

Nontuberculous mycobacteria (NTM) pathogens particularly infect patients with structural lung disorders. We previously reported novel indole-2-carboxamides (ICs) that are active against a wide panel of NTM pathogens.
KEYWORDS: MmpL3, Mycobacterium, Mycobacterium abscessus, NTM, indole-2-carboxamide, nontuberculous mycobacteria
ABSTRACT
Nontuberculous mycobacteria (NTM) pathogens particularly infect patients with structural lung disorders. We previously reported novel indole-2-carboxamides (ICs) that are active against a wide panel of NTM pathogens. This study discloses in vivo data for two lead molecules (compounds 5 and 25) that were advanced for efficacy studies in Mycobacterium abscessus-infected mouse models. Oral administration of the lead molecules showed a statistically significant reduction in the bacterial loads in lung and spleen of M. abscessus-infected mice.
INTRODUCTION
Nontuberculous mycobacteria (NTM) are environmentally prevalent opportunistic pathogens that are increasingly infecting patients who have various chronic lung diseases, such as those with chronic obstructive pulmonary disorder (COPD) and cystic fibrosis (CF) (1, 2). The Mycobacterium avium complex (MAC) and the Mycobacterium abscessus complex (MABSC) account for the vast majority of NTM infections globally (3). For adolescent CF patients, MABSC infections accelerate inflammatory lung damage, and treatment often fails (4, 5). Current treatment recommendations for MAC and MABSC infections are for at least 12 months and include multidrug therapy with combinations of intravenous and oral antibiotics accompanied, in some cases, by surgical resection (6). Therefore, there is an unmet medical need for the development of new anti-NTM compounds with novel mechanisms of action that can potentially reduce treatment time, lower the incidence of adverse drug events, and provide a more effective treatment option for CF and COPD patients.
Indole-2-carboxamides (ICs) have been identified as a novel chemical scaffold showing promising preclinical results against Mycobacterium tuberculosis and NTM pathogens (7–13). We recently disclosed a novel series of ICs with MIC values of 0.0039 to 8 µg/ml against various slow- and fast-growing NTM of clinical interest (7). These compounds were also shown to be noncytotoxic and selective for mycobacteria, acting through the essential MmpL3 transporter protein (7). In this study, we have further characterized two of those ICs (compounds 5 and 25, shown in Fig. 1) that are orally bioavailable and are efficacious in a M. abscessus-infected mouse model, giving this class of compounds high potential for future translational studies.
FIG 1.
Structures of our previously reported ICs (7).
ICs 5 and 25 target the essential mycolic acid transporter, MmpL3, in MABSC.
Previous studies have shown that ICs target the essential mycolic acid transporter MmpL3 (7, 9, 10, 12, 14). Here, we report that compounds 5 and 25 target the inner membrane transporter MmpL3 of M. abscessus, resulting in the abolition of the translocation of mycolic acids from the cytoplasm to the periplasmic space, ultimately causing cell death. The 16-fold to >128-fold increase in MICs of compounds 25 and 5, respectively, against an isogenic M. abscessus mutant harboring a missense mutation (A309P) in the MmpL3 protein of M. abscessus ATCC 19977 is further evidence that this transporter most likely serves as the primary bactericidal target for these class of compounds in M. abscessus, as is the case in M. tuberculosis (10, 12, 13) and Mycobacterium massiliense (MIC of compound 5 against the MmpL3A309P mutant, >32 μg/ml, compared to 0.25 μg/ml against wild-type M. abscessus ATCC 19977; MIC of compound 25 against the MmpL3A309P mutant, 1 μg/ml, compared to 0.063 μg/ml against wild-type M. abscessus ATCC 19977) (7).
In vivo acute toxicity study.
As an initial in vivo study, the safety of compounds 5 and 25 was assessed in an acute toxicity mouse model (maximum tolerated dose [MTD] assay) to evaluate any potential adverse effects and determine a safe dose for mouse pharmacokinetic and efficacy studies. In this acute toxicity test, healthy mice (3 per group) were given three consecutive daily doses of either compound 5 or 25 at 100, 200, and 300 mg/kg of body weight in 20% cyclodextrin and were observed at regular times for any adverse effects. Cyclodextrin was added to improve aqueous solubility. Doses higher than 300 mg/kg were not possible due to limited solubility. Mice were observed at 10 min and 1, 2, 4, and 24 h after dosing on days 1, 2, and 3, followed by twice-daily observations on days 4 and 5 and once-daily observations on days 6 to 10. Compounds 5 and 25 were well tolerated at 100, 200, and 300 mg/kg for three consecutive days of dosing, and there were no adverse effects, such as significant weight loss or unresponsiveness, noted over the course of the study period.
Detailed methods for in vivo acute toxicity study.
The in vivo acute toxicity profile was established for compounds 2 and 25 after oral (p.o.) doses of 100, 200, and 300 mg/kg administered to 6- to 8-week-old female BALB/c mice (n = 3). The compounds were given twice per day for three consecutive days by gavage, at 100 µl/mouse. Observations were recorded immediately after each dose at 10 min and 1, 2, 4, and 24 h. Beginning on the fourth day, mice were observed twice daily for 2 days and then once daily for 5 days. During the observations, humane handling practices and animal welfare regulations were strictly followed.
Pharmacokinetics for compounds 5 and 25.
The pharmacokinetic (PK) profile for compound 5 was established after a single oral (p.o.) dose of 300 mg/kg administered to BALB/c mice (n = 3/time point) and analyzed using an AB Sciex 5500 liquid chromatography-tandem mass spectrometer (LC-MS/MS) (Fig. 2). The plasma concentration-time curve was described using the WinNonLin software. The maximum drug level was 6.5 µg/ml at 50 min after oral dosing. The elimination half-life averaged 2.5 h, and the area under the plasma-concentration-time curve (AUC) averaged 29.6 µg · h/ml in this animal model. Compound 5 plasma clearance (CL/F) averaged 10.1 liters/h/kg, and the volume of distribution (V) averaged 36.6 liters/kg. Similar studies have been performed with compound 25, demonstrating similar results (10). Thus, further in vivo efficacy testing against M. abscessus-infected mice was warranted for these compounds.
FIG 2.
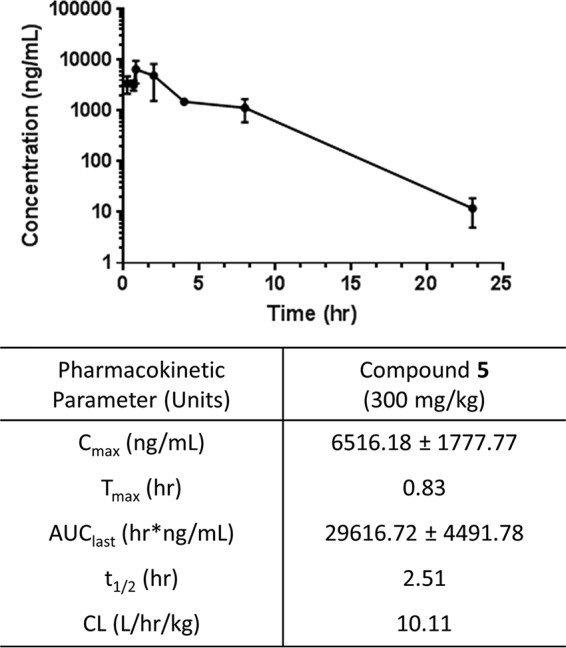
Pharmacokinetics for compound 5. In vivo plasma PK of compound 5 administered as 300 mg/kg p.o. to BALB/c mice (n = 3/time point). Plasma concentrations were taken over 24 h and analyzed using LC-MS/MS. Cmax, maximum concentration of drug in serum; Tmax, time to Cmax; AUClast, AUC calculation from time 0 to last measurable concentration; t1/2, half-life.
Detailed methods for pharmacokinetics study.
BALB/c mice (20 g each; Charles River Laboratories) were given a single dose of compound 5 at 300 mg/kg by oral gavage. Mice were dosed with a volume not greater than 10% of its body weight. At 0.25, 0.67, 0.83, 2, 4, 8, and 23 h after dose administration, mice (n = 3 per time point) were euthanized, and blood was collected by cardiac puncture. Plasma was separated using centrifugation at 2,000 rpm for 10 min at 4°C and stored at 80°C until analysis. Plasma concentrations of compound 5 were analyzed using LC-MS/MS (AB Sciex QTrap 5500 system). The mass transitions 295.2/135.2 and 309.1/163.0 were used for compound 5 and internal standard (warfarin), respectively, for quantification. A Kinetex EVO C18 column (50 by 3.0 mm, 5 µm) with isocratic mobile phase consisting of 0.1% formic acid in water (A) and acetonitrile (B) (30:70 [vol/vol]) at a flow rate of 0.50 ml/min was used for chromatographic separation. The LC-MS/MS was controlled by the Analyst 1.6.3 software. Chromatography integration and data analysis were performed using the MultiQuant 3.0.2 software. Noncompartmental pharmacokinetic analysis for compound 2 was performed using Phoenix WinNonlin 8.0 (Certara USA, Inc., NJ, USA).
ICs are efficacious in an M. abscessus-infected mouse model.
The M. abscessus efficacy study was performed similarly to previously published methods (15). The in vivo efficacy study was divided into five treatment groups. Treatment group 1 (3 mice) was an untreated control group sacrificed 1 day postinfection to determine bacterial uptake. The other four treatment groups (6 mice per group) comprised an untreated control and animals treated with either compound 5 plus 20% cyclodextrin (300 mg/kg p.o.), compound 25 plus 20% cyclodextrin (300 mg/kg p.o.), or positive control (150 mg/kg amikacin, subcutaneous [SQ]). Treatment began 2 days postinfection for nine consecutive days, and mice were sacrificed 2 days later. Mice treated with daily doses of 300 mg/kg compound 5 and 300 mg/kg compound 25 did not demonstrate any significant weight loss, and all animals survived until the day 12 endpoint of the assay.
The mean ± standard error of the mean (SEM) for lung, spleen, and liver log10 CFU for all treatment groups are summarized in Fig. 3. Lung, spleen, and liver log10 CFU for the untreated control at day 12 were 5.41 ± 0.24, 5.41 ± 0.15, and 6.17 ± 0.33, respectively. Mice treated with compound 5 resulted in lung, spleen, and liver log10 CFU counts of 3.96 ± 0.23, 3.79 ± 0.40, and 5.30 ± 0.23, respectively. Bacterial load was significantly reduced in the lung and spleen (lung, P = 0.0016; spleen, P = 0.0035; liver, P = 0.0558) compared to the untreated control group. Mice treated with compound 25 resulted in lung, spleen, and liver log10 CFU counts of 3.58 ± 0.50, 2.91 ± 0.45, and 4.90 ± 0.49, also showing a significant reduction in bacterial load in the lung and spleen (lung, P = 0.0084; spleen, P = 0.0004; liver, P = 0.0571) compared to the untreated control group. The mean ± SEM for lung, spleen, and liver log10 CFU for the positive control (amikacin) at day 12 were 3.52 ± 0.22, 3.12 ± 0.30, and 4.03 ± 0.62, respectively. Compound 5 was not significantly different (lung, P = 0.1969; spleen, P = 0.2099; liver, P = 0.0837) from the mice receiving amikacin alone. Similarly, compound 25 did not have statistically higher CFU counts than those in the positive control (lung, P = 0.9147; spleen, P = 0.7059; liver, P = 0.2967). These data demonstrate that both compounds 5 and 25 show no relevant difference in bacterial activity with the amikacin study treatment.
FIG 3.

Acute SCID treatment mouse model. Log10 CFU in the lungs (A), spleen (B), and liver (C) of M. abscessus-infected SCID mice. The SCID mice began the 9-day treatment on day 2 with saline (▲), compound 5 plus 20% 300 mg/kg cyclodextrin (red ▼), compound 25 plus 20% 300 mg/kg cyclodextrin (green ♦), and 150 mg/kg amikacin (blue ●). Results represent the average of the results from one experiment (n = 6 mice per experiment), with the bacterial load in each group expressed as average ± SEM log10 CFU. P values are shown to denote significance of differences from untreated control.
Detailed methods for IC efficacy study in M. abscessus-infected mouse model.
Six- to 8-week-old SCID female mice were ordered from Charles River. Mice were rested 1 week before infection. The acute SCID mouse model received an intravenous infection via the tail vein with 1 × 106 CFU/mouse (M. abscessus strain 103). Three mice were sacrificed 1 day postinfection to determine bacterial uptake. Whole lungs, spleens, and livers were extracted, homogenized in 4.5 ml of 1× phosphate-buffered saline (PBS), diluted, and plated on 7H11 agar plates. The plates were placed in a 37°C dry-air incubator for ∼7 days. Compounds 5 and 25 were dosed at 300 mg/kg p.o. by gavage in a volume of 100 μl per mouse, which began day 2 postinfection and continued for 8 consecutive days for a total 9-day treatment ending on day 10 postinfection. Amikacin was dosed at 150 mg/kg via s.q. injection in a volume of 100 μl per mouse per day. Six mice were used for each group (untreated, control amikacin, and compound 5- and 25-treated mice) and sacrificed 2 days after administering the last dose. Bacterial loads were determined by plating diluted lung, spleen, and liver homogenates, which were homogenized in 4.5 ml sterile PBS. Statistical analysis was performed by first converting CFU to logarithms, which were then evaluated by a one-way analysis of variance (ANOVA) followed by a multiple-comparison analysis of variance by a one-way Tukey test (GraphPad software program). Differences were considered significant at the 95% level of confidence. All procedures involving animals were approved by the Colorado State University Animal Care and Use Committee.
We and other groups have shown that ICs are active against whole-cell M. tuberculosis and NTM pathogens (7–13). In addition, IC analogs have been shown to be efficacious in M. tuberculosis-infected mouse models (10, 12). Herein, we have demonstrated further utility of the IC chemotype as an effective treatment option for M. abscessus infections. ICs were shown to be orally bioavailable and efficacious in in vivo mouse models, making them excellent candidates for future NTM preclinical translational drug development.
ACKNOWLEDGMENTS
This project was supported by the National Institutes of Health/National Institute of Allergy and Infectious Diseases research grant AI116525 and the Jack and Lois Wareham Research Award. The efficacy study was supported by the National Institutes of Health and the National Institute of Allergy and Infectious Diseases under contract no. HHSN272201100009I/HHSN27200004 A19.
The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
We thank the Department of Chemistry at Creighton University for the use of their nuclear magnetic resonance (NMR) instrument.
We declare no competing financial interests.
REFERENCES
- 1.Brown-Elliott BA, Wallace RJ Jr. 2002. Clinical and taxonomic status of pathogenic nonpigmented or late-pigmenting rapidly growing mycobacteria. Clin Microbiol Rev 15:716–746. doi: 10.1128/CMR.15.4.716-746.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Faria S, Joao I, Jordao L. 2015. General overview on nontuberculous mycobacteria, biofilms, and human infection. J Pathog 2015:809014. doi: 10.1155/2015/809014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park IK, Olivier KN. 2015. Nontuberculous mycobacteria in cystic fibrosis and non-cystic fibrosis bronchiectasis. Semin Respir Crit Care Med 36:217–224. doi: 10.1055/s-0035-1546751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Floto RA, Haworth CS. 2015. The growing threat of nontuberculous mycobacteria in CF. J Cyst Fibros 14:1–2. doi: 10.1016/j.jcf.2014.12.002. [DOI] [PubMed] [Google Scholar]
- 5.Roux AL, Catherinot E, Ripoll F, Soismier N, Macheras E, Ravilly S, Bellis G, Vibet MA, Le Roux E, Lemonnier L, Gutierrez C, Vincent V, Fauroux B, Rottman M, Guillemot D, Gaillard JL, Herrmann L-J, OMA Group. 2009. Multicenter study of prevalence of nontuberculous mycobacteria in patients with cystic fibrosis in France. J Clin Microbiol 47:4124–4128. doi: 10.1128/JCM.01257-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ryu YJ, Koh WJ, Daley CL. 2016. Diagnosis and treatment of nontuberculous mycobacterial lung disease: clinicians’ perspectives. Tuberc Respir Dis (Seoul) 79:74–84. doi: 10.4046/trd.2016.79.2.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Franz ND, Belardinelli JM, Kaminski MA, Dunn LC, Calado Nogueira de Moura V, Blaha MA, Truong DD, Li W, Jackson M, North EJ. 2017. Design, synthesis and evaluation of indole-2-carboxamides with pan anti-mycobacterial activity. Bioorg Med Chem 25:3746–3755. doi: 10.1016/j.bmc.2017.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kondreddi RR, Jiricek J, Rao SPS, Lakshminarayana SB, Camacho LR, Rao R, Herve M, Bifani P, Ma NL, Kuhen K, Goh A, Chatterjee AK, Dick T, Diagana TT, Manjunatha UH, Smith PW. 2013. Design, synthesis, and biological evaluation of indole-2-carboxamides: a promising class of antituberculosis agents. J Med Chem 56:8849–8859. doi: 10.1021/jm4012774. [DOI] [PubMed] [Google Scholar]
- 9.Kozikowski AP, Onajole OK, Stec J, Dupont C, Viljoen A, Richard M, Chaira T, Lun S, Bishai W, Raj VS, Ordway D, Kremer L. 2017. Targeting mycolic acid transport by indole-2-carboxamides for the treatment of Mycobacterium abscessus infections. J Med Chem 60:5876–5888. doi: 10.1021/acs.jmedchem.7b00582. [DOI] [PubMed] [Google Scholar]
- 10.Lun S, Guo H, Onajole OK, Pieroni M, Gunosewoyo H, Chen G, Tipparaju SK, Ammerman NC, Kozikowski AP, Bishai WR. 2013. Indoleamides are active against drug-resistant Mycobacterium tuberculosis. Nat Commun 4:2907. doi: 10.1038/ncomms3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Onajole OK, Pieroni M, Tipparaju SK, Lun S, Stec J, Chen G, Gunosewoyo H, Guo H, Ammerman NC, Bishai WR, Kozikowski AP. 2013. Preliminary structure-activity relationships and biological evaluation of novel antitubercular indolecarboxamide derivatives against drug-susceptible and drug-resistant Mycobacterium tuberculosis strains. J Med Chem 56:4093–4103. doi: 10.1021/jm4003878. [DOI] [PubMed] [Google Scholar]
- 12.Rao SP, Lakshminarayana SB, Kondreddi RR, Herve M, Camacho LR, Bifani P, Kalapala SK, Jiricek J, Ma NL, Tan BH, Ng SH, Nanjundappa M, Ravindran S, Seah PG, Thayalan P, Lim SH, Lee BH, Goh A, Barnes WS, Chen Z, Gagaring K, Chatterjee AK, Pethe K, Kuhen K, Walker J, Feng G, Babu S, Zhang L, Blasco F, Beer D, Weaver M, Dartois V, Glynne R, Dick T, Smith PW, Diagana TT, Manjunatha UH. 2013. Indolcarboxamide is a preclinical candidate for treating multidrug-resistant tuberculosis. Sci Transl Med 5:214ra168. doi: 10.1126/scitranslmed.3007355. [DOI] [PubMed] [Google Scholar]
- 13.Stec J, Onajole OK, Lun S, Guo H, Merenbloom B, Vistoli G, Bishai WR, Kozikowski AP. 2016. Indole-2-carboxamide-based MmpL3 inhibitors show exceptional antitubercular activity in an animal model of tuberculosis infection. J Med Chem 59:6232–6247. doi: 10.1021/acs.jmedchem.6b00415. [DOI] [PubMed] [Google Scholar]
- 14.Li W, Yazidi A, Pandya AN, Hegde P, Tong W, Calado Nogueira de Moura V, North EJ, Sygusch J, Jackson M. 2018. MmpL3 as a target for the treatment of drug-resistant nontuberculous mycobacterial infections. Front Microbiol 9:1547. doi: 10.3389/fmicb.2018.01547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Obregón-Henao A, Arnett KA, Henao-Tamayo M, Massoudi L, Creissen E, Andries K, Lenaerts AJ, Ordway DJ. 2015. Susceptibility of Mycobacterium abscessus to antimycobacterial drugs in preclinical models. Antimicrob Agents Chemother 59:6904–6912. doi: 10.1128/AAC.00459-15. [DOI] [PMC free article] [PubMed] [Google Scholar]