

A series of 4-amino 2-anilinoquinazolines optimized for activity against the most lethal malaria parasite of humans, Plasmodium falciparum, was evaluated for activity against other human Plasmodium parasites and related apicomplexans that infect humans and animals. Four of the most promising compounds from the 4-amino 2-anilinoquinazoline series were equally as effective against the asexual blood stages of the zoonotic P. knowlesi, suggesting that they could also be effective against the closely related P. vivax, another important human pathogen.
KEYWORDS: Plasmodium, Plasmodium falciparum, antimalarial agents, apicomplexan parasites, drug metabolism, pharmacokinetics, quinazoline
ABSTRACT
A series of 4-amino 2-anilinoquinazolines optimized for activity against the most lethal malaria parasite of humans, Plasmodium falciparum, was evaluated for activity against other human Plasmodium parasites and related apicomplexans that infect humans and animals. Four of the most promising compounds from the 4-amino 2-anilinoquinazoline series were equally as effective against the asexual blood stages of the zoonotic P. knowlesi, suggesting that they could also be effective against the closely related P. vivax, another important human pathogen. The 2-anilinoquinazoline compounds were also potent against an array of P. falciparum parasites resistant to clinically available antimalarial compounds, although slightly less so than against the drug-sensitive 3D7 parasite line. The apicomplexan parasites Toxoplasma gondii, Babesia bovis, and Cryptosporidium parvum were less sensitive to the 2-anilinoquinazoline series with a 50% effective concentration generally in the low micromolar range, suggesting that the yet to be discovered target of these compounds is absent or highly divergent in non-Plasmodium parasites. The 2-anilinoquinazoline compounds act as rapidly as chloroquine in vitro and when tested in rodents displayed a half-life that contributed to the compound’s capacity to clear P. falciparum blood stages in a humanized mouse model. At a dose of 50 mg/kg of body weight, adverse effects to the humanized mice were noted, and evaluation against a panel of experimental high-risk off targets indicated some potential off-target activity. Further optimization of the 2-anilinoquinazoline antimalarial class will concentrate on improving in vivo efficacy and addressing adverse risk.
INTRODUCTION
Malaria is caused by infection with Plasmodium parasites. In humans, Plasmodium parasites cause over 200 million infections and are responsible for more than 400,000 deaths annually (1). Five species of Plasmodium are known to infect humans. Plasmodium falciparum and P. vivax are the most lethal causes of malaria in humans and account for 70% and 25% of all infections, respectively. P. falciparum is hyperendemic in Africa and is the most lethal parasite, while P. vivax is generally localized to Southeast Asia and South America and is responsible for relapses in blood-stage infection due to the activation of dormant liver-stage hypnozoites (2). In patients infected with P. malariae and P. ovale, which cause milder symptoms of infection, death is rare, and these species are not seen as a public health threat (3). P. knowlesi, a simian zoonotic parasite, is now considered the most common cause of human malaria in Malaysia, and the emergence of high-parasitemia cases and reported deaths have raised concerns of its virulence in human hosts (4).
There is currently a strong commitment to eliminate malaria in the 21st century. Current efforts to combat malaria have focused on vector control measures and the deployment of combination drug therapies. Together, these approaches have markedly reduced malaria-induced mortality in recent years, but malaria continues to represent a global health challenge (5). The current arsenal of clinically used artemisinin combination therapies and drug candidates undergoing clinical assessment may not be sufficient to eliminate the disease, due to the threat of emerging drug resistance (6) and a general decline in malaria immunity in areas of endemicity (7). Moreover, these emerging problems underscore the importance of efforts to identify novel therapies to combat malaria infection.
To contribute to the global fight against malaria, we previously disclosed preliminary findings on the antimalarial activities of the 2-anilinoquinazoline class (8). This class was identified by analyzing publicly disclosed data from high-throughput screens against multiple life cycle stages of the Plasmodium parasite (9–12). 2-Anilinoquinazoline hits from these screens were also included in the Medicines for Malaria Venture (MMV) Malaria Box (13). The 2-anilinoquinazoline class identified had modest activity against the asexual, liver schizont, and gametocyte forms of P. falciparum, while it possessed selectivity over mammalian cell lines.
We recently described the optimization of the antimalarial activity of the 2-anilinoquinazoline class (8). In this study, we defined the structure-activity relationship to generate early lead compounds WEB-484 to WEB-487, known herein as compounds 1 to 4, respectively (Fig. 1), which possess potent antimalarial activity against P. falciparum asexual parasites in vitro comparable to that of the known antimalarials chloroquine and mefloquine. Compounds 1 to 4 were also shown to maintain activity against the P. falciparum mefloquine-resistant line W2mef, but it is not known if compounds 1 to 4 maintain activity against other Plasmodium species or other drug-resistant parasite strains. In the optimization process, the physicochemical properties of the 2-anilinoquinazoline class, such as in vitro metabolic stability and aqueous solubility, were also improved, but limited pharmacokinetic data were acquired to inform in vivo models. In a preliminary study, compound 2 demonstrated 99.8% and 95% suppression of parasitemia when dosed (20 mg/kg of body weight) intraperitoneally and orally (p.o.), respectively, in a 4-day P. berghei mouse model of malaria. It is not known whether compounds 1 to 4 possess in vivo activity against P. falciparum.
FIG 1.

Structures of the 2-anilinoquinazoline compounds used in this study.
Several apicomplexan parasites closely related to Plasmodium are Toxoplasma, Babesia, and Cryptosporidium. Toxoplasma is an obligate parasite that infects nucleated cells but, in contrast to Plasmodium, does not infect erythrocytes or utilize hemoglobin as a source of sustenance. While Toxoplasma is not as deadly as Plasmodium, it is known to cause blindness (14), congenital birth defects (15), and toxoplasmosis in immunodeficient individuals (16). Babesia, akin to the Plasmodium parasite, infects host red blood cells and is a significant issue in livestock and, on occasions, in humans, causing babesiosis (17, 18). In humans, Cryptosporidium parasites primarily infect intestinal epithelial cells and cause acute diarrhea in the immunocompetent but can be life threatening to immunodeficient individuals (19). These apicomplexan species individually represent a major public health threat, and like Plasmodium, the development of new therapies is urgently required because of emerging resistance to known therapies.
Several hit compounds from the 2-anilinoquinazoline class (MMV001041, MMV011944, MMV000963, MMV006169, and MMV000986) in the MMV Malaria Box have been screened against multiple species of human and agricultural pathogens, including the apicomplexan parasites Toxoplasma, Babesia, and Cryptosporidium (see Table S1 in the supplemental material) (20). The 2-anilinoquinazolines in the Malaria Box displayed no activity against Toxoplasma gondii (50% effective concentrations [EC50s] > 30 μM) (21). MMV011944 and MMV006169 were shown to possess respective EC50s of 3.5 and 1.5 μM in a Cryptosporidium parvum viability assay (22). The 2-anilinoquinazolines in the Malaria Box also displayed activity against Babesia bovis, B. bigemina, B. caballi, and Theileria equi (20). The compounds generally displayed EC50s of between 1 and 10 μM against both Babesia and Theileria species, but the most potent compounds were MMV000963, which displayed an EC50 of 0.06 μM against B. bovis, and MMV001041, which displayed EC50s of 0.5 and 0.1 μM against B. bovis and B. caballi, respectively. These data, aside from indicating anti-Toxoplasma activity, provide evidence that the molecular target(s) of the 2-anilinoquinazoline class is potentially conserved in apicomplexan parasites. However, it was not known whether 2-anilinoquinazoline compounds 1 to 4 optimized for activity against P. falciparum would maintain or lose activity against apicomplexan parasites compared to that of the compounds in the MMV Malaria Box.
Screening of the activities of compounds 1 to 4 against Toxoplasma, Babesia, and Cryptosporidium parasites may reveal a new utility for the 2-anilinoquinazoline compound class in treating diseases caused by these organisms. We have therefore evaluated compounds 1 to 4 against common laboratory strains of these apicomplexan parasites to determine activity conservation across the phylum. We also further evaluated early lead compounds (compounds 1 to 4) from the 2-anilinoquinazoline class against drug-sensitive and -resistant strains of the Plasmodium parasite and determined the pharmacokinetic profile and the efficacy of the most promising 2-anilinoquinazoline in a P. falciparum humanized mouse model.
RESULTS AND DISCUSSION
In vitro P. falciparum and P. knowlesi asexual-stage activity.
Compounds 1 to 4 (Fig. 1) were previously evaluated against P. falciparum (8) using the lactate dehydrogenase (LDH)-based assay format (23). To ensure the consistency of EC50 values obtained by the different assay technologies, we determined the EC50 values of the lead compounds against P. falciparum in three previously described assay formats: the LDH (10) and hypoxanthine (24) assays and SYBR green fluorescence flow cytometry (25). Within each assay, compounds 1 to 4 displayed similar EC50 values (Table 1). The EC50 values for compounds 1 to 4 indicated that they were slightly less potent than the same compounds reported previously (EC50 values ranged from 26 to 35 nM for compounds numbered 53 to 56 in reference 8). Counterscreening of the compounds indicated that they did not directly suppress LDH activity, and the assay appeared to perform accurately since the EC50 values for antimalarial drugs were at the levels generally expected for these compounds (Table 1) (8).
TABLE 1.
Evaluation of compounds against P. falciparum 3D7 and P. knowlesi YH1 parasites
| Compound | Mean (SD) EC50 (μM) |
|||
|---|---|---|---|---|
| P. falciparum LDHa | P. falciparum hypoxanthineb |
Flow cytometryc
|
||
| P. falciparum | P. knowlesi | |||
| 1 | 0.079 (0.030) | 0.081 (0.011) | 0.087 (0.001) | 0.065 (0.007) |
| 2 | 0.121 (0.009) | 0.073 (0.008) | 0.081 (0.011) | 0.059 (0.010) |
| 3 | 0.074 (0.021) | NDd | 0.088 (0.002) | 0.065 (0.011) |
| 4 | 0.090 (0.004) | 0.118 (0.024) | 0.110 (0.037) | 0.095 (0.008) |
| Chloroquine | 0.017 (0.001) | 0.034 (0.002) | 0.053 (0.007) | 0.038 (0.008) |
| Mefloquine | 0.011 (0.005) | ND | 0.018 (0.004) | 0.003 (0.001) |
Growth was quantified by lactate dehydrogenase (LDH) activity after 72 h of treatment.
Proliferation was quantified by [3H]hypoxanthine incorporation after 72 h of treatment.
Growth was quantified by flow cytometry using SYBR green-stained asexual-stage parasites of P. falciparum 3D7 (72 h) or P. knowlesi YH1 (48 h).
ND, not determined.
To determine the activity conservation of compounds 1 to 4 across Plasmodium species that infect and cause symptoms in humans, we evaluated our compounds against P. knowlesi, a Plasmodium species of simian origin that causes symptoms similar to those seen in P. falciparum infection in humans (26). We used the SYBR green flow cytometry assay to compare the potency of compounds 1 to 4 between the asexual stage of P. knowlesi and P. falciparum. The results showed that compounds 1 to 4 possessed comparable activity against the two species (Table 1; see also Fig. S1 in the supplemental material), suggesting that the molecular target of the compound series is conserved between these two species and potentially all Plasmodium species that infect humans.
In vitro P. falciparum viability kinetics.
To determine the kinetics of the loss of viability in P. falciparum asexual stages following treatment with compounds 1, 2, and 4, we followed the protocol described by Linares et al. (27). This assay measures the rate of parasite killing within 24 and 48 h of treatment. The assay proceeds by first culturing unlabeled erythrocytes infected with P. falciparum in the presence of 10× EC50 of compound for 24 and 48 h. After the compounds were washed out, parasites that had been treated for 24 and 48 h were added to erythrocytes that had been prelabeled with the cell-permeant carboxyfluorescein diacetate succinimidyl ester (CFDA-SE) fluorescent dye. The parasites were then labeled with the Hoechst 33342 stain, allowing the measurement of doubly stained infected erythrocytes, and this thus determined the ability of the compounds to eliminate viable parasites within 24 or 48 h. We evaluated compounds 1, 2, and 4 in this assay and compared the results to those for known antimalarials with different parasite killing kinetic profiles (Fig. 2). The results demonstrate that the 2-anilinoquinazolines 1, 2, and 4 display fast parasite killing kinetics comparable to those of artemisinin and chloroquine. Pyrimethamine and atovaquone showed a slower killing response in this assay, consistent with the findings described in the literature (27). This assay reconfirms existing data that the 2-anilinoquinazoline compound class kills parasites within the first 24 h of invasion of the erythrocyte (8).
FIG 2.
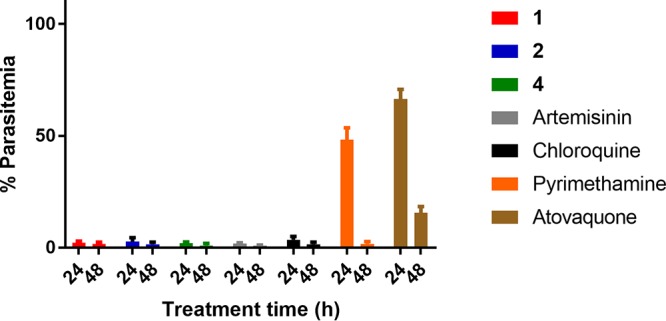
P. falciparum asexual-stage growth reduction rate assay. P. falciparum 3D7-infected erythrocytes were treated with compound (10× EC50 in Table 1, as determined by the hypoxanthine assay) for 24 or 48 h, after which the compound was washed out and the parasites were grown for another 48 h. Parasite viability was quantified by flow cytometry and is shown as the percentage of infected CFDA-SE-stained erythrocytes in drug-treated samples at 24 or 48 h using vehicle-treated parasites as a reference (100% parasitemia).
Activity against a panel of P. falciparum-resistant strains.
The early lead compounds were previously shown to display slightly less activity against the chloroquine-resistant P. falciparum strain W2mef (28) than the chloroquine-sensitive strain 3D7 (8). To ensure the effectiveness of the 2-anilinoquinazoline class against P. falciparum drug-resistant strains other than W2mef, we profiled compounds 2 and 4 against a diverse panel of resistant P. falciparum lines. These included the chloroquine-resistant strains K1 and Dd2 as well as the mefloquine-resistant strains W2mef3 (29) and Cam3 (30). Dd2 and W2 are also resistant to pyrimethamine. Cam3 R539T was resistant to artemisinin, in contrast to the Cam3 Rev artemisinin-revertant control strain. The data in Table 2 show that compounds 2 and 4 were slightly less potent against all the drug-resistant P. falciparum lines evaluated than against 3D7 by a factor of 1.4- to 2-fold. These data suggest that compounds 2 and 4 and likely the 2-anilinoquinazoline class are not susceptible to the same drug resistance mechanisms conferred by the subset of clinically used antimalarials, namely, chloroquine, mefloquine, and artemisinin, to which the parasites were 3- to 10-fold more resistant. Compounds 1 to 4 possess a chemical structure that resembles the scaffolds of known quinoline antimalarials, such as chloroquine and mefloquine, and therefore, it is pertinent that the 2-anilinoquianzoline class is not a substrate of the same transport/efflux channels. However, the degree of resistance to compounds 2 and 4 indicates that broad resistance mechanisms, such as Pfmdr1 amplifications, could be playing a role (28, 29). Previous studies of a related 2,4-diamino quinazoline series indicated that histone lysine methyltransferases could be the target of these compounds; however, recent use of photo-cross-linkable probes indicated that the compounds bound to many targets, including redox and nucleosome proteins, as well as chaperones (31–33).
TABLE 2.
Evaluation of compounds against drug-resistant P. falciparum strainsa
| Compound | Mean (SD) EC50 (μM) |
||||||
|---|---|---|---|---|---|---|---|
| 3D7b | K1 | Dd2 | W2mef | W2mef3 | Cam3 R539Tc | Cam3 Revc | |
| 2 | 0.084 (0.023) | 0.144 (0.073) | 0.150 (0.063) | 0.147 (0.039) | 0.138 (0.075) | 0.115 (0.005) | 0.162 (0.041) |
| 4 | 0.082 (0.024) | 0.167 (0.040) | 0.154 (0.047) | 0.163 (0.042) | 0.151 (0.078) | 0.133 (0.002) | 0.176 (0.049) |
| Chloroquine | 0.007 (0.008) | 0.237 (0.049) | 0.114 (0.032) | 0.115 (0.038) | 0.097 (0.055) | 0.131 (0.025) | 0.114 (0.013) |
| Mefloquine | 0.012 (0.004) | 0.006 (0.001) | 0.018 (0.002) | 0.017 (0.001) | 0.043 (0.013) | 0.041 (0.010) | 0.042 (0.013) |
| Dihydroartemisininc | 1.7% | NDd | ND | ND | ND | 31% | 10.1% |
EC50 data represent means and SDs for two biological replicates each from three technical experiments measuring the viability of P. falciparum drug-resistant asexual-stage parasites following exposure to compounds in a 10-point dilution series for 72 h. Parasitemia was quantified by flow cytometry using SYBR green.
3D7 was used as a drug-sensitive control strain.
Survival in the presence of 700 nM dihydroartemisinin was tested. Cam3 R539T was used as the artemisinin-resistant control strain, and the bottom row of data confirm its resistance phenotype relative to the phenotype of reversed strain Cam3 Rev and 3D7 (30) in the ring-stage survival assay following the previously described protocol (44).
ND, not determined.
In vitro activity of 2-anilinoquinazolines in other apicomplexan parasites.
On discovering that the activity of the 2-anilinoquinazoline class was conserved across two species of Plasmodium and potentially all species infectious to humans, we sought to determine the in vitro activity of compounds 1 to 4 across several apicomplexan parasites. The conservation of activity between Plasmodium and apicomplexan parasites may indicate that the molecular target of the 2-anilinoquinzoline class is conserved between species. Toxoplasma and Plasmodium parasites share several pathways, such as trafficking pathways (34), and express proteins with high sequence conservation, such as kinases involved in host cell invasion and remodeling (35). Toxoplasma parasites, however, have a life cycle distinct from that of Plasmodium, and most antimalarial drugs do not possess the same degree of potency against Toxoplasma, suggesting a divergence in the types of proteins expressed, their homology, or the biochemical processes occurring within the parasites.
We used a host lytic cell assay (36) adapted to measure the viability of Toxoplasma parasites when exposed to compounds 1 to 4 to determine the correlation with activity against P. falciparum. The results showed that all compounds were significantly less active against Toxoplasma than against Plasmodium according to the EC50 values observed (Table 3). Interestingly, compounds 3 and 4 (EC50s, 0.44 and 0.39 μM, respectively) were significantly more potent than compounds 1 and 2 (EC50s, >5 μM). Both compound 3 and compound 4 have an N-methyl-4-piperidine substitution in the N-4 position of the quinazoline, whereas compounds 1 and 2 have ethyl(N-methylpiperazine) in the N-4 position. The differences in activity between different substitution patterns in the N-4 position suggests divergent structure-activity relationships between the species. It is also plausible that the activity of analogues 3 and 4 observed in Toxoplasma is not related to the activity observed in Plasmodium, and therefore, it is likely that the molecular target is not conserved between species.
TABLE 3.
Evaluation of compounds against apicomplexan parasitesd
| Compound | Mean (SD) EC50 (μM) |
||
|---|---|---|---|
| Toxoplasma gondiia | Babesia bovisb | Cryptosporidium parvumc | |
| 1 | >5 | 0.84 (0.20) | 8.0 (0.65) |
| 2 | >5 | 1.60 (0.27) | 14.6 (0.24) |
| 3 | 0.44 (0.08) | 1.70 (0.38) | 20.1 (2.9) |
| 4 | 0.39 (0.23) | 1.90 (0.11) | 11.2 (1.6) |
T. gondii viability was quantified by crystal violet staining (A590) following exposure to compounds in an 8-point compound dilution series for 72 h from 3 replicates. The EC50 of control compound 3MB-PP1 was 0.20 (0.03) μM (36).
B. bovis growth assays were conducted by counting thin blood smears of three replicates following 24 h of compound treatment.
Data for C. parvum (intestinal epithelial host cell line HCT-8 was used) were derived from four replicates following 48 h of compound treatment. The EC50 of control compound nitazoxanide was 1.68 (0.33) μM.
Data for HCT-8 cell toxicity were also measured using the CellTiter Glo assay and were derived from 2 replicates following 48 h of compound treatment, and the value for all compounds was >75 μM.
Several compounds from the 2-anilinoquinazoline class found in the MMV Malaria Box have been evaluated in viability assays against B. bovis, B. bigemina, and B. caballi (20). To determine whether our series of 2-anilinoquinazolines optimized for activity against Plasmodium possessed activity against Babesia, we evaluated compounds 1 to 4 against B. bovis using a protocol adapted from that of Jackson et al. (37), measuring the parasitemia of thin blood smears after 24 h of drug treatment as the viability readout. The results indicated that compounds 1 to 4 all have EC50 values of between 0.84 and 1.90 μM (Table 3). These EC50 values indicate that these compounds are significantly less potent against Babesia species than against Plasmodium species (Table 1). The Malaria Box data and the data presented here imply that the molecular target(s) homology in Plasmodium is divergent from that in Babesia.
The Malaria Box data also show that the 2-anilinoquinazolines MMV006169 and MMV011944 (Table S1) possess activity against C. parvum, with EC50 values of 1.5 and 3.5 μM, respectively, in a viability assay (20, 22). To determine if the C. parvum and P. falciparum activities of 2-anilinoquinazolines 1 to 4 are conserved, we used a previously described high-content imaging fluorescence assay to quantify parasite viability (38). The data indicated that none of the compounds 1 to 4 was potent against C. parvum, since all had EC50 values over 10 μM (Table 3).
The 2-anilinoquinazoline class in general has micromolar activity against the apicomplexan parasites evaluated, in contrast to the concentrations at which they show activity against Plasmodium. This is likely because compounds 1 to 4 have been optimized on the basis of their activity against P. falciparum. It is possible that the protein target(s) of the 2-anilinoquinazoline class in Plasmodium is not conserved in other apicomplexan parasites or that the protein target(s) is conserved between Plasmodium and other apicomplexan parasites but that the homology of amino acids in the 2-anilinoquinazoline binding site is low.
The 2-anilinoquinazolines MMV000963 and MMV001041 from the MMV Malaria Box possess EC50 values of less than 100 nM against Babesia species (20). The differential anti-Babesia activity of the MMV Malaria Box analogues MMV000963, MMV006169, MMV001041, and MMV011944 (20) suggests that the homology of the molecular target is not highly conserved and that the development of compounds targeting a specific Babesia species may be possible. Optimization of the 2-anilinoquinazoline series for other apicomplexan parasites does not appear to be feasible because of the modest micromolar activity of the MMV Malaria Box compounds and compounds 1 to 4.
In vitro metabolism and pharmacokinetics.
Evaluation of 2-anilinoquinazolines 1 to 4 in apicomplexan parasite viability assays determined that this class was the most potent against Plasmodium. From these data, we next evaluated the in vitro metabolism and pharmacokinetics of the most promising compound from this class, WEB-485 (compound 2), to assess its suitability for administration in rodent models of malaria. Previously, the in vitro metabolic stability in mouse (and human) microsomes was established to predict intrinsic clearance in vivo (Table 4) (8). Here, we determined the in vitro metabolism and the pharmacokinetic profile of compound 2 in Sprague-Dawley rats.
TABLE 4.
Metabolic stability in liver microsomes and protein plasma binding for WEB-485 (compound 2)b
| Species | Half-life (min) | In vitro CLint (μl/min/mg protein) | In vivo predicted CLint (ml/min/kg) | Predicted EH | PPB (%) | Predicted CLblood (ml/min/kg) |
|
|---|---|---|---|---|---|---|---|
| Binding not corrected | Binding corrected | ||||||
| Rat | 42 | 42 | 72 | 0.51 | 96.9 | 35 | 4.8 |
| Humana | 77 | 23 | 19 | 0.47 | 92.2 | 10 | |
| Mousea | 47 | 37 | 95 | 0.44 | 98.8 | 53 | |
Metabolic stability parameters in human and mouse liver microsomes were reported previously (8).
CLint, intrinsic clearance; EH, hepatic extraction ratio, which was predicted based on in vitro intrinsic clearance; PPB, plasma protein binding; CLblood, clearance from blood.
In vitro metabolism was performed by incubation in rat liver microsomes. These results indicated predicted hepatic extraction ratios similar to the previously described mouse and human values (Table 4) (8). To determine the major metabolites formed, mass spectrometry (MS) was used to identify metabolite masses to predict sites of metabolism on the scaffold of compound 2. These results showed that demethylated N-methylpiperazine was the major metabolite, accounting for 60% of the total metabolite peak area determined by liquid chromatography (LC)-MS (Table 5). Two other metabolites were detected, including a mono-oxygenated product, presumably with mono-oxygenation at the 6 position of the quinazoline, and a product with dealkylation of the ethylpiperazine moiety at the N-4 position. The N-demethylated and mono-oxygenated products are metabolites previously detected in mouse liver microsome studies (8), while an N-4-dealkylated product was not detected. The previously described structure-activity relationship of the 2-aminoquinazoline series shows that the 4 position is tolerant of a diverse array of functionality (8), and therefore, the piperazine N-demethylated and N-4-dealkylated metabolites likely contribute to the in vivo antimalarial activity. Synthesis of the metabolites in future work will be required to unequivocally determine this. It was additionally determined that compound 2 was stable following incubation in rat whole blood in vitro.
TABLE 5.
Metabolite identification of WEB-485 (compound 2) from incubation in rat microsomes
| Predicted metabolite description | ΔMassa (Da) | MH+b | tR (min)c | % of total metabolite peak aread |
|---|---|---|---|---|
| WEB-485 (parent) | 415 | 2.15 | ||
| Dealkylation | −128 | 287 | 3.15 | 16 |
| Mono-oxygenation | +16 | 431 | 2.18 | 24 |
| N-demethylation | −14 | 401 | 2.14 | 60 |
ΔMass, change in mass from the parent ion.
MH+, mass detected by positive electrospray ionization.
tR, LC retention time of compound or metabolite detected.
Metabolite peak area profiles were calculated by assuming comparable response factors for each component; however, authentic standards would be required for confirmation.
To assess the pharmacokinetic properties in vivo, compound 2 was administered intravenously (i.v.) (2.9 mg/kg) and orally (p.o.) (29.5 mg/kg) to male Sprague-Dawley rats, and the plasma concentrations of compound 2 were monitored at several time points over 24 h. It was noted that there was no evidence of adverse reactions or compound-related side effects observed in any rats during the 24-h-postdose sampling period. Following i.v. administration, the plasma concentration of compound 2 remained above the analytical limit for the 24-h duration of the study (Table 6 and Fig. 3), and the blood volume distribution was high and clearance was low. The measured blood clearance was lower than that predicted using rat microsomes (Table 4), possibly due to the influence of binding to plasma and microsomal proteins, which was not factored into the in vitro predictions. There was no evidence of elimination of intact compound 2 via the urine. After oral administration, compound 2 was slowly absorbed, with maximum plasma concentrations being observed at between 2.5 and 7 h (Table 6 and Fig. 3). The slow absorption is possibly due to ionization of the dibasic piperazine functionality in the intestinal tract, resulting in a reduced rate of absorption. Additionally, compound 2 possesses moderate aqueous solubility (measured by nephelometry), possibly limiting the rate of dissolution. The apparent bioavailability (F) was high (71%), and concentrations of compound 2 greater than 0.1 μM were present at 24 h postadministration. Compound 2 was highly bound to plasma proteins (Table 4), and this likely contributed to the long half-life in rats. Given the similarity between the predicted hepatic extraction ratio and the plasma protein plasma binding of compound 2 in mice and rats (Table 4), compound 2 would be predicted to also have a good exposure profile in mice. For this reason, the efficacy of compound 2 was examined in a mouse model of P. falciparum infection.
TABLE 6.
Pharmacokinetic data on WEB-485 (compound 2) in male Sprague-Dawley ratsa
| Adminstration route | Dose (mg/kg) | Half-life (h) | Plasma CL (ml/min/kg) | Plasma Vss (liters/kg) | Blood CL (ml/min/kg)b | Blood Vss (liters/kg)b | Plasma Cmax (μM) | Tmax (h) | AUC (h·μM) | BA (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| i.v. | 2.9 (0.01) | 6.7 (0.2) | 46.1 (6.4) | 26.4 (4.0) | 15.9 (2.2) | 9.1 (1.4) | NA | NA | 2.6 (0.3) | NA |
| p.o. | 29.5 (0.2) | 8.9 (1.7) | NA | NA | NA | NA | 1.0 (0.3) | 4.8 (2.3) | 18.4 (5.8) | 71 (23) |
The compounds were administered intravenously (i.v.) and by oral gavage (p.o.) using a 0.9% saline vehicle. Data represent the averages for 3 rats; values in parentheses represent SD. CL, clearance; Vss, volume of distribution at steady state; BA, bioavailability; NA, not applicable.
The blood-to-plasma ratio measured ex vivo was 2.9.
FIG 3.
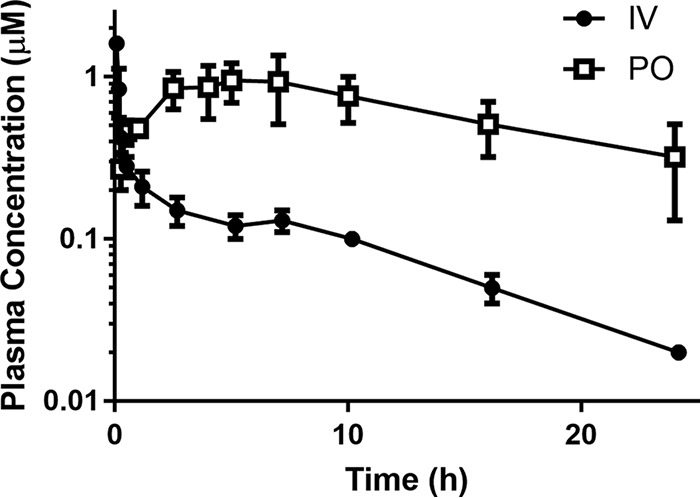
Pharmacokinetic profile of WEB-485 (compound 2) in male Sprague-Dawley rats. The compounds were administered i.v. (2.9 mg/kg) and p.o. (29.5 mg/kg) using a 0.9% saline vehicle. Data are averages for 3 rats; error bars represent SD.
Humanized P. falciparum NOD-scid IL2Rγnull 4-day mouse model.
Compounds 1 to 4 were previously shown to exhibit efficacy in a Peters 4-day mouse model of malaria using P. berghei, a rodent-specific parasite species (8), but no in vivo efficacy study has been undertaken using human parasites. We therefore determined the in vivo efficacy of the 2-anilinoquinazoline class against P. falciparum by employing compound 2 in a 4-day model using humanized NOD-scid IL2Rγnull mice (39). These immunodeficient mice, carrying a null mutation of the interleukin 2 (IL-2) receptor γ chain (IL2Rγ), lack mature natural killer cells and possess defects in their immune system, enabling them to receive daily engraftments of human erythrocytes. The 3D70087/N9 strain of P. falciparum was selected for its ability to develop reproducibly in human erythrocyte-engrafted immunodeficient mice.
For the model, NOD-scid IL2Rγnull mice were inoculated with P. falciparum-parasitized human erythrocytes on day 0. On day 3, 1% parasitemia was achieved, and then chloroquine and compound 2 were independently administered by oral (p.o.) gavage at 50 mg/kg and were then administered again on days 4, 5, and 6. At 4 h postinfection and on days 3 to 7, blood smears were taken and parasitemia was evaluated. The results displayed in Fig. 4A show that compound 2 cleared the parasitemia by day 6. The efficacy of compound 2 was comparable to that of chloroquine according to the parasite clearance rate. The plasma exposure of compound 2 in the mouse model (time to maximum concentration in plasma [Tmax], 5 h; maximum concentration in plasma [Cmax], 0.97 μM; area under the concentration-time curve [AUC] from 0 to 24 h, 17.7 h·μM) (Fig. 4B) was reasonably consistent with the pharmacokinetic profile determined in rats (Fig. 3).
FIG 4.

Efficacy profile of WEB-485 (compound 2) in the P. falciparum humanized NOD-scid IL2Rγnull 4-day mouse model of malaria. (A) Mice were infected with P. falciparum parasites (2 × 107 parasitized erythrocytes) intravenously. Compound 2 and chloroquine (50 mg/kg in a Tween 80-ethanol vehicle) and the vehicle were administered by oral gavage on day 3 after infection (day 0) and then on days 4, 5, and 6 (signified by arrows below the x axis). On days 3 to 7, blood was taken and parasitemia was evaluated. (B) Blood exposure levels of compound 2 (50 mg/kg by oral gavage) in mice following the first dose on day 3 of the mouse model. The mean values of the pharmacokinetic parameters were as follows: Tmax, 5 h; Cmax, 0.97 μM; AUC from 0 to 24 h, 17.7 h·μM. Data are averages for 2 mice; error bars represent the range.
It was noted that after four 50-mg/kg oral doses of compound 2, on day 6 of the humanized mouse study, compound 2-treated mice displayed acute toxicity symptoms, such as ataxia, decreased activity, and rough fur. Acute toxicity was not observed in compound 2-treated (29 mg/kg) rats after a single oral dose in the pharmacokinetic study and was not previously observed in P. berghei mouse models with dosing at 20 mg/kg for 4 days (8). These observations are suggestive of dose-dependent toxicity in vivo, although it is possible that the toxicity of compound 2 is specific to mice with a compromised immune system.
One plausible reason for the toxicity observed in the mouse model is hemolysis. An earlier study observed after artemisinin treatment that mice displayed toxicity symptoms associated with hemolysis, which is a consequence of the fast parasite killing ability of artemisinin (40). We postulated that compound 2 could also cause toxicity due to hemolysis since it has a parasite killing rate similar to that of artemisinin. However, toxicity was not observed in the P. berghei mouse models previously conducted with compound 2 (8), and therefore, it is unlikely that the toxicity displayed by the humanized mice in this model was not due to hemolysis.
To determine the mechanisms potentially responsible for the dose-dependent in vivo toxicity, we screened compound 2 in a panel of 87 high-risk targets known to confer adverse complications in vivo, such as G protein-coupled receptors (GPCRs), ion channels, and transporters. The panel therefore provides a reliable overview of compound promiscuity and risk. Compound 2 was screened in this panel at 10 μM, a concentration that is 10-fold higher than the plasma exposure levels attained in the humanized mouse model. The results of the screen showed that compound 2 inhibits 21 targets at greater than 50% inhibition at 10 μM (Table S2). These targets included adrenergic α subtypes, histamine H subtypes, human ether-à-go-go-related gene, muscarinic M subtypes, opiate κ, serotonin 5-hydroxytryptamine subtypes, dopamine, and norepinephrine transporters. One or a combination of these targets may be responsible for the acute toxicity observed in the humanized mouse model. The high hit rate in this panel is likely due to the lipophilicity and the dibasic piperazine functionality in the N-4 position of compound 2. Compounds with these types of properties are notorious for inhibiting GPCRs, ion channels, and transporters (41, 42). It was previously found that 2-anilinoquinazolines without the basic functionality in the N-4 position were devoid of hERG activity (unpublished data), suggesting that the high hit rate of compound 2 in the panel is primarily due to the presence of this functionality. It is also conceivable that one of the metabolites of compound 2 is responsible for the dose-dependent toxicity observed, and this will be another factor monitored in future studies. Ultimately, further optimization of the 2-anilinoquinazoline series will concentrate on addressing this issue.
Conclusions.
The 2-anilinoquinazolines 1 to 4 retained potency against the P. knowlesi blood stage, suggesting that they are likely to also be potent against all human malaria parasites, including P. vivax. The compounds were, however, considerably less effective against related apicomplexan parasites, suggesting that their unknown target(s) is Plasmodium specific or is highly divergent. It is likely the 2-anilinoquinazoline series is not susceptible to the same drug-resistant mechanisms of other clinically used antimalarial compounds, since the activities of 2-anilinoquinazolines 2 and 4 in a panel of drug-resistant P. falciparum lines were slightly reduced compared to those in the parental line. The 2-anilinoquinazoline compound 2 was able to rapidly clear P. falciparum in a humanized mouse in vivo model of infection at a rate comparable to that of the clinically used chloroquine. Identifying the mechanism of action of the 2-anilinoquinazoline series is now a priority since this will assist in further improvement of the potency of the series and help reduce off-target effects.
MATERIALS AND METHODS
Compounds.
Compounds 1 to 4 were synthesized using previously described methods (8). Known antimalarial drugs used as controls were purchased from commercial sources.
P. falciparum LDH assay.
Parasite viability assays were performed as described by Gamo et al. (10). Briefly, P. falciparum 3D7 parasites were cultured according to the procedure described by Trager and Jensen (43) in RPMI-HEPES medium supplemented with l-glutamine and AlbuMAX II lipid-rich bovine serum albumin (BSA). Early-ring-stage P. falciparum 3D7 parasites were obtained by sorbitol synchronization and incubated with compounds solubilized in dimethyl sulfoxide (DMSO; at not greater than a 0.02% final concentration to limit toxicity) in 10-point titrations. Parasitemia was determined at 72 h using a lactate dehydrogenase (LDH) readout as a percentage relative to the value for the DMSO vehicle control. Values were plotted using a 4-parameter log dose, nonlinear regression analysis with a sigmoidal dose-response (variable slope) curve fit in GraphPad Prism (version 6.05) software to generate drug curves and EC50 values.
P. falciparum and P. knowlesi flow cytometry assay.
The P. falciparum 3D7 strain and the P. knowlesi YH1 strain were cultured in RPMI-HEPES medium supplemented with 2.3 g/liter sodium bicarbonate and 5 g/liter AlbuMAX. For P. knowlesi, an additional 4 g/liter dextrose and 0.292 g/liter glutamine were added into the culture medium. P. falciparum and P. knowlesi parasites were synchronized by sorbitol and Nycodenz medium, respectively. The drug assays were performed in 50 μl in 96-well plates at 0.5% parasitemia and 2% hematocrit. The compounds were serially diluted in 1% DMSO. Parasite growth assays were performed for 72 h and 48 h for P. falciparum and P. knowlesi, respectively. Parasite cultures were fixed with 50 μl 0.25% glutaraldehyde for 30 min at room temperature. The culture supernatants were removed, and the cells were stained with 5% SYBR green for 30 min. Parasitemia was defined by the proportion of Alexa Fluor 488-A-positive cells in 50,000 recorded events. Parasite growth values were normalized to those for the vehicle control group and plotted as a dose-response curve in GraphPad Prism software.
P. falciparum hypoxanthine incorporation assay.
The hypoxanthine incorporation assay was adapted from the procedure described by de Cozar et al. (24). A culture of red blood cells parasitized with strain 3D7 at 0.5% parasitemia and 2% hematocrit in RPMI 1640 medium, AlbuMAX, and 5 μM hypoxanthine was exposed to 9-step 3-fold serial dilutions of the compounds. The plates were incubated for 48 h at 37°C in 5% CO2, 5% O2, and 90% N2. After 24 h of incubation, [3H]hypoxanthine was added, and the plates were incubated for another 24 h. After that period, the plates were harvested on a glass fiber filter using a Tomtec Cell Harvester 96 apparatus. The filters were dried, and melt-on scintillator sheets were used to determine the incorporation of [3H]hypoxanthine. Radioactivity was measured using a microbeta counter. Data were normalized using the incorporation of the positive control (parasitized red blood cells without drug). The 50% inhibitory concentration (IC50) was determined using Excel and Grafit software. Inhibition data were fit to a 2-parameter equation, y = 100/[1 + (x/IC50)]s, where s is the slope factor, the lower data limit was 0, and the upper data limit was 100.
P. falciparum drug-resistant parasite viability assays.
Ring-stage parasites at 0.5% parasitemia were grown in a 50-μl culture at 2% hematocrit in 96-well round-bottom plates (Falcon) with doubling dilutions of each drug. After incubation for 72 h, each well was fixed at room temperature for 30 min with 50 μl of 0.25% glutaraldehyde (ProSciTech) diluted in phosphate-buffered saline (PBS). Following centrifugation at 350 × g for 2 min, supernatants were discarded and trophozoite-stage parasites were stained with 50 μl of 5× SYBR green (Invitrogen) diluted in PBS. The parasitemia of each well was determined by counting 50,000 cells by flow cytometry using a Cell Lab Quanta SC-MPL flow cytometer (Beckman Coulter). Growth was expressed as a percentage of the parasitemia obtained using a drug-free control. All samples were tested in triplicate. The ring survival assay for determining the sensitivity of artemisinin-resistant strain Cam3 Rev and the artemisinin-resistant control strain, Cam3 R539T, to the compounds was followed as previously described (44).
P. falciparum killing rate assay.
The killing rate assay was adapted from the procedure described by Linares et al. (27). Briefly, P. falciparum 3D7-infected erythrocytes were incubated in the presence of compound (at a concentration of 10× IC50; the IC50 was determined from the hypoxanthine assay; Table 1) for 24 and 48 h. Drug was renewed after the first 24 h of treatment by removing old medium and replenishing with new culture medium with fresh compound. After treatment, the drug was removed and the culture was diluted (1 in 3 dilution) using fresh erythrocytes (2% hematocrit) previously labeled with the intracellular dye (carboxyfluorescein diacetate succinimidyl ester [CFDA-SE]). Following a further 48 h of incubation under standard conditions, the ability of treated parasites to establish infection in freshly labeled erythrocytes was detected by two-color flow cytometry after labeling of parasite DNA (using Hoechst 33342 stain). Cultures containing untreated parasites were used as controls. Parasite viability is shown as the percentage of infected CFDA-SE-stained erythrocytes in drug-treated samples at 24 or 48 h, using as a reference untreated samples of the initial inoculum, after 48 h of incubation with labeled erythrocytes and labeling of parasite DNA. Samples were analyzed using FACSDiva software. Quantification of double-stained (with CFDA-SE intracellular dye and Hoechst dye) erythrocytes was used to evaluate parasite viability after the drug treatments. Parasite viability was shown as the percentage of infected CFDA-SE-stained erythrocytes in drug-treated samples at 24 or 48 h, using as a reference untreated samples of the initial inoculum, after 48 h of incubation with labeled erythrocytes and labeling of parasite DNA. Chloroquine, pyrimethamine, atovaquone, and artemisinin were included as controls for comparative classification of the killing behavior of the compounds tested.
Toxoplasma growth inhibition assay.
Primary human foreskin fibroblasts (HFFs) cells were maintained in culture at 37°C in 10% CO2 with Dulbecco modified Eagle medium (DMEM) supplemented with 10% Cosmic Calf serum. Prior to inoculation, HFFs were refreshed with DMEM supplemented with 1% fetal calf serum and 1% GlutaMAX medium (Invitrogen). Parasites were inoculated onto HFFs and maintained at 37°C in 10% CO2. Compound titrations were added to the parasite culture at 1 h postinvasion. Parasites were incubated for 72 h in the presence of the compounds. Growth was assessed by crystal violet staining, and the absorbance was measured at 590 nm. Inhibition was normalized to complete lysis (0%) and measured against that for uninfected host cells (100%), performed in 3 replicate experiments. Best-fit and IC50 calculations were performed using 2-parameter equations in GraphPad Prism software. The EC50 values for the HFF toxicity of compounds 1 to 4 were determined to be greater than 5 μM.
Babesia viability assay.
B. bovis parasites (W strain [45]) were cultured following a previously described procedure (37). Briefly, bovine red blood cells were maintained under HEPES-buffered RPMI 1640 medium with l-glutamine, AlbuMAX II, hypoxanthine, and sodium bicarbonate and maintained under microaerophilic conditions. One hundred microliters of B. bovis parasite-infected red blood cells at 1% parasitemia and 5% hematocrit was added to a 96-well plate. Compounds 1 to 4 were diluted in DMSO to reach the various working concentrations and added to parasite cultures to a final DMSO concentration of 0.5%. Parasite growth assays were conducted in triplicate for 24 h, covering approximately two red blood cell life cycles. After 24 h, Giemsa-stained thin blood smears were made and parasitemia was quantified by enumerating the percentage of parasite-infected red blood cells.
Cryptosporidium viability assay.
Cryptosporidium parvum growth within the intestinal epithelial cell line HCT-8 was assayed using high-content microscopy as previously described (38). Briefly, C. parvum oocysts (Iowa strain; Bunch Grass Farms, Deary, ID) stored in phosphate-buffered saline (PBS) with 1,000 IU/ml penicillin and 1 mg/ml streptomycin were induced to excyst by treatment with 10 mM HCl (37°C, 10 min), followed by treatment with 2 mM sodium taurocholate (Sigma-Aldrich) in PBS with Ca2+ and Mg2+ (16°C, 10 min). Excysted oocysts (∼5.5 × 103/well) were used to infect >90% confluent monolayers of the human ileocecal adenocarcinoma cell line HCT-8 (ATCC) grown in 384-well culture plates. The experimental compounds or DMSO (vehicle) was added 3 h after infection, and the cultures were incubated for 48 h. The culture plates were then washed, fixed with 4% paraformaldehyde, and probed with 1.33 μg/ml fluorescein-labeled Vicia villosa lectin (Vector Laboratories) diluted in 1% bovine serum albumin (BSA)–PBS with 0.1% Tween 20 for 1 h at 37°C to label the vegetative forms of C. parvum (38, 46). Nuclei were counterstained with Hoechst 33258 dye. A Nikon Eclipse TI2000 epifluorescence microscope with a motorized stage and an EXi Blue digital camera (QImaging, Surrey, BC, Canada) were then used to acquire a three-by-three 20× field photo of the center of each well (covering approximately 13% of the surface area). Parasites and host cell nuclei were enumerated by exporting the images as .tif files into NIH ImageJ software and using the batch processing function to execute previously validated macros (38). Graphs were plotted, and the half-maximal effective concentrations (EC50s) were calculated using GraphPad Prism (version 7.01) software.
Host cell growth inhibition assay.
HCT-8 cell toxicity was measured using the CellTiter AQueous assay (Promega) (47). This colorimetric method measures the reduction of a tetrazolium compound by NADPH and NADH in metabolically active cells. HCT-8 cells were grown to confluence in 384-well plates. The growth medium was removed and replaced with fresh medium containing the experimental compounds, and the cultures were incubated for an additional 48 h, at which point the medium was discarded and fresh medium was added to all wells. The CellTiter AQueous assay was then used according to the manufacturer’s instructions. The plates were incubated for 70 min, and the absorbance was read at 490 nm with a PowerWave microplate reader (BioTek).
In vitro metabolism study using liver microsomes.
Metabolic stability was assessed by incubating the test compounds individually (1 μM) at 37°C with rat liver microsomes and protein at a 0.4-mg/ml concentration. The metabolic reaction was initiated by the addition of an NADPH-regenerating buffer system and quenched at various time points over 60 min by the addition of acetonitrile. Control samples (containing no NADPH) were included (and quenched at 2, 30, and 60 min) to monitor for potential degradation in the absence of cofactor. The relative loss of parent compound and the formation of metabolic products were monitored by LC-MS. Test compound concentration-versus-time data were fitted to an exponential decay function to determine the first-order rate constant for substrate depletion, which was used to calculate an in vitro intrinsic clearance value as described previously (48). Scaling parameters (49) were used to predict the in vivo blood clearance and hepatic extraction ratio. For metabolite identification, WEB-485 (compound 2) was incubated under the same conditions with 2 μM substrate and a 0.8-mg/ml protein concentration to increase metabolite formation. Following protein precipitation with acetonitrile, samples were centrifuged and the supernatant was removed and analyzed by LC-MS.
Protein plasma binding study using ultracentrifugation.
Human plasma (pooled; n = 3 donors) was separated from whole blood procured from the Australian Red Cross Blood Service. Rat plasma (pooled; multiple rats) was from male Sprague-Dawley rats, and mouse plasma (pooled; multiple mice) was from male Swiss outbred mice. Aliquots of plasma were spiked with DMSO-acetonitrile-water solutions of test compound to a nominal compound concentration of 1,000 ng/ml. The final DMSO and acetonitrile concentrations were 0.2% (vol/vol) and 0.4% (vol/vol), respectively. Spiked plasma was vortex mixed briefly, and aliquots (n = 4) were transferred to ultracentrifuge tubes and subjected to ultracentrifugation at 37°C for 4.2 h to separate the proteins. Additional ultracentrifuge tubes (n = 2) containing spiked plasma were maintained at 37°C for 0.5 h and 4.2 h to serve as controls for the assessment of stability and to obtain a measure of the total compound concentration in plasma. Following ultracentrifugation, an aliquot of protein-free supernatant was taken from each of the ultracentrifuge tubes to obtain measures of the unbound concentration. At specified time points, the contents of control ultracentrifuge tubes were mixed and aliquots (n = 4) were taken for measurement of the total concentration. All samples were matrix matched by addition of blank plasma to protein-free supernatant samples and phosphate-buffered saline (PBS) to noncentrifuged plasma samples to give 1:1 (vol/vol) plasma-saline. The unbound fraction (fu) of WEB-485 in human, rat, and mouse plasma was calculated using the average values for the total and unbound concentrations.
Blood-to-plasma partitioning ratio.
WEB-485 (compound 2) was spiked into fresh heparinized rat blood to a nominal concentration of 1,000 ng/ml, and the mixture was incubated at 37°C. At 30 and 120 min, 4 replicate aliquots of whole blood were collected for assessment of the blood concentration and 4 additional aliquots of blood were taken for centrifugation and collection of plasma. The whole-blood and plasma fraction samples were matrix matched (by addition of blank blood or plasma to result in a 1:1 mixture of blood and plasma for each sample) and then immediately snap-frozen in dry ice. All samples were stored frozen at −80°C until analysis by LC-MS. Concentrations were determined by comparison to a calibration curve prepared in a 1:1 mixture of blank blood and plasma, and the blood-to-plasma partitioning ratio was calculated from the blood and plasma concentrations.
Pharmacokinetic study in rats.
All animal studies were conducted using established procedures in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes, and study protocols were reviewed and approved by the Monash Institute of Pharmaceutical Sciences Animal Ethics Committee. The in vivo pharmacokinetics of WEB-485 (compound 2) were studied in overnight-fasted male Sprague-Dawley rats weighing 271 to 320 g. WEB-485 was administered intravenously as a 10-min constant-rate infusion via an indwelling jugular vein cannula (1 ml per rat, n = 3 rats) and orally by gavage (10 ml/kg per rat, n = 3 rats) in a 0.9% saline vehicle. Samples of arterial blood and total urine were collected up to 24 h postdose. Arterial blood was collected directly into borosilicate vials (at 4°C) containing heparin, COmplete (a protease inhibitor cocktail), and potassium fluoride to minimize the potential for ex vivo degradation of WEB-485. Blood samples were centrifuged, and the supernatant plasma was removed and stored frozen (−80°C). Samples were analyzed using a Waters Micromass Quattro Premier mass spectrometer coupled to a Waters Acquity ultraperformance liquid chromatograph equipped with a Supelco Ascentis Express RP amide column (50 by 2.1 mm; particle size, 2.7 μm). LC conditions used a gradient cycle time of 4 min, an injection volume of 3 μl, a flow rate of 0.4 ml/min, and an acetonitrile-water gradient with 0.05% formic acid. Calibration standards were prepared in blank plasma for quantitation of the study samples. Plasma concentration-versus-time data were analyzed using noncompartmental methods (PKSolver, version 2.0, software). Standard calculations for each pharmacokinetic parameter were used.
P. falciparum humanized NOD-scid IL2Rγnull mouse model.
The P. falciparum humanized NOD-scid IL2Rγnull mouse model was adapted from the procedure described by Jiménez-Díaz et al. (39). Briefly, NOD-scid IL2Rγnull mice, continually engrafted with human erythrocytes, were infected intravenously with 2 × 107 P. falciparum 3D70087/N9-infected erythrocytes from a humanized donor mouse on day 0. Compounds were prepared in a vehicle consisting of 70% Tween 80 and 30% ethanol, followed by a 10-fold dilution in H2O. Experimental mice (2 per cohort) were left untreated (control mice) or treated at days 3, 4, 5, and 6 postinfection with an oral dose of the WEB-485 (compound 2; 50 mg/kg) or chloroquine (catalog number C6628; Sigma). Peripheral blood samples were taken on days 3, 4, 5, and 6 postinfection, and parasitemia was measured by microscopic analysis of Giemsa-stained blood smears. Parasitemia values were averages for 2 mice per group and are expressed as percent parasitemia at days 3, 4, 5, and 6.
Plasma exposure in mouse model.
Peripheral blood samples (20 μl) were taken on day 3 from each mouse at 1, 2, 4, 6, and 24 h after the first administration, mixed with 20 μl of Milli-Q H2O, and immediately frozen on dry ice. The frozen samples were stored at −80°C until analysis. Blood from control mice was used for calibration and quality control purposes. Blood samples were processed under standard liquid-liquid extraction conditions and analyzed by LC-MS/MS using heated electrospray ionization in the positive ion mode for quantification. A noncompartmental analysis was performed for the determination of the pharmacokinetic parameters, using the Phoenix WinNonlin (version 6.3) program.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by the National Health and Medical Research Council of Australia (development grant 1113712 to B.E.S. and program grant 1092789 to A.F.C.), the Australian Research Council (to B.M.C.), The International Development Research Centre Canada (to B.M.C.), the Australian Cancer Research Foundation, the Victorian State Government Operational Infrastructure Support, and Australian Government NHMRC IRIISS. C.J.T. is an Australian Research Council Future Fellow (FT120100164), B.E.S. is a Corin Centenary Fellow, and A.F.C. is a Howard Hughes International Scholar.
We thank David Fidock for the supplying the artemisinin-resistant P. falciparum line Cam3 R539T and control line Cam3 Rev. We thank Ursula Lehmann for assistance in performing the P. falciparum SCID mouse model and Swiss BioQuant (Reinach, Switzerland) for providing the blood exposure of compound 2 in this mouse model.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01804-18.
REFERENCES
- 1.World Health Organization. 2017. World malaria report 2016. World Health Organization, Geneva, Switzerland https://www.who.int/malaria/publications/world-malaria-report-2016/report/en/. Accessed 23 December 2016.
- 2.Mendis K, Sina BJ, Marchesini P, Carter R. 2001. The neglected burden of Plasmodium vivax malaria. Am J Trop Med Hyg 64(1-2 Suppl):97–106. doi: 10.4269/ajtmh.2001.64.97. [DOI] [PubMed] [Google Scholar]
- 3.Sutherland CJ. 2016. Persistent parasitism: the adaptive biology of malariae and ovale malaria. Trends Parasitol 32:808–819. doi: 10.1016/j.pt.2016.07.001. [DOI] [PubMed] [Google Scholar]
- 4.Millar SB, Cox-Singh J. 2015. Human infections with Plasmodium knowlesi—zoonotic malaria. Clin Microbiol Infect 21:640–648. doi: 10.1016/j.cmi.2015.03.017. [DOI] [PubMed] [Google Scholar]
- 5.World Health Organization. 2015. World malaria report 2015. World Health Organization, Geneva, Switzerland: https://www.who.int/malaria/publications/world-malaria-report-2015/report/en/. [Google Scholar]
- 6.Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, Sreng S, Anderson JM, Mao S, Sam B, Sopha C, Chuor CM, Nguon C, Sovannaroth S, Pukrittayakamee S, Jittamala P, Chotivanich K, Chutasmit K, Suchatsoonthorn C, Runcharoen R, Hien TT, Thuy-Nhien NT, Thanh NV, Phu NH, Htut Y, Han K-T, Aye KH, Mokuolu OA, Olaosebikan RR, Folaranmi OO, Mayxay M, Khanthavong M, Hongvanthong B, Newton PN, Onyamboko MA, Fanello CI, Tshefu AK, Mishra N, Valecha N, Phyo AP, Nosten F, Yi P, Tripura R, Borrmann S, Bashraheil M, Peshu J, Faiz MA, Ghose A, Hossain MA, Samad R, et al. 2014. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 371:411–423. doi: 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fowkes FJ, Boeuf P, Beeson JG. 2016. Immunity to malaria in an era of declining malaria transmission. Parasitology 143:139–153. doi: 10.1017/S0031182015001249. [DOI] [PubMed] [Google Scholar]
- 8.Gilson PR, Tan C, Jarman KE, Lowes KN, Curtis JM, Nguyen W, Di Rago AE, Bullen HE, Prinz B, Duffy S, Baell JB, Hutton CA, Jousset Subroux H, Crabb BS, Avery VM, Cowman AF, Sleebs BE. 2017. Optimization of 2-anilino 4-amino substituted qinazolines into potent antimalarial agents with oral in vivo activity. J Med Chem 60:1171–1188. doi: 10.1021/acs.jmedchem.6b01673. [DOI] [PubMed] [Google Scholar]
- 9.Duffy S, Avery VM. 2013. Identification of inhibitors of Plasmodium falciparum gametocyte development. Malar J 12:408. doi: 10.1186/1475-2875-12-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gamo F-J, Sanz LM, Vidal J, de Cozar C, Alvarez E, Lavandera J-L, Vanderwall DE, Green DVS, Kumar V, Hasan S, Brown JR, Peishoff CE, Cardon LR, Garcia-Bustos JF. 2010. Thousands of chemical starting points for antimalarial lead identification. Nature 465:305–310. doi: 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- 11.Guiguemde WA, Shelat AA, Bouck D, Duffy S, Crowther GJ, Davis PH, Smithson DC, Connelly M, Clark J, Zhu F, Jimenez-Diaz MB, Martinez MS, Wilson EB, Tripathi AK, Gut J, Sharlow ER, Bathurst I, Mazouni FE, Fowble JW, Forquer I, McGinley PL, Castro S, Angulo-Barturen I, Ferrer S, Rosenthal PJ, DeRisi JL, Sullivan DJ, Lazo JS, Roos DS, Riscoe MK, Phillips MA, Rathod PK, Van Voorhis WC, Avery VM, Guy RK. 2010. Chemical genetics of Plasmodium falciparum. Nature 465:311–315. doi: 10.1038/nature09099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plouffe D, Brinker A, McNamara C, Henson K, Kato N, Kuhen K, Nagle A, Adrian F, Matzen JT, Anderson P, Nam TG, Gray NS, Chatterjee A, Janes J, Yan SF, Trager R, Caldwell JS, Schultz PG, Zhou Y, Winzeler EA. 2008. In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc Natl Acad Sci U S A 105:9059–9064. doi: 10.1073/pnas.0802982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spangenberg T, Burrows JN, Kowalczyk P, McDonald S, Wells TNC, Willis P. 2013. The open access Malaria Box: a drug discovery catalyst for neglected diseases. PLoS One 8:e62906. doi: 10.1371/journal.pone.0062906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Furtado JM, Winthrop KL, Butler NJ, Smith JR. 2013. Ocular toxoplasmosis I: parasitology, epidemiology and public health. Clin Exp Ophthalmol 41:82–94. doi: 10.1111/j.1442-9071.2012.02821.x. [DOI] [PubMed] [Google Scholar]
- 15.McAuley JB. 2014. Congenital toxoplasmosis. J Pediatric Infect Dis Soc 3:S30–S35. doi: 10.1093/jpids/piu077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Contini C. 2008. Clinical and diagnostic management of toxoplasmosis in the immunocompromised patient. Parassitologia 50:45–50. [PubMed] [Google Scholar]
- 17.Vannier E, Gewurz BE, Krause PJ. 2008. Human babesiosis. Infect Dis Clin North Am 22:469–488. doi: 10.1016/j.idc.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vannier EG, Diuk-Wasser MA, Ben Mamoun C, Krause PJ. 2015. Babesiosis. Infect Dis Clin North Am 29:357–370. doi: 10.1016/j.idc.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bouzid M, Hunter PR, Chalmers RM, Tyler KM. 2013. Cryptosporidium pathogenicity and virulence. Clin Microbiol Rev 26:115–134. doi: 10.1128/CMR.00076-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Voorhis WC, Adams JH, Adelfio R, Ahyong V, Akabas MH, Alano P, Alday A, Alemán Resto Y, Alsibaee A, Alzualde A, Andrews KT, Avery SV, Avery VM, Ayong L, Baker M, Baker S, Ben Mamoun C, Bhatia S, Bickle Q, Bounaadja L, Bowling T, Bosch J, Boucher LE, Boyom FF, Brea J, Brennan M, Burton A, Caffrey CR, Camarda G, Carrasquilla M, Carter D, Belen Cassera M, Chih-Chien Cheng K, Chindaudomsate W, Chubb A, Colon BL, Colón-López DD, Corbett Y, Crowther GJ, Cowan N, D'Alessandro S, Le Dang N, Delves M, DeRisi JL, Du AY, Duffy S, Abd El-Salam El-Sayed S, Ferdig MT, Fernández Robledo JA, Fidock DA, et al. 2016. Open source drug discovery with the Malaria Box compound collection for neglected diseases and beyond. PLoS Pathog 12:e1005763. doi: 10.1371/journal.ppat.1005763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boyom FF, Fokou PV, Tchokouaha LR, Spangenberg T, Mfopa AN, Kouipou RM, Mbouna CJ, Donfack VF, Zollo PH. 2014. Repurposing the open access malaria box to discover potent inhibitors of Toxoplasma gondii and Entamoeba histolytica. Antimicrob Agents Chemother 58:5848–5854. doi: 10.1128/AAC.02541-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bessoff K, Spangenberg T, Foderaro JE, Jumani RS, Ward GE, Huston CD. 2014. Identification of Cryptosporidium parvum active chemical series by repurposing the open access Malaria Box. Antimicrob Agents Chemother 58:2731–2739. doi: 10.1128/AAC.02641-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Makler MT, Ries JM, Williams JA, Bancroft JE, Piper RC, Gibbins BL, Hinrichs DJ. 1993. Parasite lactate dehydrogenase as an assay for Plasmodium falciparum drug sensitivity. Am J Trop Med Hyg 48:739–741. doi: 10.4269/ajtmh.1993.48.739. [DOI] [PubMed] [Google Scholar]
- 24.de Cozar C, Caballero I, Colmenarejo G, Sanz LM, Alvarez-Ruiz E, Gamo FJ, Cid C. 2016. Development of a novel high-density [3H]hypoxanthine scintillation proximity assay to assess Plasmodium falciparum growth. Antimicrob Agents Chemother 60:5949–5956. doi: 10.1128/AAC.00433-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kadjoian V, Gasquet M, Delmas F, Guiraud H, De Meo M, Laget M, Timon-David P. 1992. Flow cytometry to evaluate the parasitemia of Plasmodium falciparum. J Pharm Belg 47:499–503. [PubMed] [Google Scholar]
- 26.Singh B, Daneshvar C. 2013. Human infections and detection of Plasmodium knowlesi. Clin Microbiol Rev 26:165–184. doi: 10.1128/CMR.00079-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Linares M, Viera S, Crespo B, Franco V, Gómez-Lorenzo MG, Jiménez-Díaz MB, Angulo-Barturen Í, Sanz LM, Gamo F-J. 2015. Identifying rapidly parasiticidal anti-malarial drugs using a simple and reliable in vitro parasite viability fast assay. Malar J 14:441. doi: 10.1186/s12936-015-0962-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cowman AF, Galatis D, Thompson JK. 1994. Selection for mefloquine resistance in Plasmodium falciparum is linked to amplification of the pfmdr1 gene and cross-resistance to halofantrine and quinine. Proc Natl Acad Sci U S A 91:1143–1147. doi: 10.1073/pnas.91.3.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Foote SJ, Kyle DE, Martin RK, Oduola AM, Forsyth K, Kemp DJ, Cowman AF. 1990. Several alleles of the multidrug-resistance gene are closely linked to chloroquine resistance in Plasmodium falciparum. Nature 345:255–258. doi: 10.1038/345255a0. [DOI] [PubMed] [Google Scholar]
- 30.Straimer J, Gnädig NF, Witkowski B, Amaratunga C, Duru V, Ramadani AP, Dacheux M, Khim N, Zhang L, Lam S, Gregory PD, Urnov FD, Mercereau-Puijalon O, Benoit-Vical F, Fairhurst RM, Ménard D, Fidock DA. 2015. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science 347:428–431. doi: 10.1126/science.1260867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malmquist NA, Moss TA, Mecheri S, Scherf A, Fuchter MJ. 2012. Small-molecule histone methyltransferase inhibitors display rapid antimalarial activity against all blood stage forms in Plasmodium falciparum. Proc Natl Acad Sci U S A 109:16708–16713. doi: 10.1073/pnas.1205414109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malmquist NA, Sundriyal S, Caron J, Chen P, Witkowski B, Menard D, Suwanarusk R, Renia L, Nosten F, Jimenez-Diaz MB, Angulo-Barturen I, Santos Martinez M, Ferrer S, Sanz LM, Gamo FJ, Wittlin S, Duffy S, Avery VM, Ruecker A, Delves MJ, Sinden RE, Fuchter MJ, Scherf A. 2015. Histone methyltransferase inhibitors are orally bioavailable, fast-acting molecules with activity against different species causing malaria in humans. Antimicrob Agents Chemother 59:950–959. doi: 10.1128/AAC.04419-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lubin AS, Rueda-Zubiaurre A, Matthews H, Baumann H, Fisher FR, Morales-Sanfrutos J, Hadavizadeh KS, Nardella F, Tate EW, Baum J, Scherf A, Fuchter MJ. 2018. Development of a photo-cross-linkable diaminoquinazoline inhibitor for target identification in Plasmodium falciparum. ACS Infect Dis 4:523–530. doi: 10.1021/acsinfecdis.7b00228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coffey MJ, Jennison C, Tonkin CJ, Boddey JA. 2016. Role of the ER and Golgi in protein export by Apicomplexa. Curr Opin Cell Biol 41:18–24. doi: 10.1016/j.ceb.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 35.Lim DC, Cooke BM, Doerig C, Saeij JP. 2012. Toxoplasma and Plasmodium protein kinases: roles in invasion and host cell remodelling. Int J Parasitol 42:21–32. doi: 10.1016/j.ijpara.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lourido S, Shuman J, Zhang C, Shokat KM, Hui R, Sibley LD. 2010. Calcium-dependent protein kinase 1 is an essential regulator of exocytosis in Toxoplasma. Nature 465:359–362. doi: 10.1038/nature09022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jackson LA, Waldron SJ, Weier HM, Nicoll CL, Cooke BM. 2001. Babesia bovis: culture of laboratory-adapted parasite lines and clinical isolates in a chemically defined medium. Exp Parasitol 99:168–174. doi: 10.1006/expr.2001.4655. [DOI] [PubMed] [Google Scholar]
- 38.Bessoff K, Sateriale A, Lee KK, Huston CD. 2013. Drug repurposing screen reveals FDA-approved inhibitors of human HMG-CoA reductase and isoprenoid synthesis that block Cryptosporidium parvum growth. Antimicrob Agents Chemother 57:1804–1814. doi: 10.1128/AAC.02460-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiménez-Díaz MB, Mulet T, Viera S, Gómez V, Garuti H, Ibáñez J, Alvarez-Doval A, Shultz LD, Martínez A, Gargallo-Viola D, Angulo-Barturen I. 2009. Improved murine model of malaria using Plasmodium falciparum competent strains and non-myelodepleted NOD-scid IL2Rgammanull mice engrafted with human erythrocytes. Antimicrob Agents Chemother 53:4533–4536. doi: 10.1128/AAC.00519-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jaureguiberry S, Ndour PA, Roussel C, Ader F, Safeukui I, Nguyen M, Biligui S, Ciceron L, Mouri O, Kendjo E, Bricaire F, Vray M, Angoulvant A, Mayaux J, Haldar K, Mazier D, Danis M, Caumes E, Thellier M, Buffet P. 2014. Postartesunate delayed hemolysis is a predictable event related to the lifesaving effect of artemisinins. Blood 124:167–175. doi: 10.1182/blood-2014-02-555953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blomme EA, Will Y. 2016. Toxicology strategies for drug discovery: present and future. Chem Res Toxicol 29:473–504. doi: 10.1021/acs.chemrestox.5b00407. [DOI] [PubMed] [Google Scholar]
- 42.Hughes JD, Blagg J, Price DA, Bailey S, Decrescenzo GA, Devraj RV, Ellsworth E, Fobian YM, Gibbs ME, Gilles RW, Greene N, Huang E, Krieger-Burke T, Loesel J, Wager T, Whiteley L, Zhang Y. 2008. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg Med Chem Lett 18:4872–4875. doi: 10.1016/j.bmcl.2008.07.071. [DOI] [PubMed] [Google Scholar]
- 43.Trager W, Jensen JB. 1976. Human malaria parasites in continuous culture. Science 193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 44.Witkowski B, Khim N, Chim P, Kim S, Ke S, Kloeung N, Chy S, Duong S, Leang R, Ringwald P, Dondorp AM, Tripura R, Benoit-Vical F, Berry A, Gorgette O, Ariey F, Barale JC, Mercereau-Puijalon O, Menard D. 2013. Reduced artemisinin susceptibility of Plasmodium falciparum ring stages in western Cambodia. Antimicrob Agents Chemother 57:914–923. doi: 10.1128/AAC.01868-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bock RE, de Vos AJ, Kingston TG, Shiels IA, Dalgliesh RJ. 1992. Investigations of breakdowns in protection provided by living Babesia bovis vaccine. Vet Parasitol 43:45–56. doi: 10.1016/0304-4017(92)90047-D. [DOI] [PubMed] [Google Scholar]
- 46.Gut J, Nelson RG. 1999. Cryptosporidium parvum: synchronized excystation in vitro and evaluation of sporozoite infectivity with a new lectin-based assay. J Eukaryot Microbiol 46:56S–57S. [PubMed] [Google Scholar]
- 47.Cory AH, Owen TC, Barltrop JA, Cory JG. 1991. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun 3:207–212. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]
- 48.Obach RS. 1999. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: an examination of in vitro half-life approach and non-specific binding to microsomes. Drug Metab Dispos 27:1350–1359. [PubMed] [Google Scholar]
- 49.Ring BJ, Chien JY, Adkison KK, Jones HM, Rowland M, Jones RD, Yates JWT, Ku MS, Gibson CR, He H, Vuppugalla R, Marathe P, Fischer V, Dutta S, Sinha VK, Björnsson T, Lavé T, Poulin P. 2011. PhRMA CPCDC initiative on predictive models of human pharmacokinetics, part 3: comparative assessement of prediction methods of human clearance. J Pharm Sci 100:4090–4110. doi: 10.1002/jps.22552. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.