47).
Prepared according to the general procedure 2 using 45 (29 mg, 0.082 mmol) and (R)-3-aminotetrahydropyran HCl (15 mg, 0.11 mmol) with purification on silica gel column chromatography using CH2Cl2/MeOH (0 to 10% MeOH) to give 47 as a yellow solid in 87% yield (32 mg, 0.073 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.50 (d, J=1.5 Hz, 1H), 7.28 (s, 1H), 7.24 (dd, J=11.6, 1.7 Hz, 1H), 3.90–3.84 (m, 1H), 3.80–3.75 (m, 1H), 3.74–3.66 (m, 2H), 3.42–3.36 (m, 1H), 3.14 (dd, J=11.0, 8.5 Hz, 1H), 3.01–2.93 (m, 2H), 2.82–2.71 (m, 4H), 2.50 (s, 3H), 1.96–1.84 (m, 3H), 1.70–1.51 (m, 4H), 1.38–1.29 (m, 2H), 1.28 (t, J=7.6 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.3, 167.9, 156.8, 156.5 (J=254.4 Hz), 141.6 (J=3.0 Hz), 140.6 (J=6.4 Hz), 137.1 (J=11.7 Hz), 124.9 (J=3.0 Hz), 121.5 (J=4.0 Hz), 117.0 (J=18.1 Hz), 112.4, 73.1, 68.3, 51.9, 50.1, 48.97, 48.93, 33.0, 32.9, 30.9, 28.7, 24.8, 15.3, 11.9.
HRMS (ESI-TOF) calcd for C24H30FN5O2 [M + H]+ 440.2456; found 440.2464.
Synthesis of (S)-1-(6-ethyl-8-fluoro-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)-N-(tetrahydro-2H-pyran-3-yl)piperidin-4-amine (48).
Prepared according to the general procedure 2 using 45 (25 mg, 0.0705 mmol) and (S)-3-aminotetrahydropyran.HCl (19.4 mg, 0.141 mmol) with purification on silica gel column chromatography using CH2Cl2/MeOH (0 to 10% MeOH) to give 48 as a yellow solid in 84% yield (26 mg, 0.059 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.51 (d, J=1.5 Hz, 1H), 7.28 (s, 1H), 7.25 (dd, J=11.5, 1.8 Hz, 1H), 3.90–3.86 (m, 1H), 3.80–3.75 (m, 1H), 3.74–3.67 (m, 2H), 3.43–3.37 (m, 1H), 3.15 (dd, J=10.9, 8.5 Hz, 1H), 3.02–2.94 (m, 2H), 2.82–2.71 (m, 4H), 2.51 (s, 3H), 1.97–1.85 (m, 3H), 1.71–1.50 (m, 4H), 1.39–1.31 (m, 2H), 1.29 (t, J=7.6 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.4, 167.9, 156.9, 156.6 (J=253.4 Hz), 141.6 (J=3.1 Hz), 140.7 (J=6.4 Hz), 137.1 (J=11.7 Hz), 124.9 (J=3.0 Hz), 121.5 (J=3.9 Hz), 117.1 (J=18.1 Hz), 112.4, 73.1, 68.3, 52.0, 50.1, 49.00, 48.96, 33.0, 32.9, 31.0, 28.7, 24.8, 15.3, 11.9.
HRMS (ESI-TOF) calcd for C24H30FN5O2 [M + H]+ 440.2456; found 440.2463.
Synthesis of (R)-1-(6-ethyl-8-fluoro-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)-N-(tetrahydrofuran-3-yl)piperidin-4-amine (49).
Prepared according to the general procedure 2 using 45 (25 mg, 0.0705 mmol) and (R)-3-aminotetrahydrofuran HCl (17 mg, 0.141 mmol) with purification on silica gel column chromatography using CH2Cl2/MeOH (0 to 10% MeOH) to give 49 as a yellow solid in 77% yield (23 mg, 0.054 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.52 (d, J=1.5 Hz, 1H), 7.29 (s, 1H), 7.26 (dd, J=11.6, 1.8 Hz, 1H), 3.95–3.90 (m, 1H), 3.85 (dd, J=8.4, 5.5 Hz, 1H), 3.80–3.69 (m, 3H), 3.59–3.51 (m, 2H), 3.03–2.95 (m, 2H), 2.78–2.67 (m, 3H), 2.51 (s, 3H), 2.16–2.09 (m, 1H), 1.98–1.89 (m, 2H), 1.74–1.67 (m, 1H), 1.64–1.50 (m, 3H), 1.29 (t, J=7.6 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.3, 167.9, 156.9, 156.6 (J=253.4 Hz), 141.7 (J=2.9 Hz), 140.7 (J=6.4 Hz), 137.1 (J=11.7 Hz), 124.9 (J=2.8 Hz), 121.5 (J=3.9 Hz), 117.1 (J=18.1 Hz), 112.4, 74.0, 67.3, 55.4, 53.5, 49.02, 49.00, 33.8, 32.71, 32.69, 28.7, 15.3, 11.9.
HRMS (ESI-TOF) calcd for C23H28FN5O2 [M + H]+ 426.23; found 426.2310.
Synthesis of (S)-1-(6-ethyl-8-fluoro-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)-N-(tetrahydrofuran-3-yl)piperidin-4-amine (50).
Prepared according to the general procedure 2 using 45 (25 mg, 0.075 mmol) and (S)-3-aminotetrahydrofuran p-toluenesulfonate (36.6 mg, 0.141 mmol) with purification on silica gel column chromatography using CH2Cl2/MeOH (0 to 10% MeOH) to give 50 as a yellow solid in 78% yield (25 mg, 0.0587 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 8.52 (d, J=1.4 Hz, 1H), 7.29 (s, 1H), 7.25 (dd, J=11.6, 1.6 Hz, 1H) 3.95–3.89 (m, 1H), 3.85 (dd, J=8.5, 5.7 Hz, 1H), 3.80–3.69 (m, 3H), 3.58–3.51 (m, 2H), 3.02–2.95 (m, 2H), 2.78–2.67 (m, 3H), 2.51 (s, 3H), 2.16–2.08 (m, 1H), 1.98–1.89 (m, 2H), 1.74–1.66 (m, 1H), 1.64–1.53 (m, 2H), 1.46 (br s, 1H), 1.29 (t, J=7.6 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.3, 167.9, 156.9, 156.6 (J=253.5 Hz), 141.7 (J=3.0 Hz), 140.7 (J=6.3 Hz), 137.1 (J=11.6 Hz), 124.9 (J=2.9 Hz), 121.5 (J=3.9 Hz), 117.1 (J=18.1 Hz), 112.4, 74.0, 67.3, 55.4, 53.5, 49.01, 48.99, 33.8, 32.71, 32.69, 28.7, 15.3, 11.9.
HRMS (ESI-TOF) calcd for C23H28FN5O2 [M + H]+ 426.23; found 426.2307.
Synthesis of 4-ethyl-2-fluoro-6-iodoaniline (52).
To a mixture of 51 (9.8 g, 70.4 mmol) in acetic acid (110 mL) was added in a single portion NIS (16.63 g, 73.93 mmol) and the mixture was stirred at rt for 1h. The mixture was diluted with EtOAc (800 mL) and washed with brine (x3) and sat. aq. NaHCO3 solution (x2). The organic phase was dried over Na2SO4 and concentrated under reduced pressure. The product was purified by column chromatography using hexanes/EtOAc (0 to 20% EtOAc) as mobile phase. 52 was obtained as reddish oil in 94% yield (17.5 g, 66.04 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 7.25 (s, 1H), 6.82 (dd, J=11.6, 1.8 Hz, 1H), 3.98 (br s, 2H), 2.51 (q, J=7.6 Hz, 2H), 1.17 (t, J=7.6 Hz, 2H). 13C NMR (125 MHz, CDCl3): δ (ppm) 149.9 (J=241.7 Hz), 136.1 (J=6.3 Hz), 133.7 (J=14.1 Hz), 132.9 (J=3.0 Hz), 114.8 (J=18.7 Hz), 84.1, 27.5, 15.5.
LCMS: (M+1) m/z = 265.
Synthesis of 1-(2-amino-5-ethyl-3-fluorophenyl)ethanone (54).
A mixture of 52 (17.5 g, 66.02 mmol), 53 (35.5 mL, 396 mmol), K2CO3 (10.95 g, 79.2 mmol), DPPP (1.31 g, 3.3 mmol) and Pd(OAc)2 (161 mg, 0.66 mmol) in H2O/toluene (9:1, 200mL) was heated at 90 °C for 32h. After the mixture was cooled to rt, concentrated HCl (33 mL) was slowly added and the mixture was stirred at rt for 1h. The mixture was neutralized with sat. aq. NaHCO3 solution. The product was extracted with EtOAc (3×200mL). The organic phase was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The product was purified by column chromatography using hexanes/EtOAc (0 to 20% EtOAc) and obtained as a pale yellow solid in 16% yield (1.91 g, 10.5 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 7.30 (s, 1H), 6.99 (dd, J=12.0, 1.5 Hz, 1H), 6.14 (br s, 2H), 2.58 (s, 3H), 2.56 (q, J=7.6 Hz, 2H), 1.21 (t, J=7.6 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 200.5 (J=2.5 Hz), 151.7 (J=238.8 Hz), 137.4 (J=13.4 Hz), 130.6 (J=6.0 Hz), 125.5 (J=2.7 Hz), 119.9 (J=4.1 Hz), 118.6 (J=17.7 Hz), 28.3, 28.1, 15.8.
LCMS: (M+1) m/z = 182.
Synthesis of 5-(2-chloro-6-ethyl-8-fluoro-4-methylquinolin-3-yl)-3-methyl-1,2,4-oxadiazole (55).
A mixture of 54 (400 mg, 2.2 mmol) and 42 (344 mg, 2.42 mmol) in POCl3 (5 mL) was stirred at 80 °C for 1h. The mixture was concentrated under reduced pressure and quenched with ice/H2O. The mixture was stirred at rt for 30 min and the solid was collected by filtration. The solid was purified by column chromatography using hexanes/EtOAc (0 to 30% EtOAc) and the product was obtained as a pale yellow solid in 40% yield (271 mg, 0.886 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 7.60 (s, 1H), 7.42 (d, J=10.8 Hz, 1H), 2.87 (q, J=7.6 Hz, 2H), 2.58 (s, 3H), 2.57 (s, 3H), 1.35 (t, J=7.6 Hz, 2H). 13C NMR (125 MHz, CDCl3): δ (ppm) 172.9, 167.9, 157.3 (J=255.9 Hz), 149.2 (J=2.5 Hz), 147.7, 145.0 (J=6.8 Hz), 136.7 (J=12.4 Hz), 127.6, 120.2, 118.1 (J=4.1 Hz), 117.3 (J=17.8 Hz), 29.5, 17.2, 15.3, 12.0.
LCMS: (M+1) m/z = 306.
Synthesis of 8-(6-ethyl-8-fluoro-4-methyl-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)-1,4-dioxa-8-azaspiro[4.5]decane (56).
To a mixture of 55 (600 mg, 1.97 mmol), 21 (366 mg, 12.56 mmol) and DIPEA (687 μL, 3.94 mmol) in EtOH (10 mL) was heated at 110 °C overnight. After cooling to rt the mixture was concentrated under reduced pressure and purified by column chromatography using hexanes/EtOAc (0 to 60% EtOAc) to give ketal 56 as a yellow solid in 93% yield (750 mg, 1.82 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 7.45 (s, 1H), 7.25 (dd, J=11.3, 1.7 Hz, 1H), 3.94 (s, 4H), 3.35–3.30 (m, 4H), 2.78 (q, J=7.6 Hz, 2H), 2.54 (s, 3H), 2.50 (s, 3H), 1.72–1.67 (m, 4H), 1.30 (t, J=7.6 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.4, 167.9, 157.3, 157.2 (J=252.5 Hz), 147.8 (J=2.7 Hz), 140.3 (J=6.7 Hz), 136.4 (J=11.6 Hz), 125.2 (J=2.5 Hz), 117.9 (J=4.0 Hz), 116.1 (J=18.2 Hz), 113.0, 107.4, 64.4, 47.5, 34.8, 29.2, 16.6, 15.6, 11.9.
LCMS: (M+1) m/z = 413.
Synthesis of 1-(6-ethyl-8-fluoro-4-methyl-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)piperidin-4-one (57).
To a solution of ketal 56 (800 mg, 1.94 mmol) in THF (3 mL), 10% aq. H2SO4 (15 mL) was added. The mixture was stirred at 45 °C for 2h. After cooling to rt the mixture was neutralized with sat. aq. Na2CO3 and extracted with EtOAc (3×100ml). The combined organic phase was dried over Na2SO4 and concentrated under reduced pressure. The product was purified by column chromatography using hexanes/EtOAc (0 to 60% EtOAc) to give ketone 57 as a yellow solid in 84% yield (601 mg, 1.63 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 7.50 (s, 1H), 7.29 (dd, J=11.2, 1.7 Hz, 1H), 3.55 (t, J=6.0 Hz, 4H), 2.80 (q, J=7.6 Hz, 2H), 2.56 (s, 3H), 2.54 (s, 3H), 2.48 (t, J=5.8 Hz, 4H), 1.31 (t, J=7.6 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 208.3, 175.1, 168.0, 157.2 (J=253.2 Hz), 156.5, 148.5 (J=2.7 Hz), 141.1 (J=6.7 Hz), 136.1 (J=11.5 Hz), 125.5 (J=2.4 Hz), 118.0 (J=4.0 Hz), 116.4 (J=18.2 Hz), 112.8, 48.9, 41.2, 29.3, 16.7, 15.6, 12.0.
LCMS: (M+1) m/z = 369.
Synthesis of 1-(6-ethyl-8-fluoro-4-methyl-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)-N-(tetrahydro-2H-pyran-4-yl)piperidin-4-amine (58).
Prepared according to the general procedure 1 using 57 (800 mg, 2.17 mmol) and 4-aminotetrahydropyran (337 μL, 3.26 mmol) with purification on silica gel column chromatography using CH2Cl2/MeOH (0 to 8% MeOH) to give 58 as a yellow solid in 75% yield (740 mg, 1.63 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 7.45 (s, 1H), 7.25 (dd, J=11.3, 1.7 Hz, 1H), 3.98–3.93 (m, 2H), 3.60–3.54 (m, 2H), 3.41–3.34 (m, 2H), 2.91–2.72 (m, 6H), 2.54 (s, 3H), 2.49 (s, 3H), 1.85–1.75 (m, 4H), 1.46 (br s, 1H), 1.41–1.25 (m, 8H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.4, 167.8, 157.8, 157.1 (J=252.3 Hz), 147.8 (J=2.6 Hz), 140.4 (J=6.8 Hz), 136.4 (J=11.5 Hz), 125.3 (J=2.5 Hz), 118.0 (J=4.0 Hz), 116.1 (J=18.3 Hz), 113.3, 67.1, 51.0, 50.3, 48.6, 34.4, 33.0, 29.2, 16.6, 15.6, 12.0.
HRMS (ESI-TOF) calcd for C25H32FN5O2 [M + H]+ 454.2613; found 454.2623.
Synthesis of (R)-1-(6-ethyl-8-fluoro-4-methyl-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)-N-(tetrahydro-2H-pyran-3-yl)piperidin-4-amine (59).
Prepared according to the general procedure 2 using 57 (30 mg, 0.081 mmol) and (R)-3-aminotetrahydropyran HCl (22.4 mg, 0.162 mmol) with purification on silica gel column chromatography using CH2Cl2/MeOH (0 to 8% MeOH) to give 59 as a yellow solid in 84% yield (31 mg, 0.068 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 7.45 (s, 1H), 7.25 (dd, J=11.2, 1.7 Hz, 1H), 3.87–3.82 (m, 1H), 3.79–3.74 (m, 1H), 3.60–3.52 (m, 2H), 3.42–3.36 (m, 1H), 3.12 (dd, J=11.0, 8.4 Hz, 1H), 2.91–2.83 (m, 2H), 2.80–2.71 (m, 3H), 2.71–2.64 (m, 1H), 2.54 (s, 3H), 2.49 (s, 3H), 1.94–1.88 (m, 1H), 1.86–1.74 (m, 2H), 1.70–1.54 (m, 2H), 1.35–1.26 (m, 7H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.4, 167.8, 157.8, 157.1 (J=252.3 Hz), 147.7 (J=2.7 Hz), 140.4 (J=6.6 Hz), 136.4 (J=11.4 Hz), 125.2 (J=2.5 Hz), 118.0 (J=3.9 Hz), 116.1 (J=18.3 Hz), 113.3, 73.1, 68.3, 52.0, 50.1, 48.57, 48.50, 33.1, 33.0, 30.9, 29.2, 24.8, 16.6, 15.6, 12.0.
HRMS (ESI-TOF) calcd for C25H32FN5O2 [M + H]+ 454.2613; found 454.2625.
Synthesis of (S)-1-(6-ethyl-8-fluoro-4-methyl-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)-N-(tetrahydro-2H-pyran-3-yl)piperidin-4-amine (60).
Prepared according to the general procedure 2 using 57 (30 mg, 0.081 mmol) and (S)-3-aminotetrahydropyran.HCl (22.4 mg, 0.162 mmol) with purification on silica gel column chromatography using CH2Cl2/MeOH (0 to 8% MeOH) to give 60 as a yellow solid in 79% yield (29 mg, 0.063 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 7.45 (s, 1H), 7.25 (dd, J=11.4, 1.7 Hz, 1H), 3.87–3.82 (m, 1H), 3.80–3.74 (m, 1H), 3.61–3.52 (m, 2H), 3.42–3.36 (m, 1H), 3.12 (dd, J=11.0, 8.4 Hz, 1H), 2.91–2.83 (m, 2H), 2.80–2.64 (m, 4H), 2.54 (s, 3H), 2.49 (s, 3H), 1.94–1.88 (m, 1H), 1.86–1.74 (m, 2H), 1.70–1.54 (m, 2H), 1.36–1.25 (m, 7H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.4, 167.8, 157.8, 157.1 (J=252.5 Hz), 147.7 (J=2.5 Hz), 140.4 (J=6.7 Hz), 136.4 (J=11.3 Hz), 125.2 (J=2.5 Hz), 118.0 (J=3.8 Hz), 116.1 (J=18.3 Hz), 113.3, 73.1, 68.3, 52.0, 50.1, 48.57, 48.50, 33.1, 33.0, 30.9, 29.2, 24.8, 16.6, 15.6, 12.0.
HRMS (ESI-TOF) calcd for C25H32FN5O2 [M + H]+ 454.2613; found 454.2623.
Synthesis of (R)-1-(6-ethyl-8-fluoro-4-methyl-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)-N-(tetrahydrofuran-3-yl)piperidin-4-amine (61).
Prepared according to the general procedure 2 using 57 (30 mg, 0.081 mmol) and (R)-3-aminotetrahydrofuran HCl (20 mg, 0.162 mmol) with purification on silica gel column chromatography using CH2Cl2/MeOH (0 to 8% MeOH) to give 61 as a yellow solid in 73% yield (26 mg, 0.059 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 7.45 (s, 1H), 7.25 (dd, J=11.3, 1.7 Hz, 1H), 3.93–3.87 (m, 1H), 3.84–3.79 (m, 1H), 3.78–3.72 (m, 1H), 3.61–3.54 (m, 2H), 3.54–3.47 (m, 2H), 2.91–2.84 (m, 2H), 2.77 (q, J=7.6 Hz, 2H), 2.64–2.58 (m, 1H), 2.54 (s, 3H), 2.49 (s, 3H), 2.13–2.05 (m, 1H), 1.86–1.78 (m, 2H), 1.70–1.63 (m, 1H), 1.37–1.25 (m, 6H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.4, 167.8, 157.8, 157.1 (J=252.3 Hz), 147.8 (J=2.6 Hz), 140.4 (J=6.7 Hz), 136.4 (J=11.4 Hz), 125.2 (J=2.4 Hz), 118.0 (J=3.9 Hz), 116.1 (J=18.3 Hz), 113.3, 74.0, 67.3, 55.4, 53.5, 48.56, 48.55, 33.7, 32.8, 29.2, 16.6, 15.6, 12.0.
HRMS (ESI-TOF) calcd for C24H30FN5O2 [M + H]+ 440.2456; found 440.2467.
Synthesis of (S)-1-(6-ethyl-8-fluoro-4-methyl-3-(3-methyl-1,2,4-oxadiazol-5-yl)quinolin-2-yl)-N-(tetrahydrofuran-3-yl)piperidin-4-amine (62).
Prepared according to the general procedure 2 using 57 (20 mg, 0.054 mmol) and (S)-3-aminotetrahydrofuran p-toluenesulfonate (21 mg, 0.081 mmol) with purification on silica gel column chromatography using CH2Cl2/MeOH (0 to 8% MeOH) to give 62 as a yellow solid in 73% yield (17.5 mg, 0.0398 mmol). 1H NMR (500 MHz, CDCl3): δ (ppm) 7.45 (s, 1H), 7.25 (dd, J=11.3, 1.6 Hz, 1H), 3.92–3.87 (m, 1H), 3.83–3.79 (m, 1H), 3.77–3.71 (m, 1H), 3.60–3.53 (m, 2H), 3.52–3.46 (m, 2H), 2.90–2.83 (m, 2H), 2.76 (q, J=7.6 Hz, 2H), 2.64–2.57 (m, 1H), 2.54 (s, 3H), 2.49 (s, 3H), 2.13–2.05 (m, 1H), 1.86–1.76 (m, 2H), 1.70–1.62 (m, 1H), 1.37–1.25 (m, 6H). 13C NMR (125 MHz, CDCl3): δ (ppm) 175.4, 167.7, 157.8, 157.1 (J=252.2 Hz), 147.7 (J=2.7 Hz), 140.4 (J=6.8 Hz), 136.4 (J=11.4 Hz), 125.2 (J=2.5 Hz), 118.0 (J=3.8 Hz), 116.1 (J=18.4 Hz), 113.3, 74.0, 67.3, 55.4, 53.5, 48.55, 48.53, 33.7, 32.8, 29.2, 16.6, 15.6, 11.9.
HRMS (ESI-TOF) calcd for C24H30FN5O2 [M + H]+ 440.2456; found 440.2465.
General method for the preparation of mono-tartrate salts.
To a solution of L-(+)-tartaric acid (342 mg, 2.27 mmol) in D.I. water (3mL) was added a solution 58 (1.032g, 2.27 mmol) in EtOH (3mL). The mixture was stirred at rt for 1h and concentrated under reduced pressure. The solid was dried under reduced pressure overnight.
Acknowledgments:
We thank Ronald B. Franklin and Paul C. Anderson for insightful scientific consultation during the course of the project and for their help in preparing the manuscript. This work was supported by the National Institute of Neurological Disorders and Stroke (NINDS) of the National Institutes of Health (NIH) and BlackThorn Therapeutics. The grant is funded as part of the Blueprint Neurotherapeutics Network (BPN) of the NIH Blueprint for Neuroscience Research grant 1UH2 NS093030–01 (Edward Roberts, Hugh Rosen, Frank Porreca).
ABBREVIATIONS USED:
- ACN
acetonitrile
- AcOH
acetic acid
- ADMET
absorption, distribution, metabolism, excretion and toxicity
- AUC0-t
area under the plasma concentration-time curve from time zero to time t
- AUC0−∞
area under the plasma concentration-time curve from time 0 extrapolated to infinite time
- BH3-THF
borane–tetrahydrofuran
- Cav1.2
L-type voltage-gated calcium channel 1.2
- Cbz
carboxybenzyl
- CL
clearance
- Clint
intrinsic clearance
- cLogP
calculated logarithm of the partition coefficient between n-octanol and water
- Cmax
maximum observed plasma concentration
- CNS
central nervous system
- cpKa
calculated pKa
- CSF
cerebrospinal fluid
- CYP1A2
cytochrome P450 1A2
- CYP3A4
cytochrome P450 3A4
- CYP2C9
cytochrome P450 2C9
- CYP2D6
cytochrome P450 2D6
- DIPEA
N,N-diisopropylethylamine
- DMF
dimethylformamide
- DNA
deoxyribonucleic acid
- DOR
delta opioid receptor
- DPPP
1,3-bis(diphenylphosphino)propane
- EDCI
3-(ethyliminomethyleneamino)-N,N-dimethylpropan-1-amine
- EtOAc
ethyl acetate
- EtOH
ethanol
- F
bioavailability
- GPCRs
G protein–coupled receptors
- HCN4
potassium/sodium hyperpolarization-activated cyclic nucleotide-gated channel
- hERG
human ether-a-go-go-related gene
- HLM
human liver microsomes
- HOBt
hydroxybenzotriazole
- HPLC
high-performance liquid chromatography
- HRMS
High-resolution mass spectrometry
- HTS
high-throughput screening
- i.p.
intraperitoneal
- i.v.
intravenous dosing
- IC50
50 percent inhibitory concentration
- Ki
binding affinity
- Kir2.1
inward-rectifying voltage-gated potassium 2.1
- KOR
kappa opioid receptor
- MDD
major depressive disorder
- MeOH
methanol
- MHz
megahertz
- MLM
mouse liver microsomes
- MLSMR
Molecular Libraries-Small Molecule Repository
- MOR
mu opioid receptor
- MW
molecular weight
- mw
microwave
- Nav1.5
voltage-gated sodium channel type 5
- NDA
new drug application
- nM
nanomolar
- ORL-1
opioid receptor like
- pKa
negative base-10 logarithm of the acid dissociation constant
- p.o.
oral administration
- Pd(dppf)Cl2 DCM
[1,1’-bis(diphenylphosphino)ferrocene]dichloropalladium(II) complex with dichloromethane
- ppm
parts-per-million
- PRL
prolactin
- RLM
rat liver microsomes
- rt
room temperature
- SAR
structure–activity relationship
- SNAr
nucleophilic aromatic substitution
- S9
rat hepatic S9 fractions
- THF
tetrahydrofuran
- THP
tetrahydropyran
- TIDA
tuberoinfundibular dopaminergic
- tPSA
topological polar surface area
- t1/2
- Vss
steady-state volume of distribution
- μM
micromolar
Footnotes
Supporting Information.
Experimental details of the ADME/PK and off-target studies. Plasma protein and brain tissue binding, microsomal Stability, hepatocyte Stability, MDCK-MDR1 or Caco-2 cell permeability and efflux assay, Kinetic Solubility, Off-Target Selectivity, Safety Off-Target Panels, Ion channel inhibition and functional hERG, Cytochrome P450 (Cyp) inhibition, Screening bacterial reverse mutation tests in Salmonella typhimurium and Escherichia coli (Ames), Pharmacokinetic Analysis (brain and plasma) (DOCX)
Molecular formula strings (CSV)
REFERENCES:
- (1).Fredriksson R; Lagerström MC; Lundin LG; Schiöth HB The G-protein-coupled receptors in the human genome form five main families. phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol 2003, 63, 1256–1272. [DOI] [PubMed] [Google Scholar]
- (2).Waldhoer M; Bartlett SE; Whistler JL Opioid receptors. Annu. Rev. Biochem 2004,73, 953–990. [DOI] [PubMed] [Google Scholar]
- (3).Chavkin C; James IF; Goldstein A Dynorphin is a specific endogenous ligand of the kappa opioid receptor. Science 1982, 215, 413–415. [DOI] [PubMed] [Google Scholar]
- (4).Bruchas MR; Land BB; Chavkin C The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Res. 2010, 1314, 44–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Simonin F; Gavériaux-Ruff C; Befort K; Matthes H; Lannes B; Micheletti G; Mattéi MG; Charron G; Bloch B; Kieffer B Kappa-opioid receptor in humans: cDNA and genomic cloning, chromosomal assignment, functional expression, pharmacology, and expression pattern in the central nervous system. Proc. Natl. Acad. Sci. USA 1995, 92, 7006–7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Cross AJ; Hille C; Slater P Subtraction autoradiography of opiate receptor subtypes in human brain. Brain Res 1987, 418, 343–348. [DOI] [PubMed] [Google Scholar]
- (7).Barg J; Belcheva M; Rowinski J; Ho A; Burke WJ; Chung HD; Schmidt CA; Coscia CJ Opioid receptor density changes in Alzheimer amygdala and putamen. Brain Res. 1993, 632, 209–215. [DOI] [PubMed] [Google Scholar]
- (8).Pfeiffer A; Pasi A; Mehraein P; Herz A Opiate receptor binding sites in human brain. Brain Res. 1982, 248, 87–96. [DOI] [PubMed] [Google Scholar]
- (9).Delay-Goyet P; Zajac JM Javoy-Agid F; Agid Y; Roques BP. Regional distribution of μ, δ and κ opioid receptors in human brains from controls and parkinsonian subjects. Brain Res. 1987, 414, 8–14. [DOI] [PubMed] [Google Scholar]
- (10).Carlezon WA; Béguin C; DiNieri JA; Baumann MH; Richards MR; Todtenkopf MS; Rothman RB; Ma Z; Lee DY; Cohen BM Depressive-like effects of the kappa-opioid receptor agonist salvinorin A on behavior and neurochemistry in rats. J. Pharmacol. Exp. Ther 2006, 316, 440–447. [DOI] [PubMed] [Google Scholar]
- (11).Mague SD; Pliakas AM; Todtenkopf MS; Tomasiewicz HC; Zhang Y; Stevens WC; Jones RM; Portoghese PS; Carlezon WA Antidepressant-like effects of kappa-opioid receptor antagonists in the forced swim test in rats. J. Pharmacol. Exp. Ther 2003, 305, 323–330. [DOI] [PubMed] [Google Scholar]
- (12).Ranganathan M; Schnakenberg A; Skosnik PD; Cohen BM; Pittman B; Sewell RA; D’Souza DC. Dose-related behavioral, subjective, endocrine, and psychophysiological effects of the κ opioid agonist salvinorin A in humans. Biol. Psychiatry 2012, 72, 871–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).de Lanerolle NC; Williamson A; Meredith C; Kim JH; Tabuteau H; Spencer DD; Brines ML Dynorphin and the kappa 1 ligand [3H]u69,593 binding in the human epileptogenic hippocampus. Epilepsy Res. 1997, 28,189–205. [DOI] [PubMed] [Google Scholar]
- (14).Loacker S; Sayyah M; Wittmann W; Herzog H; Schwarzer C Endogenous dynorphin in epileptogenesis and epilepsy: anticonvulsant net effect via kappa opioid receptors. Brain 2007, 130, 1017–1028. [DOI] [PubMed] [Google Scholar]
- (15).Mathieu-Kia AM; Fan LQ; Kreek MJ; Simon EJ; Hiller JM Mu-, delta- and kappa-opioid receptor populations are differentially altered in distinct areas of postmortem brains of Alzheimer’s disease patients. Brain Res. 2001, 893, 121–134. [DOI] [PubMed] [Google Scholar]
- (16).Cohen RM; Andreason PJ; Doudet DJ; Carson RE; Sunderland T Opiate receptor avidity and cerebral blood flow in Alzheimer’s disease. J. Neurol. Sci 1997, 148,171–180. [DOI] [PubMed] [Google Scholar]
- (17).Shippenberg TS The dynorphin/kappa opioid receptor system: a new target for the treatment of addiction and affective disorders? Neuropsychopharmacology 2009, 34, 247. [DOI] [PubMed] [Google Scholar]
- (18).Mash DC; Staley JK D3 dopamine and kappa opioid receptor alterations in human brain of cocaine‐overdose victims. Ann. N. Y. Acad. Sci 1999, 877, 507–522. [DOI] [PubMed] [Google Scholar]
- (19).Van’t Veer A; Carlezon WA Role of kappa-opioid receptors in stress and anxiety-related behavior. Psychopharmacology 2013, 229, 435–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Pietrzak RH; Naganawa M; Huang Y; Corsi-Travali S; Zheng MQ; Stein MB; Henry S; Lim K; Ropchan J; Lin SF; Carson RE; Neumeister A Association of in vivo κ-opioid receptor availability and the transdiagnostic dimensional expression of trauma-related psychopathology. JAMA Psychiatry 2014, 71, 1262–1270. [DOI] [PubMed] [Google Scholar]
- (21).https://clinicaltrials.gov/ct2/show/NCT02237703 (accessed Aug 15, 2018).
- (22).Xie JY; De Felice M; Kopruszinski CM; Eyde N; LaVigne J; Remeniuk B; Hernandez P; Yue X; Goshima N; Ossipov M; King T; Streicher JM; Navratilova E; Dodick D; Rosen H; Roberts E; Porreca F. Kappa opioid receptor antagonists: a possible new class of therapeutics for migraine prevention. Cephalalgia 2017, 37, 780–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Munro TA; Berry LM; Van’t Veer A; Béguin C; Carroll FI; Zhao Z; Carlezon WA; Cohen BM Long-acting κ opioid antagonists nor-BNI, GNTI and JDTic: pharmacokinetics in mice and lipophilicity. BMC Pharmacol. 2012, 12, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Urbano M; Guerrero M; Rosen H; Roberts E Antagonists of the kappa opioid receptor. Bioorg. Med. Chem. Lett 2014, 24, 2021–2032. [DOI] [PubMed] [Google Scholar]
- (25).Verhoest PR; Basak AS; Parikh V; Hayward M; Kauffman GW; Paradis V; McHardy SF; McLean S; Grimwood S; Schmidt AW; Vanase-Frawley M; Freeman J; Van Deusen J; Cox L; Wong D; Liras S Design and discovery of a selective small molecule κ opioid antagonist (2-methyl-N-((2’-(pyrrolidin-1-ylsulfonyl)biphenyl-4-yl)methyl)propan-1-amine, PF-4455242). J. Med. Chem 2011, 54, 5868–5877. [DOI] [PubMed] [Google Scholar]
- (26).https://clinicaltrials.gov/ct2/show/study/NCT00939887 (accessed Aug 15, 2018).
- (27).Mitch CH; Quimby SJ; Diaz N; Pedregal C; de la Torre MG; Jimenez A; Shi Q; Canada EJ; Kahl SD; Statnick MA; McKinzie DL; Benesh DR; Rash KS; Barth VN Discovery of aminobenzyloxyarylamides as κ opioid receptor selective antagonists: application to preclinical development of a κ opioid receptor antagonist receptor occupancy tracer. J. Med. Chem 2011, 54, 8000–8012. [DOI] [PubMed] [Google Scholar]
- (28).Domi E; Barbier E; Augier E; Augier G; Gehlert D; Barchiesi R; Thorsell A; Holm L; Heilig M Preclinical evaluation of the kappa-opioid receptor antagonist CERC-501 as a candidate therapeutic for alcohol use disorders. Neuropsychopharmacology. 2018, 43, 1805–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Jackson KJ; Jackson A; Carroll FI; Damaj MI Effects of orally-bioavailable short-acting kappa opioid receptor-selective antagonist LY2456302 on nicotine withdrawal in mice. Neuropharmacology 2015, 97, 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Lowe SL; Wong CJ; Witcher J; Gonzales CR; Dickinson GL; Bell RL; Rorick-Kehn L; Weller M; Stoltz RR; Royalty J; Tauscher-Wisniewski S Safety, tolerability, and pharmacokinetic evaluation of single- and multiple-ascending doses of a novel kappa opioid receptor antagonist LY2456302 and drug interaction with ethanol in healthy subjects. J. Clin. Pharmacol 2014, 54, 968–978. [DOI] [PubMed] [Google Scholar]
- (31).Reed B; Butelman ER; Fry RS; Kimani R; Kreek MJ Repeated administration of opra kappa (LY2456302), a novel, short-acting, selective kop-r antagonist, in persons with and without cocaine dependence. Neuropsychopharmacology. 2018, 43, 739–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).https://ir.cerecor.com/press-releases/detail/36 (accessed Sept 6, 2018).
- (33).Rorick-Kehn LM; Witcher JW; Lowe SL; Gonzales CR; Weller MA; Bell RL; Hart JC; Need AB; McKinzie JH; Statnick MA; Suico JG; McKinzie DL; Tauscher-Wisniewski S; Mitch CH; Stoltz RR; Wong CJ Determining pharmacological selectivity of the kappa opioid receptor antagonist LY2456302 using pupillometry as a translational biomarker in rat and human. Int. J. Neuropsychopharmacol 2015, 18, pyu036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Ragguett RM; Rong C; Rosenblat JD; Ho RC; McIntyre RS Pharmacodynamic and pharmacokinetic evaluation of buprenorphine + samidorphan for the treatment of major depressive disorder. Expert Opin. Drug Metab. Toxicol 2018, 14, 475–482. [DOI] [PubMed] [Google Scholar]
- (35).Fava M; Memisoglu A; Thase ME; Bodkin JA; Trivedi MH; de Somer M; Du Y; Leigh-Pemberton R; DiPetrillo L; Silverman B; Ehrich E Opioid modulation with buprenorphine/samidorphan as adjunctive treatment for inadequate response to antidepressants: a randomized double-blind placebo-controlled trial. Am. J. Psychiatry 2016, 173, 499–508. [DOI] [PubMed] [Google Scholar]
- (36). http://phx.corporate-ir.net/phoenix.zhtml?c=92211&p=irol-corporateNewsArticle&ID=2342624.
- (37).Guerrero M; Urbano M; Brown SJ; Cayanan C; Ferguson J; Cameron M; Devi LA; Roberts E; Rosen H. Optimization and characterization of an opioid kappa receptor (OPRK1) antagonist Apr 15, 2013 [Updated Sep 18, 2014]. In: Probe Reports from the NIH Molecular Libraries Program [Internet] Bethesda (MD): National Center for Biotechnology Information (US); 2010-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK179827/ (accessed Dec 14, 2018). [PubMed] [Google Scholar]
- (38).Ghose AK; Herbertz T; Hudkins RL; Dorsey BD; Mallamo JP Knowledge-Based, Central nervous system (CNS) lead selection and lead optimization for CNS drug discovery. ACS Chem. Neurosci 2012, 3, 50–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Benigni R Structure-activity relationship studies of chemical mutagens and carcinogens: mechanistic investigations and prediction approaches. Chem. Rev 2005, 105, 1767–1800. [DOI] [PubMed] [Google Scholar]
- (40).Marco-Contelles J; Pérez-Mayoral E; Samadi A; Carreiras MC; Soriano E Recent advances in the Friedländer reaction. Chem. Rev 2009, 109, 2652–2671. [DOI] [PubMed] [Google Scholar]
- (41).Arvela RK; Pasquini S; Larhed M Highly regioselective internal heck arylation of hydroxyalkyl vinyl ethers by aryl halides in water. J. Org. Chem 2007, 72, 6390–6396. [DOI] [PubMed] [Google Scholar]
- (42).Torner L Actions of prolactin in the brain: from physiological adaptations to stress and neurogenesis to psychopathology. Front. endocrinol 2016, 7, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Bernard V; Young J; Chanson P; Binart N New insights in prolactin: pathological implications. Nat. Rev. Endocrinol 2015, 11, 265–275. [DOI] [PubMed] [Google Scholar]
- (44).Bart G; Schluger JH; Borg L; Ho A; Bidlack JM; Kreek MJ Nalmefene induced elevation in serum prolactin in normal human volunteers: partial kappa opioid agonist activity? Neuropsychopharmacology. 2005, 30, 2254–2262. [DOI] [PubMed] [Google Scholar]
- (45).Ben-Jonathan N; Hnasko R Dopamine as a prolactin (PRL) inhibitor. Endocr. Rev 2001, 22, 724–763. [DOI] [PubMed] [Google Scholar]
- (46).Butelman ER; Kreek MJ Kappa-opioid receptor agonist-induced prolactin release in primates is blocked by dopamine D(2)-like receptor agonists. Eur. J. Pharmacol 2001, 423, 243–249. [DOI] [PubMed] [Google Scholar]
- (47).Grimwood S; Lu Y,; Schmidt AW; Vanase-Frawley MA; Sawant-Basak A; Miller E; McLean S; Freeman J; Wong S; McLaughlin JP; Verhoest PR. Pharmacological characterization of 2-methyl-N-((2’-(pyrrolidin-1-ylsulfonyl)biphenyl-4-yl)methyl)propan-1-amine (PF-04455242), a high-affinity antagonist selective for κ-opioid receptors. J. Pharmacol. Exp. Ther 2011, 339, 555–566. [DOI] [PubMed] [Google Scholar]
- (48).Chang C; Byon W; Lu Y; Jacobsen LK; Badura LL; Sawant-Basak A; Miller E; Liu J; Grimwood S; Wang EQ; Maurer TS Quantitative PK-PD model-based translational pharmacology of a novel kappa opioid receptor antagonist between rats and humans. AAPS J. 2011, 13, 565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).http://www.blackthornrx.com/blackthorn-therapeutics-initiates-phase-1-study-of-btrx-335140-an-investigational-kappa-opioid-receptor-kor-antagonist/ (accessed Dec 14, 2018)
- (50).cLogP and tPSA calculated with chembiodraw ultra 12.0.
- (51).CpKa calculated with Marvin 5.7.1, 2012, http://www.chemaxon.com.