

Abstract
Purpose:
Defects in the cohesin pathway are associated with cohesinopathies, notably Cornelia de Lange Syndrome (CdLS). We aim to delineate mutations in known and candidate cohesinopathy genes from a clinical exome perspective.
Methods:
We retrospectively studied patients referred for clinical exome sequencing (CES, N=10,698). Patients with causative variants in novel or recently described cohesinopathy genes were enrolled for phenotypic characterization.
Results:
Pathogenic or likely pathogenic single nucleotide and insertion/deletion variants (SNVs/indels) were identified in established disease genes including NIPBL (N=5), SMC1A (N=14), SMC3 (N=4), RAD21 (N=2) and HDAC8 (N=8). The phenotypes in this genetically defined cohort skew towards the mild end of CdLS spectrum as compared to phenotype-driven cohorts. Candidate or recently reported cohesinopathy genes were supported by de novo SNVs/indels in STAG1 (N=3), STAG2 (N=5), PDS5A (N=1) and WAPL (N=1), and one inherited SNV in PDS5A. We also identified copy number deletions affecting STAG1 (two de novo, one of unknown inheritance) and STAG2 (one of unknown inheritance). Patients with STAG1 and STAG2 variants presented with overlapping features yet without characteristic facial features of CdLS.
Conclusion:
CES effectively identified disease-causing alleles at the mild end of the cohensinopathy spectrum and enabled characterization of candidate disease genes.
Keywords: Atypical cohesinopathies, clinical exome sequencing (CES), cohesin pathway, STAG1, STAG2
INTRODUCTION
The cohesin complex mediates sister chromatid cohesion and ensures accurate chromosome segregation, recombination-mediated DNA repair, and genomic stability during DNA replication and cell division. Accumulating evidence suggests that cohesin is also involved in regulating chromosomal looping/architecture and gene transcriptional regulation1–3.
Cohesin is a multi-subunit protein complex composed of evolutionarily conserved core components encoded by SMC1A (MIM *300040), SMC3 (MIM *606062), RAD21 (MIM *606462) and either STAG1 (MIM *604358) or STAG2 (MIM *300826) depending on the chromosomal location. Direct interaction between SMC1A, SMC3 and RAD21 form a tripartite ring structure that is used to entrap the replicated chromatin during sister chromatid cohesion (Figure 1A). STAG1/2 are the core structural component of functional cohesin and critical for the loading of cohesin onto chromatin during mitosis1,2.
Figure 1.
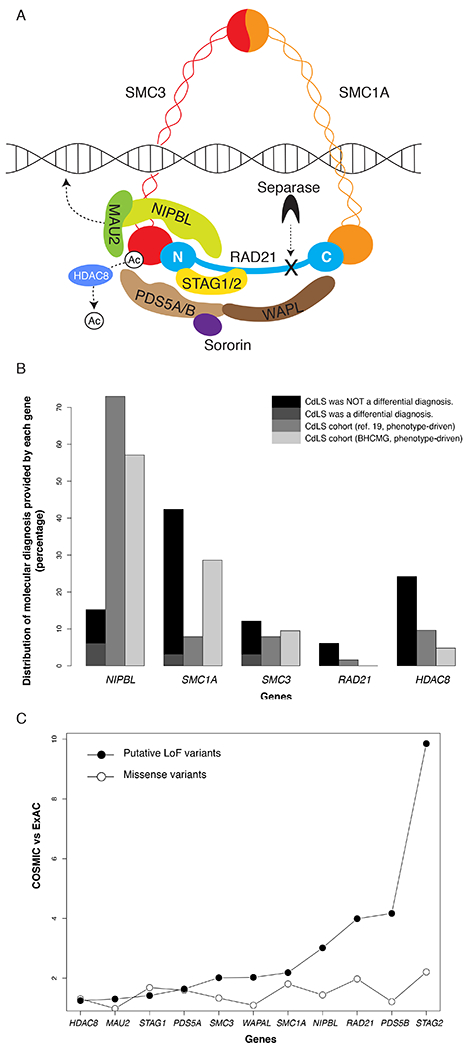
Cohesin complex and its underlying genetic variants. A. Schematic diagram of the cohesin complex. The components are represented in different color shapes labeled with protein names. B. Comparison of genic distributions between our clinical exome cohort and two phenotype-driven cohorts of clinically diagnosed CdLS patients (from ref. 19 and BHCMG, respectively) 19. Y-axis, proportion of molecular diagnosis provided by variants in each gene; x-axis, genes; black, patients without CdLS listed as differential diagnosis; dark grey, patients with CdLS as one of the differential diagnoses; grey, CdLS cohort from ref. 19; light grey, CdLS cohort from BHCMG. C. Comparison of genic variant frequencies between COSMIC and ExAC cohorts. Filled circles represent comparison between frequencies of putative LoF variants between COSMIC and ExAC; open circles represent comparison between frequencies of missense variants between COSMIC and ExAC. Y-axis, ratio bewteen frequencies of genic variants (missense or putative LoF) in COSMIC and ExAC; x-axis, genes.
In addition to the aforementioned structural components, cohesin also interacts with the regulatory factors of the cohesion cycle, including proteins encoded by NIPBL (MIM *608667), MAU2 (MIM *614560), PDS5A (MIM *613200) or PDS5B (MIM *605333), WAPL (MIM *610754), HDAC8 (MIM *300269), ESCO1 (MIM *609674), and ESCO2 (MIM *609353), to facilitate cohesin dynamics and function on chromatin (Figure 1A)1,2.
Precise orchestration of cohesin’s structural components and regulatory factors ensures faithful progression of the cohesion cycle (Figure 1A). Defects of the structural or regulatory components of cohesin lead to various multisystem malformation syndromes described as “cohesinopathies”, a collection of syndromes with shared clinical findings such as distinctive facial features, growth retardation, developmental delay/intellectual disability (DD/ID), and limb abnormalities. Clinically, the most distinguishable type of cohesinopathy is the classic Cornelia de Lange Syndrome (CdLS, MIM# 122470), with the majority of cases explained by SNVs/indels and exonic deletion copy number variants (CNVs) resulting in loss-of-function (LoF) alleles in NIPBL4–6. The traditional phenotype-driven studies that included the mild end of the CdLS spectrum led to the discovery of SMC1A, SMC3, RAD21 and HDAC8 (MIM# 122470, 300590, 610759, 614701 and 300882) as new cohesinopathy genes 4–11. The resultant CdLS phenotype is largely dependent on the genes being affected and mutation types12. Although mild forms of CdLS present with less striking phenotypes and are more clinically challenging to recognize in comparison to the classic form, they have been found in an increasing number of patients with cohesinopathies.
Here, we used a genotype-driven approach to investigate the allelic series of genes encoding cohesin components based on a large cohort of patients (N=10,698) with a variety of unselected clinical presentations who were referred for clinical exome sequencing (CES). We identified pathogenic or likely pathogenic variants in known CdLS genes (NIPBL, SMC1A, SMC3, RAD21, and HDAC8) in patients mostly without a clinical diagnosis of CdLS, representing a cohort on the mild end of the clinical presentation of cohesinopathies. By applying the same genotype-first approach in the CES cohort, we further established STAG1 and STAG2 as new cohesinopathy genes with variants that act by a putative LoF mechanism, corroborating recent reports of patients with developmental disorders carrying mutations in these two genes13–15. Additional studies of patients who had chromosome microarray analyses (CMA, N=63,127) also identified deletion CNVs affecting STAG1 and STAG2, which further supports the human disease association of these two genes via a LoF mechanism. We also provide evidence supporting the candidacy of PDS5A and WAPL as cohesinopathy disease genes. Our findings emphasize the utility of using CES to provide molecular diagnoses for disorders with extensive genetic and phenotypic heterogeneity, uncover the potential molecular etiologies of previously undiagnosed patients, and elucidate novel candidate cohesinopathy disease genes which potentially expand the genotype/phenotype characterizations of cohesinopathies.
MATERIALS AND METHODS
Samples
The study has been conducted through a collaborative effort between Baylor Genetics (BG) and Baylor-Hopkins Center for Mendelian Genomics (BHCMG), and has been approved by the Institutional Review Board of Baylor College of Medicine. Approved consents of publishing photos have been obtained. Please see Supplemental Appendix for detailed descriptions of samples in BG and BHCMG. Selected patients with STAG1, STAG2, or PDS5A variants were enrolled after obtaining informed consent for further phenotypic characterization based on clinical notes submitted along with the CES order.
CES and variant interpretation
CES was performed as previously described16,17. The variant classification and interpretation were conducted by a clinical standard based on the American College of Medical Genetics and Genomics variant interpretation guidelines 18. Details of the CES experimental procedures and sample-wise QC metrics can be found in Table S1. The possibility of mosaic variants in known CdLS genes19 was carefully evaluated. A variant is considered mosaic only if the variant read versus total read ratio is below 30% and confirmatory Sanger sequencing demonstrates a comparable mosaic fraction.
The variants identified in this study have been submitted to ClinVar (accession numbers SCV000747051 - SCV000747093).
Chromosome microarray analysis (CMA)
The experimental design and data analysis of CMA were performed according to previously described procedures 20.
X-chromosome inactivation (XCI) assay
XCI studies were performed for the patient samples with STAG2 variants based on the protocol described by Allen et al21 with modifications. Please see Supplemental Appendix for detailed protocol.
Estimation of mutation prevalence in somatic cancer samples
The datasets from the COSMIC (http://cancer.sanger.ac.uk/cosmic/download) and ExAC (The Exome Aggregation Consortium, http://exac.broadinstitute.org/) 22 databases were used for the calculation. The normalized mutation abundance per gene in cancer samples is determined by the ratio between the mutation frequencies of COSMIC versus the ExAC (y-axis in Figure 1C). Please see Supplemental Appendix for details.
RESULTS
Variants of established CdLS genes in the CES cohort
Based on a genotype-driven selection approach, we identified 33 patients with pathogenic or likely pathogenic variants in the well-recognized CdLS genes from the CES cohort. Those variants include heterozygous or hemizygous SNVs/indels in NIPBL (N=5), SMC1A (N=14, X-linked), SMC3 (N=4), RAD21 (N=2) and HDAC8 (N=8, X-linked) (Table 1). Genic variant distribution was calculated to show the per gene contribution to molecular diagnosis among the five known CdLS genes (Figure 1B). Of the 33 variants, 29 occurred de novo in the proband, three were inherited from a parent and one was of unknown inheritance (not maternally inherited, paternal sample not available, Table 1). Among the inherited variants, one variant in SMC1A was inherited from a symptomatic mother with a milder phenotype, demonstrating variable clinical presentation for X-linked dominant disorders; two variants in RAD21 were inherited from symptomatic parents with milder phenotypes, documenting variable expressivity of defects in RAD21.
Table 1.
Summary of variants in the known CdLS genes identified by Baylor Genetics clinical exome sequencing.
| Gene (Transcript) | Genomic coordinates (hg19) | Exon/Intron | Coding sequence change | Protein change | Zygosity | Inheritance | Novelty | Classification c | CdLS as a differential diagnosis? | Dual molecular diagnosis? |
|---|---|---|---|---|---|---|---|---|---|---|
| NIPBL (NM_133433.3) | Chr5: 36985760 | exon10 | c.2479_2480del | p.R827Gfs*2 | Het | de novo | Reported 38 | P | no (prenatal) | no |
| Chr5: 37017173 | exon24 | c.4829T>C | p.L1610P | Het | de novo | This cohort | LP | no | no | |
| Chr5: 37046239 | exon38 | c.6527T>C | p.L2176P | Het | de novo | This cohort | LP | yes | no | |
| Chr5: 37052580 | exon42 | c.7175G>A | p.C2392Y | Het | de novo | This cohort | LP | no | no | |
| Chr5: 36962223 | intron5 | c.459-2A>G | splicing | Het | de novo | This cohort | P | yes, among others, POC d | no | |
| SMC1A (NM_006306.2) | ChrX: 53442118 | exon2 | c.G110T | p.G37V | Het | de novo | This cohort | LP | no | no |
| ChrX: 53442112 | exon2 | c.116C>G | p.S39* | Het | de novo | This cohort | P | yes | MYH2 heterozygous c.1160C>T (p.A387V), de novo | |
| ChrX: 53442100 | exon2 | c.128A>T | p.D43V | Het | de novo | This cohort | LP | no | no | |
| ChrX: 53442088 | exon2 | c.140T>G | p.F47C | Het | de novo | This cohort | LP | no | CFTR homozygous c.1521_1523del (p.F508del) | |
| ChrX: 53441930 | exon2 | c.298G>C | p.G100R | Het | de novo | This cohort | LP | no | EFHC1 heterozygous c.1612C>T (p.R538X), paternally inherited | |
| ChrX: 53440211 | exon4 | c.586C>T | p.R196C | Hem | de novo | This cohort | LP | no | DMD hemizygous deletion exons 49-51, maternally inherited | |
| ChrX: 53440048 | exon5 | c.655del | p.A219Lfs*45 | Het | de novo | This cohort | P | no | no | |
| ChrX: 53439899 | exon5 | c.802_804del | p.K268del | Het | de novo | Reported 39 | P | no | no | |
| ChrX: 53436051 | exon9 | c.1487G>A | p.R496H | Het | de novo | Reported 8 | P | no | no | |
| ChrX: 53430523 | exon15 | c.2394dup | p.R799Tfs*4 | Het | de novo | This cohort | P | no | no | |
| ChrX: 53430498 | exon15 | c.2420G>A | p.R807H | Het | de novo | This cohort | LP | no | no | |
| ChrX: 53426525 | exon16 | c.2547del | p.I849Mfs*12 | Het | de novo | This cohort | P | no | no | |
| ChrX: 53423152 | exon18 | c.2853_2856del | p.S951Rfs*12 | Het | de novo | Reported 40 | P | no | no | |
| ChrX: 53438853 | intron7 | c.1114-2A>G | splicing | Hem | Maternal a | This cohort | P | no | no | |
| SMC3 (NM_005445.3) | Chr10: 112341720 | exon9 | c.587T>C | p.I196T | Het | de novo | This cohort | LP | no | no |
| Chr10: 112349688 | exon15 | c.1453_1455del | p.A485del | Het | de novo | This cohort | LP | no | no | |
| Chr10: 112356303 | exon19 | c.2111T>C | p.I704T | Het | de novo | This cohort | LP | no | no | |
| Chr10: 112362647 | exon27 | c.3362C>T | p.S1121F | Het | de novo | This cohort | LP | yes | CREBBP heterozygous c.6137C>T (p.A2046V), de novo | |
| RAD21 (NM_006265.2) | Chr8: 117862926 | exon12 | c.1550dupC | p.E518fs | Het | Paternal a | This cohort | P | no | no |
| Chr8: 117866483 | intron10 | c.1161+1G>A | splicing | Het | Maternal a | This cohort | P | no | no | |
| HDAC8 (NM_018486.2) | ChrX: 71787758 | exon4 | c.418G>A | p.G140R | Het | de novo | This cohort | LP | no | no |
| ChrX: 71715066 | exon5 | c.490C>T | p.R164* b | Het | de novo | Reported 7 | P | no | no | |
| ChrX: 71715066 | exon5 | c.490C>T | p.R164* b | Het | de novo | Reported 7 | P | no | no | |
| ChrX: 71715029 | exon5 | c.527A>G | p.D176G | Het | de novo | This cohort | LP | no | IRX5 compound heterozygous c.1362_1368delinsGT (p.K455fs) and c.240_242delCTC (p.S81del) | |
| ChrX: 71710823 | exon6 | c.584T>A | p.V195D | Het | de novo | This cohort | LP | no | no | |
| ChrX: 71684526 | exon8 | c.793G>A | p.G265R | Het | de novo | This cohort | LP | no | no | |
| ChrX: 71681927 | exon9 | c.932C>T | p.T311M | Het | de novo | Reported 7 | P | no | no | |
| ChrX: 71681922 | exon9 | c.937C>T | p.R313* | Het | Not maternal | This cohort | P | no | no |
Inherited variants from mildly affected parents, who were confirmed to be non-mosaic by Sanger sequencing (data not shown);
Identical pathogenic variants in unrelated patients;
classifications include pathogenic (P) and likely pathogenic (LP);
POC, product of conception
The CdLS patients in this cohort may be enriched for atypical or mild CdLS phenotypes, because those with classic CdLS presentation are more likely to be referred for specific single gene or panel testing instead of CES. We retrospectively examined the clinical notes submitted by the referral clinicians for their differential diagnoses prior to CES. CdLS was not included in the initial differential diagnoses for 60% of patients with a positive NIPBL finding, 93% with SMC1A and 75% with SMC3 variants, and all those with RAD21 or HDAC8 variants (Table 1, Figure 1B). These observations support the previous hypotheses that pathogenic variants in NIPBL have a better correlation with classic CdLS, while SMC1A and SMC3 pathogenic variants may contribute to milder CdLS features; the phenotypes caused by pathogenic variants in RAD21 and HDAC8 become more variable and sometimes present atypical CdLS features12.
As a comparison to the genic distribution of our CES cohort, we analyzed the data from a phenotype-driven cohort of CdLS patients19. Moreover, we re-examined the genic variant distribution on an independent phenotype-driven CdLS cohort (N=41) from BHCMG, in which pathogenic or likely pathogenic variants in NIPBL (N=12), SMC1A (N=6), SMC3 (N=2), and HDAC8 (N=1) were identified (Table S2). The genic variant distribution of the BHCMG CdLS cohort is overall comparable with that calculated from the phenotype-driven cohort19. However, both of these largely deviated from our CES cohort (Figure 1B). The proportion of patients with NIPBL pathogenic variants in our cohort was significantly lower in comparison to the aforementioned two phenotype-driven cohorts (Chi-squared test, both with p < 0.001). The proportion of patients with SMC1A pathogenic variants in our cohort and the BHCMG were significantly higher than the other CdLS cohorts (Chi-squared test, both with p < 0.02), indicating mild/atypical CdLS presentations in the BHCMG cohort. Therefore, the mutational spectrum in known CdLS genes in the CES cohort represent a distinct distortion and alternative perspective from phenotype-driven CdLS cohorts, where patients tend to present with classic phenotypes11.
Interestingly, 6/33 (18%) of the patients with positive findings from known CdLS genes carry a secondary diagnosis (Table 1), which is higher than the average observed fraction of patients with dual diagnoses from positive cases in the entire CES cohort (~5%)23. This is not unexpected because the predicted extent of multi-locus diagnosis can be as high as 14% under a Poisson distribution model23. The high representation of dual diagnosis and resultant blended phenotypes observed in this study may contribute to the complexity of the patients’ phenotypes, further obscuring the underlying molecular causes, making clinical diagnosis challenging without the assistance from objective molecular testing.
Candidate disease genes in the cohesin structural and regulatory components
STAG1, STAG2, PDS5A, PDS5B, WAPL and MAU2 encode close interacting factors of NIPBL, SMC3, SMC1A, RAD21, and HDAC8 in the cohesin pathway, and thus may potentially supplement the locus heterogeneity of cohesinopathies. According to the ExAC database, NIPBL, SMC3, SMC1A and RAD21 have Probability of LoF Intolerance (pLI) scores of 1.00, while HDAC8 has a pLI of 0.92. Similarly, STAG1, STAG2, PDS5A, PDS5B, WAPL and MAU2 all have pLI scores of 1.00, suggesting their intolerance to LoF variants (Table S3). In our CES cohort, we identified putative LoF (truncating/splicing) or de novo missense variants in STAG1 (3), STAG2 (2), PDS5A (2), and WAPL (1). Through collaboration with the Deciphering Developmental Disorder (DDD) study and BHCMG, three additional de novo variants in STAG2 were identified.
De novo heterozygous SNVs/indels in STAG1 (NM_005862.2), including one frameshift (c.2009_2012del [p.N670Ifs*25]) and one missense (c.1129C>T [p.R377C]), were identified in Patients 1 and 2, respectively (Figure 2A). Both patients had common clinical findings that included DD/ID, hypotonia, seizures, mild dysmorphic features and skeletal abnormalities (Table 2, Table S4). In addition, one heterozygous de novo missense SNV, c.253G>A (p.V85I) in STAG1, was identified in Patient 3 (Figure 2A) along with a heterozygous de novo c.1720-2A>G SNV (observed twice in ExAC including one potentially being mosaic) in ASXL1 (Bohring-Opitz syndrome; MIM# 605039). Patient 3 presented with global developmental delay, dysmorphic facial features, seizures, optic atrophy, mild hypotonia, skin hypopigmentation, hirsutism, possible autism spectrum disorder and structural brain abnormalities (Table 2, Table S4). The concurrent de novo variants in STAG1 and ASXL1 could possibly contribute to a dual molecular diagnosis of this patient.
Figure 2.
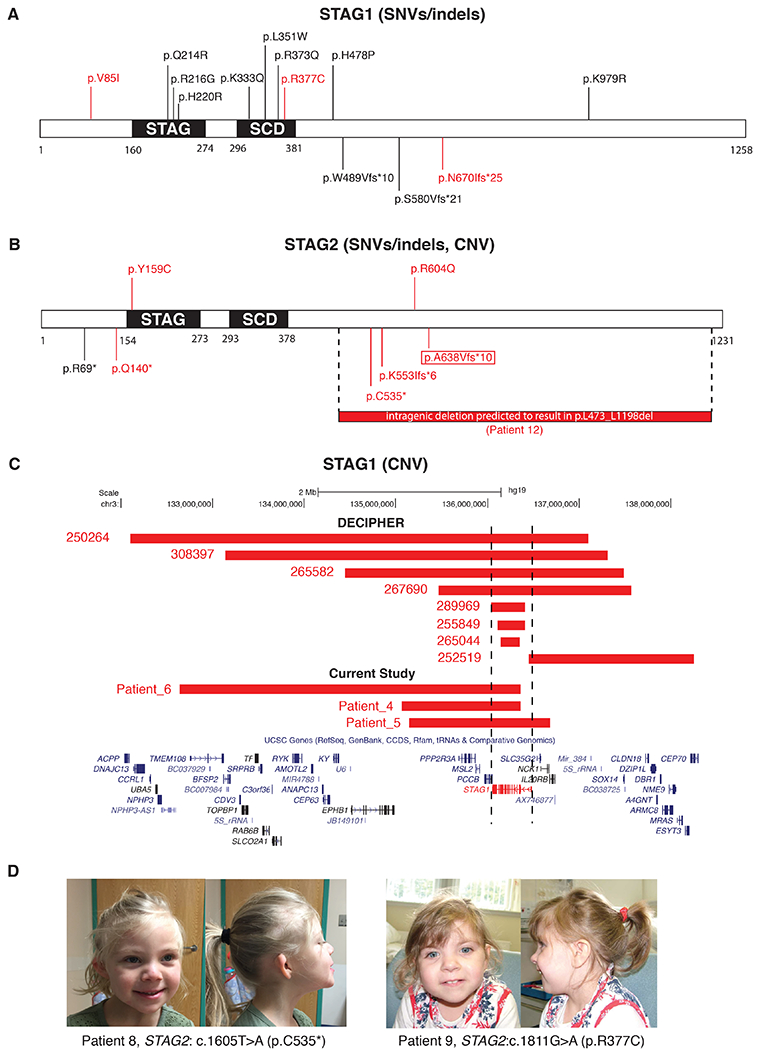
The variants in STAG1 and STAG2. A. SNVs/indels in STAG1. B. SNVs/indels and one CNV deletion in STAG2. For panels A and B, the white segment represents the full-length protein, and the black segments represent protein domains; the missense variants are annotated above the segment, while the putative LoF variants (including the CNVs deletion in STAG2) are underneath; the variants colored in red are reported in the current study. The boxed variant (p.A638Vfs*10) in panel B is reported as a research variant. C. Diagram showing the CNV deletions overlapping STAG1 reported in the DECIPHER and current study. The red segments represent the deletions, which are divided in two groups of “DECIPHER” and “Current Study”. The bottom panel shows genes in the region. STAG1 is highlighted in red. D. Photographs showing the front and side facial profiles of Patients 8 and 9 with de novo variant in STAG2. The patient numbers and variants are listed under the photograph.
Table 2.
Genotypes and phenotypes of patients with SNVs/indels in STAG1, STAG2 and PDS5A identified in current study.
| Genes | STAG1 (NM_005862.2) |
STAG2 (NM_006603.4) |
PDS5A (NM_001100399.1) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patients | Patient 1 | Patient 2 | Patient 3 | Reported in Ref. 13 (n=17) | Patient 7 | Patient 8 | Patient 9 | Patient 10 | Patient 11 | Reported in Ref 14 (n=1) | Patient 13 | |
| Age at last exam | 11 yr 5 mo | 4 yr 8 mo | 4 yr 2 mo | 30 mo to 33 yr (median 7 yr) | 3 yr 8 mo | 4 yr 6 mo | 11 yr 1 mo | 1 yr 11 mo | 5 yr 3 mo | 8 yr | 2 yr 6 mo | |
| Variant | c.2009_2012del (p.N670Ifs*25) |
c.1129C>T (p.R377C) |
c.253G>A (p.V85I): STAG1
c.1720-2A>G : ASXL1; |
CNV deletion or SNVs/indels |
c.418C>T (p.Q140*) |
c.1605T>A (p.C535*) |
c.1811G>A (p.R604Q) |
c.1658_1660delinsT (p.K553Ifs*6) |
c.476A>G (p.Y159C), hemizygous |
c.205C>T p.(Arg69*) |
c.2275G>T (p.E759*): PDS5A
c.3325A>T (p.K1109*): ASXL3; |
|
| Critical gene(s) | STAG1 | STAG1 | ASXL1, STAG1 | STAG1 and othersa | STAG2 | STAG2 | STAG2 | STAG2 | STAG2 | STAG2 | ASXL3, PDS5A | |
| Gender | F | M | F | 9M/8F | F | F | F | F | M | F | F | |
| Inheritance | de novo | de novo | both de novo | de novo or inherited | de novo | de novo | de novo | de novo | de novo | de novo | ASXL3: de novo; PDS5A: paternal | |
| Growth | IUGR | − | NR | − | 3/17 | − | + | − | − | − | − | − |
| Failure to thrive | − | + | − | 1/17 | NR | + | + | − | + | NR | + | |
| Short stature | − | + | − | 5/17 | NR | + | + | + | + | + | + | |
| Microcephaly | − | + | − | 4/17 | − | + | + | + | − | + | + | |
| Development | Intellectual disability | + | NR | + | 17/17, mild to severe | NR | + | + | + | + | + | + |
| Developmental Delay | + | + | + | 17/17 | + | + | + | + | + | + | + | |
| Autism Spectrum Disorder | NR | NR | + | 7/17 | NR | NR | NR | − | NR | − | − | |
| Neuro-behavioral | Behavioral problems | NR | + | + | NR | NR | + | NR | +,irritability | − | + | NR |
| Seizures | NR | +, during infancy | + | 7/17 with epilepsy | Myoclonic movements | NR | NR | + | − | NR | − | |
| Hypertonia | NR | − | − | NR | NR | NR | NR | − | − | NR | + | |
| Hypotonia | NR | + | + | 4/17 | NR | + | + | + | + | NR | − | |
| Craniofacial features | Brachycephaly | NR | NR | − | NR | − | + | NR | − | NR | micrognathia, ear abnormalities, wide-set eyes, beaked or prominent nose, arched eyebrows, or low-set ears, cleft/arched palate | − |
| Long curly eyelashes | + | NR | − | facial features included 14/17 with deep-set eyes, 13/17 with wide mouth, 7/17 with high nasal bridge, 8/17 with thin eyebrows, 4/17 with widely spaced central incisors | NR | + | + | − | − | NR | ||
| Synophrys | + | NR | + | NR | − | − | − | − | − | |||
| Anteverted nares | NR | NR | − | NR | NR | + | + | − | − | |||
| Depressed/broad nasal bridge | NR | + | + | − | − | + | + | + | + | |||
| Bulbous nasal tip | NR | NR | − | − | NR | + | + | + | − | |||
| Low-set ears | + | − | − | NR | NR | − | + | + | − | |||
| Dysmorphic ears | + | − | − | + | + | + | + | + | − | |||
| Long/smooth philtrum | − | NR | − | NR | + | + | − | − | − | |||
| High arched palate | NR | + | + | − | − | NR | +, narrow | NR | + | |||
| Thin upper lip | NR | NR | − | + | + | + | − | + | − | |||
| Downturned mouth | NR | NR | − | NR | NR | + | −,small | − | − | |||
| Cleft lip/palate | + | − | − | − | − | NR | − | + | − | |||
| Widely spaced teeth | NR | NR | + | NR | NR | + | NA | − | − | |||
| Micrognathia | NR | + | − | NR | + | + | + | − | − | |||
| Skin, Nails, Hair | Hypoplastic nails | − | NR | − | NR | + | NR | NR | +, with pits | − | NR | − |
| Hirsutism | NR | NR | + | NR | NR | + | NR | − | − | NR | − | |
| Hairline | NR | NR | − | NR | NR | low, posterior | NR | NR | NR | low, anterior | − | |
| Cutis marmorata | +,significant | NR | − | NR | NR | + | NR | − | − | NR | − | |
| Ocular | Strabismus | + | NR | +, exotropia | NR | NR | + | NR | NR | − | NR | − |
| Otolaryngologic | Hearing loss | − | − | − | NR | NR | +,conductive | NR | − | − | + | − |
| Cardiovascular | Congenital heart defect | − | PDA | − | 1/17 | +, Hypoplastic left heart, VSD, CA | NR | − | NR, no murmur | Minimal PFO, normal on follow-up | + | − |
| Respiratory/Thorax | Congenital diaphragmatic hernia | − | NR | − | NR | + | NR | +, right | − | NR | NR | − |
| Pulmonary hypoplasia | − | NR | − | NR | NR | NR | +, right | − | − | NR | − | |
| Gastrointestinal | Gastroesophageal reflux | NR | NR | − | 9/17 | +, Nissen and G-tube | + | + | − | − | NR | + |
| Genitourinary/Renal Anomaly | Hypoplastic male genitalia | NA | + | NA | NR | NA | NA | NA | NA | − | NR | NA |
| Cryptorchidism | NA | +, left | NA | 2/9 | NA | NA | NA | NA | − | NR | NA | |
| Structural anomalies of the renal tract | − | +, horseshoe kidney | NR, not examined | NR | − | NR | NR | − | +, Single kidney | NR | − | |
| Musculoskeletal/Extremities | Scoliosis | − | − | − | 2/17 | + | NR | NR | + | + | NR | − |
| Rib fusion | − | NR | − | NR | +, T4-5, T10-11, BL | NR | NR | + | − | NR | − | |
| Vertebral anomalies | − | NR | − | NR | +, vertebral clefts | NR | +, vertebral clefts | + | − | NR | − | |
| Arm /hand anomalies | − | + | − | NR | − | − | NR | − | NR | NR | − | |
| Limited elbow extension | + | NR | − | NR | NR | NR | NR | − | NR | NR | − | |
| Fifth finger clinodactyly | − | + | − | NR | NR | + | NR | − | NR | + | − | |
| Single transverse palmar crease | NR | + | − | NR | − | + | NR | − | NR | NR | − | |
| 2-3 toe syndactyly | + | + | − | NR | − | + | NR | − | − | + | − | |
| Studies and imaging | Abnormal Brain MRI | + | + | + | 3/17 showed atrophy; other 3/17 showed unspecific anomaly | NR | NR | NR | + | Ectopic posterior pituitary, short pituitary stalk | + | +,mild |
| Abnormal echocardiogram | − | NR | − | NR | + | NR | NR | NR | − | + | + | |
(BL bilateral; mo – months; yr-years; CA: coarctation of the aorta; IUGR - Intrauterine growth retardation; NA- not applicable; NR - no record; PDA- patent ductus arteriosus; PS - pulmonic stenosis; VSD - ventricular septal defect; PFO - patent foramen ovale)
STAG1 was affected by both CNV deletion and SNVs/indels. The deletions included 3 de novo and 1 unknown which encompassed STAG1 and PCCB, 1 intragenic which was absent in the mother, and 2 intragenic which were maternally inherited; the SNVs/indels included 8 de novo missense and 2 de novo frameshift variants of STAG1.
De novo heterozygous/hemizygous SNVs/indels in STAG2 (X-linked, NM_006603.4), including two stopgains, two missense and one frameshift, were identified in four females (Patient 7-10; Patient 7, c.418C>T [p.Q140*]; Patient 8, c.1605T>A [p.C535*]; Patient 9, c.1811G>A [p.R604Q]; Patient 10, c.1658_1660delinsT[p.K553Ifs*6]) and one male (Patient 11 (hemizygous), c.476A>G [p.Y159C]) (Figure 2B).These patients shared common clinical findings of DD/ID, hypotonia, microcephaly, dysmorphic features and skeletal abnormalities (Table 2, Table S4). Skewed X-inactivation (XCI) was observed in Patient 8, whereas XCI was non-informative for Patient 7 due to homozygosity of the marker being used for the XCI study (data not shown). In our study, truncating variants were identified in 3/4 female patients, but not in males. Although this observation is based on a limited number of patients, it is consistent with the hypothesis that truncating variants of X-linked genes may impose more severe pathogenic effect on males than females.
One heterozygous SNV, c.2275G>T (p.E759*), in PDS5A (NM_001100399.1) was identified in Patient 13 with severe developmental delay, marked hypotonia, failure to thrive, dysmorphic features, hyperextensible knees, eye anomalies and skeletal abnormalities (Table 2, Table S4). Interestingly, this patient also had a concurrent heterozygous de novo SNV, c.3325A>T (p.K1109*), in ASXL3 (Bainbridge-Ropers syndrome, MIM# 615485), which presumably explains the major phenotypes. This PDS5A variant is predicted to introduce a premature stop codon in PDS5A in the longer transcript (NM_001100399.1) but does not affect the shorter transcript (NM_001100400.1), suggesting a potential mild defect caused by this variant. However, the role of different isoforms of PDS5A in the cohesin complex is not well-established in the literature. Notably, the father of Patient 12, who shared the PDS5A p.E759* variant, had speech impediment. Although the pathogenicity of the p.E759* variant in PDS5A remains to be investigated, it may modulate the patient’s phenotype and constitute a dual diagnosis together with ASXL3. In addition, one heterozygous de novo SNV (c.654+5G>C) in PDS5A was identified in another patient with neurodevelopmental disorders. This intronic PDS5A variant was predicted to affect splicing of the major mRNA transcript of PDS5A by prediction programs including SpliceSiteFinder-like and MaxEntScan (http://www.interactive-biosoftware.com/doc/alamut-visual/2.6/splicing.html).
Finally, one de novo heterozygous SNV in WAPL (NM_015045.3), c.2192G>A (p.R731H) was identified in one patient with neurodevelopmental disorders. This observation corroborates a previous report in which a partial duplication involving WAPL was identified in a patient from a phenotype-driven CdLS cohort24, providing further evidence for WAPL as a candidate disease gene.
Each of the variants in STAG1, STAG2, PDS5A and WAPL described above were not observed in the control population databases including ExAC and ESP5400 (NHLBI Exome Sequencing Project, http://evs.gs.washington.edu/EVS/). The interpretation of deleterious effects of the de novo missense SNVs identified in this study was supported by multiple prediction algorithms (Table S5).
We identified CNV deletions affecting STAG1 and STAG2 in our clinical CMA cohort, supporting LoF as the presumed disease-contributing mechanism; no putative LoF CNVs of PDS5A, PDS5B, WAPL or MAU2 were identified. In total, we identified three CNV deletions affecting STAG1 (two de novo, one of unknown inheritance) in patients with developmental disorders (Figure 2C, Table S6). In the literature, six CNV deletions overlapping STAG1 were reported, with the smallest two deletions being intragenic (exons 2-5 and exons 13-18, respectively)13. Moreover, eight cases with neurodevelopmental disorders were reported in the DECIPHER database harboring relatively small-sized deletions (< 5 Mb) affecting STAG1 (https://decipher.sanger.ac.uk/)25 (Figure 2C, Table S6). These STAG1-overlapping deletions identified in affected patients strongly indicate that haploinsufficiency is likely to be the disease-contributing mechanism for STAG1. In addition, a 33.9 Kb CNV deletion with unknown inheritance encompassing exons 15-32 of STAG2 (predicted to result in an in-frame deletion p.L473_L1198del), was identified in Patient 12 with dysmorphic features, microcephaly and seizures (Figure 2B, Table S6). This female patient showed skewed XCI, consistent with the observation in Patient 8.
Patients with STAG1 and STAG2 variants have phenotypes overlapping the CdLS-spectrum
We evaluated the clinical phenotypes for Patient 1-2 (STAG1) and Patient 7-11 (STAG2). Patient 3 (STAG1) was excluded from the evaluation since the identification of concurrent de novo variants in ASXL1 together with STAG1 may largely complicate the STAG1-alone phenotypes.
Patients described in this paper presented for genetic evaluation due to developmental delay and/or congenital anomalies but not with classic distinctive facial features or a recognizable pattern of malformation suggestive for a particular syndrome such as CdLS (Figure 2D). The most common features among these patients with STAG1 and STAG2 variants were DD/ID, behavioral problems, hypotonia, seizures, microcephaly, failure to thrive, short stature, mild dysmorphic features, and 2-3 toe syndactyly (Table 2).
Clinical profiling suggested many overlapping features with CdLS, which include DD/ID, growth failure including short stature and microcephaly, hearing loss, synophrys, micrognathia, limb anomalies and hypoplastic male genitalia. Some other less common features of CdLS, such as cutis marmorata, myopia, congenital diaphragmatic hernia (CDH), and renal anomalies among others, were also observed in several of these patients. A more detailed characterization is described in Table 2 and Table S4.
Among the distinctive craniofacial features present in over 95% of the patients with a clinical diagnosis of CdLS11, our patients collectively had microbrachycephaly, low set ears, synophrys, long curly eyelashes, broad nasal bridge, anteverted nares, long and smooth philtrum, thin upper lip and micrognathia; however, these features were not present concurrently in a single patient. Interestingly while microcephaly is one of the most characteristic features in CdLS, only 4/7 patients (one STAG1 and three STAG2) had microcephaly. Although the numbers are small, a higher percentage of microcephaly was observed in patients with a STAG2 variant (3/5) in comparison to STAG1 (1/2). In contrast to CdLS, where mild to severe limb anomalies are common and are usually helpful to establish a clinical diagnosis, the patients in this study had common but more subtle findings in their extremities, such as fifth finger clinodactyly and syndactyly. Skeletal anomalies including scoliosis (3/7), vertebral anomalies (3/7) and rib fusion (2/7) were observed in our patients, all with variants in STAG2. Even though these skeletal anomalies can be observed in patients with classic CdLS, vertebral and rib anomalies would be considered as rare or atypical features for CdLS.
Comparing patients with STAG1 or STAG2 variants, DD/ID and mild dysmorphic features have been consistently observed, which is in line with the previous reports13–15 (Table 2). Despite the small cohort size, it seems that patients with STAG2 variants have more multisystem congenital anomalies such as CDH, congenital heart disease and vertebral anomalies. Growth failure was observed as well, but apparently more in the postnatal period than prenatally. Patients with a STAG2 variant appear to have more severe growth failure especially in weight and length parameters compared to those with STAG1 variants.
Although STAG1 and STAG2 have been implicated in cancers due to their function in the cohesin pathway and the observation of chromosomal segregation defects in defective cell lines (e.g. STAG2 as an indicator for myeloid neoplasms), onset of tumors has not been observed in our study nor in the patients reported in the literature with developmental disorders caused by constitutional pathogenic variants in STAG1 and STAG213–15. Moreover, no obvious increased risk of cancer is reported in patients with other cohesinopathies caused by defects in genes such as NIPBL, SMC1A, and SMC31. Consistent with this observation, our chromosome analysis of one patient (Patient 7) did not reveal any evidence for chromosomal segregation defects (data not shown).
DISCUSSION
In this study, we applied a genotype-driven approach to decipher the genetic causes of cohesinopathy from a CES perspective. We describe a series of disease-contributing variants in known cohesinopathy genes, and also provide molecular evidence supporting the candidacy of recently described or new disease genes.
NIPBL defects are underrepresented in this cohort likely due to ascertainment bias associated with its more clinically recognizable presentations. The scarcity of putative LoF variants for certain cohesin genes including PDS5B and MAU2 in this cohort indicates that LoF variants in these genes may exert strong pathogenic effects on early development leading to incompatibility with life. Alternatively, the lack of evidence supporting the pathogenicity of variants in PDS5B and MAU2 could reflect limitations of interpreting missense variants based on proband-only CES. HDAC8 and SMC1A are the only two well-studied X-linked genes among the cohesin components. They seem to be relatively spared from the strong selection in human development possibly due to protection of pathogenic alleles in the gene pool by XCI in females. Consistently, variants in these two genes are highly represented in the CES cohort as compared to cohorts assembled by phenotypic characterization (Figure 1B).
Patients harboring STAG1 or STAG2 variants seem to share many of the clinical features seen in the well-described CdLS phenotype. Apparently affected patients in our cohort are developmentally and intellectually as impaired as those with CdLS. However, their spectrum of growth, craniofacial and musculoskeletal features are not as severe as the spectrum of CdLS. Overall, only one patient (Patient 3 [STAG1]) fulfills the diagnostic criteria for CdLS by meeting the CdLS characteristic facial features 26. Note that the concurrent de novo variant in ASXL1 may largely contribute to the differential diagnosis of CdLS for Patient 3 (Table S7). Although the currently available clinical information we had might not be as sufficient for a diagnosis of CdLS or other cohesinopathies, a “CdLS-like” syndrome started to emerge. The STAG1/STAG2-related disorders seem to be at the mild end of the CdLS spectrum, making the clinical diagnosis for these two genes more challenging for physicians. Putting together the constellation of clinical features might help to end the diagnostic odyssey earlier, and with this series of cases awareness can be extended. Given the challenges, comprehensive genomic analysis, such as CES, should be offered to efficiently provide a molecular diagnosis for these cohesinopathy conditions.
Notably, the LoF PDS5A variant (Patient 13) was inherited from a father with speech impediment. Although the phenotypic consequence of this variant remains unclear (as discussed in the RESULTS), its potential contribution cannot be completely ruled out. Unfortunately, samples from the parental grandparents or other relatives are not available for testing. Defects in the cohesin complex, as demonstrated in the CdLS genes, are likely to be detrimental to proper organismal development, milder phenotypic consequences have been observed11. With our experience of known CdLS gene variants among 10,698 individuals, two distinct novel pathogenic variants in RAD21 as well as one novel pathogenic variant in SMC1A (X-linked) were identified in three unrelated patients with neurodevelopmental disorders, all inherited from affected parents with milder phenotypes (Table 1). Moreover, transmission of pathogenic variant between generations has been reported in STAG113. Therefore, with the reported variable expressivity of the cohesin defects, it is plausible that the reproductive potential, genetic transmission and severity of phenotype may be dependent on various factors, including the components being affected, the mutation types, the inheritance mode (e.g. X-linked or autosomal dominant) and the downstream pathways disrupted by defects in a particular component. Thus, additional genotype-phenotype correlation studies are warranted to further delineate the spectrum of cohesinopathies.
The mutational landscape of cohesin genes in somatic cancer may represent an alternative view to reflect contribution of these genes to biological processes, with minimum selection as compared to that imposed during early human development. Among cancer samples deposited to the COSMIC database subjected to genome wide screening, truncating variants were observed in all cohesin genes. While missense variants did not show any substantive difference between cohesin genes, putative LoF variants in STAG2 were highly represented in the somatic cancer cohort (Figure 1C). LoF variants in STAG2 have been significantly associated with several cancers27,28, suggesting a likely pleiotropic effect of STAG2, possibly with strong involvement in tumorigenesis. Interestingly, we have observed a patient with mosaic STAG2 LoF variant in the CES cohort. The patient does not have neurodevelopmental problems, but instead presented with hematological malignancy. Therefore, we considered the STAG2 defect in this patient as not being causal for a cohesinopathy. Consequently, caution should be taken when interpreting variants in cohesin genes by considering the possibility that they may arise as somatic changes after the critical period of early human development.
Accumulating evidence suggest that cohesin contributes to the topological organization of the genome, regulates DNA replication, and facilitates long-range gene transcription regulation2,29,30. In addition, the interactions between cohesin and other transcription machinery and chromatin remodeling complexes to recognize specific genomic loci and regulate gene transcription have aggregated these complexes into the same pathways of transcription regulation30–33. Notably, genes encoding components of chromosome remodeling and transcription regulation machineries, such as ANKRD11, AFF4, KMT2A, TAF1 and TAF6, have been associated with phenotypes reminiscent of CdLS3,19,34–36. Such findings expand the molecular mechanism underlying cohesinopathies into transcriptional regulation. Interestingly, gene expression studies of patients with elevated dosage of STAG2 reveal a dysregulated transcriptome and pinpoints altered expression levels of developmentally important genes37. Therefore, the versatility of cohesin in cohesion and transcription regulation warrants a further investigation of its downstream effectors.
In summary, the genotype-first approach focusing on a specific pathway enabled us to investigate patients with non-classic cohesinopathy phenotypes; this approach also allowed us to discover patients with variants in new or recently reported disease genes, namely STAG1, STAG2, and potentially PDS5A and WAPL, which may further expand the genetic heterogeneity underlying cohesinopathies. Future studies of cellular phenotypes, with regard to functional studies of DNA repair and transcriptome analysis, are warranted to further elucidate the mechanistic consequences due to defects in specific cohesin components, which may shed light on precision medicine efforts targeting distinct molecular pathways.
Supplementary Material
Acknowledgement
This study was supported in part by the National Human Genome Research Institute/National Heart Lung and Blood Institute (NHGRI/NHLBI) grant UM1HG006542 to the BHCMG; and National Institutes of Neurological Disorders and Stroke (NINDS) grant R35 NS105078-01 to JRL. JEP was supported by the NHGRI grant K08 HG008986. We acknowledge Dr. Sureni V. Mullegama for critical comments on this manuscript.
Footnotes
Disclosure statements
Baylor College of Medicine (BCM) and Miraca Holdings Inc. have formed a joint venture with shared ownership and governance of Baylor Genetics (BG), formerly the Baylor Miraca Genetics Laboratories (BMGL), which performs chromosomal microarray analysis and clinical exome sequencing. JR, VP, WJ, CS, WB, SWC, AMB, JLS, CE, YY, RX and PL are employees of BCM and derive support through a professional services agreement with the BG. JRL serves on the Scientific Advisory Board of the BG. JRL has stock ownership in 23andMe, is a paid consultant for Regeneron Pharmaceuticals, has stock options in Lasergen, Inc and is a co-inventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases and bacterial genomic fingerprinting. Other authors have no disclosures relevant to the manuscript. All authors read and approved the final manuscript.
This study makes use of data generated by the DECIPHER community. A full list of centres who contributed to the generation of the data is available from http://decipher.sanger.ac.uk and via email from decipher@sanger.ac.uk. Funding for the project was provided by the Wellcome Trust.
The DDD study presents independent research commissioned by the Health Innovation Challenge Fund [grant number HICF-1009-003], a parallel funding partnership between the Wellcome Trust and the Department of Health, and the Wellcome Trust Sanger Institute [grant number WT098051]. The views expressed in this publication are those of the author(s) and not necessarily those of the Wellcome Trust or the Department of Health. The study has UK Research Ethics Committee approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12 granted by the Republic of Ireland REC). The research team acknowledges the support of the National Institute for Health Research, through the Comprehensive Clinical Research Network.
References
- 1.Liu J, Krantz ID. Cornelia de Lange syndrome, cohesin, and beyond. Clin. Genet October 2009;76(4):303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Losada A Cohesin in cancer: chromosome segregation and beyond. Nat. Rev. Cancer. June 2014;14(6):389–393. [DOI] [PubMed] [Google Scholar]
- 3.Yuan B, Pehlivan D, Karaca E, et al. Global transcriptional disturbances underlie Cornelia de Lange syndrome and related phenotypes. J. Clin. Invest February 2015;125(2):636–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krantz ID, McCallum J, DeScipio C, et al. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat. Genet June 2004;36(6):631–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pehlivan D, Hullings M, Carvalho CM, et al. NIPBL rearrangements in Cornelia de Lange syndrome: evidence for replicative mechanism and genotype-phenotype correlation. Genet. Med March 2012;14(3):313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tonkin ET, Wang TJ, Lisgo S, Bamshad MJ, Strachan T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat. Genet June 2004;36(6):636–641. [DOI] [PubMed] [Google Scholar]
- 7.Deardorff MA, Bando M, Nakato R, et al. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature. September 13 2012;489(7415):313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deardorff MA, Kaur M, Yaeger D, et al. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of cornelia de Lange syndrome with predominant mental retardation. Am. J. Hum. Genet March 2007;80(3):485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deardorff MA, Wilde JJ, Albrecht M, et al. RAD21 mutations cause a human cohesinopathy. Am. J. Hum. Genet June 8 2012;90(6):1014–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Musio A, Selicorni A, Focarelli ML, et al. X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat. Genet May 2006;38(5):528–530. [DOI] [PubMed] [Google Scholar]
- 11.Deardorff MA, Noon SE, Krantz ID. Cornelia de Lange Syndrome GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2017. 2005. [PubMed] [Google Scholar]
- 12.Mannini L, Cucco F, Quarantotti V, Krantz ID, Musio A. Mutation spectrum and genotype-phenotype correlation in Cornelia de Lange syndrome. Hum. Mutat December 2013;34(12):1589–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lehalle D, Mosca-Boidron AL, Begtrup A, et al. STAG1 mutations cause a novel cohesinopathy characterised by unspecific syndromic intellectual disability. J. Med. Genet January 24 2017. [DOI] [PubMed] [Google Scholar]
- 14.Mullegama SV, Klein SD, Mulatinho MV, et al. De novo loss-of-function variants in STAG2 are associated with developmental delay, microcephaly, and congenital anomalies. Am. J. Med. Genet. A. May 2017;173(5):1319–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soardi FC, Machado-Silva A, Linhares ND, et al. Familial STAG2 germline mutation defines a new human cohesinopathy. NPJ genomic medicine. 2017;2:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med October 17 2013;369(16):1502–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. November 12 2014;312(18):1870–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med May 2015;17(5):405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ansari M, Poke G, Ferry Q, et al. Genetic heterogeneity in Cornelia de Lange syndrome (CdLS) and CdLS-like phenotypes with observed and predicted levels of mosaicism. J. Med. Genet October 2014;51(10):659–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gambin T, Yuan B, Bi W, et al. Identification of novel candidate disease genes from de novo exonic copy number variants. Genome Med September 21 2017;9(1):83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am. J. Hum. Genet December 1992;51(6):1229–1239. [PMC free article] [PubMed] [Google Scholar]
- 22.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. August 18 2016;536(7616):285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Posey JE, Harel T, Liu P, et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N. Engl. J. Med January 05 2017;376(1):21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pehlivan D, Erdin S, Carvalho CMB, et al. Evidence implicating cohesin/condensin gene non-coding CNVs in the Cornelia de Lange. Genomic Disorders 2012: The Genomics of Rare Diseases. 2012. [Google Scholar]
- 25.Firth HV, Richards SM, Bevan AP, et al. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet April 2009;84(4):524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kline AD, Krantz ID, Sommer A, et al. Cornelia de Lange syndrome: clinical review, diagnostic and scoring systems, and anticipatory guidance. Am. J. Med. Genet. A. June 15 2007;143A(12):1287–1296. [DOI] [PubMed] [Google Scholar]
- 27.Solomon DA, Kim JS, Bondaruk J, et al. Frequent truncating mutations of STAG2 in bladder cancer. Nat. Genet December 2013;45(12):1428–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Solomon DA, Kim T, Diaz-Martinez LA, et al. Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science. August 19 2011;333(6045):1039–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sofueva S, Yaffe E, Chan WC, et al. Cohesin-mediated interactions organize chromosomal domain architecture. EMBO J December 11 2013;32(24):3119–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kagey MH, Newman JJ, Bilodeau S, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. September 23 2010;467(7314):430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rubio ED, Reiss DJ, Welcsh PL, et al. CTCF physically links cohesin to chromatin. Proc. Natl. Acad. Sci. U. S. A June 17 2008;105(24):8309–8314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Allen BL, Taatjes DJ. The Mediator complex: a central integrator of transcription. Nat. Rev. Mol. Cell Biol March 2015;16(3):155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strubbe G, Popp C, Schmidt A, et al. Polycomb purification by in vivo biotinylation tagging reveals cohesin and Trithorax group proteins as interaction partners. Proc. Natl. Acad. Sci. U. S. A April 05 2011;108(14):5572–5577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Izumi K, Nakato R, Zhang Z, et al. Germline gain-of-function mutations in AFF4 cause a developmental syndrome functionally linking the super elongation complex and cohesin. Nat. Genet April 2015;47(4):338–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Rawe JA, Wu Y, Dorfel MJ, et al. TAF1 Variants Are Associated with Dysmorphic Features, Intellectual Disability, and Neurological Manifestations. Am. J. Hum. Genet December 03 2015;97(6):922–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parenti I, Gervasini C, Pozojevic J, et al. Broadening of cohesinopathies: exome sequencing identifies mutations in ANKRD11 in two patients with Cornelia de Lange-overlapping phenotype. Clin. Genet January 2016;89(1):74–81. [DOI] [PubMed] [Google Scholar]
- 37.Kumar R, Corbett MA, Van Bon BW, et al. Increased STAG2 dosage defines a novel cohesinopathy with intellectual disability and behavioral problems. Hum. Mol. Genet December 20 2015;24(25):7171–7181. [DOI] [PubMed] [Google Scholar]
- 38.Gillis LA, McCallum J, Kaur M, et al. NIPBL mutational analysis in 120 individuals with Cornelia de Lange syndrome and evaluation of genotype-phenotype correlations. Am. J. Hum. Genet October 2004;75(4):610–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu J, Feldman R, Zhang Z, et al. SMC1A expression and mechanism of pathogenicity in probands with X-Linked Cornelia de Lange syndrome. Hum. Mutat November 2009;30(11):1535–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goldstein JH, Tim-Aroon T, Shieh J, et al. Novel SMC1A frameshift mutations in children with developmental delay and epilepsy. Eur. J. Med. Genet October 2015;58(10):562–568. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.