

Abstract
Activity-dependent changes in the effective connection strength of synapses is a fundamental feature of a nervous system. This so-called synaptic plasticity is thought to underlie storage of information in memory and has been hypothesized to be crucial for the effects of cognitive behavioral therapy. Synaptic plasticity stores information in a neural network, creating a trace of neural activity from past experience. The plasticity can also change the behavior of the network so the network can differentially transform/compute information in future activations. We discuss these two related but separable functions of synaptic plasticity; one we call “item memory” as it represents and store items of information in memory, the other we call “process memory” as it encodes and stores functions such as computations to modify network information processing capabilities. We review evidence of item and process memory operations in behavior and evidence that experience modifies the brain’s functional networks. We discuss neurodevelopmental rodent models relevant for understanding mental illness and compare two models in which one model, neonatal ventral hippocampal lesion (NVHL) has beneficial adult outcomes after being exposed to an adolescent cognitive experience that is potentially similar to cognitive behavioral therapy. The other model, gestational day 17 methylazoxymethanol acetate (GD17-MAM), does not benefit from the same adolescent cognitive experience. We propose that process memory is altered by early cognitive experience in NVHL rats but not in GD17-MAM rats, and discuss how dysplasticity factors may contribute to the differential adult outcomes after early cognitive experience in the NVHL and MAM models.
Keywords: dysplasticity, hippocampus, neurodevelopment, schizophrenia, cognitive behavioral therapy, synaptic function
1.1. Introduction
Learning and memory storage in the mammalian brain is hypothesized to involve modifications of synaptic connections, termed synaptic plasticity (see review Takeuchi et al., 2013). It is thus natural to expect that experience-driven modifications of synaptic function contribute to remodeling the brain’s functional circuits as a result of experience (Bannerman et al., 1995; Inglis et al., 2013), and in particular following cognitive behavioral therapy (de Villers-Sidani et al., 2010; Froemke et al., 2013; Guic et al., 2008). Cognitive behavioral therapy (CBT) may fundamentally cause cognitive information storage by causing neurobiological synaptic plasticity to form memory for a particular skill or appropriate specific responses to specific situations. Alternatively, CBT may change the overall functioning of neural circuits that mediate cognition by causing training-induced neuroplasticity to bias particular neural circuit-mediated pathways of information flow in the brain. Neuroplasticity includes synaptic plasticity and other changes in brain function and metabolism (Voss et al., 2017) and because neuroplasticity is common to both memory storage and neural circuit function plasticity, this paper aims to conceptualize the different consequences of neuroplasticity and examine dysplasticity that may occur in mental illness. Because we can better measure neuroplasticity and mental function in animals, we will focus the discussion on animal studies, and in particular on our studies of experience-induced memory and neural circuit function, both in normal rodents, and in rodent models used to study mental illnesses thought to be of neurodevelopmental origins, such as schizophrenia and autism.
1.2. Related but distinct: item and process memory
The difference between cognitive information storage versus the formation of memory for a particular skill can be conceptualized as “item memory” and “process memory,” respectively. We will use an ambigram - a word with distinct meanings when read in both directions - to illustrate the distinction between the notions of item memory and process memory in neural network function (Fig. 1). Item memory and process memory (also called representation learning) are a pair of concepts that are used in cognitive psychology as well as computer science (Schapiro et al., 2017; Wilson and Niv, 2012) and, as applied here, may be useful for understanding the neurobiological dysfunctions that can underlie memory deficits relevant to mental illness.
Figure 1. Conceptualizing Item and Process Memory.
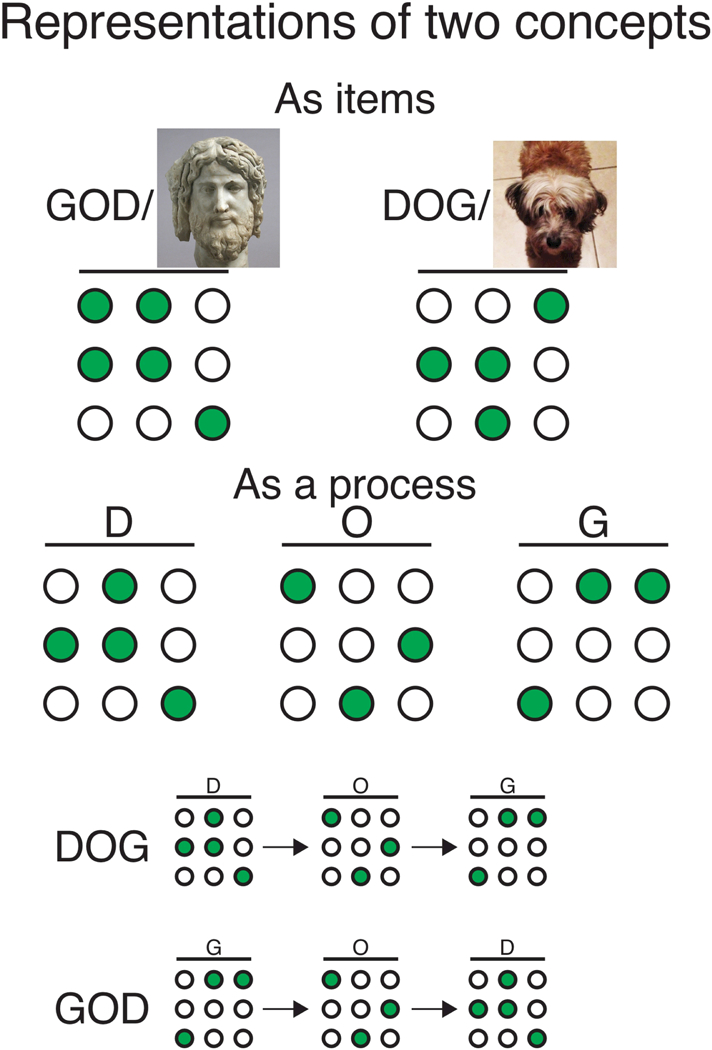
The ambigram DOG is useful for describing the distinction between the concepts of “item” and “process” memory. The circles represent a network of neurons and green indicates the active subset of neurons, which generate an activity pattern to represent information. In the case of item memory, two distinct patterns would each represent the notions of “dog” and “god.” These patterns would be different from the networks that represent the individual letters, for example, another pattern of coactivity would represent the letter “D.” The neurons may be active for multiple representations, but a unique combination of active and inactive neurons defines the pattern for each representation. In the case of process memory, the activity patterns that represent the letters “D,”“O,” and “G” would be the same for both the notions “god” and “dog” but the letter-specific patterns would activate in one temporal sequence to represent “dog” and the reverse sequence to represent “god.” Whichever activation sequence is more likely will bias representation to “dog” or “god.”
In Figure 1, the example ambigram of DOG has one meaning (dog) when read from left to right, but has a different meaning (god) when read from right to left. Item memory corresponds to the typical concept of memory. The standard view is that a subgroup of neurons, likely distributed in the brain, are coactive to represent the concept of dog and separate, perhaps partially overlapping subgroups of neurons are active to represent the concept of god. When either item is recalled, the appropriate neurons are activated. There have been dramatic examples of such neuronal activity. So-called concept cells, like ones representing the actress Halle Berry, have been recorded from the human medial temporal lobe (Gelbard-Sagiv et al., 2008) and place cells, head- direction cells and grid cells of the hippocampus and related subcortical and neocortical regions have been recorded in freely-behaving animals (Moser and Moser, 2013; Muller et al., 1996; O’Keefe, 1979; Taube, 2007; Taube et al., 1990).
Now consider the DOG ambigram within the concept of process memory. To enable activation of the concept “dog,” the left-to-right sequential process of encoding “d” then “o” then “g” must operate and a rather different right-to-left sequential process is necessary before “god” can be encoded. The same individual item representations of the letters “d”, “o” and “g” operate in each instance but the order in which the items are represented and assembled must necessarily differ to end up with the item representations of “dog” and “god”.
The notion of process memory in a more familiar context is computer hardware. While the computer stores information, like a document or image file, on hard disc, analogous to item memory, the computer also requires random access memory (RAM) and other forms of processor memory, like CPU registers, in order to read, write, compute and otherwise manipulate the information in those files and other, similar types of files that store the information about distinct items. In neurobiological terms, process memory refers to the information storage requirements in neural pathways that are needed to generate the output neuronal activity that we recognize as representing items including concepts. Unlike the computer hardware case where disk memory and RAM are mechanistically distinct, item and process memory may share the same cellular and molecular substrates, and even occur in the same brain regions.
Before proceeding, we note that long-term memory has been fractionated into several distinct, often dichotomized categories, which are in fact mapped to various regions of the brain and we now distinguish the familiar concepts of explicit (declarative) and implicit (nondeclarative) memory from the concepts of item and process memory we are proposing to use in this review (see reviews McDougall, 1923; Squire, 2004). Item memory aligns well with explicit memory in that both refer to information that relies on the temporal lobe function and that can be consciously communicated. From a neuropsychological perspective, process memory resembles the various forms of implicit/non-declarative memory such as procedural memory, priming and perceptual learning, classic conditioning and non-associative learning like habituation, all of which can be acquired and expressed unconsciously and are mapped to brain regions outside of the temporal lobe. In contrast, as described above, process memory can accompany and can even enable item memory, as in the computer analogy above. Importantly, also, both item and process memory can depend on the same memory system. As we will soon describe, distinct item and process memories depend on hippocampus in the temporal lobe, although they may involve different neurobiological mechanisms within the same memory system.
1.3. Process memory as synaptically-controlled excitation-inhibition coordination
The hippocampus is well-known as the canonical repository of explicit memory (Squire, 2004; Squire et al., 2004) but hippocampus physiology also provides numerous examples of process memory. In particular, consider the idea that networks of neurons are functionally organized in winner-take-all competitive networks, the so-called attractor dynamics of which are governed by multi-time scale excitatory and inhibitory interactions amongst the neurons (Samsonovich and McNaughton, 1997; Zhang, 1996). In such a network, the cells that are coactive to signal the same information tend to have strong, mutually excitatory synapses and strong, excitatory synapses onto inhibitory cells. This excitatory drive onto inhibitory cells in turn weakly inhibits the active cells and strongly inhibits the currently less-active cells that represent other information by their mutual activity (de Almeida et al., 2009; Dvorak et al., 2018; Hopfield, 1982). This set of interactions is cartooned in Figure 2. While these activity dynamics execute neural computations like pattern separation, pattern completion, divisive normalization, regularization (Carandini and Heeger, 2011; Marr, 1971; Richards and Frankland, 2017) and such, these dynamics are themselves realized because of the synaptic properties that govern the neural interactions, and those properties are themselves a result of experience-dependent gene expression and protein synthesis (see review Kandel, 2004). Accordingly, these operational dynamics of a neural circuit are tuned by experience, the underlying neurobiology of which involves synaptic plasticity and maintenance (Pavlowsky et al., 2017; Tsokas et al., 2016). As noted, similar, if not identical, neurobiological processes are thought to underlie the storage of memories for items of experience as well as process memory.
Figure 2. Competitive neural networks and basic network excitation-inhibition coordination.
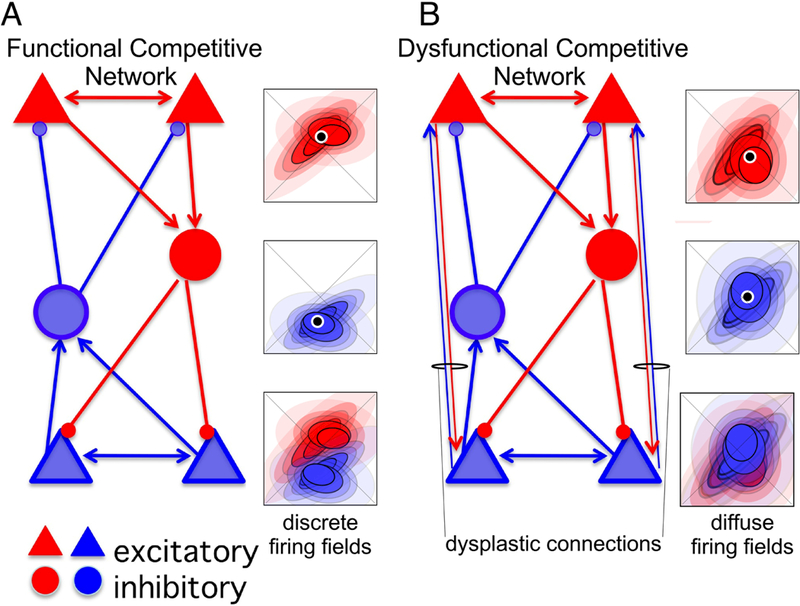
In these schemes, each cell type that is depicted, principal cell (PC) and inhibitory interneuron (IN), represents a population of the cell type, not an individual neuron. Cells that are mutually excitatory, and therefore more likely to be coactive, are color coded either red or blue; the red and blue cells will tend not to be coactive. Activity amongst the red cells, by virtue of the connectivity, will tend to recruit and enhance activity amongst red cells, and suppress activity of the blue cells. A) Left - In a properly- configured competitive network, once excitation and inhibition are appropriately balanced in the network, any bias in activity in favor of the red or blue cells will cause the more active subset to increase activation and suppress the other, competing pattern of activity. Right - Despite receiving similar inputs, the hypothetical spatial discharge of the red and blue cells would tend to occupy, and therefore represent, distinct places because of the competitive neural dynamics. The firing field will tend to be discrete. B) Left - Dysplasticity, such as by increasing connectivity between the competing rarely coactive (red versus blue) neurons can corrupt the network causing dysfunction, so that the red and blue activity patterns are less distinctive. Right - Spatial representations in the corrupted network are consequently less distinctive, generating diffuse firing fields that tend to overlap.
While item and process memory may be realized by the same molecular mechanisms, they are nonetheless functionally dissociable. A subset of place cells in the rat hippocampus appear to fire at a specific location in space, indicating what might constitute an item memory for that location (see Figure 2). A different subset of hippocampal cells also discharges as place cells but at distinct locations, presumably constituting a distinct item memory. What happens if those two locations are nearby? What causes one subset of place cells to discharge and not the other? Winner-take-all competitive networks, as schematized in Figure 2, provide the most likely answer. Consistent with such competitive network models, every CA1 principal cell seems to receive sufficient excitation to discharge in every location of an environment (Olypher et al., 2002) yet only 30–40 percent of the cells discharge at all in a given environment, and most of these as place cells (Guzowski et al., 1999; Thompson and Best, 1989). Furthermore, recent work shows that any CA1 pyramidal cell has the potential to be a place cell at any location of an environment if it receives just a few seconds of location- specific excitatory current injection (Bittner et al., 2015), which is due to coincidence- dependent synaptic plasticity that operates on the time scale of seconds (Bittner et al., 2017). Consequently, to discharge action potentials selectively at a location and not at other nearby locations, a place cell must be strongly inhibited by the subset of cells that are active at the nearby location, which is the characteristic of a competitive network (Samsonovich and McNaughton, 1997). The subset of cells that is first and most excited, as well as least inhibited, defines the sub-network of cells that will discharge together; such co-firing further lowers the probability that other cells will sufficiently depolarize to discharge until the excitation-inhibition balance changes. The excitation-inhibition balance changes periodically, the result of the dynamic, predominantly inhibitory, synaptic network activity that generates oscillations in the local field potential like ~8 Hz theta and 30–100 Hz gamma (Buzsaki et al., 2012; Royer et al., 2012). Accordingly, these synaptic and consequent oscillatory dynamics must be appropriately tuned by network-wide synaptic adjustments as a prerequisite, an experience- and synaptic plasticity-dependent process memory that permits high fidelity expression of the item information that neuronal activity may encode and recollect from memory (Dvorak et al., 2018).
1.4. Neural and cognitive behavioral consequences of excitation-inhibition discoordination
Failures of neural coordination, as might occur by improper excitation-inhibition function dynamics in hippocampus, can disrupt the information processing capabilities of the network potentially without affecting the individual place cell firing fields, perhaps analogous to disrupting process but not item memory. For some this may appear unintuitive, but it is observed, for example, in models of intellectual disability and autism (Talbot et al., 2018), and under phencyclidine (PCP) intoxication, and this dissociation is proposed to account for cognitive dysfunction in psychosis and other mental dysfunction (Phillips and Silverstein, 2003; Uhlhaas and Singer, 2012). The firing fields of individual place cells are undisturbed during PCP intoxication, although the temporal coordination of how the cells discharge relative to each other and gamma oscillations is dramatically abnormal, as is learned spatial behavior, even with intra-hippocampal administration of PCP (Kao et al., 2017). One particular form of discoordination selectively affects the neuron pairs that had initially been independently or negatively coactive on the 140-ms time scale of theta oscillations. Under PCP, this functionally uncoupled subset of cells becomes more coupled because they discharge together more. The pattern of normal place fields of individual place cells and temporal discoordination amongst the same cells is also observed in mice that have a null mutation of the Fmr1 gene (Dvorak et al., 2018; Talbot et al., 2018). Fmr1 codes for Fragile X mental retardation protein (FMRP), a key negative regulator of protein synthesis at synapses (Darnell et al., 2011) and the absence of FMRP causes activity-dependent dysfunction of both CA3→CA1 Schaffer collateral synaptic potentiation (Talbot et al., 2018) and synaptic depression (Huber et al., 2002; Huber et al., 2000), as well as synaptic dysfunction in other brain regions (Harlow et al., 2010; Mercaldo et al., 2009; Patel et al., 2013). In the case of Fmr1-null mice, temporally coordinated neural activity is excessively rigid compared to the activity in control mice at the levels of action potential spike trains within the CA1 network as well as the timing relationships of those spike trains to the oscillations in the local field potential that arise from population synaptic activity (Talbot et al., 2018). Despite normal place cell firing fields, rats treated with PCP fail to consistently express adaptive use of place memories during the PCP-induced neural discoordination, although normal expression of the place memories return as soon as the drug washes out and normal neural coordination returns (Kao et al., 2017). Although rodents lacking FMRP express normal place cell firing fields, and learning and memory perse, the cognitive behavioral consequences of their neural discoordination only manifest strongly when the animals must learn new information that contradicts what they had initially learned (Bakker et al., 1994; Bhattacharya et al., 2012; D’Hooge et al., 1997; Dvorak et al., 2018; Radwan et al., 2016; Till et al., 2015; Zhao et al., 2005). The deficit in FMRP-sensitive synaptic plasticity seems to predominantly disrupt process memory with little effect on item memory, a pattern that is grossly similar to the effects of the psychotomimetic PCP.
1.5. Impact of synaptic dysplasticity on neural network operations
To investigate how synaptic dysplasticity can influence neural information processing, we used computational and analytical neural network modeling with a Hopfield-like neural network that was configured to store two representations, analogous to representing room-defined locations and floor-defined locations in two cognitively defined spatial contexts of the same environment. Two such representations are required for effective behavior in the active place avoidance tasks we have used (Kao et al., 2017; Kelemen and Fenton, 2010; Kelemen and Fenton, 2013; Kelemen and Fenton, 2016; Talbot et al., 2018; van Dijk and Fenton, 2018). The modeling work found that the functional dynamics of the network are indifferent to random large increases or decreases of the functional connections that model synapses amongst the cells. This indifference is largely due to the fact that only a sparse number of functional connections are relevant and because the network maintained excitation-inhibition balance. However, similar to what was observed after PCP, there was neural network dysfunction when we preferentially potentiated the excitatory synapses amongst cell pairs with weak synapses (Olypher et al., 2006). This manipulation aberrantly coupled cells from the two distinctive functional subnetworks, mimicking what was observed after PCP (Kao et al., 2017) as well as after disinhibiting the hippocampus by inactivating the contralateral hippocampus with the voltage-gated sodium channel blocker tetrodotoxin (Olypher et al., 2006). This synapse-specific dysplasticity is schematized in the change from panel A to B in Figure 2. After this synaptic corruption, the network continued to represent either room locations or floor locations but could not properly switch back and forth between the two representations. Instead of switching, the network assumed a new state that did not resemble either of the learned room- or floor-activity set of activity patterns. Like the effects of PCP and FMRP loss, this modeling result is also consistent with the expectation of intact item memory but disrupted process memory.
Although such discoordination manifests as aberrantly organized spike timing amongst cells in both the modeling work and hippocampal recordings (Olypher et al., 2006), as discussed for the case of PCP and absence of FMRP, the resulting neural discoordination does not require that the individual discharge properties of single cells is abnormal. Nonetheless, as schematized in Figure 2B, excitation-inhibition discoordination can also lead to what appears as a reduction in location-specificity (item representations), when place cell spiking is summarized as a session-averaged firing rate map. Such maps assume steady-state statistics, which, as we have discussed and observed, is strongly violated during neural discoordination. Thus, individual place cells can be well-tuned to represent an animal’s position in space (an item memory), but participate in neural discoordination by aberrantly working together to achieve the appropriate collective network and behavioral outcomes (a process memory).
Indeed, inappropriately coordinated neural activity is thought to underlie mental illnesses (Phillips and Silverstein, 2003; Uhlhaas and Singer, 2012). As cited above, examples of neural discoordination are seen in rodent models used to study schizophrenia, intellectual disability, and autism, as well as diverse models of mental illness (see review Fenton, 2015). We will next discuss a long-standing hypothesis that the coordinated activity of cells is established and supported through synaptic plasticity mechanisms.
1.6. Synaptic Plasticity and Memory Hypothesis
The dominant synaptic plasticity and memory hypothesis asserts that the strength of the interactions between neurons underlies the acquisition and storage of memory (Martin et al., 2000; Takeuchi et al., 2013). By changing the synaptic weights between neurons of a network, synaptic plasticity is thought to give rise to specific patterns of neural activity that represent the memory and permit memory computations such as pattern completion (McNaughton and Morris, 1987). These synaptic changes are now recognized to include both potentiation (strengthening) and depotentiation (weakening) of the synapses between both the excitatory and inhibitory neurons (Dietz and Manahan-Vaughan, 2017; Perea et al., 2016; Ruediger et al., 2011).
Despite substantial progress, it has been difficult to gather crucial direct evidence to support the synaptic plasticity and memory hypothesis even with respect to item memory (Takeuchi et al., 2013). This is partly because the properties of the synaptic changes make validation elusive. These properties include 1) sparseness (Whitlock et al., 2006), 2) negation of detecting overall changes by increased and decreased potentiation (Bear and Malenka, 1994; Malenka and Bear, 2004), 3) the time required to form a memory, which usually occurs after repeated learning episodes, and 4) synaptic scaling to maintain homeostasis of excitability within a single neuron (Turrigiano and Nelson, 2000). Consequently, there have been relatively few demonstrations of learning- induced in vivo long-term potentiation (LTP). In the CA1 region of the dorsal hippocampus, examples include (Gruart et al., 2006; Madronal et al., 2010; Whitlock et al., 2006). After a single exposure to an inhibitory avoidance paradigm, Whitlock et al. observed experience-induced changes that included increased levels of Serine 831- phosphorylated a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPAR), the site of phosphorylation known to be specifically modified after LTP (Lee et al., 2000) as well as hippocampus CA1 field EPSPs (fEPSPs) in response to CA3 Shaffer collateral stimulation. Importantly, the fEPSP increase was sparsely distributed, detectable at −25% of topographically dispersed electrode sites, and while the potentiation could last at least 4h, the synaptic potentiation did not last as long as the memory.
Conceptually similar experiments were recently performed in mice to determine if hippocampal synaptic network function is altered by learning a spatial two-frame active place avoidance task. To accomplish the two-frame active place avoidance task, a rat or mouse on a slowly rotating arena must learn to ignore the rotating cues and use cues within the room to avoid entering a 60° shock zone that is stationary within the room (Fenton and Bures, 2003; see Video S1). Mice were trained over four days and 1 day or 30 days later memory retention was evaluated, the mice were sacrificed and acute hippocampus slices were prepared to investigate ex vivo synaptic physiology (Pavlowsky et al., 2017). A day after training, transmission at the Schaffer collateral CA3→CA1 synapse was 1) strengthened, 2) harder to potentiate, 3) easier to depress, and 4) more likely to elicit action potentials but these changes were not detected at the entorhinal layer 3 neocortical EC3→CA1 synapse. These CA3→CA1 changes persisted for at least 30 days but only in slices from mice that expressed the memory after 30 days. None of these changes in synaptic network function were detected if gamma- aminobutyric acid type A receptor (GABAA)-mediated inhibition was blocked by picrotoxin in the bath. These learning-induced changes were accompanied by increased hippocampal expression of protein kinase M zeta (ΡΚΜζ) (Hsieh et al., 2017), an atypical protein kinase C (PKC) isoform, the activity of which persistently maintains increased numbers of postsynaptic GluA2-containing AMPA receptors (Ling et al., 2006; Migues et al., 2010; Yao et al., 2008; Yu et al., 2017). ΡΚΜζ is both necessary and sufficient for the persistence of N-methyl-D-aspartate receptor (NMDAR)-induced and GluA2-mediated LTP (Ling et al., 2002) and is necessary for the maintenance of place cell firing fields (Barry et al., 2012) and the active place avoidance memory (Pastalkova et al., 2006; Tsokas et al., 2016; Wang et al., 2016), as well as other, but not all forms of long-term memory (review Sacktor, 2012; Serrano et al., 2008; Tsokas et al., 2016). While these are just a subset of the compelling data that synaptic plasticity is crucial for memory storage, crucial definitive evidence is still lacking.
Recent studies have also challenged the idea that synaptic plasticity is crucial for memory storage. These investigations used context-conditioned threat avoidance learning to genetically tag and express excitatory channelrhodopsin, preferentially in learning-activated hippocampal neurons (Garner et al., 2012; Liu etal., 2012; Ramirez et al., 2013). Subsequent optogenetic theta- or gamma-frequency stimulation of the tagged neurons is sufficient to elicit memory expression assessed as the conditioned behavior in neutral environments. Remarkably, the optogenetic stimulation remains effective in eliciting the conditioned behavior after synaptic plasticity is blocked during conditioning to produce amnesia (Ryan et al., 2015; Ryan and Tonegawa, 2016). Such optogenetic stimulation is also effective for retrieval of context-conditioned threat avoidance memory in Alzheimer’s Disease model mice that express impaired synaptic morphology and plasticity and are otherwise amnestic (Roy et al., 2016). Because effective optogenetic stimulation depends on mimicking endogenous oscillation frequencies but not synaptic plasticity, these data make a counter case that synaptic plasticity is not necessary for item memory retrieval whereas synaptic plasticity may be crucial for the appropriate processing of information in memory that is mediated by excitation-inhibition coordination amongst a sufficient subset of cells in a neural circuit.
1.7. Persistent experience-related metabolic changes in the brain
We also examined whether cognitive training has a long-term impact on brain function that could be assessed by evaluating steady state metabolic function at histological resolution. This is accomplished by imaging cytochrome oxidase activity in postmortem tissue. Critical for ATP production, cytochrome oxidase has been considered a metabolic indicator of neuronal firing (Wong-Riley, 1989). Long-Evans rats underwent cognitive training in the two-frame active place avoidance task (Bures et al., 1997; Cimadevilla et al., 2000b) or were yoked to a cognitively-trained rat such that the yoked rats experienced the identical environmental conditions, but without learning a particular location of shock (Fig. 3A). We examined cytochrome oxidase activity one week after the end of the training and yoked experiences (Fig. 3A). Behaviorally, the trained group reduced the number of entries into the shock zone, while the yoked group did not alter their entries into that same region of the space (Fig. 3B). We found no group differences in cytochrome oxidase activity in any region examined (Table 1). In contrast, there were clear differences when we investigated inter-regional fluctuations in function by measuring how much cytochrome oxidase activity levels covaried between pairs of brain regions (Fig. 3C,D).
Figure 3. Different functional networks result from two experiences of one environment in normal rats.
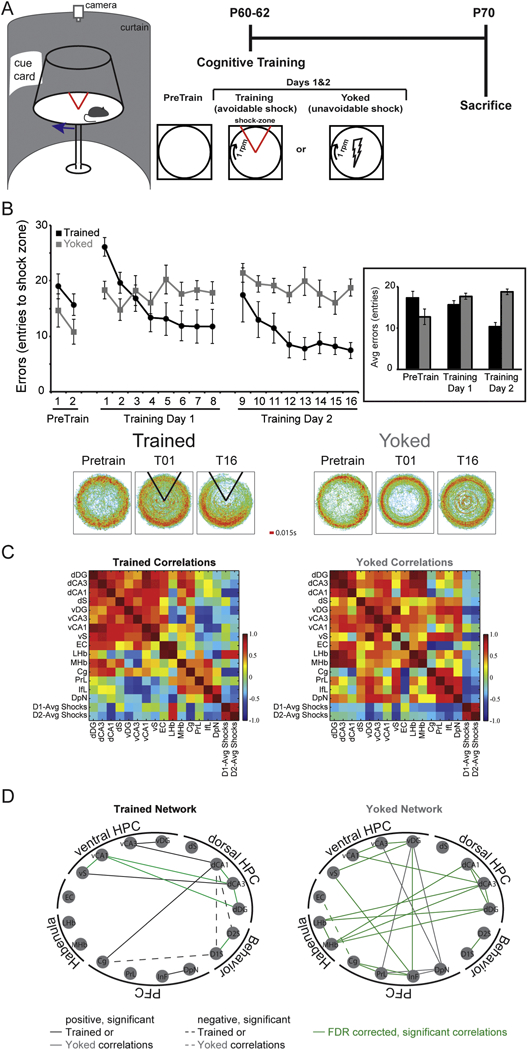
A) In the two-frame active place avoidance paradigm, a rat is placed on a rotating arena (1 rpm) and required to use the room cues to avoid a shock zone that is stationary within the room. Because the arena rotates, the cues on the arena are irrelevant for the identification of the shock zone. During training, the rats quickly learn to avoid entering the shock zone where they would receive a mild foot shock (~0.3 mA). Rats were pre-exposed (Pretrain) to the arena and room conditions on a stationary arena. Twenty-four hours later, they were trained on the rotating arena in 10-min trials separated by a 10-min inter-trial interval. Eight trials were given each day for two days. To control for the experience of being shocked, a yoked group received the same exposure to the room and arena conditions, but inescapable shocks were delivered to the yoked rats in the same temporal sequence as the trained rats. One week after training concluded, the rats were euthanized by anesthesia and decapitation. Forty pm brain sections were then histologically processed as in (O’Reilly et al., 2016) with cytochrome c and catylase to create a diaminobenzidinetetrahydrochloride (DAB) stain. Optical density readings were then taken to examine cytochrome oxidase enzyme activity. B) Trained rats learn to reduce the number of entrances they make into the shock zone. Because the shocks are inescapable and occur at random locations for the yoked rats, the number of entrances into the same location within the room is not changed over time. Average dwell maps of the time spent in each location reveal that the time spent in the shock zone is reduced in trained animals but not in yoked controls. C) Color-coded correlation matrix of the interregional cytochrome oxidase activity correlations. The hot colors represent positive correlations and cool colors negative correlations. The correlations are organized by functional brain regions and include the number of shocks received on days 1 and 2. D) Significant correlations (p < 0.05) were used to generate graph theoretical networks. Each node represents a brain region and the lines represent significant correlations. Solid lines are positively correlated and dashed lines are negatively correlated. Trained animals have brain regions that are significantly correlated with the number of shocks they received, indicative of a relationship between place learning and brain activity, while yoked animals do not have significant correlations between any brain region and the number of shocks they received. Green lines represent correlations that remain significant after false discovery rate correction (FDR corrected) with an acceptable false discovery rate of 25%. HPC = hippocampus, PFC = prefrontal cortex, dDG = dorsal dentate gyrus, dCA3 = dorsal Cornu Ammonis region 3, dCA1 = dorsal Cornu Ammonis region 1, dS = dorsal subiculum, vDG = ventral dentate gyrus, vCA3 = ventral Cornu Ammonis region 3, vCA1 = ventral Cornu Ammonis region 1, vS = ventral subiculum, EC = entorhinal cortex, LHb = lateral habenula, MHb = medial habenula, Cg = cingulate cortex, PrL = prelimbic cortex, IfL = infralimbic cortex, DpN = dorsal peduncular nucleus, D1-Avg Shocks = average number of shocks received over the eight trials on day 1 of training, D2-Avg Shocks = average number of shocks received over the eight trials on day 2 of training.
Table 1:
Relative CO activity in brain regions after experience in the two-frame spatial active place avoidance task.
| Relative CO activity/pm tissue (×10−1) | ||||||
|---|---|---|---|---|---|---|
| Trained | Yoked | |||||
| Brain Region | Ave ± SEM | N | Ave ± SEM | N | p-value | t-stat |
| Dorsal Hippocampus (dHPC) | ||||||
| dDG | 1.31 ±0.13 | 9 | 1.11 ± 0.15 | 9 | 0.34 | 0.99 |
| dCA3 | 0.73 ± 0.08 | 9 | 0.78 ± 0.08 | 9 | 0.71 | 0.37 |
| dCA1 | 0.79 ±0.11 | 9 | 0.60 ± 0.08 | 9 | 0.17 | 1.45 |
| dSubiculum (dS) | 0.92 ±0.11 | 7 | 1.02 ± 0.11 | 8 | 0.53 | 0.64 |
| Ventral Hippocampus (vHPC) | ||||||
| vDG | 1.08 ±0.07 | 7 | 1.15 ±0.07 | 7 | 0.48 | 0.72 |
| vCA3 | 1.04 ±0.08 | 7 | 1.01 ± 0.06 | 8 | 0.77 | 0.30 |
| vCA1 | 1.08 ±0.08 | 7 | 1.02 ± 0.09 | 8 | 0.66 | 0.45 |
| vSubiculum (vS) | 1.26 ±0.09 | 7 | 1.12 ± 0.12 | 7 | 0.36 | 0.94 |
| Entorhinal Cortex (EC) | 0.86 ± 0.09 | 6 | 0.97 ±0.12 | 6 | 0.48 | 0.74 |
| Habenular Complex | ||||||
| Lateral Habenula (LHb) | 1.09 ±0.06 | 6 | 1.04 ±0.10 | 9 | 0.68 | 0.42 |
| Medial Habenula (MHb) | 1.28 ±0.21 | 5 | 0.95 ±0.16 | 9 | 0.25 | 1.21 |
| Prefrontal Cortex (PFC) | ||||||
| Cingulate Cortex (Cg) | 1.29 ±0.08 | 8 | 1.22 ± 0.09 | 8 | 0.60 | 0.54 |
| Prelimbic Cortex (PrL) | 1.26 ±0.05 | 8 | 1.43 ±0.08 | 8 | 0.10 | 1.74 |
| Infralimbic Cortex (InF) | 1.09 ±0.05 | 8 | 1.25 ±0.11 | 8 | 0.21 | 1.31 |
| Dorsal Peduncular Nucleus (DpN) | 1.21 ±0.08 | 8 | 1.23 ±0.11 | 8 | 0.86 | 0.18 |
To visualize the data, the brain regions assessed were organized into functional groupings categorized according anatomy. We also calculated correlations between the metabolic activity in each brain region and the average number of shocks received on each training day to begin to assess the relationship between neuronal metabolic activity and experience (Fig. 3C,D). In the trained group, the activity in dorsal CA1 and the activity in the cingulate cortex were both significantly negatively correlated with the number of shocks that were received on the first day of training. In contrast, in the yoked group of rats that received the identical number of shocks as the counterparts from the trained group, there were no significant correlations between cytochrome oxidase activity in any brain region measured and the number of unavoidable shocks the animals received (Fig. 3C,D). The fact that there was a relationship between metabolic activity and shocks received in the trained but not the yoked group suggests that the shocks themselves did not drive the correlation, but rather that these relationships were driven by some internal variable related to the shocks, such as memory for the shock experience and/or location. It is principally possible that differences in interregional metabolic relationships between the trained and yoked groups are due to how these animals interpret the experience such that behaviorally naive and trained brains are actually similar to one another but different from yoked, or that yoked and naive are similar but different from trained. Regardless, observing significant metabolic-avoidance correlations in the trained, but not the yoked, group implies that cognitive variables can alter interregional metabolic relationships. In addition to the training-induced persistent changes of synaptic population function (Park et al., 2015; Pavlowsky et al., 2017; Talbot et al., 2018), it is clear from these metabolic coupling data that experience can impact how the brain operates, potentially providing evidence that cognitive training can cause neuroplastic changes in hippocampus circuit function. We speculate that these changes may underlie process memory and the neurobiological changes that are hypothesized to result from CBT.
1.8. Early cognitive experience can change adult behavioral outcomes in rodents
Can CBT cause persistent changes in neural circuit function? There is evidence that training experience with potential analogies to CBT is effective in overcoming dysfunction in rodents with cognitive impairments related to mental illness (Bi et al., 2006; Voss et al., 2016; Zhou et al., 2015; Zhu et al., 2014). In particular, we used a model of atypical neurodevelopment, in which postnatal day seven (PD7) Long-Evans rats underwent ventral hippocampal lesions, to evaluate whether early cognitive training could have a beneficial impact on later cognitive abilities (Lee et al., 2012). Specifically, neonatal ventral hippocampal lesion (NVHL) rats display cognitive deficits in the above mentioned (Fig. 3A) spatial two-frame active place avoidance task during adulthood, making more entrances into the stationary 60° shock zone that is identifiable by the room cues (Lee et al., 2012). The impairment of NVHL rats is developmentally- expressed because when trained to perform the two-frame active place avoidance task during adolescence, NVHL rats are indistinguishable from control rats in their ability to learn and remember the task (Lee et al., 2012). The impairment is also developmentally- sensitive to cognitive training because when these adolescent-trained (ATrain) NVHL rats are re-exposed to the same task during adulthood, their performance remains identical to that of ATrain-control rats. These findings, reported in Lee et al., demonstrate that cognitive training can lead to a lasting improvement in subsequent cognitive performance, though it is not immediately clear whether or not this lasting improvement is a straightforward consequence of the specific place memory that was conditioned.
As described above, there is the possibility that early cognitive training merely instilled an ability that was specific to the training, for instance, the ATrain-NVHL rats have a memory for the location of the shock zone that persists until adulthood. However, this explanation is unlikely because when the shock zone location was moved 180°, instead of being impaired by their earlier, apparently normal memory, ATrain-NVHL rats performed as well as ATrain-control rats, whereas these rats would have been impaired if they did not have the adolescent training (Lee et al., 2012). In fact, NVHL rats that were exposed to the training conditions without shock during adolescence (adolescent exposed, AExpose) despite this behavioral enrichment, show the same cognitive deficits as NVHL rats that had no adolescent experience. These observations suggest the benefit of early training is not merely the persistence of memory for the specific task that had been initially learned.
As with the intention of CBT, perhaps the early training was remediating and improved a general cognitive ability, the benefits of which would generalize to other similar cognitive situations. An initial indication that this may be the case is the observation that the ATrain-NVHL rats can flexibly adapt to a relocated shock zone location. More compelling is the finding that ATrain-NVHL rats also perform as well as ATrain-controls on a T-maze reversal learning task, in which AExpose-NVHL rats make more errors than AExpose-control rats only when they have to change the arm to which they previously learned to respond (Lee et al., 2012). These additional observations indicate that information processing involved in related cognitive abilities is facilitated by the early training, sufficient to overcome the deficits that emerge after protracted development. These improvements in cognitive function are difficult to understand as the result of item memories, since cognitive improvement was generalized beyond the relevance of the locations of shock. The improvements are, however, straightforward to understand as a consequence of persistent process memory.
We then wondered if the remediation effects of early cognitive training, perhaps due to process memory formation, generalize to other animal models used to study abnormal neurodevelopment associated with mental illness. We tested a distinct model of neurodevelopmental insult in which cell proliferation is disrupted at gestational day 17 (GD17), a time at which prefrontal, temporal, and paralimbic systems are particularly susceptible to neurodevelopmental insult (Lodge and Grace, 2009). To accomplish this neurodevelopmental insult, a pregnant Long-Evans dam is injected intraperitoneally with the mitotoxin methylazoxymethanol acetate (MAM), or in control rats, an equivalent amount of saline (Moore et al., 2006). The resulting offspring are tested for cognitive abilities. Similar to the experimental design used with NVHL rats, we trained adolescent MAM and control rats to perform the two-frame active place avoidance task (Fig. 4A). In contrast to adolescent NVHL rats, adolescent MAM rat behavior is impaired compared to control rats (Fig. 4B-E), resembling the adult MAM rat impairment (compare Fig. 4B-E to O’Reilly et al., 2016). As shown in Figure 5, there was an impact of early cognitive training on the rats when tested as adults. First, after just being exposed to the rotating environment during adolescence, the MAM and control-treated groups were indistinguishable when tested as adults (Fig. 5 left); recall naive adult MAM rats are impaired (O’Reilly et al., 2016). Second, cognitive training in adolescence had a lasting effect on control rats because they performed better as adults from the very first trial, showing memory persistence, whereas the training in adolescence did not have a similar effect on the adult MAM rats (Fig. 5 right). The early training conferred no apparent benefit for cognitive flexibility in the MAM or control groups when their responses to 180° relocation of the shock zone was examined. This is consistent with item memory for the shock location being established in the control rats by the adolescent training with no distinct effect on process memory for performing other tasks.
Figure 4: Adolescent GD17-MAM rats are hyperactive, and have memory impairments with normal cognitive control.
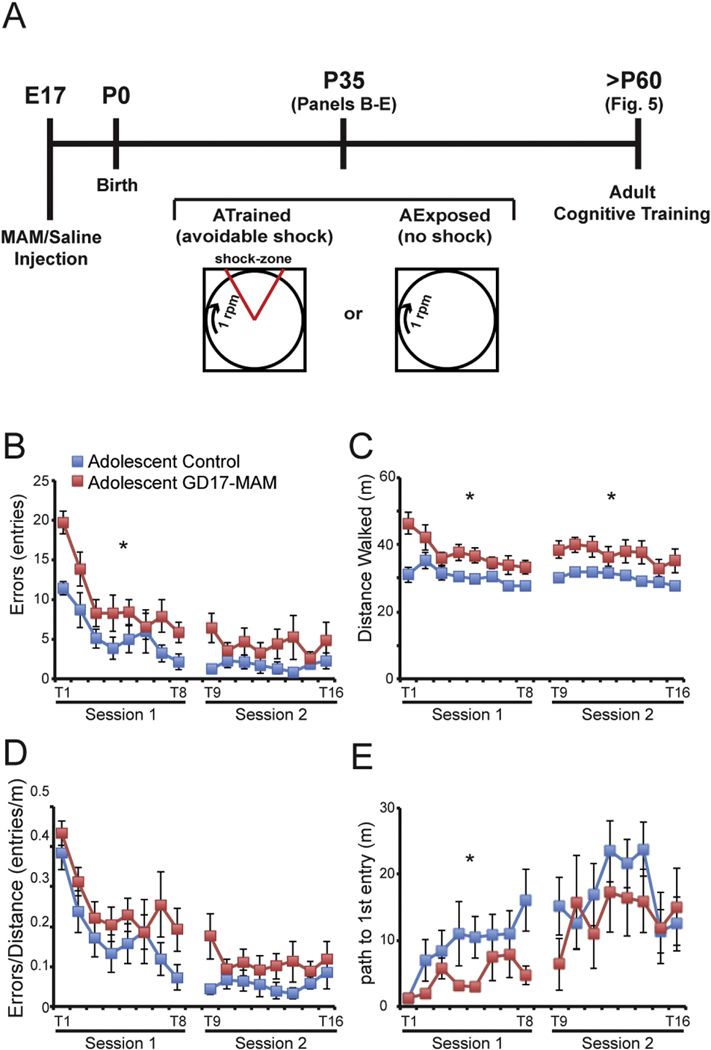
A) MAM or an equal volume of saline is administered (i.p.) to a pregnant dam at gestational day 17 (GD17). The offspring are born on P0 and trained or exposed to the learning conditions during adolescence, starting at P35. Briefly, the rats are handled for five days prior to the start of testing. On P35, the rats are allowed to explore the arena for two 10-min trials. The following two days (eight 10-min trials/day) the rats are trained on the rotating arena to avoid the 60° shock zone that is stationary within the room. All trials have at least 10 min between them. At P60–70, the rats are tested in the same paradigm to assess cognitive behavior (see Figure 5 for these results). B) GD17-MAM rats make more errors (entries into the shock zone) on the first training day (Session 1: Treatment: F1,17 = 5.74, p = 0.03; Trial: F7,11 = 25.47, p < 0.0001; Interaction: F7,11 = 3.98, p = 0.02), but perform asymptotically, similarly to control rats on the second day of training (Session 2: Treatment: F1,17 = 2.62, p = 0.12; Trial: F7,11 = 1.12, p = 0.42; Interaction: F7,11 = 0.99, p = 0.48). C) GD17-MAM rats are hyperactive during all trials on both days (Session 1: Treatment: F1,17 = 9.90, p = 0.01; Trial: F7,11 = 2.26, p = 0.11; Interaction: F7,11 = 1.33, p = 0.32. Session 2: Treatment: F1,17 = 9.94, p = 0.01; Trial: F7,11 = 1.30, p = 0.33; Interaction: F7,11 = 0.26, p = 0.96). D) As with adult GD17-MAM rats, the hyperactivity accounts for the increased number of errors in adolescent GD17-MAM rats. When the number of errors is normalized to locomotor activity, learning performance is not different between GD17-MAM and control rats (Session 1: Treatment: F1,17 = 2.25, p = 0.15; Trial: F7,11 = 10.25, p < 0.001; Interaction: F7,11 = 0.3358, p = 0.92. Session 2: Treatment: F1,17 = 2.31, p = 0.15; Trial: F7,11 = 0.87, p = 0.56; Interaction: F7,11 = 0.62, p = 0.73). E) Within session memory was measured as the ability to increase the path to first enter the shock zone from trial-to-trial with the day’s session. Adolescent GD17-MAM rats have impaired within session memory acquisition during the first session (Session 1: Treatment: F1,17 = 4.67, p = 0.05; Trial: F7,11 = 4.26, p = 0.02; Interaction: F7,11 = 1.14, p = 0.41), but are not different from controls during the second session (Session 2: Treatment: F7,11 = 0.64, p = 0.43; Trial: F7,11 = 2.41, p = 0.09; Interaction: F7,11 = 0.82, p = 0.59). Across session memory was measured as the ability to increase the path to the first shock zone entrance across sessions 1 and 2 (Trial 1 and Trial 9, respectively). GD17-MAM rats have normal long-term memory (Treatment: F7,11 = 2.20, p = 0.16; Trial: F7,11 = 10.40, p = 0.01; Interaction: F7,11 = 2.23, p = 0.15). Values are mean ± SEM. Control, n = 9. GD17-MAM, n = 10. *p < 0.05
Figure 5. The impact of early cognitive and control experience on adult cognitive ability in MAM rats.
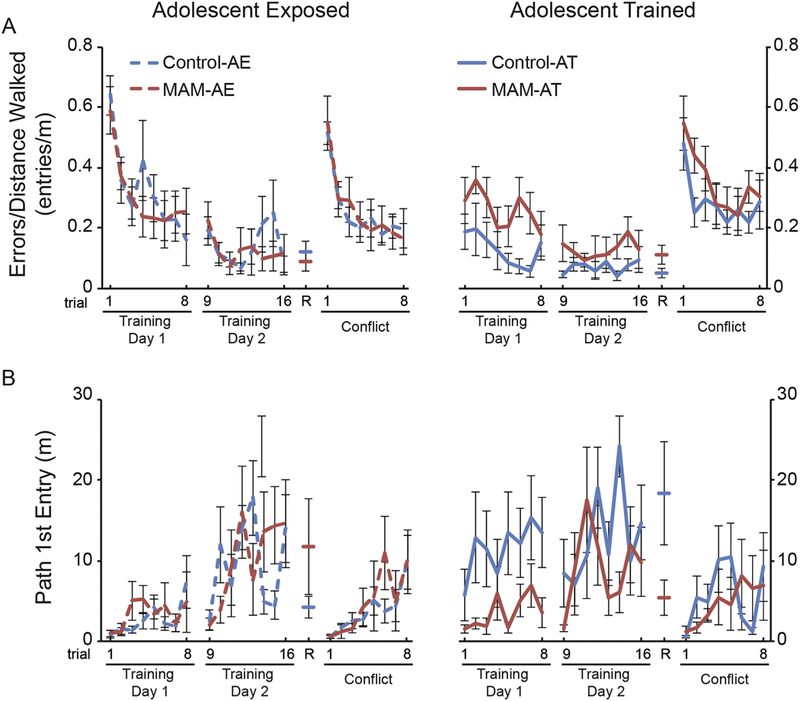
The two-frame active place avoidance task was used to assess cognitive ability and memory in adult MAM rats that were either trained to perform the task during adolescence (AT) or exposed to the environmental conditions without shock (AE). At P60–70, the rats are trained for two days (eight 10-min trials/day) to avoid the 60° shock zone. On day three of training, the rats are given a 10-min retention trial with the shock on, followed by eight trials in which the shock zone is relocated 180°. We previously found that MAM rats are hyperactive and therefore our measures of spatial cognitive behavior take the distance walked by the rats into account. We estimated place avoidance as the number of entries made into the shock zone as a function of the distance walked on the arena (errors/distance). We estimated memory as the distance (path length) walked by the rats before the first entry into the shock zone. A) There were no group differences in place avoidance on Training Day 1, however there was an interaction between adolescent experience and trial (repeated measures MANOVA comparing the two treatments, and the two types of adolescent experience, across trials: Treatment: F1,29 = 1.46, p = 0.23; Adolescent experience: F1,29 = 6.74, p = 0.01; Trial: F7,23 = 3.60, p = 0.01; Treatment x Adolescent experience: F1,29 = 2.56, p = 0.12; Treatment x Trial: F7,23 = 0.98, p = 0.47; Trial x Adolescent experience: F7,23 = 2.66, p = 0.04; Treatment x Adolescent experience x Trial: F7,23 = 1.52, p = 0.21). Because of this interaction, comparisons were made within each treatment condition (AT-control vs. AE- control and AT-MAM vs. AE-MAM). This revealed that AT-control rats performed significantly better than AE-control rats (Adolescent experience: F1,14 = 8.79, p = 0.01; Trial: F7,8 = 2.14, p = 0.15; Adolescent experience x Trial: F7,8 = 5.77, p = 0.01) but that AT-MAM and AE-MAM rats were not different (Adolescent experience: F1,15 = 0.50, p = 0.49; Trial: F7,9 = 2.84, p = 0.07; Adolescent experience x Trial: F7,9 = 0.72, p = 0.66). There was no group difference on Training Day 2, but there was a significant Treatment x Adolescent experience x Trial interaction (Treatment: F1,29 = 0.34, p = 0.57; Adolescent experience: F1,29 = 0.84, p = 0.37; Trial: F7,23 = 3.46, p = 0.01; Treatment x Adolescent experience: F1,29 = 1.28, p = 0.27; Treatment x Trial: F7,23 = 0.67, p = 0.69; Adolescent experience x T rial: F7,23 = 2.38, p = 0.06; Treatment x Adolescent experience x Trial: F7,23 = 3.89, p = 0.01). Further analysis of this interaction revealed no significant effects between AT-control and AT-MAM rats (Treatment: F1,15 = 2.58, p = 0.13; Trial: F7,9 = 0.55, p = 0.78; Treatment x Trial: F7,9 = 1.90, p = 0.18). Although it appears that there is an effect of trial between AE-control and AE-MAM rats on Training Day 2, significance was not reached (Treatment: F1,14 = 0.10, p = 0.76; Trial: F7,8 = 3.27, p = 0.06; Treatment x Trial: F7,8 = 3.19, p = 0.06). There were no group differences in the retention trial (R) (Treatment: F1,1 = 0.58, p = 0.45; Adolescent experience: F1,1 = 0.34, p = 0.56; Treatment x Adolescent experience: F1,1 = 3.77, p = 0.06) or during the conflict session (Treatment: F1,29 = 0.57, p = 0.45; Adolescent experience: F1,29 = 1.51, p = 0.23; Trial: F7,23 = 7.83, p < 0.001; Treatment x Adolescent experience: F1,29 = 0.37, p = 0.55; Treatment x Trial: F7,23 = 0.83, p = 0.66; Adolescent experience x Trial: F7,23 = 0.71, p = 0.66; Treatment x Adolescent experience x Trial: F7,23 = 1.19, p = 0.34). B) Memory was assessed as the distance walked prior to the first entry into the shock zone. Control rats displayed a better memory, influenced by the adolescent experience; their distance walked before entering the shock zone for the first time was higher on Training day 1 (Treatment: F1,29= 4.71, p = 0.04; Adolescent experience: F1,29 = 7.06, p = 0.01; Trial: F7,23 = 2.27, p = 0.07; Treatment x Adolescent experience: F1,29 = 6.49, p = 0.02; Treatment x Trial: F7,23 = 0.77, p = 0.62; Adolescent experience x Trial: F7,23 = 0.69, p = 0.68; Treatment x Adolescent experience x Trial: F7,23 = 0.53, p = 0.80). The significant interaction appears to be driven by the AT-control rats (AT-control vs. AT-MAM: Treatment: F1,15 = 6.56, p = 0.02; Trial: F7,9 = 2.20, p = 0.13; Treatment x Trial: F7,9 = 0.91, p = 0.54) whereas the AE-control and AE-MAM groups were indistinguishable: Treatment: F1,14 = 0.34, p = 0.57; Trial: F7,8 = 2.36, p = 0.13; Treatment x Trial: F7,8 = 0.97, p = 0.51). The AT-control was better than the AE-control group (Adolescent Experience: F1,14 = 7.27, p = 0.02; Trial: F7,8 = 0.72, p = 0.66; Adolescent Experience x Trial: F7,8 = 0.63, p = 0.72) but the AT-MAM and AE-MAM groups were not different (Adolescent Experience: F1,15 = 0.03, p = 0.88; Trial: F7,9 = 2.02, p = 0.16; Adolescent Experience x Trial: F7,9 = 1.62, p = 0.25). Performance was asymptotic by Day 2; there were no group differences in the path to first enter the shock zone on Training Day 2, during the retention test, or during the conflict session (Training Day 2: Treatment: F1,29 = 0.54, p = 0.47; Adolescent experience: F1,29 = 0.29, p = 0.59; Treatment x Adolescent experience: F1,29 = 0.97, p = 0.33;_Trial: F7,23 = 6.23, p = 0.0004;_Trial x Treatment: F7,23 = 1.43, p = 0.24;_Trial x Adolescent experience: F7,23 = 1.07, p = 0.41;_Trial x T reatment x Adolescent experience: F7,23 = 2.34, p = 0.06. Retention (R): Treatment: F1,29 = 0.38, p = 0.54; Adolescent experience: F1,29 = 0.77, p = 0.39; Treatment x Adolescent experience: F1,29 = 5.35, p = 0.03. Conflict: Treatment: F1,29 = 0.00, p = 0.95; Adolescent experience: F1,29 = 0.27, p = 0.61; Treatment x Adolescent experience: F1,29 = 0.38, p = 0.54; Trial: F7,23 = 3.36, p = 0.01; Trial x Treatment: F7,23 = 1.51, p = 0.21; Trial x Adolescent experience: F7,23 = 0.73, p = 0.65; Trial x Treatment x Adolescent experience: F7,23 = 1.03, p = 0.44). A post hoc Tukey’s Honest Significant Difference test on the retention test did not reveal any significant adolescent experience differences among the groups. Data are presented as mean ± SEM. AT-control n = 8; AT-MAM n = 9; AE-control n = 8; AE-MAM n = 8. Significance was set to p < 0.05.
These behavioral data suggest that MAM rats have abnormal neuroplasticity mechanisms that impair learning and the ability to retain a place avoidance item memory across weeks. Furthermore, the abnormality appears to be already present during adolescence, preventing the MAM rats with a gestational neurodevelopmental insult from benefitting from early cognitive training, in contrast to control rats and NVHL rats that received a postnatal neurodevelopmental insult (Lee et al., 2012).
1.9. Synaptic function abnormalities after MAM neurodevelopmental insult
Before investigating potentially abnormal synaptic plasticity after MAM neurodevelopmental insult, we first examined neuronal morphology and synaptic function in the dorsal hippocampus because of the role of dorsal hippocampus in learning and memory, and the reliance of the two-frame active place avoidance task on intact dorsal-hippocampus function (Cimadevilla et al., 2000a; Cimadevilla et al., 2001) and persistent synaptic plasticity (Pastalkova et al., 2006; Serrano et al., 2008). We discovered altered neuronal morphology, specifically at stratum radiatum, the intrahippocampal CA3→CA1 subcircuit where cell types were undergoing neurogenesis during the presence of the mitotoxin (O’Reilly et al., 2018). We then compared baseline in vivo synaptic network function in the dorsal hippocampus of MAM and control treated adult rats by studying the responses to stimulation of the main cortical input from the entorhinal cortex, or CA3, the intrahippocampal input (O’Reilly et al., 2018).
In both the dentate gyrus and CA1, the main input and output of the hippocampus, respectively, we observed abnormal dendrite-to-soma transfer of electrical responses to stimulation. In the dentate gyrus, the synaptic population response and population spike response to entorhinal input was unchanged, whereas the synaptic population response in the granule cell layer was enhanced. Essentially the MAM neocortical-dentate gyrus pathway is blunted, requiring a greater synaptic response to generate a particular population action potential output to the next stage of the circuit, CA3. Responses are also dampened at the output of the circuit, CA1, although the exact pattern of population synaptic and action potential response changes differ from what is observed in the dentate response. These data, together with an observed increase in dendritic spines, indicate an abnormal input-output relationship of dentate gyrus signaling in MAM rats. In CA1, the dampened responses are partly a consequence of reduced dendritic branching. Together, both at the neocortical input and the primary output, the MAM hippocampus expresses abnormal input-output relationships, and, overall, a potentially reduced output of dorsal hippocampus (O’Reilly et al., 2018). These abnormalities in synaptic structure and function may be related to the mild memory deficits observed in both adult and adolescent MAM rats, but a causal relationship has not been established and it requires further investigation to determine whether the abnormalities affect information processing and process memory independent of effects on item memory acquisition and storage.
1.10. Comparison of the NVHL and MAM rat models - relevance of the neurodevelopmental timeline
Factors that may contribute to the success of early cognitive training in NVHL rats and not in MAM rats likely include the specificity and timing of the neurodevelopmental insult and the particular neurobiological changes caused by the early cognitive experience. Success of early cognitive training in NVHL rats did not depend on the size of the hippocampal lesion (Lee et al., 2012), but the lesion must be targeted to the ventral hippocampus to produce the particular NVHL pattern of behavioral impairments (Lipska et al., 2002; Swerdlow et al., 2001). Because the hippocampus is targeted by both NVHL and MAM neurodevelopmental insults, and because many of the studies described above focus on CA1 place fields, we provide a very brief developmental overview of the hippocampus, in particular CA1. Information arriving at CA1 from multiple sources must be coordinated to generate coherent and organized CA1 output, in particular because inputs to CA1 are structurally organized along the somatodendritic axis, with CA3→CA1 projections terminating in CA1 stratum radiatum and EC→CAI projections terminating in stratum tacunosum moleculare of CA1 (Witter, 2010). The timing and coordination of these inputs and of CA1 output is likely performed by excitatory-inhibitory (E/I) coordination, and the distinct inhibitory interneuron contributors also topographically localize within the stratum radiatum and stratum lacunosum moleculare dendritic compartments of CA1 (Danglot et al., 2006). The generation of interneurons in hippocampus occurs embryonically, and interneurons arrive in the primitive hippocampus before pyramidal neurons. In the CA1 region, these interneurons establish temporary connections with CA3 in the first postnatal week in rodents (Super et al., 1998). Between the first and second postnatal week, the CA3 afferent connections transition to CA1 pyramidal cells and many GABAergic cells translocate from the stratum radiatum to other laminae (Danglot et al., 2006).
Additionally, dendritic arborization of interneurons continues until at least P20 (Lang and Frotscher, 1990). Dendritic arborization and synaptogenesis of GABAergic synapses in CA1 maximally increase in density between P7–20 in an activity dependent manner (Danglot et al., 2006). The hippocampus is heavily intra-connected along the dorsal- ventral axis (Witter, 2010). Thus, it is conceivable that denervation of the dorsal hippocampus by NVHL alters dorsal hippocampus activity, impacting the development of E/I coordination, an abnormality that can be tuned and modified by memory training (Pavlowsky et al., 2017; Ruediger et al., 2011), even within minutes of an alteration (Olypher et al., 2006). Studies need to be conducted to closely examine this possibility, but synchrony between the two dorsal hippocampal after NVHL is disrupted in adulthood and restored after adolescent cognitive training, which may further motivate such studies (Lee et al., 2012).
In contrast to the location specificity of NVHL neurodevelopmental insult, the MAM toxin exposure is not restricted to the hippocampus. MAM disrupts cell proliferation and, in the gestational day 17 model, is administered when the hippocampus, among other regions, is undergoing peak neurogenesis, especially in the CA1 and CA3 subfields (Bayer, 1980). We used the metabolic marker of neuronal activity, cytochrome oxidase, to examine the impact of the MAM neurodevelopmental insult on various brain regions thought to be disrupted in schizophrenia (O’Reilly et al., 2016). There were clear differences in covariance of neuronal metabolism between the ventral hippocampus and prefrontal cortex, which were increased in MAM rats, and in general, the MAM brains appeared to be hypercorrelated (see Fig. 6; O’Reilly et al., 2016). Although the brainwide impact of the NVHL insult has not been examined, it is discussed in the literature that the impact would be localized to structures directly connected to the ventral hippocampus (Lipska and Weinberger, 2002), while there is global disruption from the MAM insult, as is described by these cytochrome oxidase studies.
Figure 6. GD17-MAM rats have altered functional connectivity and networks of connectivity.
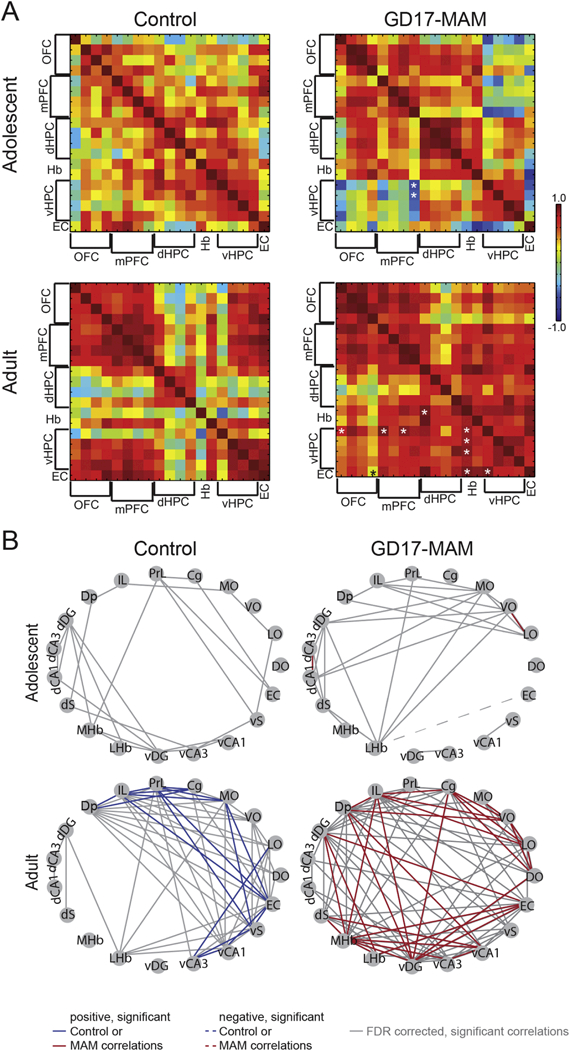
Cytochrome oxidase activity was measured from the orbitofrontal, prefrontal, and entorhinal cortices, the dorsal and ventral portions of the hippocampus, and the habenula as described for Figure 4. A) The correlation matrices are organized from top to bottom and left to right as follows: OFC = DLO, LO, VO, MO; PFC = Cg, Prl, IL, Dp; dHPC = dDG, dCA3, dCA1, dS; Hb = MHb, LHb; vHPC = vDG, vCA3, vCA1, vS; EC. Normal adolescent to adult maturation involves specialization of brain regions, which can be seen as “islands” of high correlations within brain regions, surrounded by low correlations between brain regions in the adult brain (lower, left matrix). Adult GD17- MAM rats have altered functional connectivity to the ventral hippocampus (lower, right matrix) that is evident already in adolescence (upper, right matrix). Values are Pearson Product Correlations. See Table 1 for details. Correlations significantly different (p < 0.05) from corresponding age group. B) Networks were generated from the significant correlations (p < 0.05). Gray lines are correlations that did not reach significance after false discovery rate correction. Network properties of the dorsal hippocampus undergo maturation in both control and GD17-MAM rats from adolescence to adulthood. However, while the control dorsal hippocampus loses edges from adolescence to adulthood, the GD17-MAM dorsal hippocampus gains edges. Subregions are as follows: OFC = orbitofrontal cortex, PFC = prefrontal cortex, dHPC and vHPC = dorsal and ventral hippocampus, respectively, Hb = habenula, EC = entorhinal cortex. DO, LO, VO, and MO = dorsolateral, lateral, ventrolateral, and medial orbitofrontal cortex, respectively. Cg, PrL, IL = cingulate, prelimbic, and infralimbic cortex, respectively. Dp = dorsal peduncular nucleus. dDG and vDG = dorsal and ventral dentate gyrus, respectively. dCA3 and vCA3 = dorsal and ventral Cornu Ammonis 3, respectively. dCA1 and vCA1 = dorsal and ventral Cornu Ammonis 1, respectively. dS and vS = dorsal and ventral subiculum, respectively. mHb and LHb = medial and lateral habenula, respectively.
Based on the behavioral impairments that adolescent MAM rats express, we hypothesized that abnormal brain function would also be evident during adolescence. We extended the cytochrome oxidase studies of adult MAM rats to include the orbitofrontal cortex and habenular complex and also assessed metabolic activity during adolescence (Table 2). While there are few significant group differences in interregional correlations during adolescence, significantly lower correlations between the ventral hippocampus and prefrontal cortex of adolescent GD17-MAM rats identify the presence of abnormalities already at this age. This contrasts with excessive correlations in adult GD17-MAM rats (Fig. 6). Together with the adolescent behavioral studies, these data suggest neural processing is already altered in GD17-MAM rats by the time the early cognitive training occurs. In contrast, the behavior of the adolescent NVHL rats implies that neural processing is not yet critically disrupted, or at least not as severely disrupted, in adolescent NVHL rats, allowing the early cognitive training to confer benefits, presumably by neuroplasticity mechanisms. The extent to which synaptic dysplasticity has a role in the dysfunction is unknown, but if we assume abnormal neural function promotes synaptic dysplasticity, then dysplasticity may be greater in MAM rats due to the non-specificity of the insult. Alternatively, synaptic dysfunction may be reversible in adolescent NVHL rats because the disruption in ventral hippocampus was post-natal and destroyed the tissue, whereas in MAM rats the abnormalities result from disrupting neurogenesis. Clearly, these possibilities should be examined.
Table 2.
Average CO activity by brain region between GD17-MAM and control rats during adolescence and adulthood.
| Relative CO activity/pm tissue (×10−1) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Adolescent | Adult | |||||||||||
| Brain region | P35 Control |
N | P35 MAM |
N | p- value |
t- stat |
P70 Control |
N | P70 MAM |
N |
p- value |
t- stat |
| Orbitofrontal Cortex | ||||||||||||
| DLO | 7.78 ±0.74 | 8 | 8.57 ±0.35 | 7 | 0.36 | 0.94 | 7.53 ±0.69 | 8 | 7.99 ±0.75 | 8 | 0.96 | 0.05 |
| LO | 8.28 ± 0.93 | 8 | 8.78 ± 0.53 | 7 | 0.65 | 0.46 | 8.81 ± 0.49 | 8 | 8.53 ± 0.54 | 8 | 0.46 | 0.76 |
| VO | 8.47 ± 1.16 | 8 | 9.55 ± 0.40 | 7 | 0.41 | 0.85 | 8.76 ± 0.73 | 8 | 8.70 ± 0.75 | 8 | 0.59 | 0.55 |
| MO | 9.23 ± 0.66 | 8 | 8.06 ± 0.97 | 7 | 0.34 | 0.99 | 9.34 ± 0.90 | 8 | 8.24 ± 0.84 | 8 | 0.49 | 0.70 |
| Prefrontal Cortex | ||||||||||||
| Cg | 10.21 ± 0.78 | 8 | 11.18 ± 0.67 | 7 | 0.36 | 0.94 | 11.23 ± 0.79 | 8 | 10.39 ± 1.13 | 8 | 0.55 | 0.61 |
| PrL | 10.29 ± 0.83 | 8 | 10.92 ± 0.64 | 7 | 0.56 | 0.60 | 10.65 ± 0.88 | 8 | 9.78 ± 0.64 | 8 | 0.44 | 0.80 |
| IL | 8.70 ± 0.74 | 8 | 8.85 ± 0.45 | 7 | 0.86 | 0.17 | 10.11 ± 0.77 | 8 | 9.21 ± 0.65 | 8 | 0.39 | 0.89 |
| Dp | 8.64 ± 0.65 | 8 | 9.10 ± 0.46 | 7 | 0.57 | 0.58 | 8.70 ± 0.63 | 8 | 9.29 ± 0.82 | 8 | 0.57 | 0.57 |
| Dorsal Hippocampus | ||||||||||||
| dDG | 12.79 ± 0.74 | 8 | 12.04 ± 1.08 | 8 | 0.58 | 0.56 | 12.42 ± 0.84 | 8 | 12.82 ± 1.19 | 8 | 0.79 | 0.28 |
| dCA3 | 8.10 ± 0.70 | 8 | 7.59 ± 0.41 | 8 | 0.55 | 0.62 | 7.66 ± 0.46 | 8 | 7.27 ± 0.52 | 8 | 0.71 | 0.39 |
| dCA1 | 7.49 ± 0.75 | 8 | 7.84 ± 0.58 | 8 | 0.72 | 0.36 | 7.35 ± 0.56 | 8 | 8.21 ± 0.85 | 8 | 0.41 | 0.85 |
| Dorsal Subiculum | 7.93 ± 0.63 | 8 | 8.78 ± 0.75 | 8 | 0.40 | 0.87 | 7.79 ± 0.62 | 8 | 8.31 ± 0.88 | 8 | 0.63 | 0.49 |
| Habenular Complex | ||||||||||||
| MHb | 9.73 ± 0.92 | 8 | 8.40 ± 0.47 | 7 | 0.23 | 1.25 | 9.23 ± 0.79 | 8 | 8.81 ± 0.74 | 8 | 0.71 | 0.39 |
| LHb | 11.78 ± 0.88 | 8 | 11.72 ± 0.85 | 7 | 0.96 | 0.05 | 12.34 ± 0.88 | 8 | 11.41 ± 0.75 | 8 | 0.43 | 0.81 |
| Ventral Hippocampus | ||||||||||||
| vDG | 10.22 ± 0.64 | 8 | 9.17 ± 0.80 | 8 | 0.32 | 1.02 | 10.47 ± 0.39 | 8 | 10.08 ± 0.94 | 8 | 0.72 | 0.37 |
| vCA3 | 11.48 ± 0.37 | 8 | 10.27 ± 0.86 | 8 | 0.23 | 1.26 | 12.02 ± 0.79 | 8 | 13.80 ± 0.89 | 8 | 0.16 | 1.49 |
| vCA1 | 8.66 ± 0.64 | 8 | 9.63 ± 0.52 | 8 | 0.26 | 1.18 | 10.18 ± 0.79 | 8 | 10.03 ± 0.74 | 8 | 0.89 | 0.14 |
| Ventral Subiculum | 11.98 ± 0.77 | 8 | 11.52 ± 0.64 | 8 | 0.65 | 0.46 | 12.43 ± 0.98 | 8 | 13.50 ± 1.33 | 8 | 0.53 | 0.64 |
| Entorhinal | 9.81 ± | 7 | 9.25 ± | 7 | 0.65 | 0.46 | 9.89 ± | 8 | 8.80 ± | 8 | 0.42 | 0.82 |
| Cortex | 0.57 | 1.06 | 1.01 | 0.84 | ||||||||
Shaded regions indicated values reported in (O’Reilly et al., 2016).
1.11. Conclusions
We have reviewed our work on synaptic function and plasticity in the context of neural circuit function and memory, focusing on the hippocampus and spatial cognitive behavior as model systems for investigating the potential relationships between this neurobiology and mental function and dysfunction. We have proposed there is utility to distinguish between two concepts of memory. The concept of item memory, related to declarative memory content, concerns the explicit information thought to be stored by synaptic plasticity mechanisms. The concept of process memory, related to information processing ability, is also acquired and modified by experience, and stored by synaptic plasticity mechanisms, in the same anatomical regions that are crucial for item memories. Process memory confers information processing capabilities on a neural circuit that extend beyond particular items of information in explicit memory. Cognitive dysfunction in animal models of mental illness is associated with neural discoordination in the timing of how relatively normal individual neural signals interact. These signals include single cell spike trains and oscillations in local field potentials of population synaptic origin (Fenton, 2015). While causality has not been established, this neural discoordination appears to be tightly coupled to particular forms of synaptic dysfunction at particular connections within a neural circuit (Dvorak et al., 2018). The fact that such dysfunction is circuit-specific, synapse-specific, and even mechanism-specific highlights the formidable challenge to correcting or remediating cognitive symptoms for patients suffering from mental illness. On the other hand, this challenge highlights the urgent need for translating to therapeutic approaches the novel genetically-targeting neurobiological tools that currently operate at specific circuits and synaptic junctions (Boyden, 2015; Chow and Boyden, 2013; Gradinaru et al., 2010; Knopfel et al., 2010). This challenge also highlights the potential value of pursuing systems-level analysis and therapeutic strategies such as CBT, transcranial stimulation and pharmacology that target restoration of function rather than reversal or correction of the molecular or genetic etiology of mental illness and disease (Coffman et al., 2014; Lee et al., 2014; Najib et al., 2011; Oberman et al., 2010).
Supplementary Material
Acknowledgments
Role of the funding source: AAF is supported by NIH grants R01MH099128, R01NS105472 and R21NS091830.
2.1 References
- Bakker CE, Verheij C, Willemsen R, Vanderhelm R, Oerlemans F, Vermey M, Bygrave A, Hoogeveen AT, Oostra BA, Reyniers E, DeBoulle K, Dhooge R, Cras P, Van Velzen N, Nagels G, Martin JJ, Dedeyn PP, Darby JK, Willems PJ, 1994. Fmr1 knockout mice: a model to study fragile X mental retardation. The Dutch-Belgian Fragile X Consortium. Cell 78(1), 23–33. [PubMed] [Google Scholar]
- Bannerman DM, Good MA, Butcher SP, Ramsay M, Morris RG, 1995. Distinct components of spatial learning revealed by prior training and NMDA receptor blockade. Nature 378(6553), 182–186. [DOI] [PubMed] [Google Scholar]
- Barry JM, Rivard B, Fox SE, Fenton AA, Sacktor TC, Muller RU, 2012. Inhibition of protein kinase Mzeta disrupts the stable spatial discharge of hippocampal place cells in a familiar environment. J Neurosci 32(40), 13753–13762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer SA, 1980. Development of the hippocampal region in the rat. I. Neurogenesis examined with 3H-thymidine autoradiography. The Journal of comparative neurology 190(1), 87–114. [DOI] [PubMed] [Google Scholar]
- Bear MF, Malenka RC, 1994. Synaptic plasticity: LTP and LTD. Curr Opin Neurobiol 4(3), 389–399. [DOI] [PubMed] [Google Scholar]
- Bhattacharya A, Kaphzan H, Alvarez-Dieppa AC, Murphy JP, Pierre P, Klann E, 2012. Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron 76(2), 325–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi GQ, Bolshakov V, Bu G, Cahill CM, Chen ZF, Collingridge GL, Cooper RL, Coorssen JR, El-Husseini A, Galhardo V, Gan WB, Gu J, Inoue K, Isaac J, Iwata K, Jia Z, Kaang BK, Kawamata M, Kida S, Klann E, Kohno T, Li M, Li XJ, MacDonald JF, Nader K, Nguyen PV, Oh U, Ren K, Roder JC, Salter MW, Song W, Sugita S, Tang SJ, Tao Y, Wang YT, Woo N, Woodin MA, Yan Z, Yoshimura M, Xu M, Xu ZC, Zhang X, Zhen M, Zhuo M, 2006. Recent advances in basic neurosciences and brain disease: from synapses to behavior. Mol Pain 2, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittner KC, Grienberger C, Vaidya SP, Milstein AD, Macklin JJ, Suh J, Tonegawa S, Magee JC, 2015. Conjunctive input processing drives feature selectivity in hippocampal CA1 neurons. Nat Neurosci 18(8), 1133–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittner KC, Milstein AD, Grienberger C, Romani S, Magee JC, 2017. Behavioral time scale synaptic plasticity underlies CA1 place fields. Science 357(6355), 1033–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden ES, 2015. Optogenetics and the future of neuroscience. Nat Neurosci 18(9), 1200–1201. [DOI] [PubMed] [Google Scholar]
- Bures J, Fenton AA, Kaminsky Y, Rossier J, Sacchetti B, Zinyuk L, 1997. Dissociation of exteroceptive and idiothetic orientation cues: effect on hippocampal place cells and place navigation. Philos Trans R Soc Lond B Biol Sci 352(1360), 1515–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsaki G, Anastassiou CA, Koch C, 2012. The origin of extracellular fields and currents--EEG, ECoG, LFP and spikes. Nat Rev Neurosci 13(6), 407–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carandini M, Heeger DJ, 2011. Normalization as a canonical neural computation. Nat Rev Neurosci 13(1), 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow BY, Boyden ES, 2013. Optogenetics and translational medicine. Sci Transl Med 5(177), 177ps175. [DOI] [PubMed] [Google Scholar]
- Cimadevilla JM, Fenton AA, Bures J, 2000a. Functional inactivation of dorsal hippocampus impairs active place avoidance in rats. Neurosci Lett 285(1), 53–56. [DOI] [PubMed] [Google Scholar]
- Cimadevilla JM, Kaminsky Y, Fenton A, Bures J, 2000b. Passive and active place avoidance as a tool of spatial memory research in rats. J Neurosci Methods 102(2), 155–164. [DOI] [PubMed] [Google Scholar]
- Cimadevilla JM, Wesierska M, Fenton AA, Bures J, 2001. Inactivating one hippocampus impairs avoidance of a stable room-defined place during dissociation of arena cues from room cues by rotation of the arena. Proc Natl Acad Sci U S A 98(6), 3531–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffman BA, Clark VP, Parasuraman R, 2014. Battery powered thought: enhancement of attention, learning, and memory in healthy adults using transcranial direct current stimulation. Neuroimage 85 Pt 3, 895–908. [DOI] [PubMed] [Google Scholar]
- D’Hooge R, Nagels G, Franck F, Bakker CE, Reyniers E, Storm K, Kooy RF, Oostra BA, Willems PJ, De Deyn PP, 1997. Mildly impaired water maze performance in male Fmr1 knockout mice. Neuroscience 76(2), 367–376. [DOI] [PubMed] [Google Scholar]
- Danglot L, Triller A, Marty S, 2006. The development of hippocampal interneurons in rodents. Hippocampus 16(12), 1032–1060. [DOI] [PubMed] [Google Scholar]
- Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, Licatalosi DD, Richter JD, Darnell RB, 2011. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146(2), 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida L, Idiart M, Lisman JE, 2009. A second function of gamma frequency oscillations: an E%-max winner-take-all mechanism selects which cells fire. J Neurosci 29(23), 7497–7503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Villers-Sidani E, Alzghoul L, Zhou X, Simpson KL, Lin RC, Merzenich MM, 2010. Recovery of functional and structural age-related changes in the rat primary auditory cortex with operant training. Proc Natl Acad Sci U S A 107(31), 13900–13905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz B, Manahan-Vaughan D, 2017. Hippocampal long-term depression is facilitated by the acquisition and updating of memory of spatial auditory content and requires mGlu5 activation. Neuropharmacology 115, 30–41. [DOI] [PubMed] [Google Scholar]
- Dvorak D, Radwan B, Sparks FT, Talbot ZN, Fenton AA, 2018. Control of recollection by slow gamma dominating mid-frequency gamma in hippocampus CA1. PLoS Biol 16(1), e2003354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton AA, 2015. Excitation-inhibition discoordination in rodent models of mental disorders. Biol Psychiatry 77(12), 1079–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton AA, Bures J, 2003. Navigation in the moving world, in: Jeffery K (Ed.), The Neurobiology of Spatial Behaviour Oxford University Press, Oxford. [Google Scholar]
- Froemke RC, Carcea I, Barker AJ, Yuan K, Seybold BA, Martins AR, Zaika N, Bernstein H, Wachs M, Levis PA, Polley DB, Merzenich MM, Schreiner CE, 2013. Long-term modification of cortical synapses improves sensory perception. Nat Neurosci 16(1), 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner AR, Rowland DC, Hwang SY, Baumgaertel K, Roth BL, Kentros C, Mayford M, 2012. Generation of a synthetic memory trace. Science 335(6075), 1513–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelbard-Sagiv H, Mukamel R, Harel M, Malach R, Fried I, 2008. Internally generated reactivation of single neurons in human hippocampus during free recall. Science 322(5898), 96–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradinaru V, Zhang F, Ramakrishnan C, Mattis J, Prakash R, Diester I, Goshen I, Thompson KR, Deisseroth K, 2010. Molecular and cellular approaches for diversifying and extending optogenetics. Cell 141(1), 154–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruart A, Munoz MD, Delgado-Garcia JM, 2006. Involvement of the CA3-CA1 synapse in the acquisition of associative learning in behaving mice. J Neurosci 26(4), 1077–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guic E, Carrasco X, Rodriguez E, Robles I, Merzenich MM, 2008. Plasticity in primary somatosensory cortex resulting from environmentally enriched stimulation and sensory discrimination training. Biol Res 41(4), 425–437. [PubMed] [Google Scholar]
- Guzowski JF, McNaughton BL, Barnes CA, Worley PF, 1999. Environment- specific expression of the immediate-early gene Arc in hippocampal neuronal ensembles. Nat Neurosci 2(12), 1120–1124. [DOI] [PubMed] [Google Scholar]
- Harlow EG, Till SM, Russell TA, Wijetunge LS, Kind P, Contractor A, 2010. Critical period plasticity is disrupted in the barrel cortex of FMR1 knockout mice. Neuron 65(3), 385–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfield JJ, 1982. Neural networks and physical systems with emergent collective computational abilities. Proc Natl Acad Sci U S A 79(8), 2554–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh C, Tsokas P, Serrano P, Hernandez AI, Tian D, Cottrell JE, Shouval HZ, Fenton AA, Sacktor TC, 2017. Persistent increased PKMzeta in long-term and remote spatial memory. Neurobiol Learn Mem 138, 135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF, 2002. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A 99(11), 7746–7750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Kayser MS, Bear MF, 2000. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science 288(5469), 1254–1257. [DOI] [PubMed] [Google Scholar]
- Inglis J, Martin SJ, Morris RG, 2013. Upstairs/downstairs revisited: spatial pretraining-induced rescue of normal spatial learning during selective blockade of hippocampal N-methyl-d-aspartate receptors. Eur J Neurosci 37(5), 718–727. [DOI] [PubMed] [Google Scholar]
- Kandel ER, 2004. The molecular biology of memory storage: a dialog between genes and synapses. Biosci Rep 24(4–5), 475–522. [DOI] [PubMed] [Google Scholar]
- Kao HY, Dvorak D, Park E, Kenney J, Kelemen E, Fenton AA, 2017. Phencyclidine discoordinates hippocampal network activity but not place fields. J Neurosci 37, 12031–12049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelemen E, Fenton AA, 2010. Dynamic grouping of hippocampal neural activity during cognitive control of two spatial frames. PLoS Biol 8(6), e1000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelemen E, Fenton AA, 2013. Key features of human episodic recollection in the cross-episode retrieval of rat hippocampus representations of space. PLoS Biol 11(7), e1001607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelemen E, Fenton AA, 2016. Coordinating different representations in the hippocampus. Neurobiol Learn Mem 129, 50–59. [DOI] [PubMed] [Google Scholar]
- Knopfel T, Lin MZ, Levskaya A, Tian L, Lin JY, Boyden ES, 2010. Toward the second generation of optogenetic tools. J Neurosci 30(45), 14998–15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang U, Frotscher M, 1990. Postnatal development of nonpyramidal neurons in the rat hippocampus (areas CA1 and CA3): a combined Golgi/electron microscope study. Anatomy and embryology 181(6), 533–545. [DOI] [PubMed] [Google Scholar]
- Lee H, Dvorak D, Fenton AA, 2014. Targeting Neural Synchrony Deficits is Sufficient to Improve Cognition in a Schizophrenia-Related Neurodevelopmental Model. Front Psychiatry 5, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Dvorak D, Kao HY, Duffy AM, Scharfman HE, Fenton AA, 2012. Early cognitive experience prevents adult deficits in a neurodevelopmental schizophrenia model. Neuron 75(4), 714–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL, 2000. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 405(6789), 955–959. [DOI] [PubMed] [Google Scholar]
- Ling DS, Benardo LS, Sacktor TC, 2006. Protein kinase Mzeta enhances excitatory synaptic transmission by increasing the number of active postsynaptic AMPA receptors. Hippocampus 16(5), 443–452. [DOI] [PubMed] [Google Scholar]
- Ling DS, Benardo LS, Serrano PA, Blace N, Kelly MT, Crary JF, Sacktor TC, 2002. Protein kinase Mzeta is necessary and sufficient for LTP maintenance. Nat Neurosci 5(4), 295–296. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Aultman JM, Verma A, Weinberger DR, Moghaddam B, 2002. Neonatal damage of the ventral hippocampus impairs working memory in the rat. Neuropsychopharmacology 27(1), 47–54. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Weinberger DR, 2002. A neurodevelopmental model of schizophrenia: neonatal disconnection of the hippocampus. Neurotox Res 4(5–6), 469–475. [DOI] [PubMed] [Google Scholar]
- Liu X, Ramirez S, Pang PT, Puryear CB, Govindarajan A, Deisseroth K, Tonegawa S, 2012. Optogenetic stimulation of a hippocampal engram activates fear memory recall. Nature 484(7394), 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA, 2009. Gestational methylazoxymethanol acetate administration: a developmental disruption model of schizophrenia. Behav Brain Res 204(2), 306–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madronal N, Gruart A, Sacktor TC, Delgado-Garcia JM, 2010. PKMzeta inhibition reverses learning-induced increases in hippocampal synaptic strength and memory during trace eyeblink conditioning. PLoS One 5(4), e10400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF, 2004. LTP and LTD: an embarrassment of riches. Neuron 44(1), 5–21. [DOI] [PubMed] [Google Scholar]
- Marr D, 1971. Simple memory: a theory for archicortex. Philos Trans R Soc Lond B Biol Sci 262(841), 23–81. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RG, 2000. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci 23, 649–711. [DOI] [PubMed] [Google Scholar]
- McDougall W, 1923. Outline of psychology. Scribners, New York. [Google Scholar]
- McNaughton BL, Morris RGM, 1987. Hippocampal synaptic enhancement and information storage within a distributed memory system. Trends in Neuroscience 10(10), 409–415. [Google Scholar]
- Mercaldo V, Descalzi G, Zhuo M, 2009. Fragile X mental retardation protein in learning-related synaptic plasticity. Mol Cells 28(6), 501–507. [DOI] [PubMed] [Google Scholar]
- Migues PV, Hardt O, Wu DC, Gamache K, Sacktor TC, Wang YT, Nader K, 2010. PKMzeta maintains memories by regulating GluR2-dependent AMPA receptor trafficking. Nat Neurosci. [DOI] [PubMed] [Google Scholar]
- Moore H, Jentsch JD, Ghajarnia M, Geyer MA, Grace AA, 2006. A neurobehavioral systems analysis of adult rats exposed to methylazoxymethanol acetate on E17: implications for the neuropathology of schizophrenia. Biol Psychiatry 60(3), 253–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser EI, Moser MB, 2013. Grid cells and neural coding in high-end cortices. Neuron 80(3), 765–774. [DOI] [PubMed] [Google Scholar]
- Muller RU, Ranck JB Jr., Taube JS, 1996. Head direction cells: properties and functional significance. Curr Opin Neurobiol 6(2), 196–206. [DOI] [PubMed] [Google Scholar]
- Najib U, Bashir S, Edwards D, Rotenberg A, Pascual-Leone A, 2011. Transcranial brain stimulation: clinical applications and future directions. Neurosurg Clin N Am 22(2), 233–251, ix. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Keefe J, 1979. A review of the hippocampal place cells. Prog Neurobiol 13(4), 419–439. [DOI] [PubMed] [Google Scholar]
- O’Reilly KC, Levy ERJ, Patino AV, Perica MI, Fenton AA, 2018. Sub-circuit alterations in dorsal hippocampus structure and function after global neurodevelopmental insult. Brain Struct Funct. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly KC, Perica MI, Fenton AA, 2016. Memory deficits with intact cognitive control in the methylazoxymethanol acetate (MAM) exposure model of neurodevelopmental insult. Neurobiol Learn Mem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberman L, Ifert-Miller F, Najib U, Bashir S, Woollacott I, Gonzalez-Heydrich J, Picker J, Rotenberg A, Pascual-Leone A, 2010. Transcranial magnetic stimulation provides means to assess cortical plasticity and excitability in humans with fragile x syndrome and autism spectrum disorder. Front Synaptic Neurosci 2, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olypher AV, Klement D, Fenton AA, 2006. Cognitive disorganization in hippocampus: a physiological model of the disorganization in psychosis. J Neurosci 26(1), 158–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olypher AV, Lansky P, Fenton AA, 2002. Properties of the extra-positional signal in hippocampal place cell discharge derived from the overdispersion in location- specific firing. Neuroscience 111(3), 553–566. [DOI] [PubMed] [Google Scholar]
- Park EH, Burghardt NS, Dvorak D, Hen R, Fenton AA, 2015. Experience-Dependent Regulation of Dentate Gyrus Excitability by Adult-Born Granule Cells. J Neurosci 35(33), 11656–11666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastalkova E, Serrano P, Pinkhasova D, Wallace E, Fenton AA, Sacktor TC, 2006. Storage of spatial information by the maintenance mechanism of LTP. Science 313(5790), 1141–1144. [DOI] [PubMed] [Google Scholar]
- Patel AB, Hays SA, Bureau I, Huber KM, Gibson JR, 2013. A target cell-specific role for presynaptic Fmr1 in regulating glutamate release onto neocortical fast- spiking inhibitory neurons. J Neurosci 33(6), 2593–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlowsky A, Wallace E, Fenton AA, Alarcon JM, 2017. Persistent modifications of hippocampal synaptic function during remote spatial memory. Neurobiol Learn Mem 138, 182–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Gomez R, Mederos S, Covelo A, Ballesteros JJ, Schlosser L, Hernandez-Vivanco A, Martin-Fernandez M, Quintana R, Rayan A, Diez A, Fuenzalida M, Agarwal A, Bergles DE, Bettler B, Manahan-Vaughan D, Martin ED, Kirchhoff F, Araque A, 2016. Activity-dependent switch of GABAergic inhibition into glutamatergic excitation in astrocyte-neuron networks. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips WA, Silverstein SM, 2003. Convergence of biological and psychological perspectives on cognitive coordination in schizophrenia. Behav Brain Sci 26(1), 65–82; discussion 82–137. [DOI] [PubMed] [Google Scholar]
- Radwan B, Dvorak D, Fenton A, 2016. Impaired cognitive discrimination and discoordination of coupled theta-gamma oscillations in Fmr1 knockout mice. Neurobiol Dis(88), 125–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez S, Liu X, Lin PA, Suh J, Pignatelli M, Redondo RL, Ryan TJ, Tonegawa S, 2013. Creating a false memory in the hippocampus. Science 341(6144), 387–391. [DOI] [PubMed] [Google Scholar]
- Richards BA, Frankland PW, 2017. The Persistence and Transience of Memory. Neuron 94(6), 1071–1084. [DOI] [PubMed] [Google Scholar]
- Roy DS, Arons A, Mitchell TI, Pignatelli M, Ryan TJ, Tonegawa S, 2016. Memory retrieval by activating engram cells in mouse models of early Alzheimer’s disease. Nature 531(7595), 508–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer S, Zemelman BV, Losonczy A, Kim J, Chance F, Magee JC, Buzsaki G, 2012. Control of timing, rate and bursts of hippocampal place cells by dendritic and somatic inhibition. Nat Neurosci 15(5), 769–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruediger S, Vittori C, Bednarek E, Genoud C, Strata P, Sacchetti B, Caroni P, 2011. Learning-related feedforward inhibitory connectivity growth required for memory precision. Nature 473, 514–518. [DOI] [PubMed] [Google Scholar]
- Ryan TJ, Roy DS, Pignatelli M, Arons A, Tonegawa S, 2015. Memory. Engram cells retain memory under retrograde amnesia. Science 348(6238), 1007–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan TJ, Tonegawa S, 2016. Rehebbilitating Memory. Neuropsychopharmacology 41(5), 1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacktor TC, 2012. Memory maintenance by PKMzeta--an evolutionary perspective. Mol Brain 5, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samsonovich A, McNaughton BL, 1997. Path integration and cognitive mapping in a continuous attractor neural network model. J Neurosci 17(15), 5900–5920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapiro AC, Turk-Browne NB, Botvinick MM, Norman KA, 2017. Complementary learning systems within the hippocampus: a neural network modelling approach to reconciling episodic memory with statistical learning. Phil. Trans. R. Soc. B 372, 20160049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano P, Friedman EL, Kenney J, Taubenfeld SM, Zimmerman JM, Hanna J, Alberini C, Kelley AE, Maren S, Rudy JW, Yin JC, Sacktor TC, Fenton AA, 2008. PKMzeta maintains spatial, instrumental, and classically conditioned long-term memories. PLoS Biol 6(12), 2698–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squire LR, 2004. Memory systems of the brain: a brief history and current perspective. Neurobiol Learn Mem 82(3), 171–177. [DOI] [PubMed] [Google Scholar]
- Squire LR, Stark CE, Clark RE, 2004. The medial temporal lobe. Annu Rev Neurosci 27, 279–306. [DOI] [PubMed] [Google Scholar]
- Super H, Martinez A, Del Rio JA, Soriano E, 1998. Involvement of distinct pioneer neurons in the formation of layer-specific connections in the hippocampus. The Journal of neuroscience : the official journal of the Society for Neuroscience 18(12), 4616–4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Halim N, Hanlon FM, Platten A, Auerbach PP, 2001. Lesion size and amphetamine hyperlocomotion after neonatal ventral hippocampal lesions: more is less. Brain research bulletin 55(1), 71–77. [DOI] [PubMed] [Google Scholar]
- Takeuchi T, Duszkiewicz AJ, Morris RG, 2013. The synaptic plasticity and memory hypothesis: encoding, storage and persistence. Philos Trans R Soc Lond B Biol Sci 369(1633), 20130288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot ZN, Sparks FT, Dvorak D, Curran BM, Alarcon JM, Fenton AA, 2018. Normal CA1 Place Fields but Discoordinated Network Discharge in a Fmr1-Null Mouse Model of Fragile X Syndrome. Neuron 97(3), 684–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taube JS, 2007. The head direction signal: origins and sensory-motor integration. Annu Rev Neurosci 30, 181–207. [DOI] [PubMed] [Google Scholar]
- Taube JS, Muller RU, Ranck JB Jr., 1990. Head-direction cells recorded from the postsubiculum in freely moving rats. I. Description and quantitative analysis. J Neurosci 10(2), 420–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson LT, Best PJ, 1989. Place cells and silent cells in the hippocampus of freely-behaving rats. J Neurosci 9(7), 2382–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till SM, Asiminas A, Jackson AD, Katsanevaki D, Barnes SA, Osterweil EK, Bear MF, Chattarji S, Wood ER, Wyllie DJ, Kind PC, 2015. Conserved hippocampal cellular pathophysiology but distinct behavioural deficits in a new rat model of FXS. Hum Mol Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsokas P, Hsieh C, Yao Y, Lesburgueres E, Wallace EJ, Tcherepanov A, Jothianandan D, Hartley BR, Pan L, Rivard B, Farese RV, Sajan MP, Bergold PJ, Hernandez AI, Cottrell JE, Shouval HZ, Fenton AA,Sacktor TC, 2016. Compensation for PKMzeta in long-term potentiation and spatial long-term memory in mutant mice. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB, 2000. Hebb and homeostasis in neuronal plasticity. Curr Opin Neurobiol 10(3), 358–364. [DOI] [PubMed] [Google Scholar]
- Uhlhaas PJ, Singer W, 2012. Neuronal dynamics and neuropsychiatric disorders: toward a translational paradigm for dysfunctional large-scale networks. Neuron 75(6), 963–980. [DOI] [PubMed] [Google Scholar]
- van Dijk MT, Fenton AA, 2018. On How the Dentate Gyrus Contributes to Memory Discrimination. Neuron 98(4), 832–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss P, Thomas M, Chou YC, Cisneros-Franco JM, Ouellet L, de Villers-Sidani E, 2016. Pairing Cholinergic Enhancement with Perceptual Training Promotes Recovery of Age-Related Changes in Rat Primary Auditory Cortex. Neural Plast 2016, 1801979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss P, Thomas ME, Cisneros-Franco JM, de Villers-Sidani E, 2017. Dynamic Brains and the Changing Rules of Neuroplasticity: Implications for Learning and Recovery. Front Psychol 8, 1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Sheng T, Ren S, Tian T, Lu W, 2016. Distinct Roles of PKCiota/lambda and PKMzeta in the Initiation and Maintenance of Hippocampal Long-Term Potentiation and Memory. Cell Rep 16(7), 1954–1961. [DOI] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, Bear MF, 2006. Learning induces long-term potentiation in the hippocampus. Science 313(5790), 1093–1097. [DOI] [PubMed] [Google Scholar]
- Wilson RC, Niv Y, 2012. Inferring relevance in a changing world. Front Hum Neurosci 5, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witter MP, 2010. Connectivity of the hippocampus, in: Cutsuridis V, Graham B, Cobb S, Vida I (Eds.), Hippocampal microcircuits: a computational modeler’s rescource book. Springer, New York, pp. 5–26. [Google Scholar]
- Wong-Riley MT, 1989. Cytochrome oxidase: an endogenous metabolic marker for neuronal activity. Trends Neurosci 12(3), 94–101. [DOI] [PubMed] [Google Scholar]
- Yao Y, Kelly MT, Sajikumar S, Serrano P, Tian D, Bergold PJ, Frey JU, Sacktor TC, 2008. PKM zeta maintains late long-term potentiation by N- ethylmaleimide-sensitive factor/GluR2-dependent trafficking of postsynaptic AMPA receptors. J Neurosci 28(31), 7820–7827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu NK, Uhm H, Shim J, Choi JH, Bae S, Sacktor TC, Hohng S, Kaang BK, 2017. Increased PKMzeta activity impedes lateral movement of GluA2-containing AMPA receptors. Mol Brain 10(1), 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, 1996. Representation of spatial orientation by the intrinsic dynamics of the head-direction cell ensemble: a theory. J Neurosci 16(6), 2112–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao MG, Toyoda H, Ko SW, Ding HK, Wu LJ, Zhuo M, 2005. Deficits in trace fear memory and long-term potentiation in a mouse model for fragile X syndrome. J Neurosci 25(32), 7385–7392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Lu JY, Darling RD, Simpson KL, Zhu X, Wang F, Yu L, Sun X, Merzenich MM, Lin RC, 2015. Behavioral training reverses global cortical network dysfunction induced by perinatal antidepressant exposure. Proc Natl Acad Sci U S A 112(7), 2233–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Wang F, Hu H, Sun X, Kilgard MP, Merzenich MM, Zhou X, 2014. Environmental acoustic enrichment promotes recovery from developmentally degraded auditory cortical processing. J Neurosci 34(16), 5406–5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.