

The spread of multidrug or extensively drug-resistant Gram-negative bacteria is a serious public health issue. There are too few new antibiotics in development to combat the threat of multidrug-resistant infections, and consequently the rate of increasing antibiotic resistance is outpacing the drug development process.
KEYWORDS: FosA, Gram negative, fosfomycin
ABSTRACT
The spread of multidrug or extensively drug-resistant Gram-negative bacteria is a serious public health issue. There are too few new antibiotics in development to combat the threat of multidrug-resistant infections, and consequently the rate of increasing antibiotic resistance is outpacing the drug development process. This fundamentally threatens our ability to treat common infectious diseases. Fosfomycin (FOM) has an established track record of safety in humans and is highly active against Escherichia coli, including multidrug-resistant strains. However, many other Gram-negative pathogens, including the “priority pathogens” Klebsiella pneumoniae and Pseudomonas aeruginosa, are inherently resistant to FOM due to the chromosomal fosA gene, which directs expression of a metal-dependent glutathione S-transferase (FosA) that metabolizes FOM. In this study, we describe the discovery and biochemical and structural characterization of ANY1 (3-bromo-6-[3-(3-bromo-2-oxo-1H-pyrazolo[1,5-a]pyrimidin-6-yl)-4-nitro-1H-pyrazol-5-yl]-1H-pyrazolo[1,5-a]pyrimidin-2-one), a small-molecule active-site inhibitor of FosA. Importantly, ANY1 potentiates FOM activity in representative Gram-negative pathogens. Collectively, our study outlines a new strategy to expand FOM activity to a broader spectrum of Gram-negative pathogens, including multidrug-resistant strains.
INTRODUCTION
There is a significant clinical and public health burden associated with the increasing prevalence and spread of multidrug-resistant (MDR) or extensively drug-resistant (XDR) Gram-negative bacteria such as carbapenem-resistant and extended-spectrum β-lactamase (ESBL)-producing Enterobacteriaceae. These pathogens have been designated a critical priority for antibiotic research and development as strains are emerging worldwide that cannot be treated with any of the currently available antibiotics. However, a recent report from the World Health Organization highlighted the lack of new potential therapeutic options in the clinical pipeline for multidrug-resistant Gram-negative pathogens (1). Indeed, nearly all of the agents currently in development are modifications of existing antibiotic classes and are active only against specific pathogens or a limited set of resistant strains (1). As such, there is an urgent need to identify more innovative products with no cross- or coresistance to existing classes of antibiotics.
Fosfomycin (FOM), a broad-spectrum antibiotic with an extensive track record of safety in humans, exerts its bactericidal activity by covalent attachment to UDP-(N-acetyl)glucosamine-3-enolpyruvyl transferase (MurA) (2), the enzyme which catalyzes the first step in cell wall biosynthesis (Fig. 1a). FOM is highly active against Escherichia coli, including strains producing ESBL (3). In the United States, a tromethamine FOM formulation is approved as a single-dose, orally administered treatment for acute uncomplicated cystitis. In several European and Asian countries, an intravenous disodium formulation is available and is used to treat bacteremia, pneumonia, pyelonephritis, osteomyelitis, and central nervous system infections, usually in combination with another active agent (4). In contrast to E. coli, many other Gram-negative pathogens, including Klebsiella pneumoniae, Klebsiella oxytoca, Serratia marcescens, Enterobacter aerogenes, Enterobacter cloacae, Pseudomonas aeruginosa, and Morganella morganii, are inherently resistant to FOM (5). This inherent resistance is conferred by a chromosomal fosA gene, which encodes a dimeric K+- and Mn2+-dependent glutathione S-transferase ([GST] FosA) that catalyzes the nucleophilic addition of glutathione to carbon-1 in the epoxide ring of FOM, rendering the antibiotic inactive (Fig. 1b) (6). Of note, plasmid-borne fosA variants (e.g., fosA3) are also emerging as a transferable mechanism by which E. coli, which naturally lacks fosA as a species, acquires FOM resistance in the clinic (3).
FIG 1.
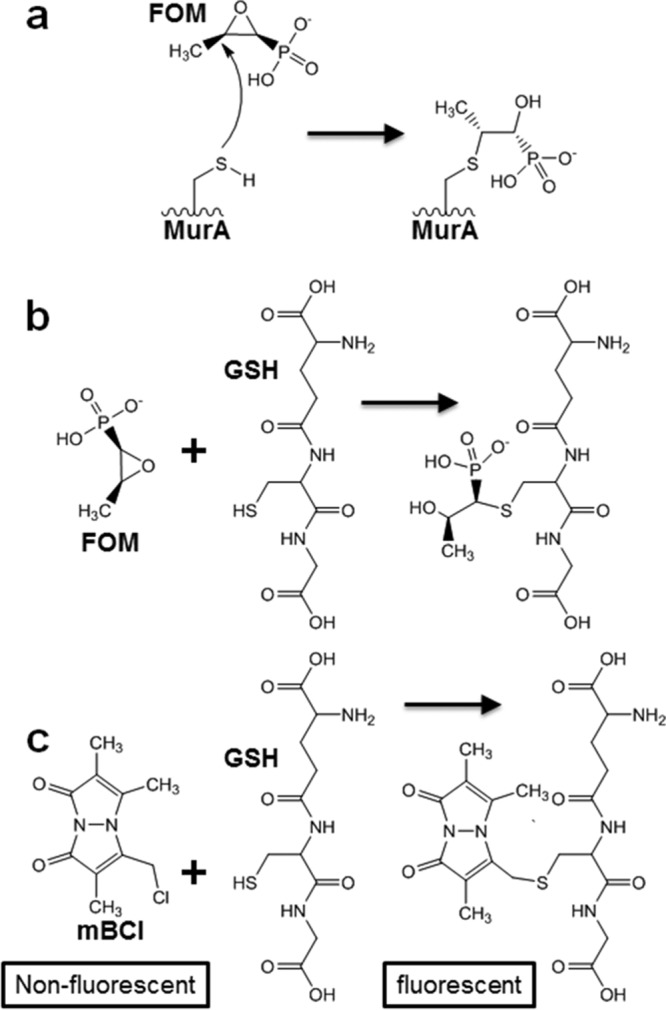
Reaction schemes. (a) Covalent modification of the MurA active-site cysteine residue by FOM. (b) FosA-mediated nucleophilic addition of glutathione (GSH) to carbon-1 in the epoxide ring of FOM. (c) Fluorescence quantification of glutathione via conjugation with mBCl.
Inhibition of FosA activity may provide a novel approach to expand the use of FOM to Gram-negative species that produce FosA. A similar approach to expand the use of β-lactam antibiotics has been clinically implemented for many years following the development and approval of β-lactamase inhibitors such as clavulanic acid, tazobactam, avibactam, and vaborbactam. We postulate that FosA is an excellent target for drug discovery for two reasons: (i) deletion of chromosomal fosA in S. marcescens (5) or transposon-mediated disruption of fosA in K. pneumoniae or P. aeruginosa eliminates intrinsic FOM resistance; and (ii) clinically achievable concentrations of foscarnet, a pyrophosphate analog that inhibits DNA polymerases and also FosA (7), reduces FOM MICs by >4-fold among representative K. pneumoniae, E. cloacae, and P. aeruginosa clinical strains and leads to a bacteriostatic or bactericidal effect in time-kill assays (8).
While foscarnet is approved for the treatment of cytomegalovirus retinitis and refractory mucocutaneous herpes simplex virus infections, its use is associated with significant side effects including nephrotoxicity, hypocalcemia, and seizures. Therefore, there is a need to identify and develop selective small-molecule inhibitors of bacterial FosA. In this study, we describe the discovery and characterization of a first-in-class, competitive small-molecule inhibitor of FosA which significantly potentiates FOM activity against Gram-negative pathogens that harbor the fosA gene.
RESULTS
Discovery of ANY1.
FosA catalyzes the Mn2+- and K+-dependent conjugation of glutathione to carbon-1 of FOM (Fig. 1b) (6). To quantify FosA activity, we developed an endpoint fluorescence-based high-throughput screening (HTS) assay which quantifies glutathione consumption using the thiol-reactive dye monochlorobimane (Fig. 1c). The assay is sensitive and robust with a signal-to-noise ratio of 8 and a Z′-factor of 0.52. To identify inhibitors of FosA, we screened the ApexScreen library from TimTec (Newark, DE), which contains 5,040 small molecules enriched for chemical diversity and Lipinski rule parameters (see Table S1 in the supplemental material). Recombinant purified K. pneumoniae FosA (FosAKP) was used in the screen. We identified 40 hits with >50% inhibition (0.8% hit rate). Upon further validation, including dose-response assays, this number decreased to 12. Of these, ANY1 (3-bromo-6-[3-(3-bromo-2-oxo-1H-pyrazolo[1,5-a]pyrimidin-6-yl)-4-nitro-1H-pyrazol-5-yl]-1H-pyrazolo[1,5-a]pyrimidin-2-one) (Fig. 2a) was the most potent (50% inhibitory concentration [IC50], 5.1 ± 2.2 μM). The dissociation constant for ANY1 binding to FosAKP, measured by isothermal titration calorimetry (ITC) (Fig. 2b), was 180 ± 30 nM with a binding stoichiometry of 1:1 for an ANY1/FosAKP monomer (or 2 molecules of ANY1 for each FosA dimer). In steady-state kinetic assays under near-saturating glutathione concentrations, ANY1 binding increased the Michaelis constant (Km) for FOM without affecting its maximum rate of reaction (Vmax) (Fig. 2c), whereas under saturating FOM concentrations, ANY1 did not impact the Km for glutathione binding but decreased the Vmax (Fig. 2d). This pattern of inhibition is consistent with ANY1 acting as a competitive inhibitor of FOM binding and a noncompetitive inhibitor of glutathione binding.
FIG 2.

Thermodynamic and steady-state kinetic characterization of ANY1 binding to FosAKP. (a) Chemical structure of ANY1, highlighting the pyrazolopyrimidine and pyrazole moieties. (b) Representative run of ANY1 binding to FosAKP as measured by ITC. The upper panel represents the isotherms measured for 3,860 s at 230-s injection intervals. The lower panel shows a sigmoidal curve from an individual heat flow as a function of the total molar ratio (ANY1/FosAKP monomer) in the calorimeter cell. Binding isotherms were performed in triplicate and corrected for heats of dilution. ΔH, enthalpy; ΔS, entropy. (c) Michaelis-Menten plot of FosA activity, in the absence or presence of 5 μM, 10 μM, or 20 μM ANY1, in which the glutathione (GSH) concentration was held constant (25 mM) whereas the FOM concentration ranged from 0.5 to 25 mM. Data are shown as the means ± standard deviations from three separate biological replicates. (d) Michaelis-Menten plot of FosA activity, in the absence or presence of 5 μM, 10 μM, or 20 μM ANY1, in which the FOM concentration was held constant (20 mM) whereas the GSH concentration ranged from 1 to 25 mM. Data are shown as the means ± standard deviations from three separate biological replicates.
Crystal structures of FosAKP and FosA3 in complex with ANY1.
To understand how ANY1 interacts with FosA, we solved X-ray crystal structures of ANY1 bound to FosAKP (3.1 Å) and E. coli FosA3 (3.5 Å) (Table 1). Consistent with prior structures (9), both FosAKP and FosA3 display a three-dimensional domain-swapped arrangement of the paired βαβββ-motifs. The amino acid sequence identity between FosA3 and FosAKP is 79%, and superimposition of the two enzymes reveals that the overall structure is largely conserved, with a Cα root mean square deviation of less than 0.5 Å (Fig. 3a). The basic architecture of the active sites in both FosA proteins, including the essential divalent cation, is also maintained. We found that ANY1 binds at both active sites of FosAKP and FosA3 (Fig. 3b) and, consistent with the kinetic data which showed that it was a competitive inhibitor of FOM (Fig. 2c and d), its binding site overlaps that of FOM (Fig. 3c). Despite the moderate resolution of the data sets, we observed clear electron density for all of the bound ANY1 molecules (Fig. 3d). Furthermore, anomalous bromine signal and high-contour electron density maps allowed us to unambiguously assign the locations of both bromine atoms in each of the bound ANY1 molecules (Fig. S1). While ANY1 and FOM share contacts with multiple amino acid residues in FosA (T9, W46, Y65, and R122), ANY1 makes unique contacts with additional residues that form the putative glutathione channel (S36, Y39, W46, and Y131) (Fig. 3b). All of these residues, shared and unshared, are highly conserved across all FosA enzymes (Table S2), and introduction of alanine or phenylalanine substitutions at positions 9, 34, 39, 46, 65, and 131 in FosA3 either eliminated or reduced enzyme activity (Fig. S2). Using protein fluorescence quenching, we measured ANY1 binding to the mutant FosA3 proteins (Table 2; Fig. S2b). The T9A, W34A, S36A, W46A, and Y131A substitutions all significantly decreased the affinity of ANY1 for FosA3, further highlighting their role in binding.
TABLE 1.
Data collection and refinement statistics
| Parametera | Value for the proteinb
|
||
|---|---|---|---|
| FosAKP-ANY1 | FosA3-ANY1 | FosAKP-ANY2 | |
| Data collection | |||
| Resolution range (Å) | 36.28–3.178 (3.292–3.178) | 28.51–3.502 (3.627–3.502) | 37.53–1.85 (1.916–1.85) |
| Space group | C2221 | P43212 | P21 |
| Unit cell dimensions | |||
| a, b, c (Å) | 120.399, 197.615, 117.027 | 73.181, 73.181 123.818 | 44.765, 68.837, 90.312 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90.464, 90 |
| Total no. of reflections | 98,891 (9,776) | 61,983 (5,911) | 154,817 (15,655) |
| No. of unique reflections | 23,338 (2,128) | 4,588 (417) | 45,290 (4,448) |
| Multiplicity | 4.2 (4.3) | 13.5 (13.7) | 3.4 (3.5) |
| Completeness (%) | 89 (98) | 95 (100) | 95 (97) |
| Mean I/σ(I) | 4.45 (1.23) | 12.54 (1.66) | 9.01 (1.76) |
| Wilson B factor (Å2) | 47.47 | 125.71 | 17.25 |
| Rmerge (%) | 0.359 (1.25) | 0.179 (1.95) | 0.127 (0.829) |
| Rmeas (%) | 0.408 (1.42) | 0.186 (2.02) | 0.151 (0.982) |
| CC1/2 | 0.953 (0.497) | 0.998 (0.652) | 0.995 (0.661) |
| CC* | 0.988 (0.815) | 0.999 (0.888) | 0.999 (0.892) |
| Refinement | |||
| No. of reflections used in refinement | 21,260 (2119) | 4,366 (417) | 44,418 (4437) |
| No. of reflections used for Rfree | 1,105 (104) | 222 (20) | 2,280 (223) |
| Rwork (%) | 0.221 (0.294) | 0.214 (0.389) | 0.185 (0.277) |
| Rfree (%) | 0.262 (0.318) | 0.261 (0.439) | 0.212 (0.294) |
| CCwork | 0.923 (0.766) | 0.954 (0.652) | 0.962 (0.786) |
| CCfree | 0.895 (0.814) | 0.898 (0.445) | 0.962 (0.772) |
| No. of nonhydrogen atoms | 8,654 | 2,058 | 5,073 |
| No. of macromolecules | 8,406 | 1,996 | 4,273 |
| No. of ligands | 248 | 62 | 86 |
| No. of protein residues | 1094 | 271 | 551 |
| No. of water molecules | 0 | 0 | 163 |
| RMSD | |||
| Bond length (Å) | 0.002 | 0.003 | 0.003 |
| Bond angle (°) | 0.49 | 0.69 | 0.64 |
| Avg B factor (Å2) | |||
| Macromolecules | 41.88 | 123.29 | 20.11 |
| Ligands | 41.91 | 118.56 | 28.10 |
CC, Pearson correlation coefficient; CC1/2, the CC value between two random half data sets; CC*, estimate of the value of CCtrue, based on a finite size sample; CCwork, the standard correlation of the experimental intensity with the intensity calculated from the refined molecular model; CCfree, the cross-validated correlation of the experimental intensity with the intensity calculated from the refined molecular model; RMSD, root mean square deviation.
Overall values are reported, with those of the highest-resolution shell in parentheses.
FIG 3.

Three-dimensional structures of FosAKP or FosA3 in complex with ANY1 or ANY2. (a) Overlay of FosAKP and FosA3, both in complex with ANY1, as indicated. Two adjacent FosA dimers are shown. (b) Residues in FosAKP and FosA3 that interact with ANY1. Yellow dashed lines represent contacts shared between FosA and ANY1/FOM, while black dashed lines represent contacts unique to FosA and ANY1. The pyrazolopyrimidine moiety (pink) of the ANY1 molecule from the adjacent FosA molecule is also shown. (c) Overlay of FosAKP in complex with ANY1 and in complex with FOM, as indicated (PDB accession number 5V3D). Yellow dashed lines represent contacts shared between FosA and ANY1/fosfomycin, while black dashed lines represent contacts unique to FosA and FOM. (d) Structure of FosAKP highlighting electron density surrounding two ANY1 molecules that link adjacent FosAKP active sites. Electron density was generated from composite omit maps, contoured to 1.5 σ and carved to 2 Å from ANY1. (e) Overlay of FosAKP in complex with ANY1 or ANY2, as indicated.
TABLE 2.
Dissociation constant determined for ANY1 binding to wild-type and mutant FosA3 measured by fluorescence quenching
| FosA3 | Kd (nM)a | Fold change in Kd (P value)b |
|---|---|---|
| Wild type | 440 ± 50 | |
| T9A | 1,580 ± 100 | 3.6 (< 0.05) |
| W34A | 2,620 ± 200 | 6.0 (<0.05) |
| S36A | 1,360 ± 130 | 3.1 (<0.05) |
| Y39F | 870 ± 200 | 2.0 |
| W46A | 2,480 ± 110 | 5.6 (<0.05) |
| Y65F | 780 ± 80 | 1.8 |
| Y131A | 4,880 ± 770 | 11.1 (<0.05) |
Kd, dissociation constant.
Relative to the wild-type value.
We also observed drug-drug and drug-protein interactions between two adjacent FosA molecules (Fig. 3b and d). Despite the fact that ANY1 crystallized with FosA3 and FosAKP under different conditions and in unrelated space groups, these same interactions were observed in both cases. Specifically, there are π-π stacking interactions between the pyrazolopyrimidine rings of two ANY1 molecules bound to adjacent FosA proteins. Additionally, S36 can form hydrogen bonds to either the pyrazole group of the ANY1 molecule in its own active site or to the pyrazolopyrimidine group of the ANY1 molecule in an adjacent active site (Fig. 3b). In order to determine the importance of these drug-drug and drug-adjacent protein interactions, we solved a 1.9-Å resolution crystal structure of FosAKP bound to 3-bromo-6-(4-nitro-1H-pyrazol-5-yl)-1H-pyrazolo[1,5-a]pyrimidin-2-one (ANY2) (Fig. S3a), which lacks the pyrazolopyrimidine group that mediates these interactions. As expected, ANY2 binding overlaps ANY1 binding without forming the same drug-drug or drug-adjacent protein interactions (Fig. 3e and Fig. S1c); however, its binding affinity, as measured by ITC, was ∼22-fold weaker (Fig. S3b), thereby suggesting that the drug-drug and drug-adjacent protein interactions observed for ANY1 contribute significantly to its binding affinity and activity. Additional structure-activity relationship studies of ANY1 revealed that the pyrazole moiety and the bromines on the pyrazolopyrimidines were required for inhibition of FosA (Fig. S4).
HDX-MS of FosAKP in the absence or presence of FOM, ANY1, and ANY2.
Since X-ray crystallography provides only a static snapshot of ligand binding, we sought to validate our models and explore how substrate and drug binding affects FosA structure and dynamics in solution. To do so, we performed hydrogen-deuterium exchange-mass spectrometry (HDX-MS) of FosAKP in the presence and absence of FOM, ANY1, and ANY2. HDX-MS relies on the exchange of hydrogen with deuterium on peptide backbone amides and provides information on backbone solvent accessibility and dynamics because residues that are more frequently exposed to solvent will undergo faster deuteration. By subtracting the percent deuteration for ligand-bound and unbound FosAKP peptides, we determined which regions of the protein displayed statistically significant protection as a result of ligand binding. These regions were then mapped onto the structure(s) of FosAKP (Fig. 4). As reported previously, FOM binding decreases deuteration throughout the enzyme although this is particularly prominent in the K+ binding loop, glutathione channel, and dimer-interface loop (Fig. 4a) (9). Overall, FOM and ANY1 display a largely similar HDX protection footprint, consistent with their similar modes of binding (Fig. 4a and b). However, in contrast to FOM, ANY1 binding does not impact the K+ binding loop, consistent with our X-ray crystal structures. The HDX protection of the glutathione binding channel is also different in that FOM provides greater protection to the β6-α3 loop and α3-helix (residues 115 to 126), whereas ANY1 provides greater protection to the C terminus of the α3-helix (residues 125 to 135) and the β2-β3 sheets and loop (residues 31 to 46). This phenomenon could be explained by R122 forming stronger ionic interactions with the phosphate group on FOM, which is more negatively charged than the pyrazolopyrimidine group on ANY1, while S36 and Y131 form interactions with ANY1 but not FOM. Finally, FOM and ANY1 differentially impact W46. FOM appears to destabilize W46, as demonstrated by the increase in deuterium uptake, whereas ANY1 stabilizes W46, as indicated by a decrease in deuterium uptake, highlighting the potential importance of the π-halogen bond formed between this residue and ANY1 (Fig. 4c). A comparison of the protection mediated by ANY1 versus ANY2 further supports the potential importance of the additional pyrazolopyrimidine group on ANY1, which may mediate drug-drug and drug-protein interactions between ANY1 and adjacent active sites (Fig. 4d). While there are no observable interactions between FosA and this additional pyrazolopyrimidine group on the ANY1 molecule in its active site, ANY1 nonetheless offers more protection than ANY2. Furthermore, this protection occurs in the region where the crystal structures predict that ANY1 forms an ∼240-Å2 protein interface, raising the possibility that the additional pyrazolopyrimidine group on ANY1 mediates the formation of this interface in solution.
FIG 4.

Hydrogen-deuterium exchange mass spectrometry of FosAKP alone or in complex with FOM, ANY1 or ANY2. (a to d) Differences in deuteration (%D) between peptides from FosAKP and FosAKP-FOM (a), from FosAKP and FosAKP-ANY1 (b), from FosAKP-ANY1 and FosAKP-FOM (c), and from FosAKP-ANY1 and FosAKP-ANY2 (d). For all panels, peptides that contain residues from the glutathione (GSH) channel, dimer interface, and K+ binding loop are highlighted. Differences in deuteration at individual time points are plotted as colored lines. The 98% confidence intervals for individual time points are plotted as dotted lines. The cartoon representations on the right-hand side illustrate the FOM-, ANY1-, or ANY2-induced changes in hydrogen-deuterium exchange of FosAKP. For each pair (Apo versus FOM, Apo versus ANY1, ANY1 versus FOM, and ANY1 versus ANY2), regions where the second member of the pair demonstrates statistically significant differences in relative deuteration are colored as follows: dark blue, region with relative decreases in deuteration at the earliest time point (10 s); light blue, regions with relative decreases in deuteration observed only at later time points; dark red, regions with relative increases in deuteration at 10 s; light red, regions with relative increases in deuteration at later time points. The colors in the titles correspond to the relative increase in protection mediated by that member.
Antibacterial activity of ANY1 alone and in combination with FOM.
We evaluated the antimicrobial activity of ANY1, alone and in combination with 32, 64, or 128 μg/ml FOM, against the carbapenemase-producing (KPC) clinical strains K. pneumoniae I1, E. cloacae YDC612, S. marcescens YDC760-2, and P. aeruginosa 75B2 by bacterial growth curve analysis. Each of these clinical strains carries a chromosomal copy of the fosA gene. We also assessed activity against E. coli clinical isolate YD472, a FOM-resistant, ESBL-producing clinical strain that carries a copy of the fosA3 gene on a plasmid (10). Growth curves were modeled using a modified three-parameter Gompertz equation, as described previously (11), which facilitated quantification of the lag time (in minutes), growth rate (in optical density [OD] units/minute), and maximum growth (in OD units) (Fig. S5). Both the growth rate and maximum growth decreased with increasing concentrations of FOM, which facilitated determination of the FOM concentration required to decrease bacterial growth by 50% (i.e., IC50) (Fig. S5c and d).
We found that bacterial growth of K. pneumoniae I1 was significantly attenuated in a dose-dependent manner when FOM was combined with ANY1 (Fig. 5a). ANY1 alone had no effect on the growth of K. pneumoniae I1 (Fig. 5b), a finding which is consistent with its mechanism of action. However, it potentiated FOM activity in a dose-dependent manner, resulting in a ∼6-fold increase in activity at the highest concentration tested (Fig. 5b). Similar to growth of K. pneumoniae I1, the bacterial growth curves of P. aeruginosa 75B2, E. cloacae YDC612, S. marcescens YDC760-2, and E. coli YD472 were significantly attenuated in a dose-dependent manner when 32 μg/ml FOM was combined with ANY1 (Fig. 5c). We next evaluated the effect of a single concentration of ANY1 (112 μg/ml) on FOM activity against K. pneumoniae I1, using growth curve analysis as described above. ANY1 reduced by ∼6-fold the concentration of FOM required to decrease K. pneumoniae I1 maximum growth by 50% (P < 0.05); the IC50 values for FOM were 144.1 ± 16.5 μg/ml and 23.2 ± 4.3 μg/ml in the absence and presence of ANY1, respectively (Fig. 6a). Time-kill experiments confirmed that ANY1 significantly increased FOM activity (Fig. 6c to e). In these time-kill experiments, bacterial regrowth was observed at the 24-h time point when 112 μg/ml ANY1 was combined with 64 or 128 μg/ml FOM (Fig. 6c and d) but not when it was combined with 256 μg/ml FOM (Fig. 6e). The mechanisms involved in resistance to combinations of FOM and ANY1 have yet to be characterized, but we suspect that they most likely involve mutations in glycerol-3-phosphate (GlpT) or glucose-6-phosphate (UhpT) transporters that are known to be rapidly selected by FOM in vitro (12). They are, however, unlikely to be clinically significant due to concomitant loss of fitness (13). In contrast to FOM, ANY1 did not alter the activity of gentamicin, an antimicrobial agent with a mechanism of action distinct from that of FOM and, thus, used as a control here (Fig. 6b). Importantly, 112 μg/ml of ANY1 also significantly decreased the IC50 of FOM for E. cloacae YDC612 (23-fold, P < 0.05; the IC50 values for FOM were 243.3 ± 41.1 μg/ml and 10.4 ± 2.9 μg/ml in the absence and presence of ANY1, respectively) (Fig. 6f), S. marcescens YDC760-2 (>100-fold, P < 0.05; the IC50 values for FOM were >1,000 μg/ml and 16.3 ± 3.4 μg/ml in the absence and presence of ANY1, respectively) (Fig. 6g), and FosA3-producing E. coli YD472 (1.8-fold, P = 0.04; the IC50 values for FOM were 102.6 ± 5.3 μg/ml and 56.8 ± 2.4 μg/ml in the absence and presence of ANY1, respectively) (Fig. 6h).
FIG 5.

Bacterial growth curves of K. pneumoniae I1, P. aeruginosa 75B2, E. cloacae YDC612, S. marcescens YDC760-2 or E. coli YD472 in the presence of a fixed concentration of FOM and different concentrations of ANY1 (0 to 224 μg/ml). (a) Growth curve of K. pneumoniae I1 in the presence of 32, 64, or 128 μg/ml FOM and different concentrations of ANY1 (0 to 224 μg/ml). (b) Analysis of the data shown in panel a illustrating changes in the maximum growth asymptote (OD units) as a function of FOM (red dotted lines) and ANY1 with or without FOM at the concentrations indicated in the legend on the figure. (c) Growth curve of P. aeruginosa 75B2, E. cloacae YDC612, S. marcescens YDC760-2, and E. coli YD472 in the presence of 32, 64, or 128 μg/ml FOM and different concentrations of ANY1 (0 to 224 μg/ml).
FIG 6.

Bacterial growth curves of K. pneumoniae I1, E. cloacae YDC612, S. marcescens YDC760-2, and E. coli YD472 in the in the presence of a fixed concentration of ANY1 (112 μg/ml) and various concentrations of FOM (0 to 1,024 μg/ml). (a) Maximum growth values for K. pneumoniae I1 in the absence or presence of 112 μg/ml ANY1 and various concentrations of FOM, as indicated. The concentration of FOM that yielded 50% inhibition (i.e., IC50) was determined by fitting data to a hyperbolic ligand binding curve in Sigma Plot and is reported in the figure. (b) Maximum growth values for K. pneumoniae I1 in the absence or presence of 112 μg/ml ANY1 and various concentrations of gentamicin (0 to 64 μg/ml), as indicated. (c to e) Time-kill analysis of K. pneumoniae I1 in the absence or presence of 112 μg/ml ANY1 and/or 64 μg/ml FOM (c), and/or 128 μg/ml FOM (d), and/or 256 μg/ml (e). (f) Maximum growth values for E. cloacae YDC612 in the absence (black circles) or presence (white triangles) of 112 μg/ml ANY1 and various concentrations of FOM (0 to 1,024 μg/ml). (g) Maximum growth values for S. marcescens YDC760-2 in the absence or presence of 112 μg/ml ANY1 and various concentrations of FOM (0 to 1,024 μg/ml). (h) Maximum growth values for E. coli YD472 in the absence or presence of 112 μg/ml ANY1 and various concentrations of FOM (0 to 1,024 μg/ml). All data presented in this figure are shown as the means ± standard deviations from at least three independent biological replicates.
Toxicity.
To evaluate potential cellular toxicity, we assessed the effect of various concentrations of ANY1 (0 to 224 μg/ml) on the viability of the human-derived kidney epithelial cell line HK2 (Fig. 7a) and of human peripheral blood mononuclear cells (PBMC) (Fig. 7b). HK2 cells were included in this study as the FOM concentrations in urine are exceedingly high (1,000 to 4,000 μg/ml) following a 3-mg oral dose, and therefore we sought to assess toxicity in a kidney cell line. In contrast, the PBMC provide some insight into potential toxicity in the blood. Our results show that ANY1, or combinations of FOM and ANY1, had minimal impact on cell viability in both cell types even at the highest concentrations tested.
FIG 7.

ANY1 toxicity toward HK2 cells and peripheral blood mononuclear cells (PBMC). (a) HK2 cell viability following 24 h of exposure to 1 mg/ml FOM or various concentrations of ANY1 (0 to 224 μg/ml). Dimethyl sulfoxide ([DMSO] 10%) was used as a positive control for toxicity. *, P < 0.05, for comparison of results to those with the medium control. (b) PBMC viability following 24 h of exposure to 1 mg/ml FOM or various concentrations of ANY1 (0 to 224 μg/ml) and 112 μg/ml ANY1 in combination with different concentrations of FOM. Dimethyl sulfoxide (10%) was used as a positive control for toxicity. *, P < 0.05, for comparison of results to those with the medium control. All data presented in this figure are shown as the means ± standard deviations from at least three independent biological replicates.
DISCUSSION
The spread of MDR or XDR Gram-negative bacteria is a serious public health issue (14). FOM has a strong track record of safety in humans with no cross-resistance to other antibiotics and is one of the drugs that have been proposed as part of combination regimens for the treatment of K. pneumoniae carbapenemase (KPC)-producing K. pneumoniae infections (15). However, in comparison to E. coli, many Gram-negative bacteria, including K. pneumoniae, exhibit intrinsic resistance to FOM due to inherent expression of FosA (5), an enzyme that catalyzes the nucleophilic addition of glutathione to the carbon-1 of the epoxide ring of FOM, rendering the antibiotic inactive. In this regard, inhibition of FosA could help to expand the activity of FOM against Gram-negative bacteria that inherently express this enzyme. In support of this hypothesis, deletion of chromosomal fosA in S. marcescens (5) or transposon-mediated disruption of fosA in K. pneumoniae and P. aeruginosa eliminates intrinsic FOM resistance (see Table S3 in the supplemental material).
Using an in vitro biochemical HTS assay, we identified ANY1, which binds to the active site of the enzyme and inhibits FOM metabolism. A key feature of ANY1 is that it exhibits antibacterial activity against representative Gram-negative pathogens, including K. pneumoniae, when it is combined with FOM (Fig. 5) but not gentamicin (Fig. 5f), highlighting its specificity as a FOM potentiator. In contrast, most in vitro HTS campaigns have failed to identify small-molecule inhibitors with antibacterial activity largely due to their poor penetration into Gram-negative pathogens (16). In this regard, Richter et al. recently described a set of physicochemical properties that enable small molecules to accumulate in Gram-negative bacteria (17). Interestingly, ANY1 harbors many of these described properties, including the presence of an amine, an amphiphilic and rigid structure, and low globularity. In Fig. 5 and 6, we show that ANY1 potency varies across different pathogens, with potent activity against K. pneumoniae I1, E cloacae YD612, and S. marcescens YD6760-2 (Fig. 5a, 5c, 6a, 6f, and 6g) and weaker activity against E. coli YD472 (Fig. 5c and 6h). Given that the ANY1 binding and inhibition constants are similar for purified FosAKP and FosA3, these differences in antibacterial potency are likely driven by differences in the intracellular penetration of ANY1 into the different Gram-negative pathogens. Importantly, the crystal structures of FosA in complex with ANY1 described in this study provide a platform for structure-guided drug design to potentially improve inhibitor binding affinity. For example, ANY1 could be modified such that it interacts with residues that form the K+ binding loop (K93, S97, and Y103) which are critical for FOM binding and enzyme function (Fig. 3c) (18).
ANY1 binds in the active site of FosA and interacts with amino acid residues which are highly conserved throughout the FosA superfamily (Table S2), thus suggesting a high genetic barrier to resistance for a FOM/ANY1 drug combination. However, FOM resistance can also be conferred by mechanisms other than fosA, including mutation of the conserved cysteine residue in the active site of MurA or the development of mutations in the bacterial glycerol-3-phosphate (GlpT) or glucose-6-phosphate (UhpT) transporters, resulting in reduced FOM permeability (3). Mutation of the active-site cysteine in MurA has been documented only in vitro for clinically relevant Gram-negative bacteria (12, 13) and has not been observed clinically. Mutations in GlpT and UhpT are known to be a mechanism of de novo resistance to FOM in vitro (20), but they are unlikely to be clinically significant due to a concomitant loss of fitness (21).
In conclusion, in this study we describe the discovery and characterization of a novel, competitive, small-molecule inhibitor of FosA, which significantly potentiates FOM activity in representative Gram-negative pathogens. However, additional studies focused on the pharmacology, pharmacokinetics, and resistance development of ANY1 are needed to comprehensively assess the therapeutic potential of this compound. Nonetheless, this study shows that combination of a FosA inhibitor, such as ANY1, and FOM provides a new strategy to expand FOM activity to a broader spectrum of Gram-negative pathogens, including MDR and XDR strains.
MATERIALS AND METHODS
Protein expression and purification.
The fosAKP and fosA3 genes (including mutants) were synthesized with the inclusion of a C-terminal His6 tag by GenScript (Piscataway, NJ, USA) and cloned into pET-22(b+) for protein expression and purification, as described previously (9).
FosA assays.
FOM-dependent glutathione conjugation was detected spectrophotometrically using monochlorobimane (Sigma-Aldrich) to detect unreacted free glutathione (Fig. 1c). Assays were carried out in a volume of 50 μl at 25°C in 0.1 M sodium phosphate buffer, pH 8.0, containing 50 mM KCl, 25 μM MnCl2, 30 mM glutathione, and various concentrations of fosfomycin (0 to 50 mM). FosA (100 nM) was used to initiate the reaction, which was quenched after 20 min. A no-enzyme control was also performed. Reactions were quenched by the addition of 150 μl of methanol for 30 min, and then mixtures were diluted 100-fold in 0.1 M sodium phosphate buffer, pH 8.0, containing 1 mM EDTA. Following the addition of 500 μM monochlorobimane (1.7-fold molar excess of monochlorobimane to glutathione) and a 2-h incubation period, the concentration of glutathione was established by fluorescence spectroscopy using a SpectraMax M2 plate reader (Molecular Devices). Excitation and emission wavelengths of 390 nm and 478 nm, respectively, were used. A standard curve was prepared using 0 to 750 μM glutathione. Data were fit to Michaelis-Menten equations using SigmaPlot (Systat Software, Inc., San Jose, CA).
Isothermal titration calorimetry.
Experiments were performed using an iTC200 instrument (GE Healthcare), using a syringe loaded with ligand (250 to 500 μM) and a cell loaded with FosAKP (15 to 30 μM) in 75 mM NaCl plus 10 mM Tris, pH 7.8. Titrations were performed at 25°C with 16 injections of 2.42-μl aliquots, with 230-s intervals between injections. All runs were performed in triplicate, and heats of dilutions were measured and subtracted from each data set. All data were analyzed using Origin, version 7.0, software. Uncertainty in ANY1/ANY2 concentrations led to binding ratios that varied from 0.8 to 0.9 (ligand/(FosAKP monomer), which were subsequently fixed to equal 1, based on X-ray crystal structures.
Protein crystallization.
FosA3 was concentrated to 12 mg/ml and combined with 2.5 mM ANY1, 6 mM MnCl2, and 100 mM KCl. This mixture was centrifuged (19,150 × g for 10 min), and 250 nl of the supernatant was combined with 250 nl of mother liquor (0.2 M magnesium formate, 20% [wt/vol] polyethylene glycol 3350) in sitting drops. FosAKP was concentrated to 12 mg/ml, combined with 2.5 mM ANY1, 6 mM MnCl2, and 100 mM KCl and centrifuged (19,150 × g for 10 min). One microliter of supernatant was combined in hanging drops with 1 μl of mother liquor (0.02 M CaCl2, 0.1 M sodium acetate, pH 4.6, 30% [vol/vol] 2-methyl-2,4-pentanediol). Finally, 1.25 mM ANY2 and 500 μM MnCl2 were added to an 11.5-mg/ml stock solution of FosAKP and centrifuged (19,150 × g for 10 min), and 1 μl of the supernatant was combined in hanging drops with 1 μl of mother liquor (0.2 M ammonium sulfate, 0.1 M bis-Tris, pH 5.5, 20% [wt/vol] polyethylene glycol 3350). Resulting crystals were improved by streak seeding. Crystals were harvested and flash cooled with liquid nitrogen in mother liquor (ANY1-FosAKP) or mother liquor supplemented with 20% (vol/vol) glycerol as cryoprotectant (ANY2-FosAKP and ANY1-FosA3).
X-ray diffraction, data processing, structure determination, and refinement.
X-ray diffraction data for FosA3 in complex with ANY1 were collected using a Dectris Eiger X 16M detector on Advanced Photon Source (APS) beamline 23-ID-B. Data sets were collected at native wavelength (1.0332 Å) as well as at the K-edge of bromine (0.91 Å) to facilitate construction of anomalous maps. Data for FosAKP in complex with ANY1 were collected using a Dectris PILATUS3 S 6M detector on Stanford Synchrotron Radiation Lightsource (SSRL) beamline 12-2. Data for FosAKP in complex with ANY2 were collected using a Dectris PILATUS3 S 6M detector on SSRL beamline 9-2. All data sets were processed using XDS, scaled in AIMLESS, and solved by molecular replacement using Phenix-MR with apo FosA3 (PDB accession number 5VB0) (9) as the search model for the ANY1-FosA3 structure and using fosfomycin-bound FosAKP (PDB accession code 5V3D) (9) as the search model for the ANY1-FosAKP and ANY2-FosAKP structures. The ANY1-FosAKP data set was significantly anisotropic along one of the axes between 3.2 and 5 Å, resulting in higher than normal Rmerge values. Rmerge values could be restored to typical ranges (∼0.18) by cutting the resolution to 5 Å; however, loss in map clarity led us to include the higher-resolution data. Models were further built and refined using Coot and Phenix, respectively. The locations of the heavy atoms (zinc and bromine) were determined from the ANY1-FosA3 data set by removing all ligands from the structure and using Phenix Phaser-EP MR-SAD to find the location of zinc and bromine atoms. Two zinc and four bromine atoms were found, with a figure of merit of 0.747 and log-likelihood gain of 565, with locations matching as expected.
HDX-MS.
HDX-MS was performed as previously described (9), with the following modifications. For ANY1, 3 μl of 50 μM FosAKP plus 100 μM ANY1 in 20 mM Tris (pH 7.8), 150 mM KCl, and 50 μM MnCl2 was deuterated with 27 μl of 20 mM Tris, 99.99% D2O (pD 7.8), 150 mM KCl, and 50 μM MnCl2 prior to quenching. For ANY2, 3 μl of 50 μM FosAKP plus 100 μM ANY1 in 20 mM Tris (pH 7.8), 150 mM KCl, and 50 μM MnCl2 was deuterated with 27 μl of 20 mM Tris, 99.99% D2O (pD 7.8), 150 mM KCl, 50 μM MnCl2, and 125 μM ANY2. Statistical confidence was determined as previously described (8). Briefly, confidence intervals for plots of the percent difference in deuteration (Δ%D) were determined using the method outlined by Houde et al. (19).
Fluorescence binding assays.
ANY1-dependent quenching of the wild-type and mutant FosA protein fluorescence was detected using an FP-8500 spectrofluorometer (Jasco, Easton, MD). Assays were carried out in a total volume of 600 μl at 25°C in 0.1 M sodium phosphate buffer (pH 8.0) containing 100 nM FosA and various concentrations of ANY1 (0 to 5 μM). Excitation and emission wavelengths of 280 nm and 311 nm, respectively, were used. To correct for inner-filter and dilution effects, quenching data were adjusted based on control assays that were performed using a solution of l-(−)-tryptophan (Acros Organics) that was diluted to approximately match initial fluorescence of the protein solutions. Adjusted fluorescence quenching data were fit to a simple hyperbolic binding equation using SigmaPlot (Systat Software, Inc., San Jose, CA)
Bacterial growth curve analysis.
Overnight culture of the clinical isolates K. pneumoniae I1, P. aeruginosa 75B2, E. cloacae YDC612, S. marcescens YDC760-2, and E. coli YD472 were grown in Mueller-Hinton broth at 37°C and 150 rpm. E. coli YD472 is a previously reported strain that produces plasmid-encoded FosA3 along with CTX-M-65 ESBL. K. pneumoniae I1 and E. cloacae YDC612 are carbapenem-resistant strains that produce KPC-type carbapenemases and were isolated from blood and a hematoma, respectively. P. aeruginosa 75B2 is a carbapenem-resistant strain from a urine culture. All strains were from clinical specimens obtained from patients at the University of Pittsburgh Medical Center. The following day, the culture was diluted such that its OD at 600 nm (OD600) was 0.2. The diluted culture was allowed to grow at 37°C for 1 h. Following this, the culture was further diluted into 96-well, round-bottom plates such that the OD600 was 0.1. Various concentrations of ANY1 (0 to 224 μg/ml) or FOM (0 to 1,024 μg/ml) were added in addition to 25 μg/ml glucose-6-phosphate. The plate was incubated at 37°C for up to 300 min. The OD600 was assessed every 30 min. Data were analyzed using the following modified 3-parameter Gompertz equation: y = A exp{−exp [(μme/A)(λ − t) + 1]}, where μm is the growth rate, λ is the lag time, and A is the asymptote (10). Data were fitted using SigmaPlot software (Systat Software, Inc., San Jose, CA). Time-kill experiments were carried out as described previously (9).
Toxicity.
Cytotoxicity in HK2 cells, which are an immortalized proximal tubule epithelial cell line from normal adult human kidney (American Type Culture Collection [ATCC], Manassas, VA), and in human peripheral blood mononuclear cells was assessed using a CellTiter-Glo luminescent cell viability assay (Promega). For the peripheral blood mononuclear cells, blood from three separate healthy donors was purchased from the Central Blood Bank (Pittsburgh, PA). The University of Pittsburgh Institutional Review Board deemed that this study did not involve human subjects, according federal regulations, and was therefore classified as exempt (IRB number PRO1793975). To assess cytotoxicity, cells were seeded at 5 × 103 to 5 × 104 cells/well in 96-well cell culture plates containing ANY1 for 24 h before cell viability was measured.
Accession number(s).
The atomic coordinates have been deposited in the Protein Data Bank (www.pdb.org) under accession numbers 6C3U, 5WEW, and 5WEP.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by a research grant from the National Institutes of Health (grant number R21AI123747). M.L.E. was supported by grants T32DK061296 and TL1TR001858, and E.H.K. was supported by T32AI095190.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01524-18.
REFERENCES
- 1.The World Health Organization. 2017. Antibacterial agents in clinical development. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Kahan FM, Kahan JS, Cassidy PJ, Kropp H. 1974. The mechanism of action of fosfomycin (phosphonomycin). Ann N Y Acad Sci 235:364–386. [DOI] [PubMed] [Google Scholar]
- 3.Sastry S, Doi Y. 2016. Fosfomycin: resurgence of an old companion. J Infect Chemother 22:273–280. doi: 10.1016/j.jiac.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grabein B, Graninger W, Rodríguez Baño J, Dinh A, Liesenfeld DB. 2017. Intravenous fosfomycin - back to the future. Systematic review and meta-analysis of the clinical literature. Clin Microbiol Infect 23:363–372. doi: 10.1016/j.cmi.2016.12.005. [DOI] [PubMed] [Google Scholar]
- 5.Ito R, Mustapha MM, Tomich AD, Callaghan JD, McElheny CL, Mettus RT, Shanks RMQ, Sluis-Cremer N, Doi Y. 2017. Widespread fosfomycin resistance in Gram-negative bacteria attributable to the chromosomal fosA gene. mBio 8:e00749-17. doi: 10.1128/mBio.00749-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bernat BA, Laughlin LT, Armstrong RN. 1997. Fosfomycin resistance protein (FosA) is a manganese metalloglutathione transferase related to glyoxalase I and the extradiol dioxygenases. Biochemistry 36:3050–3055. doi: 10.1021/bi963172a. [DOI] [PubMed] [Google Scholar]
- 7.Rigsby RE, Rife CL, Fillgrove KL, Newcomer ME, Armstrong RN. 2004. Phosphonoformate: a minimal transition state analogue inhibitor of the fosfomycin resistance protein, FosA. Biochemistry 43:13666–13673. doi: 10.1021/bi048767h. [DOI] [PubMed] [Google Scholar]
- 8.Ito R, Tomich AD, McElheny CL, Mettus R, Sluis-Cremer N, Doi Y. 2017. Inhibition of fosfomycin resistance protein FosA by phosphonoformate (foscarnet) in multidrug-resistant Gram-negative pathogens. Antimicrob Agents Chemother 61:e01424-17. doi: 10.1128/AAC.01424-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klontz EH, Tomich AD, Günther S, Lemkul JA, Deredge D, Silverstein Z, Shaw JF, McElheny C, Doi Y, Wintrode PL, MacKerell AD Jr, Sluis-Cremer N, Sundberg EJ. 2017. Structure and dynamics of FosA-mediated fosfomycin resistance in Klebsiella pneumoniae and Escherichia coli. Antimicrob Agents Chemother 61:e01572-17. doi: 10.1128/AAC.01572-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alrowais H, McElheny CL, Spychala CN, Sastry S, Guo Q, Butt AA, Doi Y. 2015. Fosfomycin resistance in Escherichia coli, Pennsylvania, USA. Emerg Infect Dis 21:2045–2047. doi: 10.3201/eid2111.150750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zwietering MH, Jongenburger I, Rombouts FM, van 't Riet K. 1990. Modeling of the bacterial growth curve. Appl Environ Microbiol 56:1875–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim DH, Lees WJ, Kempsell KE, Lane WS, Duncan K, Walsh CT. 1996. Characterization of a Cys115 to Asp substitution in the Escherichia coli cell wall biosynthetic enzyme UDP-GlcNAc enolpyruvyl transferase (MurA) that confers resistance to inactivation by the antibiotic fosfomycin. Biochemistry 35:4923–4928. doi: 10.1021/bi952937w. [DOI] [PubMed] [Google Scholar]
- 13.Zhu JY, Yang Y, Han H, Betzi S, Olesen SH, Marsilio F, Schönbrunn E. 2012. Functional consequence of covalent reaction of phosphoenolpyruvate with UDP-N-acetylglucosamine 1-carboxyvinyltransferase (MurA). J Biol Chem 287:12657–12667. doi: 10.1074/jbc.M112.342725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.The World Health Organization. 2014. Antimicrobial resistance: global report on surveillance. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 15.Michalopoulos A, Virtzili S, Rafailidis P, Chalevelakis G, Damala M, Falagas ME. 2010. Intravenous fosfomycin for the treatment of nosocomial infections caused by carbapenem-resistant Klebsiella pneumoniae in critically ill patients: a prospective evaluation. Clin Microbiol Infect 16:184–186. doi: 10.1111/j.1469-0691.2009.02921.x. [DOI] [PubMed] [Google Scholar]
- 16.Tommasi R, Brown DG, Walkup GK, Manchester JI, Miller AA. 2015. ESKAPEing the labyrinth of antibacterial discovery. Nat Rev Drug Discov 14:529–542. doi: 10.1038/nrd4572. [DOI] [PubMed] [Google Scholar]
- 17.Richter MF, Drown BS, Riley AP, Garcia A, Shirai T, Svec RL, Hergenrother PJ. 2017. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 545:299–304. doi: 10.1038/nature22308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beharry Z, Palzkill T. 2005. Functional analysis of active site residues of the fosfomycin resistance enzyme FosA from Pseudomonas aeruginosa. J Biol Chem 280:17786–17791. doi: 10.1074/jbc.M501052200. [DOI] [PubMed] [Google Scholar]
- 19.Houde D, Berkowitz SA, Engen JR. 2011. The utility of hydrogen/deuterium exchange mass spectrometry in biopharmaceutical comparability studies. J Pharm Sci 100:2071–2086. doi: 10.1002/jps.22432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Silver LL. 2017. Fosfomycin: mechanism and resistance. Cold Spring Harb Perspect Med 7:a025262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nilsson AI, Berg OG, Aspevall O, Kahlmeter G, Andersson DI. 2003. Biological costs and mechanisms of fosfomycin resistance in Escherichia coli. Antimicrob Agents Chemother 47:2850–2858. doi: 10.1128/AAC.47.9.2850-2858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.