

Abstract
Pulmonary arterial hypertension (PAH) is a complex degenerative disorder marked by aberrant vascular remodeling associated with hyperproliferation and migration of endothelial cells (ECs). Previous reports implicated bone morphogenetic protein antagonist Gremlin 1 in this process; however, little is known of the molecular mechanisms involved. The current study was designed to test whether redox signaling initiated by NADPH oxidase 1 (Nox1) could promote transcription factor CREB activation by redox factor 1 (Ref-1), transactivation of Gremlin1 transcription, EC migration, and proliferation. Human pulmonary arterial EC (HPAECs) exposed in vitro to hypoxia to recapitulate PAH signaling displayed induced Nox1 expression, reactive oxygen species (ROS) production, PKA activity, CREB phosphorylation, and CREB:CRE motif binding. These responses were abrogated by selective Nox1 inhibitor NoxA1ds and/or siRNA Nox1. Nox1-activated CREB migrated to the nucleus and bound to Ref-1 leading to CREB:CRE binding and Gremlin1 transcription. CHiP assay and CREB gene-silencing illustrated that CREB is pivotal for hypoxia-induced Gremlin1, which, in turn, stimulates EC proliferation and migration. In vivo, participation of Nox1, CREB, and Gremlin1, as well as CREB:CRE binding was corroborated in a rat PAH model. Activation of a previously unidentified Nox1-PKA-CREB/Ref-1 signaling pathway in pulmonary endothelial cells leads to Gremlin1 transactivation, proliferation and migration. These findings reveal a new signaling pathway by which Nox1 via induction of CREB and Gremlin1 signaling contributes to vascular remodeling and provide preclinical indication of its significance in PAH.
Keywords: Nox1, CREB, Proliferation, Gremlin1, Ref-1, Migration
Graphical abstract
1. Introduction
Pulmonary arterial hypertension (PAH) is a complex and progressive disease characterized by elevated pulmonary vascular resistance (PVR) and pulmonary arterial pressure (PAP) leading to an increase in the right ventricle overload, which ultimately promotes right heart maladaptive hypertrophy and failure [1], [2], [3]. PAH is often initiated with vascular endothelial cell (ECs) injury followed by changes intrinsically associated with vascular remodeling including fast and aggressive hyperproliferation of apoptotic-resistant ECs, cell death, cell migration, and cell differentiation [4], [5], [6], [7]. EC dysfunction is known to yield increases in the levels of various endothelial vasoactive mediators, i.e., endothelin-1, nitric oxide, serotonin, and prostacyclin, which promote vascular smooth muscle cells (VSMCs) migration and proliferation [5], [8]. Disordered EC and VSMC proliferation, when hyperproliferative, elicit the formation of plexiform lesions, glomeruloid-like vascular structures originating from pulmonary arterioles [9], [10], [11]. These processes culminate in a common pathological feature of partial and obliterative occlusions and increased pulmonary vascular resistance in patients with PAH [10], [11]. To date, however, the underlying mechanisms central to EC proliferation in PAH are not well understood.
NADPH oxidase (Nox) is a major source of reactive oxygen species (ROS), including superoxide anion (O2•-), and hydrogen peroxide (H2O2), in the vasculature [12], [13], [14]. Studies have implicated an essential role of Nox-derived ROS in the progression and development of PAH [15]. Specifically, Nox1 oxidase is comprised of 2 membrane-bound components, Nox1 and p22phox and normally distinct organizing and activating subunits NoxO1 and NoxA1 [13], [16]. For the purposes of this study, we focus on Nox1 which was demonstrated by our group to be associated with the bone morphogenetic protein receptor antagonist Gremlin1-driven pulmonary endothelial cell proliferation and PAH [13]. However, the mechanisms by which Nox1-induced cell signaling promotes Gremlin1 transcription, and, in turn, EC hyperplasia and migration in PAH are not known.
PAH is triggered by several stimuli whose mechanism of action mimics changes caused by chronic hypoxia (CH) [17]. CH exposure is well known to induce changes in the structure of pulmonary arteries via shifts in cellular phenotype involving a variety of factors both genomic and non-genomic [18], [19]. Essential to this process is the activation of transcription factors that promote hyperplasia, migration and vascular remodeling [20]. Among these, cAMP response element-binding protein (CREB) is known to be activated by hypoxia [21]. Phosphorylation at serine 133 of CREB promotes its translocation to the nucleus - regulating gene transcription by binding at the cAMP response element (CRE) on CREB-regulated genes [22], [23]. The basic leucine zip domain (bZIP) of CREB plays a key role in promoting its binding at the CRE motif [22], [23], which is a conserved eight-base-pair palindromic sequence TGACGTCA [24]. In this way, Goren et al. demonstrated that reduction of cysteine 300 and 310 residues in the bZIP domain of CREB enhances its binding efficiency to the CRE motif and consequently promotes activation of CREB-regulated genes [25].
Furthermore, it has been proposed that redox factor 1 (Ref-1), via its reducing potential, enhances the activation of a variety of transcription factors including CREB [26], [27]. Thus, we postulated that Nox1 mediates CREB and Ref-1 interaction, and activation of CREB, leading to an increase in CREB DNA binding at the CRE motif of human Gremlin1, Gremlin1 transcription, and EC activation and PAH. Indeed, a causal relationship between Nox1, CREB, Ref-1, Gremlin1 and ECs in PAH is entirely unknown.
2. Methods and materials
2.1. Reagents
Catalase, SOD and propidium iodide were purchased from Sigma–Aldrich (St. Louis, MO, U.S.A.). Protease and phosphatase inhibitor cocktail tablets were purchased from Roche Diagnostics GmbH (Mannheim, Germany). Silencer select siRNA against Nox1 (s25728), CREB (s3489), Ref-1 (s1446) were purchased from Thermo Fisher (Walthan, MA, U.S.A). Antibodies for phospho CREB (87G3) and total CREB (48H2), PKARI (D54D9), and Histone H3 (9715) were purchased from Cell Signaling Technology (Danvers, MA, U.S.A.). Nox1 (ab131088), Nox2 (ab80508), Nox4 (ab154244), Ref-1 (ab194) and Gremlin1 (ab140010) antibodies were purchased from Abcam (Cambridge, MA, U.S.A.). PCNA (sc-9857) and β-actin (sc-47778) were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, U.S.A.). Rabbit (925–68070), mouse (925–68071), and goat (925–68074) secondary antibodies were purchased from LI-COR Biosciences (Lincoln, NE, U.S.A.). Nuclear extract kit (Cat. 40010) and pCREB TransAM© transcription factor ELISA kit (Cat. 43096) were purchased from Active Motif (Carlsbad, CA, U.S.A.). PKA activity kit (Cat. EIAPKA) was purchased from Thermo Fisher Scientific (Waltham, MA, U.S.A.). EZ Chromatin immunoprecipitation kit (EZ-CHiP assay kit, Cat. 17-371) was purchased from Millipore-Sigma (Burlington, MA, U.S.A). CBA (Coumarin 7-Boronic Acid) (Cat. 1357078–03-5) was purchased from Cayman Chemical (Ann Arbor, Michigan, U.S.A.). HPr+ (Hydropropidine) was a generous gift from Dr. Jacek Zielonka (Department of Biophysics, Medical College of Wisconsin, U.S.A.).
2.2. Cell culture and treatment
Human pulmonary arterial endothelial cells (HPAECs – CC2530; Lonza, Walkersville, MD, U.S.A.) were grown in EBM-2 medium containing EGM-2 bullet kit components (CC-3182, Lonza, Walkersville, MD, U.S.A.). Cells between passages 3 and 6 were used in all the experiments. Cells were incubated in either normoxia (21% oxygen) or hypoxia (1% oxygen) for 24 h and subjected to either homogenization in ice-cold disruption buffer (RIPA buffer containing 0.1 mM protease and phosphatase inhibitor) or trypsinized for whole cell analysis. HPAECs were grown on 6-well plates to 70–80% confluence and subjected to Nox1 (10 nM), CREB (10 nM), Ref-1 (10 nM), and scrambled control (10 nM) siRNA for 24 h (Silencer select - Life Technologies) using Lipofectamine 3000 transfection reagent (Life Technologies). Cells were synchronized in serum-reduced media (0.2% FBS) for 16 h. Media was removed and replaced with complete serum medium (2% FBS) before hypoxia or normoxia treatment. Gene silencing was confirmed using immunoblotting (detailed below), and knockdown was normalized by comparison to siRNA scrambled control.
2.3. Sugen 5416/hypoxia rat model
All animal studies were performed under a protocol approved by the IACUC of the University of Pittsburg. To induce PAH, the SU5416 (Sugen)/hypoxia model was employed as previously described [28], [29] with modifications. Male rats (8 per group; 250–275 g; Charles River Laboratories, Cambridge, MA) were injected with a single subcutaneous bolus of SU5416 (20 mg/kg) (S8442, Sigma, St. Louis, MO) and subsequently subjected to hypoxia (10% O2) for 3 weeks, followed by 1 week of normoxia (21% O2). Lung tissue was obtained, protein concentrations were measured, and samples were processed for Western blot analysis for protein expression.
2.4. Western blot
Western blot experiments were performed as described previously [30], [31]. Briefly, HPAECs seeded into six-well tissue culture dishes were transfected with scrambled, and/or Nox1, CREB, Ref-1 siRNA, and serum deprived (0.2% FBS) for 16 h. After the requisite incubation time, serum-deprived media was replaced for complete media (2% FBS), and cells were exposed to hypoxia (1% O2, 24 h). Total protein (30–45 μg) from cell lysates was added to Tris-glycine SDS sample buffer, boiled, resolved with SDS/PAGE, and transferred onto Trans-blot nitrocellulose membranes (Bio-Rad). Membranes were blocked with the Odyssey Blocking Buffer (LI-COR Biosciences, Lincoln, NE) and incubated with rabbit anti-Nox1 (1:1000), rabbit anti-Grem1 (1:1000), mouse anti-Ref-1 (1:2000), rabbit anti-Nox2 (1:1000), rabbit ant-Nox4 (1:1000), rabbit anti-phosphorylated and total CREB (1:1000), goat anti-PCNA (1:1000), mouse anti-β-actin (1:5000), or rabbit ant-histone H3 (1:2000). Membranes were probed with anti-rabbit, anti-mouse, or anti-goat secondary antibodies (1:5000 dilution, LI-COR Biosciences). In some cases blots were cut in strips to probe for more than one antibody at the same time. Digital images were captured using the Odyssey Infrared Imaging system (LI- OR Biosciences). Optical density (OD) of protein-of-interest bands were quantitated and normalized to β-actin or histone H3 using ImageJ software (NIH, U.S.A.).
2.5. Co-immunoprecipitation (Co-IP)
HPAEC were seeded in 100 mm dishes to 80% confluence and serum-deprived (0.2% FBS) for 16 h. After the requisite incubation time, serum-deprived media was replaced for complete media (2% FBS), and cells were exposed to hypoxia (1% O2, 24 h). Next, cells were washed with ice-cold PBS/phosphatase inhibitors and collected for further nuclear protein extraction. The nuclear extract kit was employed according to the manufacturer’s instructions (TransAm; Active Motif, Carlsbad, CA, USA). For the Co-IP, rabbit anti-pCREB antibody (87G3, Cell Signaling Technology) was complexed to protein A/G plus agarose (sc-2003, Santa Cruz Biotechnology) by incubation for 10 h/4 °C, and incubated with 250 µg of nuclear protein sample overnight at 4 °C. Antibody/protein complex was then subjected to Western Blot using mouse anti-Ref-1 antibody (ab194, Abcam). Digital images were captured using the Odyssey Infrared Imaging system (LI- OR Biosciences). Optical density (OD) of protein-of-interest bands were quantitated and normalized to Ponceau S using ImageJ software (NIH, U.S.A.). Data are presented as a fold change from normoxia control.
2.6. Detection of PKA oxidation
For PKA oxidation detection, HPAECs grown on 6-well plates to 70–80% confluence and serum-deprived (0.2% FBS) for 16 h and were treated with 100 µM of hydrogen peroxide (H2O2) for 10 min. Cells were quickly washed with PBS and subjected to homogenization in ice-cold disruption buffer (RIPA buffer containing protease and phosphatase cocktail inhibitors – Easy Pack – Roche, Basel, Switzerland), incubated for 20 min on ice, centrifuged for 15 min at 10,000g, and whole-cell protein lysate was obtained. Next, a reducing and non-reducing SDS/PAGE followed by Western blot was performed. Reducing conditions identify total PKA, and non-reducing conditions allow for the detection of both the oxidized and dimerized PKA protein as well as the monomeric form [32]. Membranes were probed with rabbit anti-PKA RI (1:1000) overnight on 4 °C and anti-rabbit secondary antibody (1:5000) for 1h. Digital images were captured using the Odyssey infrared imaging system and quantitated by Image J software as described in the Western Blotting section.
2.7. PKA activity
For PKA activity, a protein kinase A colorimetric kit assay was employed according to the manufacturer’s instructions (Thermo Fisher). Briefly, total cell extracts from HPAECs transfected with scrambled or Nox1 siRNA and subjected to hypoxia (1% O2, 24 h) or normoxia were incubated for 90 min with an immobilized PKA substrate bound to a microtiter plate. Samples containing active PKA will, in the presence of the supplied ATP, phosphorylate the immobilized PKA substrate. Next, samples were washed, and this was followed by a second incubation for 60 min with a goat-anti-rabbit IgG HRP-conjugated and phospho PKA substrate antibody in each well. Next, after washing, a third incubation with TMB substrate for 30 min was performed and followed by stop solution. Recombinant PKA is provided to generate a standard curve for the assay, and all values were interpolated from a standard curve. OD was determined by spectrophotometry at 450 nm and 650 nm, and the intensity of the absorbance signal was proportional to PKA activity.
2.8. Immunofluorescence
Immunofluorescence analysis was done on HPAECs grown on glass coverslips subjected to normoxia or hypoxia (1% O2, 24 h). Samples were antigen-retrieved, fixed in 2% paraformaldehyde, permeabilized with 0.1% Triton X-100 and washed with PBS. Sections were blocked in 2% BSA/PBS solution for 1 h at room temperature. Slides were then incubated with anti-phospho CREB antibody (1:100) and anti-Ref-1 antibody (1:100) overnight at 4 °C followed by goat anti-rabbit Cy3- and goat anti-mouse 488-conjugated secondary antibody (Life Technologies Inc., 1:1000). Slides were stained for nuclei (DAPI), and cover-slipped using gelvatol mounting media (polyvinylalcohol, glycerol, H2O, sodium azide and Tris pH 8.5). Nonspecific rabbit IgG (5 μg/mL) was used instead of primary antibody as a negative control. Confocal images were captured using a Nikon A1 spectral confocal microscope. For each experiment, 3 images per treatment group were captured. Three independent experiments were performed.
2.9. Cell proliferation assay (crystal violet and cell cycle)
The proliferation of HPAECs was measured by the cell cycle using flow cytometry, and by Crystal Violet method. For cell cycle assay, flow cytometry assay was conducted as described. Briefly, HPAECs seeded into 6-well tissue culture dishes were transfected with scrambled and/or CREB siRNA, and serum deprived (0.2% FBS) for 16 h. After the requisite period, serum-deprived media was replaced for complete media (2% FBS), and cells were exposed to hypoxia (1% O2, 24 h). Cells were trypsinized, fixed in ice-cold 70% ethanol/PBS, treated with ribonuclease A (0.2 mg/mL), and DNA content was stained with propidium iodide (50 mg/mL) at room temperature for 45 min. Cell cycle analysis was performed using a BD LSRFortessa flow cytometer and analyzed using FlowJo software (version 10). A minimum of 10,000 events was recorded for each sample. The data were tabulated as the sum of cells in S and G2-M phase and normalized as a fold change from normoxic controls. For the crystal violet assay, HPAECs transfected with scrambled and/or CREB siRNA were exposed normoxia or hypoxia (1% O2, 24 h) on a 96-well plate. After the requisite time period, cells were washed twice with PBS and stained with crystal violet for 20 min; cells were washed with PBS and allowed to dry for 2 h and incubated with methanol for 20 min for cells solubilization. The absorbance was measured at 595 nm. Data are presented as a fold change from normoxic controls.
2.10. Cell migration- wound healing assay
Scratch assay was performed as described by Liang et al. [33] with minor modification. Briefly, HPAECs seeded onto 6-well tissue culture dishes were transfected with scrambled and/or CREB siRNA, and serum deprived (0.2% FBS) for 16 h. Each well was marked on its bottom with ink bisecting the well into two halves. Serum-deprived media was replaced with complete media (2% FBS), the cell monolayer was disrupted with a P1000 pipette tip with a stroke that passed through both halves of the well, cells were photographed at time 0 h, and cells were exposed to normoxia and hypoxia (1% O2, 24 h). After 24 h, cells were again photographed to measure cell migration. ImageJ software was used to calculate the change in distance between the cell fronts as compared with the time 0 photograph. Data are presented as a fold change from normoxic controls.
2.11. ROS measurements
Methodology was adopted and modified as indicated in a previous report by Zielonka and coworkers[34]. For hydrogen peroxide detection by CBA (Coumarin- 7 Boronate Acid) and for extracellular superoxide detection by Hpr+ (Hydropropidine), HPAECs were seeded in a 96-well plate with clear bottoms and black walls and serum starved in 0.2% FBS media overnight before being subjected to 24 h hypoxia or normoxia. Cells were washed twice with PBS and added 80 µL of HBSS supplemented with 25 mM HEPES, 1% BSA, 10 µM DTPA, 100 µM L-NAME and 1 mM Taurine (L-NAME and taurine were added to prevent peroxynitrite formation, and scavenge hypochlorous acid, respectively, both which could react with CBA). Additionally, cells were treated with Nox1Ads or scrambled peptide (2 µM), a Nox1 inhibitor, or 1 KU/mL bovine liver catalase (CBA assay) and 250 U/mL SOD (Hpr+ assay) as controls. Cells were returned to hypoxia or normoxia for an additional 30 min. Plates were then removed, and CBA (0.5 mM) or Hpr+ 0.5 mg/mL (in salmon sperm DNA) was added, placed in a plate reader preheated to 37 °C and read kinetically (every minute for 3 h) at λex= 350 nm and λem= 450 nm for CBA and λex= 400 nm and λem= 574 nm for HPr+. Finally, cells were washed with PBS, lysed, and protein concentration was determined by Bradford. The average rate of fluorescence per mg protein was determined and normalized to samples with catalase for CBA and SOD for HPr+, and then normalized to normoxic control.
2.12. Chromatin immunoprecipitation (CHiP) assay
CHiP assays were performed using a commercially available EZ-CHiP kit from Millipore Sigma (Merck KGaA, Darmstadt, Germany – Millipore Sigma), as recommended by the manufacturer. Briefly, HPAECs exposed to hypoxia (1% O2, 24 h) were cross-linked with 1% formaldehyde for 10 min, and then cells were treated with glycine for an additional 5 min, scraped and collected for sonication. Cells were sonicated 5 times for 10 s (20 AMP) with 20 s intervals on ice using a MISONIX sonicator. The average fragment size was 300–500 bp under these conditions. Chromatin DNA was incubated with 1–10 µg of either anti-pCREB antibody (Cell Signaling Technology) or rabbit IgG for pulldown at 4 °C overnight. IP samples were collected after incubation with agarose beads. The IP/agarose beads complex were washed, protein-DNA cross-links were reversed, and DNA was purified. A CREB binding site (CRE) within the human Gremlin1 promoter (Genome Browser, UCSC) was identified by bioinformatics analysis within the geneXplain platform [35] using known DNA-binding motifs described in the TRANSFAC database [36], [37]. Primer sequences flanking the CRE motif within Gremlin1 promoter were as follows: Forward- GGA-GCG-GAA-AAT-GTG-ATT-TGC and Reverse- CCC-GCA-CCC-CTC-CTG-CTT-CCC. Primers for GAPDH (Glyceraldehyde 3-phosphate dehydrogenase) were used as a negative control as follows: Forward- TAC-TAG-CGG-TTT-TAC-GGG-CG and Reverse- TCG-AAC-AGG-AGG-AGC-AGA-GAG-CGA. The EZ-CHiP kit already provides primer control which are specific to the promoter region of the GAPDH gene. Purified DNA from input and IP samples were subjected to PCR using the above primer sets. Standard PCR reactions using 2 µL of the immunoprecipitated DNA were performed. PCR products were separated by electrophoresis through 1% agarose gels and visualized by Gel Star™ Nucleic acid gel stain [16]
2.13. CREB : CRE DNA-binding analysis by ELISA
CREB DNA binding analysis was applied to investigate activated (phosphorylated ser 133) CREB binding to the consensus CRE (CREB response element) sequence. An ELISA-based approach was employed according to the manufacturer’s instructions (TransAm; Active Motif, Carlsbad, CA, USA). Briefly, nuclear extracts from HPAECs were transfected with scrambled or Nox1/Ref-1 siRNA, subjected to normoxia and hypoxia (1% O2, 24 h), and total lung lysates from animals subjected to Sugen 5416/hypoxia treatment were incubated in 96-well plates pre-coated with an oligonucleotide containing the CRE consensus motif (5′-TGACGTCA-3′). Following the capture of the transcription factor by the oligonucleotide, a primary antibody to phosphorylated CREB was added. After washing, an HRP-conjugated secondary antibody was added followed by substrate solution. OD was determined by spectrophotometry at 450 nm, and the intensity of the absorbance signal deemed proportional to pCREB DNA binding. Data are presented as a fold change from normoxic controls.
2.14. Hemodynamic procedure
Animals were weighed and placed in an anesthesia induction chamber that was supplied with 5% isoflurane. Once the animal was non-responsive (verified by toe pinch), it is placed on a warming pad and restrained. A nose cone that delivers 2.5% isoflurane was placed over the animal’s muzzle. A tracheotomy was performed inserting a 14-gauge intravenous catheter into the trachea which was secured with suture. The anesthesia cone was removed, and the animal was placed on a ventilator that had been set to 70–80 beats per minute (weight dependent), and a stroke tidal volume of 2.5 cc. A thoracotomy was then performed, and the rib cage was retracted to expose the inferior vena cava (IVC), the heart and the large vessels. Silk suture was loosely placed around the IVC. A 25 gauge needle was used to pierce the RV. The needle was removed, and a 1.9 French-Fogarty catheter was then introduced to the apical region of the RV. The catheter was adjusted as needed until the optimal right ventricle (RV) maximum pressure was obtained. The RV was then dissected from the LV and septum. The Fulton index (RV/LV+Septum) was used as a measure of RV hypertrophy.
3. Statistical analysis
Results are reported as mean ± S.E.M. Two-way ANOVA followed by Sidak’s post-hoc test was applied in most cases. Statistical analyses were also performed using the unpaired Student’s t-test when appropriate. For all analyses, p < 0.05 was deemed statistically significant. GraphPad Prism software was used for data analyses (GraphPad Software v7).
4. Results
4.1. 1- Nox1-mediates PKA activation, CREB phosphorylation, and CREB : DNA binding at the CRE motif
Human pulmonary arterial endothelial cells (HPAECs) were transfected with scrambled or Nox1 siRNA for 24 h before hypoxia treatment (1% O2, 24 h), and evaluated for Nox1 expression, PKA activity, CREB phosphorylation, and pCREB transcriptional factor activity. HPAECs were also treated with scrambled or NoxA1ds, a selective canonical Nox1 inhibitor [38], for O2•- and H2O2 production measurement. Fig. 1A shows a 40% increase in Nox1 protein levels in response to hypoxia. However, no differences were observed in Nox2 and Nox4 protein levels (Fig. S1). Nox1 gene-silencing using Nox1 siRNA reduced hypoxia-induced Nox1 to basal levels (Fig. 1A). HPAECs were subjected to hypoxia for 24 h, treated with NoxA1ds for an additional 30 min in hypoxia, and were assessed for O2•- and H2O2 production by HPr+ (Hydropropidine) and CBA (Coumarin-7 boronic acid) probes [34], [39], respectively. Fig. 1B shows a significant increase in Nox1-derived O2•- compared to normoxia (1.7 ± 0.16- compared with 1 ± 0.1-fold relative to O2•- production/mg protein, **p < 0.01), which was abolished in the presence of NoxA1ds (1.15 ± 0.1- compared with 1.7 ± 0.16-fold relative to O2•- production/mg protein, *p < 0.05) compared to scrambled control (Fig. 1B). Fig. 1C shows a significant increase in hypoxia-induced H2O2 production compared to normoxia (2.2 ± 0.20-compared with 1 ± 0.14-fold relative rate of H2O2 production/mg protein, ****p < 0.0001), and the presence of NoxA1ds reverted hypoxia-induced H2O2 production (0.9 ± 0.13- compared with 2.2 ± 0.20-fold relative rate of H2O2 production/mg protein, ****p < 0.0001) to normal levels compared to scrambled control (Fig. 1C). No significant differences in superoxide levels were observed under normoxic conditions between normoxic vehicle control and NoxA1ds-treated cells (n = 3; data not shown).
Fig. 1.

Nox1 oxidase mediates hypoxia-induced ROS production, PKA activation, CREB phosphorylation, and CREB: DNA binding in HPAECs. (A) HPAECs transfected with scrambled or Nox1 siRNA were exposed to hypoxia for 24 h, and immunoblotting was performed for Nox1 (n = 5–13); *p < 0.05 and * **p < 0.001. For (B and C), HPAECs were exposed to normoxia or hypoxia for 24 h, and hypoxia-treated cells were treated for the final 30 min with 2 µM scrambled (Scrambled) or selective Nox1 peptide inhibitor NoxA1ds. (B) Evaluation of extracellular O2•- was made using HPr+ probe (n = 8–12) * p < 0.05 and * *p < 0.01; and (C) cellular H2O2 was determined using CBA probe (n = 6–10); * *p < 0.01 and * ** *p < 0.0001. For subsequent experiments, HPAECs were transfected with scrambled or Nox1 siRNA and exposed to 24 h hypoxia. (D) Analysis of PKA activity by ELISA (n = 3–5); *p < 0.05. (E) Analysis by Western blot of pCREB and tCREB protein levels (n = 6–13), data show comparisons of pCREB/tCREB ratios; * *p < 0.01. (F) Evaluation of CRE-specific DNA binding of pCREB by TransAM© pCREB transcription factor ELISA kit (n = 3–4); **p < 0.01. All data are shown as means ± S.E.M. Two-way ANOVA followed by Sidak’s post-test was performed for statistical analyses.
Inasmuch as PKA is a key modulator of CREB and previous reports in cardiomyocytes support ROS-induced protein kinase A (PKA) oxidation and activation [32], we postulated that hypoxia-induced Nox1 would lead to PKA activation in HPAECs. As proof-of-principle, HPAECs were treated with H2O2. While there was no change in total protein, H2O2 treatment (100 µM, 10 min) resulted in a significant increase in oxidized PKA (PKA dimer) (3.3 ± 0.14-fold compared to 0 µM H2O2, ***p < 0.001), accompanied by a reduction in non-oxidized PKA (PKA monomer) (0.81 ± 0.02-fold compared to 0 µM H2O2, *p < 0.05) relative to total PKA form on a reducing Western blot (Fig. S2). Importantly, Fig. 1D shows a significant increase in hypoxia-induced PKA activity (260 ± 17.4 compared with 157 ± 23.1 of U/mg protein, *p < 0.05), a response that was abrogated by siRNA Nox1 (260 ± 14.4 compared with 131 ± 16.7 of U/mg protein, *p < 0.05). To interrogate a link between Nox1 and PKA effector CREB, we evaluated whether CREB phosphorylation is upregulated in pulmonary EC. HPAECs subjected to hypoxia showed an increase in CREB phosphorylation (1.70 ± 0.2-fold compared with normoxia, **p < 0.01) (Fig. 1E). Importantly, siRNA against Nox1 abolished this increase, demonstrating that Nox1-derived ROS participates in hypoxia-induced PKA activity promoting CREB phosphorylation (Fig. 1E). CREB phosphorylation is the signal for its translocation to the cell nucleus and DNA binding at the CRE motif in the regulation of gene expression [23]. Therefore, we explored whether Nox1-induced CREB phosphorylation is associated with an increase in transcription factor activation (CREB binding at the CRE motif) under hypoxic conditions [21]. HPAECs were transfected with scrambled or Nox1 siRNA, subjected to normoxia or hypoxia (1% O2, 24 h), and assayed for CREB transcription factor activity by ELISA [40], [41], [42]. Fig. 1F demonstrates that hypoxia induced a significant increase in CREB binding on the CRE motif (1.94 ± 0.05 compared to scrambled normoxia, **p < 0.01). Importantly, Nox1 gene-silencing ablated hypoxia-induced CREB DNA binding (Fig. 1F).
4.2. 2- Hypoxia stimulates CREB and Ref-1 nuclear translocation, CREB/Ref-1 binding, and Ref-1-mediated CREB : DNA binding at the CRE motif
To gain further insight into the mechanism on CREB/Ref-1 pathway enhancing CREB: DNA binding, we first sought to verify the localization of both proteins in HPAECs subjected to hypoxia (1% O2, 24 h). Under hypoxia, an increase in the nuclear pCREB (1.24 ± 0.003-fold compared to normoxia, ****p < 0.0001) and Ref-1 (1.51 ± 0.084-fold compared to normoxia, ***p < 0.001) was observed (Figs. 2A & 2B), respectively, consistent with CREB interaction with Ref-1 in the nucleus for proper binding of CREB to DNA. Nuclear co-localization of pCREB and Ref-1 were supported by immunofluorescence. Fig. 2C illustrates (images on the left; quantifications to upper right of images) significant elevation in nuclear Ref-1 intensity (500 ± 30.7 intensity compared to 372.5 ± 19.3 normoxia, *p < 0.0001) and nuclear pCREB intensity (363 ± 25.5 intensity compared to 261.2 ± 22.7 normoxia, *p < 0.001) (Fig. 2C). To explore whether hypoxia-induced pCREB and Ref-1 co-translocation to the nucleus enhances their association, co-immunoprecipitation (Co-IP) was performed. Fig. 2D shows an increase in pCREB/Ref-1 binding (1.812 ± 0.02-fold compared to normoxia, *p < 0.05) (Fig. 2D). Ponceau S-stained blot is shown to display consistency of well loading and was used to normalize pCREB density values. To test whether Ref-1 is critical for pCREB activity (i.e., binding to CRE), HPAECs were transfected with scrambled or Ref-1 siRNA, subjected to hypoxia (1% O2, 24 h), and assayed for nuclear Ref-1 protein levels by Western blotting and CREB transcription factor activity by ELISA [40], [41], [42]. Fig. 2E shows that Ref-1 gene silencing indeed prevents an increase in hypoxia-promoted Ref-1 expression in the nucleus (67% reduction compared to scrambled hypoxia, ****p < 0.0001) (Fig. 2E). In fact, siRNA to Ref-1 causes a significant reduction in Ref-1 levels under normoxic conditions. Importantly, it also significantly impaired hypoxia-induced CREB DNA binding at the CRE motif (1.78 ± 0.18-fold scrambled hypoxia compared to 0.87 ± 0.22-fold siRNA Ref-1 hypoxia, **p < 0.01) (Fig. 2F).
Fig. 2.

Hypoxia induces nuclear co-translocation of CREB and Ref-1, nuclear CREB:Ref-1 binding and CREB:DNA-binding in HPAECs. HPAECs were exposed to hypoxia for 24 h. (A) Western blot analysis (nuclear lysate) of pCREB (n = 3); * ** *p < 0.0001, and (B) Ref-1 (n = 6); * **p < 0.001. (C) Confocal immunofluorescence images of nuclear pCREB (green staining) (n = 3); *p < 0.05, and nuclear Ref-1 (red staining) (n = 3); bar graph shows relative intensity in arbitrary units, *p < 0.05. White staining (DAPI) represents nuclei. (D) Analysis of nuclear CREB/Ref-1 binding in HPAECs exposed to hypoxia for 24 h was performed by co-immunoprecipitation (Co-IP). For the Co-IP, anti-pCREB antibody was used for immunoprecipitation, and anti-Ref-1 antibody was used for immunoblotting, and PC= Ref-1 positive control, and Ponceau image was use to experiment normalization (n = 3–4); *p < 0.05. Nuclear fractions were prepared from HPAECs transfected with scrambled or Ref-1 siRNA exposed to normoxia or hypoxia for 24 h, and (E) Western blot for Ref-1 protein levels, (n = 3–6); *p < 0.05 and * ** *p < 0.0001; and (F) CRE-specific DNA binding of pCREB by TransAM© pCREB transcription factor ELISA kit were performed, (n = 3–4); *p < 0.05. All data are shown as means ± S.E.M. Two-way ANOVA followed by Sidak’s post-test or Student t-test was performed.
4.3. 3- Hypoxia elicits Gremlin1 expression and pulmonary EC proliferation and migration in a CREB-dependent manner
We identified a CREB binding site (CRE) within the human Gremlin1 promoter (Genome Browser, UCSC) by bioinformatics analysis within the geneXplain platform [35] using known DNA-binding motifs described in the TRANSFAC database [36], [37]. We examined the binding of CREB to the revealed CRE site in the human Gremlin1 promoter in HPAECs under normoxic and hypoxic conditions using chromatin immunoprecipitation assay (ChIP). GAPDH (Glyceraldehyde 3-phosphate dehydrogenase) was used as a negative control. Fig. 3A shows a distinct band corresponding to the CRE motif within the Gremlin1 promoter under normoxic conditions indicating binding of CREB to Cre under basal conditions. An elevated level of CREB binding on the identified CRE motif was observed in hypoxic cells (Fig. 3A). No binding to GAPDH was observed. Indeed, it has been shown that CREB occupancy on the CRE motif tightly correlates with gene expression [21]. To explore whether CREB mediates hypoxia-induced Gremlin1 expression, HPAECs were transfected with scrambled or CREB siRNA, subjected to hypoxia (1% O2, 24 h) and assayed for Gremlin1 protein levels. Fig. 3B shows that CREB siRNA significantly downregulates CREB protein levels (65% reduction vs. scrambled control, ***p < 0.001) (Fig. 3B). Fig. 3C shows an increase in Gremlin1 expression induced by hypoxia (1.75 ± 0.1-fold increase compared to scrambled siRNA normoxia, ****p < 0.0001), a response that was abolished by CREB knockdown (58% reduction compared to scrambled hypoxia, ****p < 0.0001) (Fig. 3C), demonstrating that CREB is necessary for Gremlin1 transcription. A marked ability of siCREB to suppress Gremlin1 under normoxic conditions was also observed.
Fig. 3.

Hypoxia induces Nox1-controlled CREB binding to Gremlin1 Cre and CREB- and Ref-1-dependent Gremlin1 expression. (A) Analysis of CREB binding at the CRE motif within the human GREM1 promoter in HPAECs exposed to hypoxia for 24 h; pCREB antibody was used for chromatin immunoprecipitation (CHiP); NTC-Non-template control, IgG= rabbit IgG, Nor= Normoxia, Hy= Hypoxia (n = 2–3). (B) Western blot analysis of CREB in HPAECs following transfection with scrambled or CREB siRNA (n = 3); * **p < 0.001. (C) HPAECs transfected with scrambled or CREB siRNA were exposed to normoxia or hypoxia for 24 h, and Western blot was performed for Gremlin1 protein levels, (n = 5–11); * *p < 0.01 and * ** *p < 0.0001. (D) HPAECs transfected with scrambled or Ref-1 siRNA were exposed to normoxia or hypoxia for 24 h, and Western blotting was conducted for Gremlin1 expression, (n = 3); *p < 0.05; and (E) PCNA expression, (n = 3); *p < 0.05. All data are shown as means ± S.E.M. Two-way ANOVA followed by Sidak’s post-hoc test or Student t-test was performed.
As shown in Figs. 2D and 2F, Ref-1 is instrumental in the CREB:CRE binding. To interrogate whether Ref-1 is causally linked to Gremlin1 expression and pulmonary EC proliferation, Ref-1 siRNA was transfected into HPAECs and subjected to hypoxia (1% O2, 24 h). Fig. 3D shows that Ref-1 siRNA abolished hypoxia-induced Gremlin1 expression (1.85 ± 0.2-fold increase hypoxia vs. scrambled normoxia, *p < 0.05), causing a complete ablation of the response (scrambled hypoxia compared to siRef-1 hypoxia, *p < 0.05) (Fig. 3D). Fig. 3E shows a 2-fold increase in hypoxia-induced cell proliferation marker PCNA. We observed a substantial numerical decrease in PCNA with siRNA against Ref-1 indicating a trend toward a decrease although this change did not reach statistical significance (Fig. 3E).
To test whether hypoxia-induced CREB expression and DNA binding activity results in a proliferative response, HPAECs were transfected with scrambled or CREB siRNA, subjected to hypoxia (1% O2, 24 h) and cell proliferation was evaluated by crystal violet absorbance [43], [44] and cell cycle changes [45]. A significant increase in EC proliferation was found following hypoxia (1.4 ± 0.050-fold increase relative to normoxic control, ***p < 0.001), a response that was abolished by CREB siRNA (Fig. 4A). An increase in hypoxia-induced EC proliferation was also observed by a shift in cell cycle to S+G2-M phase (1.27 ± 0.09 fold increase relative to scrambled normoxia, *p < 0.05), which was eliminated by CREB gene-silencing, *p < 0.05 (Fig. 4B). These findings were extended to cell migration as suggested by the wound healing assay. Fig. 4C indicates an increase in hypoxia-induced HPAEC migration at 24 h (1.68 ± 0.09-fold increase relative to scrambled normoxia, **p < 0.01), a response that was abrogated by CREB siRNA (Fig. 4C).
Fig. 4.

Hypoxia induces CREB-mediated EC proliferation and migration. (A) and (B) HPAEC proliferation following transfection with vehicle-scrambled or CREB siRNA under 24 h normoxia or hypoxia treatment was assayed by crystal violet absorbance, (n = 3–11) * **p < 0.001; and by cell cycle flow cytometry analysis comparing numbers of cells in S + G2-M phases across groups, (n = 4–6) *p < 0.05, respectively. (C) HPAEC migration following transfection with scrambled control or CREB siRNA under 24 h hypoxia vs. normoxia was assayed by wound injury (scratch assay) (n = 3); * *p < 0.01. Vertical black lines delineate cell fronts and residual wound sizes. Two-way ANOVA followed by Sidaks post-hoc test was performed.
4.4. 4- Nox1, CREB, Gremlin1, and CREB DNA binding at the CREB motif are upregulated in lungs of rats with PAH
Our results provide detailed mechanistic insight into a role for Nox1-induced CREB activation in the regulation of hypoxia-induced Gremlin1 expression, EC proliferation, and migration. To interrogate preclinical relevance and investigate if this previously unidentified pathway is functional in an animal model of pulmonary hypertension, we assessed differences in Nox1, CREB, and Gremlin1 in lungs from hypoxic vs. normoxic rats. Nox1, CREB, and Gremlin1 protein levels were quantified in the established rat Sugen-hypoxia (SuHy) model of occlusive and angioproliferative PAH [28], [29]. Male rats were injected with VEGF receptor 2 antagonist Sugen5416 (20 mg/Kg) to initiate the injury, followed by 3 weeks of hypoxia. Right ventricle (RV) maximum pressure and RV hypertrophy (hallmark indicators of PAH) were evaluated by open chest hemodynamics and the Fulton Index, respectively. SuHy caused a marked elevation in RV maximum pressure (62.8 ± 4.35 mmHg SuHy compared with 22.5 ± 0.7 mmHg normoxia, ****p < 0.0001), and RV hypertrophy (0.62 ± 0.02 SuHy compared to 0.27 ± 0.01 normoxia, ****p < 0.0001) (Fig. 5A and B). These results verify that SuHy promoted PAH. Fig. 5 C, D and E show an increase in Nox1 (1.75 ± 0.12-fold increase compared to normoxia, *p < 0.05), CREB (1.67 ± 0.25-fold increase, *p < 0.05), and Gremlin1 (2.4 ± 0.25-fold elevation, **p < 0.01), respectively, in rat PAH lung homogenates vs. normoxic controls (Fig. 5C–E). To test the CREB : DNA binding capacity in homogenates of lungs from rats subjected to SuHy versus normoxia, CREB transcription factor activity by ELISA [40], [41], [42] was determined. Fig. 5F shows a significant increase in SuHy-induced CREB : CRE binding compared to normoxia group (1.23 ± 0.05 fold increase relative to normoxia, *p < 0.05) (Fig. 5F).
Fig. 5.

Rat PAH is associated with increased Nox1, CREB, CREB:Cre binding and Gremlin1 expression. Rats were treated with a single injection of VEGF receptor (VEGFr) inhibitor SU5416 (20 mg/Kg) followed by 3 weeks in a hypoxia ventilated chamber (10% O2) or were subjected to 3 wks normoxia. VEGFr inhibition exacerbates chronic-hypoxia induced PAH. (A) RV maximum pressure, and (B) RV hypertrophy as measured by Fulton index (RV/left ventricule+septum weight ratio), (n = 8); * ** *p < 0.0001. Western blotting of (C) Nox1, (D) CREB, and (E) Gremlin1 protein levels in lung homogenates from Su-Hy-treated rats compared with normoxic controls, (n = 4); *p < 0.05 and * *p < 0.01. Protein expression was normalized to β-actin protein expression. (F) Evaluation of CRE-specific DNA binding of pCREB by TransAM© pCREB transcription factor ELISA kit (n = 3–4); *p < 0.05. Absorbance (OD at 450 nM) was quantified and expressed as a function of normoxic controls. All data are shown as means ± S.E.M. Student’s t-test was performed.
5. Discussion
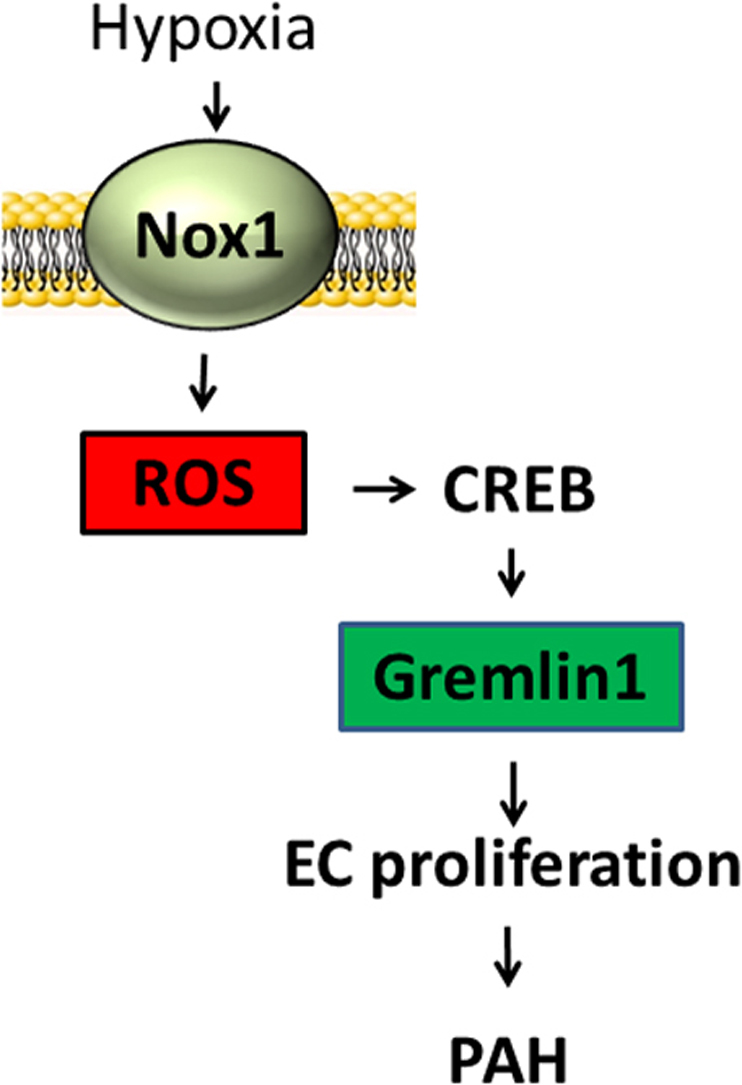
In the current study, we aimed to delineate downstream mechanisms by which Nox1 mediates human pulmonary arterial endothelial cell (HPAECs) proliferation and migration, hallmark phenotypic changes operant in PAH. Herein, we report for the first time that Nox1 induces protein kinase A-dependent, cAMP response element binding protein (CREB) phosphorylation, translocation of CREB to the nucleus and critical interaction with the redox factor-1 (Ref-1). Moreover, our findings illustrate a key novel interaction between CREB and Ref-1 permitting CREB binding to the promoter region of human bone morphogenetic protein (BMP) antagonist Gremlin1 and its transcription. Employing a variety of techniques, we display instrumental interactions at the post-translational and genome level leading to enhanced Gremlin1 expression, pulmonary arterial EC proliferation, migration, and PAH (Fig. 6). Indeed, while the assay employed for monitoring migration is not exclusive of a partial role for proliferation in the wound closure, it is widely used as an indicator of migration [33], [46], [47], [48], [49], [50], [51] and the current findings are consistent with another report showing a pro-migratory effect of hypoxia on ECs [52]. These findings are unique and highly significant in that they describe a previously unidentified, tightly regulated signaling pathway propelled by Nox1 in control of Gremlin1 that promotes pulmonary endothelial cell proliferation and migration. Our findings are novel at multiple tiers of control and are corroborated by changes observed in rat PAH.
Fig. 6.

A signaling model for CREB/Ref-1 pathway-induced EC proliferation. Hypoxia exposure of pulmonary endothelial cells leads to Nox1-derived ROS production, which in turn activates the protein kinase A, leading to CREB phosphorylation and its association with Ref-1. Ref-1 mediates CREB bZIP domain reduction, which increases CREB DNA binding to the CRE motif and promotes Gremlin1 expression, EC proliferation/migration, and PAH.
We demonstrate here for the first time that Nox1 modulates endothelial PKA activity and, in turn, CREB phosphorylation and Gremlin1 promoter binding. Control of PKA by Nox1 in any cell type, no less endothelial cells or HPAECs, has ever been reported to our knowledge. Previous reports have suggested that H2O2 can induce PKA activity through oxidation in cardiomyocytes cells and alveolar type II cells [32], [53], [54]. We show for the first time that treatment of HPAECs with H2O2 enhances the oxidized form (dimer) and decreases the reduced form (monomer) of PKA (Supplementary Fig. 2) indicating its oxidation and activation. More mechanistically, H2O2 reportedly induces dimerization of PKA’s regulatory subunits which enhance PKA-mediated phosphorylation [53]. Since Nox1 oxidase is a known generator of O2•- [38], [55], [56] which is rapidly dismuted to H2O2 [57], we examined whether hypoxia-induced Nox1 in HPAECs could mediate elevations in PKA. Though we were not able to observe an increase in PKA oxidation at 24 h, an increased PKA oxidation, however, was observed at 1 h of hypoxia in HPAEC (not shown) whose effects could elicit a prolonged PKA activity. Most importantly in that regard, our findings revealed that hypoxia elicited a near doubling of PKA activity that was abolished by silencing of Nox1 (Fig. 1D). Incidentally, this coincided with a marked rise in Nox1 and O2•- and H2O2 in response to hypoxia that was effectively ablated by NoxA1ds, a selective inhibitor of canonical Nox1 oxidase (Fig. 1A-C) [38]. These data strongly support that Nox1-derived ROS initiate a redox-mediated signaling pathway whose immediate downstream effector is PKA, a widely-reported kinase implicated in the phosphorylation of CREB [23]. Indeed, loss of Nox1 attenuated PKA oxidation and activity, and CREB phosphorylation, which demonstrate that Nox1 mediates this signaling pathway (Fig. 1 D&E).
Phosphorylation of CREB is required for its cytosol to nucleus translocation where its action as a transcription factor ensues [23]. Once localized in the nucleus, CREB is known to bind to the promoter region of CREB-regulated genes to mediate transcription [23]. Importantly, we were able to identify a requisite CRE motif in the human Gremlin1 promoter using the geneXplain platform [35] of known DNA-binding motifs described in the TRANSFAC®database [36], [37]. Indeed, our findings show that pCREB binding to the CRE motif (ELISA) revealed in the Gremlin1 promoter was increased in response to hypoxia, and that siRNA against Nox1 abolished this binding. In aggregate, these data are consistent with a causal role of Nox1 in CREB transactivation of Gremlin1 as a consequence of Nox1-induced PKA and CREB phosphorylation.
Delving deeper into the mechanism, it was important to determine the mechanisms surrounding pCREB activation of Gremlin1 and proliferation in our model. Indeed, our findings show that hypoxia induces nuclear levels of phospho CREB. Coinciding with this increase, a parallel elevation in nuclear redox factor 1 (Ref-1) was observed (Fig. 2A–D). Xanthoudakis et al. demonstrated that Ref-1 stimulates transcription factor DNA binding through reduction of specific cysteine residues in their bZIP domain [27]. Along those lines, Goren et al. also demonstrated that reduction of cysteine 300 and 310 residues in the CREB bZIP domain enhances its binding efficiency to the CRE motif and transcription of CREB-regulated genes, aka activation by reducing conditions [25], as mediated via Ref-1 [26], [58]. Our results showing a rise in nuclear pCREB and Ref-1 are consistent with this transformative and permissive interaction. Indeed, we observed an increased pCREB/Ref-1 nuclear co-localization and binding as supported by cellular imaging and co-immunoprecipitation. This, taken with our findings demonstrating that loss of Ref-1 attenuates hypoxia-induced CREB : DNA binding, supports the interaction of CREB-Ref-1 as causal in this response and is highly consistent with CREB reduction and attendant activation by Ref-1 [27]. Further studies confirming specific reduction at cysteines 300 and 310 mediated by Ref-1 are in progress and currently outside the scope of the current study.
Employing chromatin immunoprecipitation and using primers for the flanking sequences of the herein identified CRE domain in the human Gremlin1 promoter, we were able to show that hypoxia enhances binding of pCREB to Gremlin1 Cre motif required for the promotion of Gremlin1 expression. Using siRNA to effectively knockdown CREB, we observed a significant loss in Gremlin1 under both normoxic and hypoxic conditions. These data importantly indicate that CREB is pivotal to hypoxia-induced Gremlin1. The data from cells under normoxic conditions also suggest that basal expression of Gremlin1 is under the control of CREB. Moreover, we demonstrated for the first time that hypoxia-induced HPAEC proliferation, cell cycle progression, and migration are regulated by Nox1-induced CREB. Supporting the latter, CREB is one of the transcription factors reportedly upregulated in microvascular EC [21] and hypoxia which has been described as a stimulus for cellular proliferation in PAH [2], [6], [19].
In aggregate, this response is consistent with Nox1-controlled CREB playing a vital role in regulating hypoxia-induced EC proliferation & cell progression and migration [59], [60], [61]. Indeed, hypoxia-induced EC proliferation and migration appear to be dependent on CREB-regulated Gremlin1 expression, the latter of which has been implicated in the development of PAH [62]. Indeed, previous publications by our group and others have demonstrated that pulmonary arterial EC exposed to hypoxia, as well as lung samples from PAH patients, showed an upregulation of Gremlin1 [62], [63], [64], [65], but did not examine a modulatory role for CREB in this pathway or what links Nox1 to CREB or CREB to Gremlin1 and ensuing proliferation. Closing the loop, our findings prove causality (or a permissive effect) of Ref-1 in hypoxia-induced Gremlin1 and therefore are consistent with Ref-1 playing a role in a Gremlin1-mediated proliferative response. Moreover, the data are supportive of a trend for Ref-1 involvement in PCNA expression, consistent with Ref-1's role in HPAEC proliferation. The underwhelming response of siRef-1 on PCNA could be explained by insufficient power of the study or the involvement of a host of factors other than PCNA in the proliferative response. Either way, Ref-1 does indeed modulate Gremlin1 and thus, in turn, proliferation.
Our data suggest for the first time that hypoxia-induced Nox1, and, in turn, PKA, through PTM control of CREB phosphorylation effects previously unidentified, multi-tiered downstream signaling involving Ref-1 and transactivation of Gremlin1, which regulates cell cycle progression and migration. Thus, our findings are novel on multiple levels. This is not to even consider the potential role of Nox1 in modulation of cAMP:PKA-mediated or genomic control of CREB in the context of hypoxia. Combined genomic and post-translational control of CREB in this context would represent a synergized, exacerbated, pro-proliferative and pro-migratory loop that is a hallmark of aberrant vascular remodeling in PAH. It is widely held that EC proliferation, in combination with vascular smooth muscle proliferation, combined with augmented migration of both cell types leads to aberrant vascular remodeling, vascular occlusions, and RV hypertrophy-induced RV failure [1], [5], [8], [19].
We were, in fact, successful at demonstrating that redox control could proceed from the oxidation of PKA and are tantalized by the potential for redox control of Gremlin1 or any one of its modulatory factors in this pathway. For certain, interrogation of the redox modifications not just of PKA, but of CREB, Ref-1, and Gremlin1 are ripe for study. To interrogate whether Nox1, CREB, Gremlin1, and CREB DNA binding are involved in the pathophysiology of PAH, a rat animal model of PAH was employed in parallel to in vitro findings. The current studies illustrate a rise in Nox1, CREB, and Gremlin1 expression, as well as increased CREB DNA binding at the CRE motif in lungs of rats with PAH. The animal PAH model was corroborated by a rise in the RV maximum pressure and RV hypertrophy [62], [65]. One limitation of these studies might be the use of lung homogenates to extrapolate changes occurring at the pulmonary vascular endothelium. That notwithstanding, we have previously shown that alterations in lung tissue, in general, do, in fact, parallel changes occurring at the vascular endothelium [63].
In conclusion, our findings identify a previously uncharacterized mechanism whereby Nox1-induced PKA activity leads to nuclear translocation of CREB, and in association with Ref-1, promotes Gremlin1 transcription. This sets in motion EC proliferative and migratory responses which are critical in overt vascular remodeling in rat PAH. In vivo evidence described herein supports the activation of this pathway and thus underscores a number of potential therapeutic interventions at any one or a combination of nodes in this novel pathway.
Acknowledgments
We thank Dr. Jacek Zielonka for providing the HPr+ probe for superoxide measurement. We are very grateful to members of the Pagano laboratory for critical discussions and comments on the manuscript.
Acknowledgments
Funding
This work was supported by NIH Grants R01HL112914, R01HL079207, P01HL103455, and T32 GM008424 (to P.J.P.); 2R01HL098032, 1R01HL125886-01, P01HL103455, T32 HL110849, and T32 HL007563 (to M.T.G.), and AHA grant 19POST34370043 (to D.S.J.). This study was also supported by the Institute for Transfusion Medicine and the Hemophilia Center of Western Pennsylvania.
Author contributions
D.S.J. and P.J.P. conceptualized the experiments for this manuscript. D.S.J., E.D., Y.L., M.F., and D.G. performed experiments and analysis. S.S., B.K., M.T.G., and P.J.P. provided the resources. D.S.J. and P.J.P. wrote the original draft of the manuscript.
Author disclosure statement
P.J.P. is named on a filed patent (US patent no. 9,187,528 B2) for NoxA1ds. All other authors declare that they have no conflicts of interest as pertains to the contents of this article.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2019.101138.
Appendix A. Supplementary material
Fig. S1.

Hypoxia does not change Nox2 and Nox4 protein expression in HPAEC. HPAECs were exposed to hypoxia for 24 h, and Nox2 (A) and Nox4 (B) protein levels were evaluated by Western blot. No statistical difference was obsereved for Nox2 (n = 7), and Nox4 (n = 9). Student’s t-test was performed.
Fig. S2.

H2O2induces PKA oxidation (PKA dimer formation) in HPAECs. Western blot analysis of PKA dimer, PKA monomer, and total PKA in HPAECs exposed to 0 or 100 µM of H2O2 for 10 min, (n = 3); *p < 0.05 and ***p < 0.001. All data are shown as means ± S.E.M. by Student’s t-test. Under non-reducing conditions dimerized PKA (oxidized) and monomer PKA (non-oxidized) appears whereas under reducing conditions, samples were supplemented with 2-mercaptoethanol (5%), total PKA appears. The ratio of total PKA to β-actin was determined for reduced samples, and the ratio of dimerized and monomer PKA to total PKA was determined for non-reduced samples.
References
- 1.McLaughlin V.V., Archer S.L., Badesch D.B. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation. 2009;119(16):2250–2294. doi: 10.1161/CIRCULATIONAHA.109.192230. [DOI] [PubMed] [Google Scholar]
- 2.Mandegar M., Fung Y.C., Huang W., Remillard C.V., Rubin L.J., Yuan J.X. Cellular and molecular mechanisms of pulmonary vascular remodeling: role in the development of pulmonary hypertension. Microvasc. Res. 2004;68(2):75–103. doi: 10.1016/j.mvr.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 3.Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J. Clin. Investig. 2012;122(12):4306–4313. doi: 10.1172/JCI60658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Masri F.A., Xu W., Comhair S.A. Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007;293(3):L548–554. doi: 10.1152/ajplung.00428.2006. [DOI] [PubMed] [Google Scholar]
- 5.Sakao S., Tatsumi K., Voelkel N.F. Endothelial cells and pulmonary arterial hypertension: apoptosis, proliferation, interaction and transdifferentiation. Respir. Res. 2009;10:95. doi: 10.1186/1465-9921-10-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stenmark K.R., Fagan K.A., Frid M.G. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ. Res. 2006;99(7):675–691. doi: 10.1161/01.RES.0000243584.45145.3f. [DOI] [PubMed] [Google Scholar]
- 7.White K., Dempsie Y., Caruso P. Endothelial apoptosis in pulmonary hypertension is controlled by a microRNA/programmed cell death 4/caspase-3 axis. Hypertension. 2014;64(1):185–194. doi: 10.1161/HYPERTENSIONAHA.113.03037. [DOI] [PubMed] [Google Scholar]
- 8.Budhiraja R., Tuder R.M., Hassoun P.M. Endothelial dysfunction in pulmonary hypertension. Circulation. 2004;109(2):159–165. doi: 10.1161/01.CIR.0000102381.57477.50. [DOI] [PubMed] [Google Scholar]
- 9.Tuder R.M., Chacon M., Alger L. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: evidence for a process of disordered angiogenesis. J. Pathol. 2001;195(3):367–374. doi: 10.1002/path.953. [DOI] [PubMed] [Google Scholar]
- 10.Abe K., Toba M., Alzoubi A. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation. 2010;121(25):2747–2754. doi: 10.1161/CIRCULATIONAHA.109.927681. [DOI] [PubMed] [Google Scholar]
- 11.Jonigk D., Golpon H., Bockmeyer C.L. Plexiform lesions in pulmonary arterial hypertension composition, architecture, and microenvironment. Am. J. Pathol. 2011;179(1):167–179. doi: 10.1016/j.ajpath.2011.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ardanaz N., Pagano P.J. Hydrogen peroxide as a paracrine vascular mediator: regulation and signaling leading to dysfunction. Exp. Biol. Med. 2006;231(3):237–251. doi: 10.1177/153537020623100302. [DOI] [PubMed] [Google Scholar]
- 13.Drummond G.R., Sobey C.G. Endothelial NADPH oxidases: which NOX to target in vascular disease? Trends Endocrinol. Metab. 2014;25(9):452–463. doi: 10.1016/j.tem.2014.06.012. [DOI] [PubMed] [Google Scholar]
- 14.Frazziano G., Champion H.C., Pagano P.J. NADPH oxidase-derived ROS and the regulation of pulmonary vessel tone. Am. J. Physiol. Heart Circ. Physiol. 2012;302(11):H2166–2177. doi: 10.1152/ajpheart.00780.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bowers R., Cool C., Murphy R.C. Oxidative stress in severe pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2004;169(6):764–769. doi: 10.1164/rccm.200301-147OC. [DOI] [PubMed] [Google Scholar]
- 16.Drummond G.R., Selemidis S., Griendling K.K., Sobey C.G. Combating oxidative stress in vascular disease: nadph oxidases as therapeutic targets. Nat. Rev. Drug Discov. 2011;10(6):453–471. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cahill E., Rowan S.C., Sands M. The pathophysiological basis of chronic hypoxic pulmonary hypertension in the mouse: vasoconstrictor and structural mechanisms contribute equally. Exp. Physiol. 2012;97(6):796–806. doi: 10.1113/expphysiol.2012.065474. [DOI] [PubMed] [Google Scholar]
- 18.Machado R.D., Eickelberg O., Elliott C.G. Genetics and genomics of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2009;54(1 Suppl):S32–S42. doi: 10.1016/j.jacc.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rowan S.C., Keane M.P., Gaine S., McLoughlin P. Hypoxic pulmonary hypertension in chronic lung diseases: novel vasoconstrictor pathways. Lancet Respir. Med. 2016;4(3):225–236. doi: 10.1016/S2213-2600(15)00517-2. [DOI] [PubMed] [Google Scholar]
- 20.Gorski D.H., Walsh K. The role of homeobox genes in vascular remodeling and angiogenesis. Circ. Res. 2000;87(10):865–872. doi: 10.1161/01.res.87.10.865. [DOI] [PubMed] [Google Scholar]
- 21.Leonard M.O., Howell K., Madden S.F. Hypoxia selectively activates the CREB family of transcription factors in the in vivo lung. Am. J. Respir. Crit. Care Med. 2008;178(9):977–983. doi: 10.1164/rccm.200712-1890OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carlezon W.A., Jr., Duman R.S., Nestler E.J. The many faces of CREB. Trends Neurosci. 2005;28(8):436–445. doi: 10.1016/j.tins.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 23.Mayr B., Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2001;2(8):599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 24.Comb M., Birnberg N.C., Seasholtz A., Herbert E., Goodman H.M. A cyclic AMP- and phorbol ester-inducible DNA element. Nature. 1986;323(6086):353–356. doi: 10.1038/323353a0. [DOI] [PubMed] [Google Scholar]
- 25.Goren I., Tavor E., Goldblum A., Honigman A. Two cysteine residues in the DNA-binding domain of CREB control binding to CRE and CREB-mediated gene expression. J. Mol. Biol. 2001;313(4):695–709. doi: 10.1006/jmbi.2001.5064. [DOI] [PubMed] [Google Scholar]
- 26.Ray P.D., Huang B.W., Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012;24(5):981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xanthoudakis S., Miao G., Wang F., Pan Y.C., Curran T. Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J. 1992;11(9):3323–3335. doi: 10.1002/j.1460-2075.1992.tb05411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taraseviciene-Stewart L., Kasahara Y., Alger L. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001;15(2):427–438. doi: 10.1096/fj.00-0343com. [DOI] [PubMed] [Google Scholar]
- 29.Gomez-Arroyo J., Mizuno S., Szczepanek K. Metabolic gene remodeling and mitochondrial dysfunction in failing right ventricular hypertrophy secondary to pulmonary arterial hypertension. Circ. Heart Fail. 2013;6(1):136–144. doi: 10.1161/CIRCHEARTFAILURE.111.966127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Al Ghouleh I., Rodriguez A., Pagano P.J., Csanyi G. Proteomic analysis identifies an NADPH oxidase 1 (Nox1)-mediated role for actin-related protein 2/3 complex subunit 2 (ARPC2) in promoting smooth muscle cell migration. Int. J. Mol. Sci. 2013;14(10):20220–20235. doi: 10.3390/ijms141020220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frazziano G., Al Ghouleh I., Baust J., Shiva S., Champion H.C., Pagano P.J. Nox-derived ROS are acutely activated in pressure overload pulmonary hypertension: indications for a seminal role for mitochondrial Nox4. Am. J. Physiol. Heart Circ. Physiol. 2014;306(2):H197–205. doi: 10.1152/ajpheart.00977.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brennan J.P., Bardswell S.C., Burgoyne J.R. Oxidant-induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. J. Biol. Chem. 2006;281(31):21827–21836. doi: 10.1074/jbc.M603952200. [DOI] [PubMed] [Google Scholar]
- 33.Liang C.C., Park A.Y., Guan J.L. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007;2(2):329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 34.Zielonka J., Cheng G., Zielonka M. High-throughput assays for superoxide and hydrogen peroxide: design of a screening workflow to identify inhibitors of NADPH oxidases. J. Biol. Chem. 2014;289(23):16176–16189. doi: 10.1074/jbc.M114.548693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koschmann J., Bhar A., Stegmaier P., Kel A.E., Wingender E. "Upstream Analysis": an integrated promoter-pathway analysis approach to causal interpretation of microarray data. Microarrays. 2015;4(2):270–286. doi: 10.3390/microarrays4020270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wingender E. The TRANSFAC project as an example of framework technology that supports the analysis of genomic regulation. Brief. Bioinform. 2008;9(4):326–332. doi: 10.1093/bib/bbn016. [DOI] [PubMed] [Google Scholar]
- 37.Kel A.E., Gossling E., Reuter I., Cheremushkin E., Kel-Margoulis O.V., Wingender E. MATCH: a tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 2003;31(13):3576–3579. doi: 10.1093/nar/gkg585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ranayhossaini D.J., Rodriguez A.I., Sahoo S. Selective recapitulation of conserved and nonconserved regions of putative NOXA1 protein activation domain confers isoform-specific inhibition of Nox1 oxidase and attenuation of endothelial cell migration. J. Biol. Chem. 2013;288(51):36437–36450. doi: 10.1074/jbc.M113.521344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zielonka J., Kalyanaraman B. Small-molecule luminescent probes for the detection of cellular oxidizing and nitrating species. Free Radic. Biol. Med. 2018 doi: 10.1016/j.freeradbiomed.2018.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Corsini M., Moroni E., Ravelli C. Cyclic adenosine monophosphate-response element-binding protein mediates the proangiogenic or proinflammatory activity of gremlin. Arterioscler. Thromb. Vasc. Biol. 2014;34(1):136–145. doi: 10.1161/ATVBAHA.113.302517. [DOI] [PubMed] [Google Scholar]
- 41.Ramesh V., Nair D., Zhang S.X. Disrupted sleep without sleep curtailment induces sleepiness and cognitive dysfunction via the tumor necrosis factor-alpha pathway. J. Neuroinflamm. 2012;9:91. doi: 10.1186/1742-2094-9-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Venkatachalam K., Venkatesan B., Valente A.J. WISP1, a pro-mitogenic, pro-survival factor, mediates tumor necrosis factor-alpha (TNF-alpha)-stimulated cardiac fibroblast proliferation but inhibits TNF-alpha-induced cardiomyocyte death. J. Biol. Chem. 2009;284(21):14414–14427. doi: 10.1074/jbc.M809757200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feoktistova M., Geserick P., Leverkus M. Crystal violet assay for determining viability of cultured cells. Cold Spring Harb. Protoc. 2016;2016(4) doi: 10.1101/pdb.prot087379. (pdb prot087379) [DOI] [PubMed] [Google Scholar]
- 44.Sarenac T., Trapecar M., Gradisnik L., Rupnik M.S., Pahor D. Single-cell analysis reveals IGF-1 potentiation of inhibition of the TGF-beta/Smad pathway of fibrosis in human keratocytes in vitro. Sci. Rep. 2016;6:34373. doi: 10.1038/srep34373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sahoo S., Meijles D.N., Al Ghouleh I. MEF2C-MYOCD and Leiomodin1 Suppression by miRNA-214 promotes smooth muscle cell phenotype switching in pulmonary arterial hypertension. PLoS One. 2016;11(5):e0153780. doi: 10.1371/journal.pone.0153780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Y., Kong H., Zeng X. Activation of NLRP3 inflammasome enhances the proliferation and migration of A549 lung cancer cells. Oncol. Rep. 2016;35(4):2053–2064. doi: 10.3892/or.2016.4569. [DOI] [PubMed] [Google Scholar]
- 47.Hulkower K.I., Herber R.L. Cell migration and invasion assays as tools for drug discovery. Pharmaceutics. 2011;3(1):107–124. doi: 10.3390/pharmaceutics3010107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang S., Li X., Parra M., Verdin E., Bassel-Duby R., Olson E.N. Control of endothelial cell proliferation and migration by VEGF signaling to histone deacetylase 7. Proc. Natl. Acad. Sci. USA. 2008;105(22):7738–7743. doi: 10.1073/pnas.0802857105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reinhart-King C.A. Endothelial cell adhesion and migration. Methods Enzymol. 2008;443:45–64. doi: 10.1016/S0076-6879(08)02003-X. [DOI] [PubMed] [Google Scholar]
- 50.Zhang Y., Dong X., Shirazi J., Gleghorn J.P., Lingappan K. Pulmonary endothelial cells exhibit sexual dimorphism in their response to hyperoxia. Am. J. Physiol. Heart Circ. Physiol. 2018 doi: 10.1152/ajpheart.00416.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qian N., Li X., Wang X., Wu C., Yin L., Zhi X. Tryptase promotes breast cancer angiogenesis through PAR-2 mediated endothelial progenitor cell activation. Oncol. Lett. 2018;16(2):1513–1520. doi: 10.3892/ol.2018.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Williams P.A., Stilhano R.S., To V.P., Tran L., Wong K., Silva E.A. Hypoxia augments outgrowth endothelial cell (OEC) sprouting and directed migration in response to sphingosine-1-phosphate (S1P) PLoS One. 2015;10(4):e0123437. doi: 10.1371/journal.pone.0123437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Johnston A.S., Lehnart S.E., Burgoyne J.R. Ca(2+) signaling in the myocardium by (redox) regulation of PKA/CaMKII. Front. Pharmacol. 2015;6:166. doi: 10.3389/fphar.2015.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barlow C.A., Kitiphongspattana K., Siddiqui N., Roe M.W., Mossman B.T., Lounsbury K.M. Protein kinase A-mediated CREB phosphorylation is an oxidant-induced survival pathway in alveolar type II cells. Apoptosis. 2008;13(5):681–692. doi: 10.1007/s10495-008-0203-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Selemidis S., Sobey C.G., Wingler K., Schmidt H.H., Drummond G.R. NADPH oxidases in the vasculature: molecular features, roles in disease and pharmacological inhibition. Pharmacol. Ther. 2008;120(3):254–291. doi: 10.1016/j.pharmthera.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 56.Al Ghouleh I., Meijles D.N., Mutchler S. Binding of EBP50 to Nox organizing subunit p47phox is pivotal to cellular reactive species generation and altered vascular phenotype. Proc. Natl. Acad. Sci. USA. 2016;113(36):E5308–5317. doi: 10.1073/pnas.1514161113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Babior B.M. NADPH oxidase: an update. Blood. 1999;93(5):1464–1476. [PubMed] [Google Scholar]
- 58.Xanthoudakis S., Curran T. Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J. 1992;11(2):653–665. doi: 10.1002/j.1460-2075.1992.tb05097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Z.X., Zhang W.N., Sun Y.Y., Li Y.H., Xu Z.M., Fu W.N. CREB promotes laryngeal cancer cell migration via MYCT1/NAT10 axis. Onco Targets Ther. 2018;11:1323–1331. doi: 10.2147/OTT.S156582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hussain M.A., Porras D.L., Rowe M.H. Increased pancreatic beta-cell proliferation mediated by CREB binding protein gene activation. Mol. Cell Biol. 2006;26(20):7747–7759. doi: 10.1128/MCB.02353-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chae H.D., Mitton B., Lacayo N.J., Sakamoto K.M. Replication factor C3 is a CREB target gene that regulates cell cycle progression through the modulation of chromatin loading of PCNA. Leukemia. 2015;29(6):1379–1389. doi: 10.1038/leu.2014.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cahill E., Costello C.M., Rowan S.C. Gremlin plays a key role in the pathogenesis of pulmonary hypertension. Circulation. 2012;125(7):920–930. doi: 10.1161/CIRCULATIONAHA.111.038125. [DOI] [PubMed] [Google Scholar]
- 63.Ghouleh I.A., Sahoo S., Meijles D.N. Endothelial Nox1 oxidase assembly in human pulmonary arterial hypertension; driver of Gremlin1-mediated proliferation. Clin. Sci. 2017;131(15):2019–2035. doi: 10.1042/CS20160812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wellbrock J., Harbaum L., Stamm H. Intrinsic BMP antagonist Gremlin-1 as a novel circulating marker in pulmonary arterial hypertension. Lung. 2015;193(4):567–570. doi: 10.1007/s00408-015-9735-5. [DOI] [PubMed] [Google Scholar]
- 65.Ciuclan L., Sheppard K., Dong L. Treatment with anti-gremlin 1 antibody ameliorates chronic hypoxia/SU5416-induced pulmonary arterial hypertension in mice. Am. J. Pathol. 2013;183(5):1461–1473. doi: 10.1016/j.ajpath.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]