

Abstract
Camptothecin (CPT), a natural product and its synthetic derivatives exert potent anticancer activity by selectively targeting DNA Topoisomerase I (Top1) enzyme. CPT and its clinically approved derivatives are used as Top1 poisons for cancer therapy suffer from many limitations related to stability and toxicity. In order to envisage structurally diverse novel chemical entity as Top1 poison with better efficacy, Ligand-based-pharmacophore model was developed using 3D QSAR pharmacophore generation (HypoGen algorithm) methodology in Discovery studio 4.1 clients. The chemical features of 29 CPT derivatives were taken as the training set. The selected pharmacophore model Hypo1 was further validated by 33 test set molecules and used as a query model for further screening of 1,087,724 drug-like molecules from ZINC databases. These molecules were subjected to several assessments such as Lipinski rule of 5, SMART filtration and activity filtration. The molecule obtained after filtration was further scrutinized by molecular docking analysis on the active site of Top1 crystal structure (PDB ID: 1T8I). Six potential inhibitory molecules have been selected by analyzing the binding interaction and Ligand-Pharmacophore mapping with the validated pharmacophore model. Toxicity assessment TOPKAT program provided three potential inhibitory ‘hit molecules’ ZINC68997780, ZINC15018994 and ZINC38550809. MD simulation of these three molecules proved that the ligand binding into the protein-DNA cleavage complex is stable and the protein-ligands conformation remains unchanged. These three hit molecules can be utilized for designing future class of potential topoisomerase I inhibitor.
Keywords: Topoisomerase I, Camptothecin, Anticancer agent, Inhibitor, Pharmacophore, Virtual screening, ZINC database, Molecular dynamics, Toxicity, ADMET
Graphical Abstract
1. Introduction
Topoisomerases are ubiquitous enzymes essential for resolving the topological problems associated with DNA supercoiling during replication and transcription which can be one of the important targets for anticancer and antibacterial drugs development [1,2]. To reduce the torsional stress of the supercoiled DNA, human Topoisomerase I (Top1) cleaves the phosphodiester bond on a single DNA strand by nucleophilic attack on catalytic tyrosine 723 and form a ‘cleavage complex’ in which 3′ end of the broken DNA strand is covalently linked to the enzyme [[3], [4], [5]]. Top1 is one of the important targets for cancer chemotherapy as it relives the DNA torsional strain and stops the cell division process. As in post mitotic cells, gene transcription and DNA replication and repairing process are regulated predominantly by Top1 enzyme. Inhibition of Top1 can be a choice for controlling the nuclear process in cancer cell growth regulation [[6], [7], [8]]. The religation process of DNA by Top1 occurs much faster than the rate of DNA cleavage and the process is assured by the total concentration of the covalent 3′-phosphotyrosyl Top1-DNA duplex complex [2]. There are two types of Top1 inhibitors; suppressor and poison. Suppressor inhibits topoisomerase by interacting with the protein and not allowing it to function properly. Whereas Top1 poisons stabilize the DNA-Top1 ‘cleavable complex’ and prevent the religation of DNA strand and make a ternary complex of DNA-Top1-poison, resulting in the apoptosis of the cancer cell. Camptothecin (CPT) is a cytotoxic quinoline alkaloid that was isolated from stem and bark of Camptothecin acuminate [4],which selectively targets topoisomerase IB by binding and stabilizing the cleavage complex of the top1-DNA reaction. Various analog of CPT, such as Topotecan and Irinotecan [Fig. 1] are clinically used as Top1 poison for the cancer therapy [10,11]. CPT and its analogs suffer from certain limitations such as instability of the hydroxy lactone ring [2], dose-limiting side effect, multidrug resistance, solubility and severe side effects [6]. Thus, there is a scope for the discovery of structurally novel Top1 poison with improved efficacy.
Fig. 1.

Camptothecin and its clinically approved analogs.
The main objective of the present study was to discover new molecular entity that acts as a Top1 poison with improved efficacy. We initiated our study by pharmacophore modeling and virtual screening technique. A study by Malgorzata et al. [7] revealed two pharmacophore structures by both structure-based and ligand-based approach, where 27 CPT derivatives had been considered in training set. The pharmacophore model was not validated by any test set molecules. National Cancer Institute (NCI) database containing only 265,242 numbers of molecules was used for virtual screening on Discovery Studio with only Lipinski's Rules of 5 as filtration criteria. It would have been judicious to use more filters as well as determine ‘hit molecules’ from the database containing a molecular library with diverse biological activity to identify novel chemotypes that can inhibit Top1. In the present manuscript, we have initiated the study with structurally diverse molecules from the ZINC database with various biological activities. Also, we've subjected the molecular library through a rigorous set of filters.
In a separate study by Sanal et al. [8] pharmacophore had been built up by ‘Common Features Pharmacophore’ techniques where the common features are present only in the active compounds. The limited number of training set molecules mostly active and moderately active compounds are taken into consideration. The technique has its own limitations with the uptake of a limited number of molecules apart from the biological IC50 values which should be validated under same assay condition. In the present manuscript, we have developed our pharmacophore by utilizing 3D-QSAR Pharmacophore (HypoGen algorithm) technique by summarizing the structural features of total 62 CPT derivatives with diverse molecular structural patterns with a basic CPT scaffold. The 62 CPT derivatives are designed in seven different classes of molecules like 7-Aryliminomethyl CPT derivatives, 7-Cycloalkyl CPT derivatives, 7-Alkynyl CPT derivatives,7-Ethyl-9-Alkyl CPT derivatives; Nitrogen based CPT derivatives, 7-alkenyl CPT derivatives and phosphodiester and phosphotriester derivatives. The biological activities of the input 62 ligands were screened in a single cancer cell line (A549). The correlation between estimated activity and experimental activity was 0.917678 for the training set and for test set it was 0.874718. The selected pharmacophore (Hypo1) had been taken as a 3D Query for the subsequent virtual screening against drug-like molecules from the ZINC database containing 1,087,724 molecules. For subsequent filtration, three conditions had been employed; a) Lipinski's Rules of five where druggability of the compounds and ADME was set as a primary filtration criteria of the screened hit molecules, b) SMART filtration was applied where unrequired functional groups were filtered out and c) Next, filtration criteria was restricted to estimated activities not be >1.0 μM. The study puts forth six prospective molecules through extensive molecular docking analysis and meticulous visual inspection of the receptor protein (PDB ID: 1T8I) co-crystalized with CPT. Toxicity assessment by TOPKAT program provided three potential ‘hit molecules’. Through molecular dynamics (MD) simulation, we validated the stability of the ligand binding mode and the protein-ligands conformation. These three hit molecules ZINC68997780, ZINC15018994 and ZINC38550809 can be utilized for designing future class of potential topoisomerase I inhibitor.
2. Materials and Method
Computational drug design involves structure-based drug design and ligand-based drugs design. One of the important ligand-based pharmacophore modeling approaches is three dimensional (3D) QSAR strategy [15,16]. The availability of the vast molecular library and their corresponding IC50 values in various cancer cell lines have enabled us to focus on 3D-QSAR based ligand pharmacophore modeling. The 3D-QSAR strategy is different from the Common Feature Pharmacophore approach as there is no limitation on the number of training set compounds and strategy does not require experimental biological activity values in similar bioassay condition. Based on the previously published literature, libraries of 62 molecules with Top1 inhibitory activity were extracted [[17], [18], [19], [20], [21], [22]] for the generation of primary data-set of the 3D QSAR pharmacophore modeling study. Compounds were divided into test set and training set based on distribution of biological activities and chemical features. In order to achieve a significant pharmacophore model, the following criteria was maintained during the selection of test set and training set compounds. 1) All 62 compounds having a good range of experimental activities against A549 cancer cell lines should bind on the active site of Top1 protein-DNA cleavage complex. 2) The widely populated dataset was classified into four categories according to biological activity data as most active, active, moderately active and inactive. These molecules were distributed in the training set and test set. The IC50 values restraint for the most active set are <0.1 μM, active sets consist of compounds with IC50 values ranging between 0.1 μM to 1.0 μM, moderately active set of molecules have IC50 values ranges between 1.0 μM to 10.0 μM and rest of the compounds were placed in inactive category [11]. 3) A maximum number of most active and active compounds along with few moderately active and inactive compounds were taken into the training set compounds. The rest of the molecules were taken into test set for validation. 4) All the biologically relevant data were obtained from a homogeneous procedure against a single cancer cell line. 5) Experimental inhibitory activities of all 62 compounds mentioned in the data sets were collected from the same biological assays and biological assessment [12,13]. 4) Chemical substitution pattern on A, B and E ring of camptothecin derivatives were also considered for composition of the two sets. To ensure the statistical relevance, a training set of 29 diverse compounds [[17], [18], [19], [20], [21], [22]] (Fig. 2) with the experimental IC50 values between 0.003 μM to 11.4 μM were selected from the above mentioned categories. For the validation of the generated pharmacophore, the rest 33 compounds were clustered as a test set [17,18,[20], [21], [22]] (Supplementary Fig. S2) [14].
Fig. 2.


Training set molecules along with their corresponding IC50values.
2.1. Compound Preparations
The two dimensional (2D) structures of the selected molecular datasets were drawn using ChemDraw Ultra and sequentially converted into their three dimensional (3D) form by Accelrys Discovery Studio 4.1 (DS). Any further structural errors had been minimized by incorporating them in Marvin view application. Using the Clean-2D and Clean-3D approaches we recalculated the co-ordinates of the atom and bond order to the most appropriate location and saved in Sybyl Mol2 format. Hydrogen was added to the training and test set molecules and optimized using the CHARMM force fields. Every compound was further minimized using the smart minimizer executing 2000 steps of steepest descent along with conjugate gradient algorithms which maintained the RMS gradient of 0.001 kcal mol−1. A diverse set of maximum 255 different conformers was developed for each compound within an energy range of 20 kcal mol−1 above the global energy minimum. These conformers were utilized for pharmacophore hypothesis generation, fitting of the compound into the model hypothesis and for predicting the activity of the newly found compounds [[25], [26], [27]].
2.2. Generation of Pharmacophore Models
Two types of methodologies are reported in the literature for the generation of ligand-based pharmacophore model [16]. Common Features Pharmacophore Modeling [7] utilizes the common chemical features present on the most active compounds and 3D QSAR pharmacophore Modeling [17] is based on the chemical features present on the most active and inactive compounds along with their corresponding biological activity. In the present manuscript, we have performed 3D QSAR pharmacophore methodologies to generate the pharmacophore models which can be correlated with the specific chemical features of the molecules significant for the necessary biological activity. Feature Mapping protocol in DS was employed for seeking the different chemical features present on the training set molecules. The analysis of the training set compounds by Create 3D-Fingerprints protocol in DS revealed that Hydrogen Bond Acceptor (HBA), Hydrogen Bond Donor (HBD), Positive Ionizable (PI), Negative Ionizable (NI), Hydrophobic (HY) and Ring Aromaticity (RA) features admirably mapped on the structural/chemical features of all training set molecules [29,30]. Based on the results, NI was found to be significantly less mapped on the selected structures when compared to the other features. Thus, HBA, HBD, HY, PI and RA were selected for the 3D-QSAR Pharmacophore Generation protocol. The IC50 values of individual training set compounds were selected as an active property and energy threshold was maintained at 20 kcal mol−1 during the pharmacophore generation. Minimum Interfeature Distance was changed from 2.97 to 2.0 [19] and maximum excluded volume was set to zero. The uncertainty value was set at two, which is defined by DS as a ratio of the reported value to the minimum and maximum values [11]. The uncertainty value implies that the model can accommodate variation in experimental IC50 values and the predicted IC50 values up to two times. The developed pharmacophore model was selected from the 10 different hypotheses based on the highest correlation coefficient, lowest total cost and root mean square deviation (RMSD).
2.3. Pharmacophore Validation
Cost analysis, Test set analysis, Fischer's randomization test are the three methods utilized for validation of the resultant pharmacophore. The quality of the model is described in terms of fixed cost, total cost and null cost under cost analysis methodological aspect. Null cost and fixed cost designated in bit units are considered as a key parameter for the quality of the pharmacophore model. Null cost signifies the maximum cost value of the training set compounds. The Fixed cost is also been referred to as the ideal model cost. For developing a robust pharmacophore model, total cost should be close to the fixed cost and more distant from a null cost. The best model was assigned in light of the distinction between two cost values; such as null cost and total cost. The cost distinction >60 bits infers significant correlation. The model should fall in 70–90% prediction range if their cost difference is under 40 to 60 regions and ultimately if the cost difference is <40 bits then it will be problematic to evaluate the model [9].
Fischer's randomization technique acts as a fundamental role in making a correlation between the structural and biological activity in training set compounds. The validation of the selected pharmacophore hypothesis in this randomization technique is carried out by selecting 95% confidence levels and it produced 19 random spreadsheets [15,32]. The validation was done by randomizing the training set compounds. For effective pharmacophore generation, the randomized data set should produce comparable or better cost values, better RMSD and significant correlations [15]. The third approach applied to validate the pharmacophore model is the test set analysis method. In this method, it effectively anticipates the activity of the test set molecules having a decent correlation coefficient with cross-validating 95% confidence level. We used the Ligand Pharmacophore Mapping protocol with flexible search alternative in DS for overlapping the validated pharmacophore with the active molecules [20] and also predicted the estimated activity (Table 3) that should be as close to the experimental biological activity of the molecules. The validated pharmacophore model could be used for searching structurally diverse molecules from the molecular library database.
Table 3.
Experimental and estimated activity of individual test set compounds.
| Comp no. | IC50 value (μM) |
Errorsa | Fit valueb | Activity scalec |
||
|---|---|---|---|---|---|---|
| Experimental | Estimated | Experimental | Estimated | |||
| 1 | 0.12 | 0.14 | 1.2 | 7.28 | +++ | +++ |
| 2 | 0.015 | 0.14 | 9.6 | 7.28 | ++++ | +++ |
| 3 | 0.071 | 0.16 | 2.3 | 7.23 | ++++ | +++ |
| 4 | 0.039 | 0.17 | 4.2 | 7.22 | ++++ | +++ |
| 5 | 0.056 | 0.17 | 3 | 7.22 | ++++ | +++ |
| 6 | 0.051 | 0.17 | 3.3 | 7.21 | ++++ | +++ |
| 7 | 0.01 | 0.0099 | −1 | 8.44 | ++++ | ++++ |
| 8 | 0.046 | 0.028 | −1.6 | 7.99 | ++++ | ++++ |
| 9 | 0.094 | 0.17 | 1.8 | 7.21 | ++++ | +++ |
| 10 | 0.28 | 0.17 | −1.7 | 7.21 | +++ | +++ |
| 11 | 0.088 | 0.17 | 1.9 | 7.21 | ++++ | +++ |
| 12 | 1 | 0.17 | −6 | 7.22 | +++ | +++ |
| 13 | 1 | 0.17 | −5.7 | 7.2 | +++ | +++ |
| 14 | 0.49 | 2 | 4 | 6.14 | +++ | ++ |
| 15 | 4.7 | 5.5 | 1.2 | 5.7 | ++ | ++ |
| 16 | 3.4 | 1.9 | −1.8 | 6.16 | ++ | ++ |
| 17 | 2.7 | 1.4 | −1.9 | 6.29 | ++ | ++ |
| 18 | 0.39 | 1.3 | 3.4 | 6.32 | +++ | ++ |
| 19 | 21 | 12 | −1.7 | 5.37 | + | + |
| 20 | 0.81 | 1.3 | 1.6 | 6.34 | +++ | ++ |
| 21 | 2.5 | 1.3 | −2 | 6.34 | ++ | ++ |
| 22 | 0.49 | 1.4 | 2.9 | 6.3 | +++ | ++ |
| 23 | 5.3 | 0.91 | −5.8 | 6.48 | ++ | +++ |
| 24 | 29 | 9.2 | −3.2 | 5.48 | + | ++ |
| 25 | 8.1 | 22 | 2.7 | 5.1 | ++ | + |
| 26 | 16 | 22 | 1.4 | 5.11 | + | + |
| 27 | 49 | 8.2 | −6 | 5.53 | + | ++ |
| 28 | 1.2 | 1.9 | 1.6 | 6.16 | ++ | ++ |
| 29 | 49 | 16 | −3 | 5.22 | + | + |
| 30 | 4.8 | 8.6 | 1.8 | 5.51 | ++ | ++ |
| 31 | 3.1 | 1.9 | −1.7 | 6.17 | ++ | ++ |
| 32 | 0.047 | 0.15 | 3.2 | 7.26 | ++++ | +++ |
| 33 | 3.2 | 0.2 | −16 | 7.15 | ++ | +++ |
Error factor calculated as the ratio of the measured activity to the estimated activity; positive value indicates that the estimated IC50 is higher than the experimental IC50; a negative value indicates that the estimated IC50 is lower than the experimental IC50 value.
Fit value indicates how well the features in the pharmacophore map with the chemical features present in the compound.
Activity scale: ++++, IC50 ≤ 0.1 μM (most active); +++, IC50 0.1 to 1.0 μM (active); ++, IC50 1.0 to 10.0 μM (moderately active); +, IC50 > 10.0 μM (inactive).
2.4. Database Screening
Structurally novel and potential lead molecules identification from the diverse chemical database could be possible by virtual screening based on the generated pharmacophore [10]. We initiated the identification of novel scaffolds as topoisomerase I inhibitor by the pharmacophore-based virtual screening. Validated Pharmacophore Hypo1 was utilized as a 3D query for screening Zinc Database containing 1,087,724 molecules [18]. We assumed that the possible ‘hit molecules’ should fit with all the possible features of query pharmacophore. To retrieve prospective lead molecules from the database, a Search 3D database protocol had been followed along with best/flexible search option. The identified hit compounds were further subjected to various constraints such as Lipinski's rule of five, which consists of molecules with molecular weight <500D, HBD <5, HBA <10 and an octanol/water partition coefficient (LogP) value <5 [19]. Thereafter, SMART protocol had been applied for filtering out the molecules with unwanted functional groups. Finally, the filtered molecules were screened by estimated activity <1.0 μM to achieve the possible set of potent molecules.
2.5. Molecular Docking Study
Topoisomerase I (PDB ID: 1T8I) carries a narrow rectangular cavity as an active site where the standard molecule CPT was pre-bonded as a co-crystal in a planar geometry. Our objective was to evaluate whether the filtered hit molecules bind in the active site in a similar manner with requisite geometry as observed with CPT. The active site on 1T8I consists of Arg364, Asp533 amino acids [23] and nucleotide DC112, DA113, DT10 [20] as important residues. The filtered molecules were subjected to docking in that active site by LibDock protocol on DS 4.1. Protein preparation protocol had been followed for preparing the protein and the filtered molecules were docked into the active sites on the prepared protein. During protein preparation, the water molecules were removed along with the addition of hydrogen atoms by satisfying MMFF force fields [22]. The active sites were calculated by Ligplot analysis program. The coordinates of the active sites were: 17.2665, −0.172, and 26.737. The active site was defined as a sphere of 9.5 Å from the geometric centroid of the co-crystallized ligand. Libdock protocols generated 10 different poses for each filtered molecules by maintaining the docking tolerance at 0.25. During docking CAESAR conformational method had been followed by maintaining a maximum 255 conformations of each compound within an energy range of 20 kcal mol−1 above the global energy minimum threshold (Supplementary Table S2). On each docking 100 hotspots were generated. During the analysis, scoring function and binding interaction for every single conformational pose were chosen as a selection criterion [21]. The molecules which show interactions with the important active site residues with requisite geometry along with high docking score were selected as more desirable hit molecules (Table 4 and Table 5). The following formula [11] was used to calculate the docking score:
Table 4.
Docking interaction of Camptothecin with Topoisomerase1.
| Compound Name | H-bond monitoring | H-bond distance (Å) | Docking score |
|---|---|---|---|
| Camptothecin | A:ARG364:HH22 –L:Camptothecin:N20 | 2.15109 | 107.235 |
| L:Camptothecin:H27 - A:THR718:OG1 | 2.24314 | ||
| L:Camptothecin:H31 - A:ASP533:OD2 | 1.81828 |
Table 5.
Docking interaction of virtually screened hit compounds.
| Compound name | H-bond interacting groups | H-bond monitoring | H-bond distance (Å) |
|---|---|---|---|
| Zinc Drug Like Database | |||
| ZINC68997780 | Arg364, Arg488, DC112, DA113, Lys532, Asp533 | A:ARG364:NH2 – L:ZINC68997780:O1 | 2.51833 |
| A:ARG364:NH2 – L:ZINC68997780:N11 | 2.67677 | ||
| A:ARG488:NH1 – L:ZINC68997780:O27 | 3.348 | ||
| L:ZINC68997780:O1 - D:DA113:O4’ | 2.8055 | ||
| L:ZINC68997780:N28 - A:LYS532:O | 2.49234 | ||
| L:ZINC68997780:N28 - A:ASP533:OD2 | 3.19981 | ||
| L:ZINC68997780:O1 - D:DC112:O2 | 3.299 | ||
| ZINC38550809 | Arg364, Asp533, Thr718 | A:ARG364:HH22 - L:ZINC38550809:N9 | 2.56414 |
| L:ZINC38550809:H45 - A:ASP533:OD2 | 1.54933 | ||
| L:ZINC38550809:H39 - A:THR718:OG1 | 2.82579 | ||
| ZINC38550756 | Arg364, Asp533 | A:ARG364:HH22 - L:ZINC38550756:N19 | 2.08367 |
| L:ZINC38550756:H37 - A:ASP533:OD2 | 1.66704 | ||
| ZINC15018994 | Arg364, Asp533, Thr718 | A:ARG364:HH22 - L:ZINC15018994:O20 | 2.15411 |
| L:ZINC15018994:H41 - A:ASP533:OD2 | 2.11482 | ||
| L:ZINC15018994:H32 - A:THR718:OG1 | 2.54903 | ||
| ZINC08832860 | Arg364, Asp533, DG12 | L:ZINC08832860:H44 - A:ASP533:OD2 | 2.31329 |
| L:ZINC08832860:S32 - C:DG12:O4’ | 3.28234 | ||
| A:ARG364:HH22 - L:ZINC08832860:N11 | 2.55816 | ||
| ZINC43932053 | Arg364, Lys532, Asp533, Thr718 | A:ARG364:HH22 – L:ZINC43932053:N18 | 2.15339 |
| L:ZINC43932053:H24 - A:LYS532:O | 2.21510 | ||
| L:ZINC43932053:H24 - A:ASP533:OD2 | 3.06726 | ||
| L:ZINC43932053:H32 - A:THR718:OG1 | 2.86654 | ||
Docking Score (force fields) = − ((receptor interaction energy / ligand) + ligand internal energy).
2.6. ADMET and Toxicity Prediction
The prediction of drug toxicity and ADME properties are major filtration criterion for the drug design process. As a result adsorption, distribution, metabolism, excretion and toxicity (ADMET) which is related to pharmacokinetics are important parameters considered during the drug development process [[26], [27], [28]]. Various mathematical predictive ADMET pharmacokinetic parameters such as blood-brain-barrier penetration, human intestinal absorption, aqueous solubility, cytochrome P450 2D6 inhibition, hepatotoxicity, plasma protein binding were calculated quantitatively for the selected six ligands using ADMET modules in Discovery Studio v4.1 client. Thereafter, the selected molecules were subjected to various toxicity screening models such as carcinogenicity, mutagenicity, skin irritancy, biodegradability using DS_TOPKAT module of Discovery Studio 4.1 client. Finally, based on toxicity assessment we have selected three lead molecules ZINC68997780, ZINC38550809 and ZINC15018994 for MD simulation studies.
2.7. Molecular Dynamics Study
To validate the structural stability and conformational flexibility of the binding of the lead molecules into the protein-DNA cleavage complex, protein-ligand complex molecular dynamics was performed as described by Musyoka et al. [29]. GROMACS AMBER99SB force field had been selected for parameterization of protein-nucleic acid dimer complex. Since DNA was not in the intact form in the duplex structure, performing an all atoms protein-DNA-Ligand ternary complex simulation was challenging. The 5 prime end of the broken DNA has a nucleotide DT10 that does not contain one oxygen atom (O3’) attached to tetrahydrofuran which was refined by adding the oxygen atom at 110o placements. Another modification was performed on TGP residue at 3 prime end of the broken helix as it is not recognized by AMBER99SB force field. TGP is a modified form of the guanine residue where O5’ atom is replaced with S5’. After proper modification of atom, TGP is renamed to guanine. AnteChamber Python Parser interface (ACPYPE) [30] was applied to parameterize the required topologies, atomic types and charges for the small molecules. The output files of the ligand and the generated GROMACS compatible files for the proteins were then merged using an ad hoc Python script and taken as a protein-DNA-ligand complex. The individual protein-DNA duplex system and protein-DNA-ligand complex were taken as an initial structure for all atoms MD simulation. Extended Simple Point Charge (SPC/E) water model was utilized to solvate the system. The whole system had been placed at the center of a cubic box filled with water molecules followed by further charge stabilization using Cl− and Na+. The whole systems were further neutralized by adding nineteen Na+ atoms. Energy minimization was carried out for 0.1 ns (ns) using a maximum force ≥10.0 kJ/mol to attain the stable state of the system [31]. Energy minimization graph (Fig. 3) certified that the resultant structure was solvent saturated and geometrically stabilized.
Fig. 3.

Energy minimization curve of protein after 0.1 ns steps run.
Thereafter, the whole systems were equilibrated in two phases under isothermal and isochoric ensemble (NVT) at a constant 300 K temperature and under NPT ensemble to maintain the stabilized pressure (0 bar) inside the system in a sequential manner. Both the NVT and NPT systems would follow 50,000 steps that were corresponding to 0.1 ns. The well equilibrated system with constant pressure and temperature was released for position restrained and involved for 10 ns production run with integrator time steps of 0.002 ps utilizing leap-frog algorithms. All hydrogen bonds were constrained during equilibration by applying LINC algorithms [32] whereas, Particle Mesh Ewald module had been applied for long range ionic interaction [33] with Fourier grid spacing of 0.16 Å. The entire trajectories were saved for analysis at a frequency of 0.002 ps during the simulation run.
3. Result and Discussion
3.1. Pharmacophore Model Generation
Structurally diverse 29 training set molecules containing active and moderately active compounds with their corresponding IC50 values ranging from 0.003 μM to 11.4 μM were selected to generate a pharmacophore model (Table 1). Features Mapping protocol was performed on given training set compounds where the features like hydrogen bond acceptor (HBA), hydrogen bond donor (HBD), ring aromatic (RA), hydrophobic (HYD), positive ionizable (PI), negative ionizable (NI) were well distributed on the selected molecules. For pharmacophore model generation, we used the HypoGen algorithm 3D-QSAR Pharmacophore protocol. The statistical parameters such as total cost, cost differences, RMSD and correlation coefficient of generated pharmacophore were enlisted in Table 2. A set of 10 pharmacophore models was generated which contains two HBA and one RA features. The generated pharmacophore models have total cost range from 127.392 to 136.137, null cost 239.969 and fixed cost 101.975 bits. The cost difference was calculated by the difference between the null cost and total cost. For significant pharmacophore model, it is necessary that the cost difference must be greater and should be in between total cost and fixed cost value [9,12,13,34]. Among the 10 generated pharmacophore models, the first model (Hypo1) scored the highest cost difference 112.58 bits and total cost value is much closer to fixed cost when compared to other models. The highest cost difference value of Hypo1 signifies that it can predict the experimental IC50 value of training set compounds with >90% statistical significance. Other parameters that are considered to evaluate the generated pharmacophore models are correlation coefficient and RMSD value. The Hypo1 has the highest correlation coefficient value 0.917678 and lowest RMS deviation of 1.30358. Large correlation coefficient and low RMSD suggest that Hypo1 has a better ability to predict the experimental activity of training set compounds. All the molecules along with their IC50 values were selected from previously published research articles were divided into four groups of magnitude according to their experimental activity value (IC50). Four groups of magnitude are categorized in most active (≤ 0.1 μM, ++++), active (0.1 to 1.0 μm, +++), moderately active (1.0 to 10.0 μm, ++) and inactive (>10.0 μm, +). The predictive ability of training set compounds were shown in Table 1. In training set compound 4 (Fig. 5A), compound 5 were estimated as most active molecules as they are nicely mapped with all the essential features of the pharmacophore, whereas in case with other molecules some of the essential features are not mapped. Mapping of Hypo1 with least active molecules (compound 19) is shown in Fig. 5B. All the 10 theoretical pharmacophores were subjected to further evaluation for their capability to predict the activity of the training set molecules. Hypo1 has been selected as a signified pharmacophore model (Fig. 4) over all others pharmacophores since it has the lowest total score, highest cost difference, low RMSD value and high correlation coefficient. The selected hypo1 consists of two HBA and one RA features.
Table 1.
Experimental and estimated activity of individual training set compounds.
| Comp No. | IC50 value (μM) |
Errorsa | Fit valueb | Activity scalec |
||
|---|---|---|---|---|---|---|
| Experimental | Estimated | Experimental | Estimated | |||
| 1 | 0.05 | 0.042 | −1.2 | 6.68 | ++++ | ++++ |
| 2 | 0.066 | 0.042 | −1.6 | 6.68 | ++++ | ++++ |
| 3 | 0.024 | 0.042 | +1.7 | 6.68 | ++++ | ++++ |
| 4 | 0.36 | 0.042 | −8.7 | 6.68 | +++ | ++++ |
| 5 | 0.11 | 0.043 | −2.6 | 6.67 | +++ | ++++ |
| 6 | 0.003 | 0.042 | +14 | 6.68 | ++++ | ++++ |
| 7 | 0.04 | 0.043 | +1.1 | 6.67 | ++++ | ++++ |
| 8 | 0.011 | 0.042 | +3.8 | 6.68 | ++++ | ++++ |
| 9 | 0.031 | 0.042 | +1.3 | 6.68 | ++++ | ++++ |
| 10 | 0.092 | 0.066 | −1.4 | 6.48 | ++++ | ++++ |
| 11 | 0.055 | 0.14 | +2.6 | 6.14 | ++++ | +++ |
| 12 | 0.088 | 0.16 | +1.9 | 6.09 | ++++ | +++ |
| 13 | 0.255 | 0.14 | −1.8 | 6.15 | +++ | +++ |
| 14 | 3.25 | 1.4 | −2.3 | 5.15 | ++ | ++ |
| 15 | 2.62 | 2.3 | −1.1 | 4.94 | ++ | ++ |
| 16 | 0.431 | 2.3 | +5.3 | 4.94 | +++ | ++ |
| 17 | 4.38 | 2.3 | −1.9 | 4.94 | ++ | ++ |
| 18 | 3.15 | 4.2 | +1.3 | 4.67 | ++ | ++ |
| 19 | 11.40 | 4.9 | −2.3 | 4.61 | + | ++ |
| 20 | 11.39 | 7.6 | −1.5 | 4.42 | + | ++ |
| 21 | 2.84 | 4.9 | +1.7 | 4.61 | ++ | ++ |
| 22 | 2.30 | 4.9 | +2.1 | 4.61 | ++ | ++ |
| 23 | 4.7 | 2.9 | −1.6 | 4.84 | ++ | ++ |
| 24 | 2.9 | 2.9 | −1 | 4.84 | ++ | ++ |
| 25 | 4.6 | 2.9 | −1.6 | 4.84 | ++ | ++ |
| 26 | 1.1 | 0.042 | −1.9 | 6.68 | ++ | ++++ |
| 27 | 0.8 | 0.42 | −1.9 | 5.68 | +++ | +++ |
| CPT | 0.047 | 0.043 | −1.1 | 6.67 | ++++ | ++++ |
| TPT | 0.036 | 0.042 | +1.2 | 6.68 | ++++ | ++++ |
Error factor calculated as the ratio of the measured activity to the estimated activity; positive value indicates that the estimated IC50 is higher than the experimental IC50; a negative value indicates that the estimated IC50 is lower than the experimental IC50 value.
Fit value indicates how well the features in the pharmacophore map with the chemical features present in the compound.
Activity scale:++++, IC50 ≤ 0.1 μM (most active); +++,IC50 0.1 to 1.0 μM (active); ++, IC50 1.0 to 10.0 μM (moderately active); +, IC50 > 10.0 μM (inactive).
Table 2.
Statistical results of the top 10 pharmacophore hypotheses generated by HypoGenalgorithm.
| Hypo. no. | Total cost | Cost difference | RMSD | Correlation | Max. Fit | Features |
|---|---|---|---|---|---|---|
| 1 | 127.392 | 112.58 | 1.30358 | 0.917678 | 7.12926 | HBA, HBA, RA |
| 2 | 127.773 | 112.20 | 1.3012 | 0.91812 | 7.43089 | HBA, HBA, RA |
| 3 | 131.285 | 108.68 | 1.42158 | 0.901054 | 8.13593 | HBA, HBA, HYD, HYD |
| 4 | 131.876 | 108.09 | 1.43586 | 0.898944 | 8.13208 | HBA, HYD, HYD, RA |
| 5 | 132.748 | 107.22 | 1.44849 | 0.897126 | 9.01033 | HBA, HBA, HYD, HYD |
| 6 | 135.327 | 104.64 | 1.50916 | 0.887736 | 7.02354 | HBA, HYD, HYD, RA |
| 7 | 135.394 | 104.57 | 1.49535 | 0.889964 | 6.29567 | HBA, HBA, HYD, RA |
| 8 | 135.4 | 104.57 | 1.51813 | 0.886273 | 5.89767 | HBA, HYD, RA |
| 9 | 136.05 | 103.92 | 1.53266 | 0.883943 | 5.84977 | HBA, HYD, RA |
| 10 | 136.137 | 103.83 | 1.52657 | 0.885001 | 6.78031 | HBA, HYD, RA |
Null cost = 239.969, *Fixed cost = 101.975, *Best record in pass = 5, *configuration cost = 16.6621. (RA- Ring aromatic, HBA-Hydrogen bond acceptor, HYD- Hydrophobic).
Fig. 5.

Pharmacophore mapping of the most active, less active compounds in the training set. (A) Hypo 1 mapped on to the most active compound 4, (B) Hypo 1 mapped on to the least active compound 19.
Fig. 4.

The best HypoGen Pharmacophore model, Hypo1. (A) Chemical features present in Hypo 1 (B) 3D spatial arrangement and the distance constraints between the chemical features. Green colour represents HBA, Brown colour represents Ring aromatic. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.2. Pharmacophore Validation
The best pharmacophore model (Hypo1) has been validated by three distinct methods; a) Cost analysis, b) Test set analysis and c) Fischer's randomization test.
3.2.1. The Cost Analysis Method
The three cost values such as total cost, fixed cost, and null cost were produced by the HypoGen algorithm in DS. The hypo1 model has the cost difference 112.58; correlation coefficient value 0.917678 and the RMSD value 1.30358 bits (Table 2).
3.2.2. Test Set Analysis
The significance of the selected pharmacophore model depends on its ability to predict the biological activity of test set compounds along with the training set molecules. Test set containing 33 structurally diverse molecules with most active, active, moderately active and inactive molecules. To verify the predictability power of pharmacophore model we used the Ligand Pharmacophore Mapping protocol in DS to map the test set molecules. Estimated activities were calculated for individual test set compounds in order to correlate them with their experimental activities by using simple regression analysis.
The obtained correlation coefficient value for test set compounds is 0.874718 and for training set compounds is 0.917678 (Fig. 6). Test set of 33 compounds was mapped properly with the generated pharmacophore. Out of the 33 compounds, the best active compound 6 is nicely mapped on the three essential features. Notably, the very least active molecule 31 did not map with the two essential HBA features, which signifies the robustness of the pharmacophore model (Fig. 7).
Fig. 6.

Correlation graph between experimental and estimated activities in logarithmic scale for training and test set compounds based on Hypo1.
Fig. 7.

Pharmacophore mapping of the most active, less active and inactive compounds in the test set along with their fit score. (A) Hypo 1 mapped on to the most active compound 6 (B) Hypo 1 mapped on to the least active compound 31. The features are colour-coded with Green, hydrogen bond acceptor; Brown Ring aromatic features. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.2.3. Fischer Randomization Test
The experimental activity of training set compounds was scrambled randomly and used in pharmacophore generation with 95% confidence level put forth 19 random spreadsheets, which were compared with the original generated Pharmacophore (Hypo1). We found that none of the random generated pharmacophores scored the good statistical value than Hypo1 [35]. The difference in correlations and costs values between the HypoGen and Fischer randomizations was shown in Fig. 8. The three validation strategies employed exhibited that Hypo1 model has great certainty and can be chosen for further screening of chemical library with diverse structural entity as Top1 poison.
Fig. 8.

(A) & (B): The difference in correlation and total cost values of hypotheses between a Hypo1 spreadsheet and 19 random spreadsheets.
3.3. Database Screening
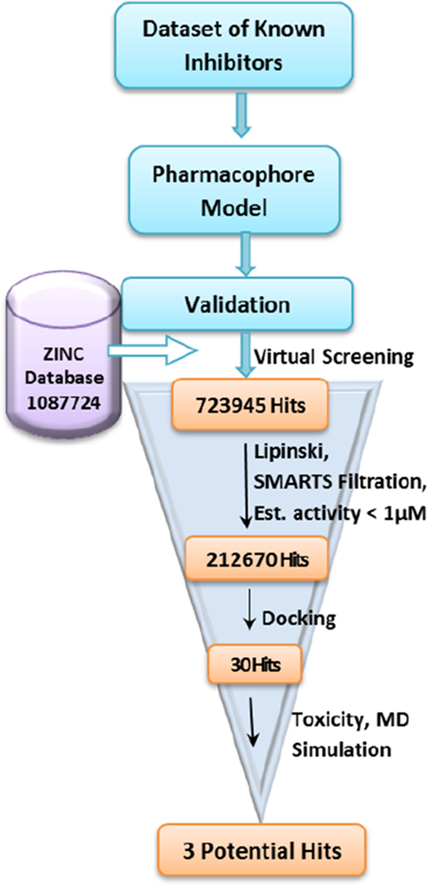
The validated Hypo1 model was utilized as a 3D query for virtual screening of ZINC database with diverse drug-like chemical structure. ZINC database containing 1,087,724 molecules was selected with BEST search option to identify the potential hit molecules. To our satisfaction, 723,945 molecules were nicely mapped on all features present in the Hypo1 model. These molecules were additionally scrutinized using Lipinski's rules of five which provided 705,531 molecules. SMARTS filtration reduced the number of molecules to 702,411. Non-essentials fragments like peroxide, sulfonyl halide, sulfonate ester, cyanide etc. which may lead to toxicity were filtered out by utilizing SMARTS properties filtration. As the filtered molecules had a huge range of activity scale, further screening using Estimated activity <1.0 μM provided 212,670 molecules. These molecules were identified by prioritizing the estimated activity <1.0 μM and subjected to molecular docking analysis using the LibDock algorithm in DS client. Schematic representation of the pharmacophore generation and virtual screening process has been depicted in Fig. 9.
Fig. 9.

Schematic representation of the virtual screening process implemented in the identification of Top1 inhibitors.
3.4. Binding Site Prediction
Based on the co-crystal structure of camptothecin bound to TopI-DNA complex (PDB ID 1T8I), the binding site of the ligand was predicted by Ligplot analysis through PDBsum website (https://www.ebi.ac.uk/pdbsum). Ligplot analysis program revealed that the quinoline nitrogen of CPT formed a conserved hydrogen bond with Arg364. Another hydrogen bonding was observed between the hydroxyl group of lactone and Asp533 (Fig. 10). Atomic co-ordinates of the quinoline nitrogen and lactone hydroxyl groups are obtained from the Ligplot analysis. The docking was performed using Libdock algorithm utilizing the active sites atomic co-ordinates i.e.17.2665, −0.172, and 26.737 from Ligplot analysis.
Fig. 10.

Ligplot analysis result of 1T8I.
3.5. Molecular Docking
The Top1 DNA duplex protein (PDB ID 1T8I) has furnished with three chains, A, B and C where chain A symbolizes the whole protein and chain B and C denote the bound DNA. The filtered drug-like compounds were docked into the active site of the Top1 protein. The active site was defined based on the co-crystallized inhibitor, EHD990 (CPT) (Fig. 11A). The docking was performed using LibDock module implemented in Discovery studio 4.1 Client. Molecules were ranked according to their docking score, planarity, rigidity, hydrogen bond interactions and estimated activity. The binding modes, molecular interactions with the active site, binding energy and docking scores were considered as important components in selecting the best poses of the docked compounds. The standard molecule CPT was also docked on the same active site of the Top1 protein.
Fig. 11.

(A) Binding interaction of the standard molecule campotethecin on 1T8I, where blue dotted line indicates the hydrogen bond and magenta dotted line indicates the π-π hydrophobic interaction. (B) Molecular overlay of experimental binding pose of CPT and predicted binding pose of CPT after docking. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Analysis of the docking results and binding pattern of all filtered molecules put forth 30 molecules which were found to be more active at binding site in DNA Top1 protein. The docking score and docking interaction of CPT were enlisted in the Table 4. The docking score of the camptothecin was 107.235. CPT was able to form 3 hydrogen bonding with Arg364, Asp533, Thr718 and one π-π hydrophobic interaction with DC112 which is shown in Fig. 11A. The molecules that showed better docking score than that of CPT, forming conserved hydrogen bonding with important amino acid residues along with attaining requisite molecular geometry were considered as the potential ‘hit’ topoisomerase I poison. Although many compounds showed proper interaction with the crucial amino acids were not considered for further analysis as they did not fulfill the planarity criteria. Overall 30 compounds were found to satisfy all the selected criteria. Meticulous visual inspection and analysis of the binding poses revealed 6 compounds with better overall docking profile compared to CPT which were listed in Table 5 and Supplementary Table 2. The estimated activity of selected the 6 lead molecules ZINC68997780, ZINC38550809, ZINC38550756, ZINC15018994, ZINC08832860 and ZINC43932053 were 0.065 μM, 0.273 μM, 0.278 μM, 0.37 μM, 0.58 μM and 0.838 μM respectively (Supplementary Table 2). The hit molecule ZINC68997780 (1-(1-(8-hydroxyquinoline-2-yr)piperidine-4-carbonyl)piperidine-4-carboxamide) with the docking score of 140.198 was able to form six hydrogen bond interactions with amino acids Arg364, Arg488, Lys532, Asp533 and nucleotide DC112, DA113 (Fig. 12A). The binding mode of the compound in the active site cavity was mapped on the Hypo1 pharmacophoric features (Fig. 13A). The terminal carbonyl group which mapped as HBA was able to make three hydrogen bonds with Arg488, Lys532 Asp533 and quinoline moiety which was mapped as RA made two hydrogen bonding with DA113 and Arg364 and hydrophobic interactions with DC112. ZINC38550809 (4-(4-ethylbenzo[d]thiazol-2-yl)-N-(3-methoxyphenyl)piperazine-1-carboxamide) with the docking score of 126.907 was able to make three hydrogen bonding with Arg364, Asp533 and Thr718 and one π-π hydrophobic interactions with nucleotide DC112 (Fig. 12B). The binding conformation of ZINC38550809 and pharmacophore overlay was performed where benzthiazole nitrogen is nicely mapped on one HBA and the sulfur atom was mapped as another HBA (Fig. 13B). ZINC38550756 (N-(3-methoxyphenyl)-4-(6-nitrobenzo[d]thiazol-2-yl)piperazine-1-carboxamide) with the docking score of 125.689 was able to make two hydrogen bonding with Arg364, Asp533 and one hydrophobic interaction with nucleotide DC112 (Fig. 12C). The binding pose of ZINC38550756 nicely mapped on the Hypo1 pharmacophore shown in Fig. 13C. ZINC15018994 (2-((5-(benzofuran-2-yl)-4-methyl-4H-1,2,4-triazol-3-yl)thio)-N-((1R,2S)-2-methylcyclohexyl)acetamide) had the docking score of 124.684 was able to make three hydrogen bond interactions with Arg364, Asp533, Thr718 (Fig. 12D). The pharmacophore overlaying study on the binding pose of the compound (Fig. 13D) showed the carbonyl function was mapped with one HBA that could make a hydrogen bond interactions with Thr718 and phenyl ring of benzofuran moiety mapped on RA was able to make a hydrophobic interaction with DC112. ZINC08832860 (N-((2-(3-chloro-4-methylphenyl)benzo[d]oxazol-5-yl)carbamothioyl)-2,3 dihydrobenzo[b][1,4]dioxine-6-carboxamide) with the docking score of 110.721 was able to build three hydrogen bonding with Arg364, Asp533, DG12 (Fig. 12E). The binding pose of ZINC08832860 was well mapped on the developed Hypo1 pharmacophore (Fig. 13E). The dioxane part with two oxygens was well mapped on the two HBA. Benzoxazole part of the molecules overlapped on RA where nitrogen part of that core made hydrogen bonding with Arg364. ZINC43932053 ((S)-2-((1-((4-(benzo[d]oxazol-2-yl)phenyl)amino)vinyl)amino)propan-1-ol) had docking score of 109.541 was able to form four hydrogen bonding with Arg364, Lys532, Asp533 and Thr718 (Fig. 12F). The binding mode of ZINC43932053 at the active site showed that HBA mapped on carbonyl urea linkage and terminal carbonyl group was also mapped on another HBA features (Fig. 13F). The terminal carbonyl group was able to make a hydrogen bond with Arg488 and Lys532 and the carbonyl group of urea linkage formed hydrogen bond interaction with Thr718; whereas phenyl moiety of the benzoxazole group was able to form hydrophobic interaction with DC112.
Fig. 12.

Binding interaction of the hit compounds (A) ZINC68997780, (B) ZINC38550809, (C) ZINC38550756, (D) ZINC15018994, (E) ZINC08832860, (F) ZINC43932053 in the active site of human Top1 protein. Blue dotted line indicates the hydrogen bond and magenta dotted line indicates the π-π hydrophobic interaction. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 13.

Overlay of lead molecules on the pharmacophore Hypo1. (A) ZINC68997780, (B) ZINC38550809, (C) ZINC38550756, (D) ZINC15018994, (E) ZINC08832860, (F) ZINC43932053.
The collective evolutional study of a pharmacophore, virtual screening and molecular docking study successfully provided six prospective molecules which were further evaluated through ADMET and toxicity prediction followed by molecular dynamics simulation studies (Fig. 14).
Fig. 14.

2D structures of identified lead molecules with their docking score.
3.6. ADMET and Toxicity Prediction
Poor pharmacokinetic profile and toxicity complications are often responsible for the dropouts of the lead molecule during preclinical and clinical trials. Thus, from an economic point of view, it would be beneficial if these issues have been traced at early stages. In view of this, application of the in-silico methodology for prediction of the possible toxicity and pharmacokinetic parameters of the hit molecules would be judicious for the identification of lead molecules [26,27]. Keeping this in mind, the six molecules obtained after virtual screening were subjected to various toxicity and ADMET modules. The toxicity results of such were enlisted in Table 6. Toxicity risk assessment results showed that compound ZINC08832860 might have carcinogenic property against male mouse while ZINC15018994, ZINC38550809, ZINC38550756, ZINC43932053 and ZINC68997780 are non-carcinogenic in nature. Interestingly, none of the compounds were found to be carcinogenic against the female mouse. Similarly, NTP carcinogenicity prediction had been carried out on both female and male rats. ZINC08832860 and ZINC38550756 are carcinogenic in nature on the female rat. Ames mutagenicity had been performed against all the six potential hits. ZINC38550756 is mutagenic in nature where the compound ZINC08832860, ZINC15018994, ZINC38550809, ZINC43932053 and ZINC68997780 are non-mutagenic. Any skin irritancy had not been shown by any these potential hits. Except for ZINC15018994 and ZINC38550756, all the hit compounds produced developmental reproductive toxicity. No potential hits were biologically degradable under aerobic condition. Rat oral maximum lethal dose was also calculated for individual hit compounds those are enlisted in the Table 6. The toxicity associated with ZINC08832860 may be due to the chelation property associated with carbamothioyl acetamide features present in the molecules. Whereas, the aromatic nitro group present in ZINC38550756 is responsible for the mutagenic and carcinogenic property. During the drug development, these structural features can be modulated to overcome these toxicity-related issues.
Table 6.
Toxicity Predictions of the lead molecules by TOPKAT.
| Toxicity Parameters | ZINC08832860 | ZINC15018994 | ZINC38550809 | ZINC38550756 | ZINC43932053 | ZINC68997780 |
|---|---|---|---|---|---|---|
| NTP carcinogenicity male Rat | Non-Carcinogen | Non-Carcinogen | Non-Carcinogen | Carcinogen | Non-Carcinogen | Non-Carcinogen |
| NTP carcinogenicity female Rat | Carcinogen | Non-Carcinogen | Non-Carcinogen | Carcinogen | Non-Carcinogen | Non-Carcinogen |
| NTP carcinogenicity Call (Male mouse) | Carcinogen | Non-Carcinogen | Non-Carcinogen | Non-Carcinogen | Non-Carcinogen | Non-Carcinogen |
| NTP carcinogenicity Call (Female mouse) | Non-Carcinogen | Non-Carcinogen | Non-Carcinogen | Non-Carcinogen | Non-Carcinogen | Non-Carcinogen |
| AMES Mutagenicity | Non-Mutagen | Non-Mutagen | Non-Mutagen | Mutagen | Non-Mutagen | Non-Mutagen |
| Developmental Toxicity Potential (DTP) | Toxic | Non-Toxic | Toxic | Non-Toxic | Toxic | Toxic |
| Rat oral LD50(in g/kg) | 0.584905 | 0.553094 | 0.625799 | 0.562206 | 1.08561 | 0.154681 |
| Skin irritation | None | None | None | None | None | None |
| Probability of Bio- degradability | Non-Degradable | Non-Degradable | Non-Degradable | Non-Degradable | Non-Degradable | Non-Degradable |
Based on the toxicity profiling, ZINC68997780, ZINC38550809, and ZINC15018994 were assessed by ADMET studies in Discovery Studio client [24]. The three molecules were further analyzed by molecular dynamics (MD) studies to determine ligand binding stability in the Top1-DNA cleavage complex. The results of the evaluation were summarized in Table 8 and discussed along with their corresponding biplot curve (Fig. 15). Results also revealed that ZINC68997780, ZINC15018994, ZINC38550809 maintain Lipinski's rule of five for possible oral bioavailability (Table 7).
Table 8.
ADMET descriptors of the selected lead candidates.
| Molecule | AbsorptionLevel | Solubility | Solubility level | Hepato toxicity | PSA | ADMET_BBB | CYP2D6 | AlogP98 |
|---|---|---|---|---|---|---|---|---|
| ZINC68997780 | 0 | −5.665 | 2 | 1.04041 | 99.923 | −1.168 | −8.93326 | 1.836 |
| ZINC15018994 | 0 | −3.343 | 3 | 0.192557 | 78.536 | 0.041 | −5.35195 | 4.663 |
| ZINC38550809 | 0 | −5.991 | 2 | −1.54694 | 57.007 | 0.385 | −10.527 | 4.24 |
Fig. 15.

ADMET biplot curve showing the 95% and 99% confidence limit ellipse corresponding to the blood-brain barrier and intestinal absorption model.
Table 7.
Drug likeness properties of selected lead for oral bioavailability by Lipinski's rule of five.
| Molecule | Molecular weight (g/mol) | Log P | H-bond donors | H-bond acceptors | Rule of 5 violations |
|---|---|---|---|---|---|
| ZINC68997780 | 382.464 | 1.75 | 3 | 7 | 0 |
| ZINC15018994 | 384.505 | 4.20 | 1 | 6 | 0 |
| ZINC38550809 | 396.516 | 4.74 | 1 | 6 | 0 |
The biplot curve consisted of two ellipses containing of 95% and 99% confidence levels for blood-brain barrier penetration and human intestinal absorption models (Fig. 15). The polar surface area (PSA) has an important role for human intestinal absorption and membrane permeability [27]. The curve showed that PSA has an inverse relationship with intestinal absorption and membrane crossing. Due to higher PSA, ZINC68997780 and ZINC15018994 have a high tendency towards more intestinal absorption and very low blood-brain barrier penetration in comparison to ZINC38550809. ZINC15018994 have good aqueous solubility whereas ZINC68997780 and ZINC38550809 are less soluble in water. Importantly, all the hit molecules are not inhibitors of cytochrome P450 2D6.
3.7. Molecular Dynamics Simulation
Molecular dynamics is the pivotal theoretical approach which can be utilized to gain molecular insight into the stability of the binding pose of the screened molecules in the active site. To determine the quality of the ligand binding towards the Top1-DNA cleavage complex after docking, the selected molecules ZINC68997780, ZINC38550809 and ZINC15018994 have been further processed by all atoms molecular dynamics simulation by GROMACS 5.1.5 in Intel(R) Xeon(R) CPU E5-2650 v4. The following 3 compounds along with the protein-DNA duplex were further simulated for 10 ns all atoms MD run for the analysis of RMSD, RMSF, radius of gyration and hydrogen bonding analysis. As the co-crystal ligand, camptothecin in PDB structure 1T8I forms H-bond interaction with Arg364, Asp533 and π-π hydrophobic interaction with DC112. MD study was initiated with preferred orientations of the individual ligands as obtained from docking studies. The planner orientation of the selected three ligands on that domain had been taken as an initial structure for the MD studies.
After the binding of the compound on the protein-DNA duplex model, a little fluctuation on the backbone residues had been observed by Root Means Square Fluctuations (RMSF) on protein residues. The RMSF values were collected by least square fitting to a starting structure as a reference frame over 10 ns trajectory run. The RMSF values of the Top1 protein residues had been compared with that when no ligand was bound (Fig. 16). The binding of ZINC15018994 and ZINC38550809 into the active site domain causes more fluctuations with respect to only protein structure fluctuations. Basically, those fluctuations were mainly observed on the loop, 7th to 9th helix and 22nd to 25th helical regions (Fig. 19). It can be concluded that during the binding of the ligand, the main protein backbone conformation does not change and moreover the active binding domain was approximately on 7th to 9th helix region. Most importantly a sharp peak is observed on near residue 650–720. Due to the attachment of the DNA with the protein, the amino acids which were missing were modified. Due to this missing portion, more fluctuations are obtained. However, the fluctuations obtained due to missing portion did not interfere with the ligand binding. This was also proved by protein backbone curve which indicates that without the ligand the fluctuations were similar at that regions. Thus, it can be concluded that the binding of three selected ligands do not change protein conformation.
Fig. 16.

Root mean square fluctuations of protein and selected leads for 10 ns production run.
Fig. 19.

Sequence structural pattern overlapping.
Cα atoms of the protein backbone were fixed by fixing translational and rotational spinning to the corresponding initial structure for molecular dynamics run during the RMSD calculations of the complex protein [36] (Fig. 17). Although the RMSD value of protein was found to be high for ZINC38550809 in comparison to ZINC68997780 and ZINC15018994, but the deviation was not significant with respect to the actual Top1 native protein backbone deviation. The pattern of the Top1 protein and compounds trajectories were pretty similar (Fig. 17) during the entire period of 10 ns MD simulations. Thus, it can be stated that the reference structure of the Top1-DNA complex does not change after binding with the lead compounds. Also, there were no significant conformational changes observed of all three compounds before and after bindings [37] (Fig. 17).
Fig. 17.

Root mean square deviations of protein and selected leads for 10 ns production run.
Apart from MD simulation, hydrogen bond stability was determined for further validation of the compound stability. Hydrogen bond stability has been calculated between all the possible donor and acceptor into the active site domain of Top1-DNA duplex. The geometric criteria for hydrogen bond formation between the acceptor and donor were kept ≤3.5 Å and the angle between them were set to 30o. Fig. 18 depicts the formation of hydrogen bond between the active residues and compounds are properly maintained during the whole scale of production MD run trajectories. Maximum five hydrogen bonds had been observed for ZINC68997780 with Top1 protein-DNA duplex (Fig. 18A). Compound ZINC15018994 also maintained three hydrogen bonds within the active site's domain (Fig. 18C).
Fig. 18.

Hydrogen bond stabilization over 10 ns production run. A) Hydrogen bonding for ZINC68997780. B) Hydrogen bonding for ZINC38550809. C) Hydrogen bonding for ZINC15018994.
Along with the stable hydrogen bond formation, RMSD of these two compounds ZINC68997780 and ZINC15018994 had maintained less RMSD near to 1.5 nm in comparison to the ZINC38550809, the reason for that is the binding of these two compounds to active amino acids was located on the mostly loop region and some of that present on the 17th and 25th helix (Fig. 19).
Thereafter, we evaluated the radius of gyration (Rg) for all three compounds along with the Top1 protein (Fig. 20). The result indicates the harmonious swirl of the protein-ligand complex system throughout the trajectories. All the three compounds were having fewer swirls in comparison to the protein itself. This clearly indicates that all the interactions between active residues and compounds are conserved in their conformational docking poses throughout the whole series of trajectories.
Fig. 20.

Radius of gyration of the protein and compounds over the 10 ns dynamics run.
Molecular dynamics trajectory analysis clearly revealed that binding pose of the individual ligand retained similar to their initial pose throughout the whole 10 ns dynamics trajectories (Fig. 21). It is noteworthy to mention that after dynamics run the binding pockets of the compounds remains same as before. The other residues of the binding pockets are also important for making the binding cavities as well as the non-bonded interaction (Fig. 21A, B, D, E, G and H).
Fig. 21.

MD simulation status of lead compounds ZINC68997780, ZINC38550809 and ZINC15018994 after 10 ns dynamics run. (A) and (B) 2D conformational pose at before and after MD simulation of compound ZINC68997780 in the binding pocket respectively. (C) Molecular overlay of compound ZINC68997780 before (green) and at the end (brown) of the simulation run. (D) and (E) 2D conformational pose before and after MD simulation run of compound ZINC38550809 in the binding pocket respectively. (F) Molecular overlay of compound ZINC38550809 before (green) and at the end (brown) of the simulation run. (G) and (H) 2D conformational pose before and after MD simulation run of compound ZINC15018994 in the binding pocket respectively. (I) Molecular overlay of compound ZINC15018994 before (green) and at the end (brown) of the simulation run. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
4. Conclusion
In conclusion, the study provided development of ligand-based pharmacophore model by 3D-QSAR Pharmacophore Generation protocol in Discovery Studio 4.1 client. For training set, 29 diverse CPT derivatives were considered for the development of the new pharmacophores model. The best quantitative pharmacophore (Hypo1) has been selected from 10 other pharmacophores based on the highest cost difference (112.58) and best correlation coefficient (0.917678), lowest total cost value (127.392). The Hypo1 model consists of two HBA and one RA. The selected pharmacophore has been cross-validated by test set predictions, Fischer randomization test and cost analysis. The test set modules containing 33 various CPT derivatives have been utilized for evaluating the predictive ability of Hypo1 model. The resulting correlation coefficient (R2) between the estimated activity and experimental activity for the 33 test set compounds was observed as 0.874718 and for 29 training set compounds, R2 value was 0.917678. The validated Hypo1 model was used as a 3D query for the virtual screening of 1,087,724 drug-like molecules from ZINC database. By applying various constraint number of molecules funnel down to 212,670 hits, which were docked on the active sites of Topo1 (PDB ID: 1T8I) by LibDock protocol on DS. Depending on the molecular interaction, molecular planarity and structural rigidity the molecules were selected. Finally, six molecules were selected by extensive molecular docking analysis through meticulous visual inspection on the receptor protein (PDB ID: 1T8I). ZINC68997780 (140.198), ZINC38550809 (126.907), ZINC38550756 (125.689), ZINC15018994 (124.684), ZINC08832860 (110.721), ZINC43932053 (109.541) are the selected ZINC compounds along with their docking score and their interaction with the active site residues were compared with the standard CPT ligand (Fig. 10). The six molecules were subjected to toxicity assessment studies under TOPKAT program. Based on the toxicity results ZINC68997780, ZINC15018994 and ZINC38550809 were selected for further ADMET studies and MD simulation. Based on the RMSD and RMSF curve of MD analysis, these selected compounds were found to be stable and the protein-ligands conformation remains unchanged (Fig. 16, Fig. 17, Fig. 21). The stability of the binding mode was validated by determining hydrogen bond stability. Hydrogen bonding distribution over 10 ns run also concluded the stability of rigid bindings. Based on our findings, the three hit molecules ZINC68997780, ZINC15018994 and ZINC38550809 can be utilized for designing future class of potential Top1 inhibitor.
The following are the supplementary data related to this article.
Video of MD simulations of compound ZINC68997780
Video of MD simulations of compound ZINC38550809
Video of MD simulations of compound ZINC15018994
Supplementary material
Conflict of Interest
Authors declare no conflict of interest.
Acknowledgment
S.P. would like to acknowledge ICMR for fellowship. V·K, M.P.R, N·P would like to acknowledge NIPER Kolkata. B·K would like to acknowledge UGC for fellowship. D.B would like to acknowledge DST-INSPIRE for fellowship. A.T for internal CSIR-IICB fund.
References
- 1.Postow L., Crisona N.J., Peter B.J., Hardy C.D., Cozzarelli N.R. vol. 98. 2001. Topological challenges to DNA replication: conformations at the fork. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kizisar D., Subasi N.T., Eroksuz S., Demir A.S., Mert O. Investigation of the stabilization of camptothecin anticancer drug via PSA-PEG polymeric particles. Anadolu Univ J Sci Technol Appl Sci Eng. 2016;17 221–31. [Google Scholar]
- 3.Tsao Y.-P., Wu H.-Y., Liu L.F. Transcription-driven supercoiling of DNA: direct biochemical evidence from in vitro studies. Cell. 1989;56 doi: 10.1016/0092-8674(89)90989-6. 111–8. [DOI] [PubMed] [Google Scholar]
- 4.Yang S.-Y. vol. 15. 2010. Pharmacophore modeling and applications in drug discovery: challenges and recent advances. [DOI] [PubMed] [Google Scholar]
- 5.Bomgaars L., Berg S.L., Blaney S.M. vol. 6. 2001. The development of camptothecin analogs in childhood cancers. [Review] [90 refs] [DOI] [PubMed] [Google Scholar]
- 6.Keszthelyi A., Minchell N.E., Baxter J. vol. 7. 2016. The causes and consequences of topological stress during DNA replication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drwal M.N., Agama K., Wakelin L.P.G., Pommier Y., Griffith R. Exploring DNA topoisomerase I ligand space in search of novel anticancer agents. PLoS One. 2011;6:1–12. doi: 10.1371/journal.pone.0025150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dev S., Dhaneshwar S., Mathew B. Discovery of Camptothecin based topoisomerase I inhibitors: identification using an atom based 3D-QSAR, pharmacophore modeling, virtual screening and molecular docking approach. Comb Chem High Throughput Screen. 2016;19:752–763. doi: 10.2174/1386207319666160810154346. [DOI] [PubMed] [Google Scholar]
- 9.John S., Thangapandian S., Arooj M., Hong J.C., Kim K.D., Lee K.W. Development, evaluation and application of 3D QSAR Pharmacophore model in the discovery of potential human renin inhibitors. BMC Bioinforma. 2011;12(Suppl. 1) doi: 10.1186/1471-2105-12-S14-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luo Y., Yu S., Tong L., Huang Q., Lu W., Chen Y. Synthesis and biological evaluation of new homocamptothecin analogs. Eur J Med Chem. 2012;54:281–286. doi: 10.1016/j.ejmech.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 11.Wang J., Champoux J., Ia T., Gellert M., An P. The P. DNA topoisomerases 5.1. Reactions. 1971:125–144. [Google Scholar]
- 12.Niu M.M., Qin J.Y., Tian C.P., Yan X.F., Dong F.G., Cheng Z.Q. Tubulin inhibitors: pharmacophore modeling, virtual screening and molecular docking. Acta Pharmacol Sin. 2014;35:967–979. doi: 10.1038/aps.2014.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.John S., Thangapandian S., Sakkiah S., Lee K.W. BACE (3) Potent bace-1 inhibitor design using pharmacophore modeling, in silico screening and molecular docking studies. BMC Bioinforma. 2011;12:1–11. doi: 10.1186/1471-2105-12-S1-S28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ran X., Zhang G., Li S., Wang J. Characterization and antitumor activity of camptothecin from endophytic fungus Fusarium solani isolated from Camptotheca acuminate. Afr Health Sci. 2017;17:566–574. doi: 10.4314/ahs.v17i2.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schuster D., Laggner C., Steindl T.M., Palusczak A., Hartmann R.W., Langer T. Pharmacophore modeling and in silico screening for new P450 19 (aromatase) inhibitors. J Chem Inf Model. 2006;46:1301–1311. doi: 10.1021/ci050237k. [DOI] [PubMed] [Google Scholar]
- 16.Willey C.D., Bonner J.A. 2012. Interaction of chemotherapy and radiation; pp. 65–82. Gunderson LL, Tepper JEBT-CRO (Third E, editors. Clin. Radiat. Oncol. 3rd Editio, Philadelphia: W.B. Saunders) [Google Scholar]
- 17.Pommier Y., Leo E., Zhang H., Marchand C. vol. 17. Elsevier Ltd; 2010. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao D., Wang H., Lian Z., Han D., Jin X. Pharmacophore modeling and virtual screening for the discovery of new fatty acid amide hydrolase inhibitors. Acta Pharm Sin B. 2011;1:27–35. [Google Scholar]
- 19.Shahin R., Swellmeen L., Shaheen O., Aboalhaija N., Habash M. Identification of novel inhibitors for Pim-1 kinase using pharmacophore modeling based on a novel method for selecting pharmacophore generation subsets. J Comput Aided Mol Des. 2016;30:39–68. doi: 10.1007/s10822-015-9887-7. [DOI] [PubMed] [Google Scholar]
- 20.Dube D., Periwal V., Kumar M., Sharma S., Singh T.P., Kaur P. 3D-QSAR based pharmacophore modeling and virtual screening for identification of novel pteridine reductase inhibitors. J Mol Model. 2012;18:1701–1711. doi: 10.1007/s00894-011-1187-0. [DOI] [PubMed] [Google Scholar]
- 21.Fei J., Zhou L., Liu T., Tang X.Y. Pharmacophore modeling, virtual screening, and mo-lecular docking studies for discovery of novel Akt2 inhibitors. Int J Med Sci. 2013;10:265–275. doi: 10.7150/ijms.5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nogara P.A., Saraiva R.D.A., Caeran Bueno D., Lissner L.J., Lenz Dalla Corte C., Braga M.M. Virtual screening of acetylcholinesterase inhibitors using the lipinski's rule of five and ZINC databank. Biomed Res Int. 2015:2015. doi: 10.1155/2015/870389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Staker B.L., Feese M.D., Cushman M., Pommier Y., Zembower D., Stewart L. Structures of three classes of anticancer agents bound to the human topoisomerase I-DNA covalent complex. J Med Chem. 2005;48:2336–2345. doi: 10.1021/jm049146p. [DOI] [PubMed] [Google Scholar]
- 24.Singh J., Kumar M., Mansuri R., Sahoo G., Deep A. Inhibitor designing, virtual screening, and docking studies for methyltransferase: a potential target against dengue virus. J Pharm Bioallied Sci. 2016;8:188. doi: 10.4103/0975-7406.171682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Madhavi Sastry G., Adzhigirey M., Day T., Annabhimoju R., Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 26.Ponnan P., Gupta S., Chopra M., Tandon R., Baghel A.S., Gupta G. 2D-QSAR, docking studies, and in Silico ADMET prediction of Polyphenolic acetates as substrates for protein Acetyltransferase function of glutamine synthetase of mycobacterium tuberculosis. ISRN Struct Biol. 2013;2013:1–12. [Google Scholar]
- 27.Gaur R., Cheema H.S., Kumar Y., Singh S.P., Yadav D.K., Darokar M.P. vol. 5. 2015. In vitro antimalarial activity and molecular modeling studies of novel artemisinin derivatives. [Google Scholar]
- 28.Pradeepkiran J.A., Kumar K.K., Kumar Y.N., Bhaskar M. Modeling, molecular dynamics, and docking assessment of transcription factor rho: a potential drug target in brucella melitensis 16M. Drug Des Devel Ther. 2015;9:1897–1912. doi: 10.2147/DDDT.S77020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Musyoka T.M., Kanzi A.M., Lobb K.A., Tastan Bishop Ö. Structure based docking and molecular dynamic studies of plasmodial cysteine proteases against a south African natural compound and its analogs. Sci Rep. 2016;6:1–12. doi: 10.1038/srep23690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Project H. 2005. Pecifications TATES; pp. 8–9. T 2 – t s. [Google Scholar]
- 31.Nath O., Singh A., Singh I.K. In-silico drug discovery approach targeting receptor tyrosine kinase-like orphan receptor 1 for cancer treatment. Sci Rep. 2017;7:1–10. doi: 10.1038/s41598-017-01254-w. /631/114/2248 /631/114/2248/631/114/2248/631/535/1267/631/535/1267/119/118 article. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hess B., Bekker H., Berendsen H.J.C., Fraaije J.G.E.M. LINCS.pdf. J Comput Chem. 1997;18:1463–1472. [Google Scholar]
- 33.Petersen H.G. Accuracy and efficiency of the particle mesh Ewald method. J Chem Phys. 1995;103:3668–3679. [Google Scholar]
- 34.Kandakatla N., Ramakrishnan G. Ligand based pharmacophore modeling and virtual screening studies to design novel HDAC2 inhibitors. Adv Bioinforma. 2014;2014 doi: 10.1155/2014/812148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rohrer S.G., Baumann K. Impact of benchmark data set topology on the validation of virtual screening methods: exploration and quantification by spatial statistics. J Chem Inf Model. 2008;48:704–718. doi: 10.1021/ci700099u. [DOI] [PubMed] [Google Scholar]
- 36.Debnath U., Verma S., Singh P., Rawat K., Gupta S.K., Tripathi R.K. Synthesis, biological evaluation and molecular modeling studies of new 2,3-diheteroaryl thiazolidin-4-ones as NNRTIs. Chem Biol Drug Des. 2015;86:1285–1291. doi: 10.1111/cbdd.12591. [DOI] [PubMed] [Google Scholar]
- 37.Sharma N., Suresh S., Debnath A., Jha S. Trigonella seed extract ameliorates inflammation via regulation of the inflammasome adaptor protein, ASC. Front Biosci Elit. 2017;9:246–257. doi: 10.2741/E799. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video of MD simulations of compound ZINC68997780
Video of MD simulations of compound ZINC38550809
Video of MD simulations of compound ZINC15018994
Supplementary material