

Summary
It remains unknown when, where, and how the site of abscission is generated during cytokinesis. Here, we show that the sites of constriction, i.e., the sites of future abscission, are initially formed at the ends of the intercellular bridge during early midbody stage, and that these sites are associated with the non-muscle myosin-IIB (not myosin-IIA), actin filaments, and septin 9 until abscission. The ESCRT-III component CHMP4B localizes to the midbody and “spreads” to the site of abscission only during late midbody stage. Strikingly, inhibition of myosin-II motor activity by a low dose of Blebbistatin completely abolishes the formation of the constriction sites, resulting in the localization of all the above-mentioned components to the midbody region. These data strongly suggest that a secondary actomyosin ring provides the primary driving force for the thinning of the intercellular bridge to allow ESCRT-mediated membrane fission.
Subject Areas: Cell Biology, Functional Aspects of Cell Biology, Molecular Biology
Graphical Abstract
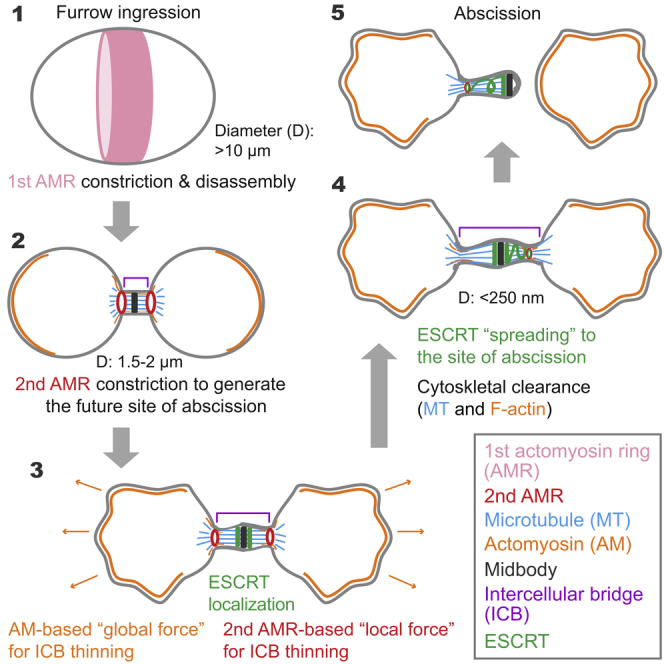
Highlights
-
•
Myosin-II motor activity is required for the generation of an abscission site
-
•
Myosin-II, F-actin, and septin 9 are associated with the site of abscission
-
•
ESCRT-III cannot generate an abscission site independently of myosin-II motor activity
-
•
Different myosin-II isoforms display distinct localization patterns during abscission
Cell Biology; Functional Aspects of Cell Biology; Molecular Biology
Introduction
Mammalian cytokinesis occurs via two major steps, ingression of the cleavage furrow followed by abscission (Bhutta et al., 2014, Mierzwa and Gerlich, 2014, D'avino et al., 2015). Ingression is driven by contraction of a cortical actomyosin ring (AMR) that disassembles at the midbody stage, whereas abscission, i.e., the final cut of a narrow intercellular bridge (ICB) between sibling cells, requires endosomal sorting complex required for transport-III (ESCRT-III)-mediated membrane fission (Hurley and Hanson, 2010, Guizetti et al., 2011, Elia et al., 2011, Agromayor and Martin-Serrano, 2013). Abscission also requires generation and release of tension as well as cytoskeletal clearance at the ICB (Burton and Taylor, 1997, Yang et al., 2008, Lafaurie-Janvore et al., 2013, Fremont et al., 2017a, Fremont et al., 2017b). However, it is unclear how each mechanism operates at the molecular level and how these distinct mechanisms are spatiotemporally coordinated to enable abscission. More importantly, it remains unknown when, where, and how the site of abscission (SOA) is formed during cytokinesis and what drives the thinning of the ICB from its initial size of ∼1.5–2.0 μm (midbody diameter) (Mullins and Biesele, 1973, Mullins and Biesele, 1977) to ∼100–300 nm to allow ESCRT filament assembly and function in abscission (Henne et al., 2012, Agromayor and Martin-Serrano, 2013, Chiaruttini et al., 2015, Alonso et al., 2016). In this study, we found that the motor activity of non-muscle myosin-II (NM-II) is required for the generation of the site of constriction (SOC), which becomes the future SOA, and that different isoforms of NM-II play distinct roles in this process. In addition, our study suggests that NM-II motor activity may spatiotemporally coordinate the various mechanisms involved in abscission.
Results
NM-II Isoforms Display Distinct Localization Patterns and Dynamics during Cytokinesis
We began the study by asking the key question whether different isoforms of NM-II carry out distinct roles in mammalian cytokinesis. We chose to address the question in HeLa-Kyoto cells because the expression of all three isoforms (IIA, IIB, and IIC) has been quantitatively assessed by RNA sequencing (IIA, 100.00%; IIB, 3.78%; IIC, 0.02%) and mass spectrometry (IIA, 100.00%; IIB, 6.20%; IIC, 0.74%) (Maliga et al., 2013). Thus IIA is the major isoform expressed in these cells, approximately 16- and 135-fold higher than IIB and IIC, respectively.
To determine whether the HeLa-Kyoto cells are appropriate for our functional analysis, we first assessed the overall requirement of NM-II activity for cytokinesis in these cells. When treated with 25 μM Blebbistatin, a specific inhibitor for NM-II (Straight et al., 2003, Limouze et al., 2004, Kovacs et al., 2004), all dividing cells displayed the stereotypical behavior of furrow initiation, followed by regression, leading to the formation of binucleated cells (Figure 1A). Thus NM-II activity is essential for furrow ingression in HeLa-Kyoto cells.
Figure 1.

Distinct Localization Patterns and Dynamics of NM-IIA, NM-IIB, and NM-IIC during Cytokinesis
(A) The motor activity of NM-II is essential for furrow ingression. Cells were treated with 25 μM Blebbistatin. Scale bar, 5 μm.
(B) Localization pattern of NM-IIA during cytokinesis. See also Video S1, left. Scale bar, 5 μm.
(C) Localization pattern of NM-IIB during cytokinesis. See also Video S1, right. Scale bar, 5 μm. See also Figure S1A for the localization pattern of NM-IIC0 during cytokinesis. Scale bar, 5 μm. See also Video S2, left.
(D and E) FRAP analysis of NM-IIA and NM-IIB during cytokinesis. See also Figures S2A and S2B and Video S3. Data for fluorescence recovery at the bleached region of all cells examined for each strain are presented as mean ± SD (D) or as individual curves (E). See also Figures S1B, S1C, and S2C, and Video S2 (right) for FRAP analysis of NM-IIC0 during cytokinesis.
We next directly compared the localization patterns and dynamics of the isoforms using cells stably transformed with the chromosome marker H2B-mCherry and GFP-tagged IIA (human), IIB (human), or IIC0 (mouse). NM-II isoforms were expressed from the same promoter on the same plasmid vector. All three isoforms (IIA–IIC) began to enrich at the cell equator after the onset of anaphase (Figures 1B, 1C, and S1A). The accumulation of IIA at the division site was strong during the early stage of furrowing, but gradually decreased toward the midbody stage (29 of 29 cells) (Figure 1B). In some cells, IIA was slightly enriched near the ends of the ICB (Video S1, left). In contrast, IIB localized at the division site throughout cytokinesis, including a prominent localization at the ICB during midbody stage (17 of 17) (Figure 1C; Video S1, right). The localization of IIC was similar to that of IIB throughout cytokinesis (13 of 13) (Figure S1A; Video S2, left).
The turnover rates of all three isoforms during cytokinesis were determined by fluorescence recovery after photobleaching (FRAP). Significant differences among the isoforms were observed. A region of GFP-IIA at the division site was photobleached, and recovery at that region was followed over time (Figure S2A). Consistent with a previous report (Kondo et al., 2011), IIA was very dynamic during cytokinesis (Figures 1D and 1E; Video S3, left). In contrast, IIB was much less dynamic than IIA (Figures 1D, 1E, and S2B; Video S3, right). Furthermore, IIB appeared to display more pronounced cell-to-cell variation (Figures 1D and 1E), with most cells exhibiting a slower turnover rate (5 of 8) and the rest resembling IIA (Figure 1E). The dynamics of IIC during cytokinesis was similar to that of IIB, although the turnover rate was further reduced (Figures S1B, S1C, and S2C; Video S2, right). The larger range of IIB behavior could be explained by the following considerations: IIA and IIB are able to self-assemble and co-assemble into homotypic and heterotypic filaments in stress fibers as well as in the contractile ring (Beach et al., 2014). Because IIA is the dominant isoform, in cells expressing a lower level of GFP-IIB, most of the protein could be in the form of IIA-IIB heterotypic filaments that might exhibit similar dynamics as IIA. The differential turnover rates of IIA and IIB observed during cytokinesis are similar to those reported during cell migration (Sandquist and Means, 2008), suggesting that this is an isoform-specific property.
Collectively, these data indicate that different NM-II isoforms display distinct localization patterns and dynamics during cytokinesis.
A Tail Fragment of NM-IIA Causes a Defect at the Terminal Stage of Cytokinesis
To determine the roles of different NM-II isoforms in cytokinesis, an RNA interference approach was used to knock down the levels of IIA and IIB. IIC was not included in this study due to the lack of reliable detection of its low-level expression by western blot. Small interfering RNA (siRNA) specific to IIA or IIB was transfected individually or in combination. Under this condition, fluorescently labeled control RNA was transfected at ∼100% efficiency. Western blot showed that IIA and IIB were reduced by more than 80% and 60%, respectively (Figure S3). Despite the significant knockdown in both IIA and IIB levels, there was no obvious increase in the number of binucleated cells at the population level. Few cells appeared to show a defect in abscission. These data suggest that the residual activity of IIA and IIB, perhaps coupled with the extremely low level of IIC, is largely sufficient to drive furrow ingression and enable abscission in HeLa-Kyoto cells.
As an alternative approach to dissect the roles of different isoforms in cytokinesis, we designed expression constructs with the potential to act in a dominant-negative manner. A short tail fragment containing the “assembly competent domain” (ACD) of an NM-II isoform is known to interact with and compete for bipolar filament assembly of the endogenous protein. Such a tail fragment for IIA or for the budding yeast myosin-II heavy chain has been shown to localize to the division site in a manner that depends on the presence of the endogenous protein (Beach and Egelhoff, 2009, Fang et al., 2010). We tried this approach on IIB first, as it displayed a prominent localization at the division site during the midbody stage, which suggests a possible role in abscission. Previous work showed that a tail fragment containing the ACD of IIB (residues 1672–1976) could inhibit enucleation of human erythroblasts (Ubukawa et al., 2012). Thus, we fused GFP to the N terminus of a 348-amino-acid fragment (residues 1629–1976) of the C-terminal tail of IIB (GFP-IIB-348-tail) and expressed this after transfection into HeLa-Kyoto cells from the cytomegalovirus (CMV) promoter (Figure 2A). In interphase cells, GFP-IIB-348-tail formed bright punctae in the cytoplasm, and during mitosis and cytokinesis, it localized weakly to the division site (Figure S4; Video S4). This weak localization suggests that the “furrow-targeting signal,” which is defined as the ability of a fragment to target to the division site in the absence of the endogenous or full-length protein, is missing in the IIB ACD construct. This is a reasonable possibility, as IIA is known to contain a furrow-targeting signal and the ACD near the middle and C terminus of its tail, respectively (Beach and Egelhoff, 2009). A similar arrangement of the furrow-targeting signals and a putative ACD occurs in the tail of NM-II in budding yeast (Fang et al., 2010). The weak localization of the GFP-IIB-348-tail is also consistent with the low-level expression of endogenous IIB (Nagaraj et al., 2011, Maliga et al., 2013) and suggests that that the IIB tail fragment interacts only with the full-length IIB, and not IIA, at the division site. No apparent defect in cytokinesis was observed for the GFP-positive cells, either at the population level by counting binucleated cells or at the individual cell level by time-lapse microscopy.
Figure 2.

A Tail Fragment of NM-IIA Causes a Defect at the Terminal Stage of Cytokinesis
(A) Diagram of the GFP-tagged IIA and IIB tail fragments used in this study.
(B) Cells expressing the GFP-IIA-349-tail fragment are defective in abscission (top) in contrast to the control cells not expressing the fragment (bottom). Cells were imaged 24–48 h after transfection. Scale bar, 10 μm.
(C) Quantification of binucleated cells caused by GFP-IIA-349-tail. Imaging data acquired in (B) were used for the count.
We then constructed a plasmid expressing GFP-IIA-349-tail (residues 1612–1960) (Figure 2A). In interphase cells, GFP-IIA-349-tail associated with stress fibers, and during mitosis and cytokinesis, it localized to the cell cortex and cleavage furrow (Figure 2B). All GFP-positive cells (n = 50) underwent normal furrow ingression, but, strikingly, 62.5% paused at the midbody stage. This pause was accompanied by the enrichment of the tail fragment at the division site for up to 2 h, which was followed by furrow regression to form binucleated cells (Figure 2C). Some cells paused at the midbody stage for 6 h or more before they underwent furrow regression. This phenotype could in principle be due to dominant negative effects on endogenous IIA. However, the phenotype could also be caused by combined disruptions of all three isoforms, because a similar tail fragment of IIA (residues 1633–1960) was previously shown to interact with the full-length IIA or IIB at the division site (Beach and Egelhoff, 2009) and it is known that full-length IIA can co-assemble with full-length IIB or IIC into heterotypic filaments (Beach et al., 2014).
Taken together, these data indicate that the major isoform IIA, possibly in combination with the minor isoforms IIB and IIC, play an essential role at the terminal stage of cytokinesis in HeLa-Kyoto cells.
Inhibition of NM-II Motor Activity by a Low Dose of Blebbistatin Causes Delayed Removal of Actin and Myosin Filaments from the Division Site at the Midbody Stage
Treatment of cells with 25 μM Blebbistatin completely blocked cytokinesis at the furrowing stage (Figure 1A), whereas transfection of cells with the IIA tail fragment caused a defect at the terminal stage of cytokinesis (Figure 2). These observations raised the possibility that an incomplete inactivation of NM-II might cause a defect at the midbody stage before abscission. To test this possibility, we treated HeLa-Kyoto cells with different concentrations of Blebbistatin and found by time-lapse analysis that in the presence of 7.5 μM Blebbistatin, cells underwent furrow ingression that was often followed by regression (Figure 3A). Furrowing had reached the midbody stage, as indicated by the cell-cycle-controlled localization of GFP-CEP55 at the division site in all drug-treated cells (n = 16) (Figure 3B) (Bastos and Barr, 2010). These data indicate that NM-II motor activity plays a role at the midbody stage during cytokinesis.
Figure 3.

Inhibition of NM-II Motor Activity by a Low Dose of Blebbistatin Causes Delayed Removal of Actin and Myosin Filaments from the Division Site at the Midbody Stage
(A) Treatment of cells by 7.5 μM Blebbistatin results in furrow ingression, followed by furrow regression. Cells were treated with DMSO or 7.5 μM Blebbistatin and observed by time-lapse microscopy. Scale bar, 10 μm.
(B) CEP55 localizes to the midbody region in cells treated with 7.5 μM Blebbistatin. Cells transfected with the plasmid carrying GFP-CEP55 and treated with 7.5 μM Blebbistatin were analyzed by time-lapse microscopy. Scale bar, 10 μm.
(C) Quantification of binucleated cells caused by 7.5 μM Blebbistatin. Cells in the presence of DMSO or 7.5 μM Blebbistatin were imaged by time-lapse microscopy and then scored for the mono- versus binucleated phenotype.
(D) IIA is enriched and prolonged at the ICB in cells treated with 7.5 μM Blebbistatin. Cells transfected with the plasmid carrying GFP-IIA in the presence of DMSO (left) or 7.5 μM Blebbistatin (right) were analyzed by time-lapse microscopy. Scale bar, 10 μm.
(E) The duration of IIB at the ICB is prolonged in cells treated with 7.5 μM Blebbistatin. Cells transfected with the plasmid carrying GFP-IIB in the presence of DMSO (left) or 7.5 μM Blebbistatin (right) were analyzed by time-lapse microscopy. Scale bar, 10 μm.
(F) Actin filaments are enriched and prolonged at the ICB in cells treated with 7.5 μM Blebbistatin. Cells infected with Adenovirus carrying GFP-LifeAct in the presence of DMSO (left) or 7.5 μM Blebbistatin (right) were analyzed by time-lapse microscopy. Scale bar, 10 μm.
See also Video S5.
To determine whether the Blebbistatin treatment affects the localization of actin and myosin filaments at the division site, we first monitored the NM-II behavior in cells transfected with GFP-tagged full-length IIA or IIB in the presence of 7.5 μM Blebbistatin or DMSO (a final concentration of 0.44%) by time-lapse analysis. For the non-transfected cells treated with Blebbistatin, 79% (n = 62) became binucleated 2 h after furrow ingression (Figure 3C). This number is likely an underestimate, as some cells could undergo regression 6 h or more after the midbody stage. In contrast, only 3.3% (n = 61) of the non-transfected cells treated with DMSO became binucleated within the same imaging period (Figure 3C). Strikingly, Blebbistatin treatment prolonged the localization of IIA and IIB at the ICB. As expected, IIA localized to the cleavage furrow, but disappeared at or near the midbody stage in DMSO-treated cells (Figure 3D, left). However, in the presence of 7.5 μM Blebbistatin, IIA was clearly enriched at the ICB with a duration of 81 ± 24 min (n = 13) (Figure 3D, right). IIB localized to the cleavage furrow and the ICB in DMSO-treated cells, with a duration at the ICB of 56 ± 21 min (n = 14) (Figure 3E, left). Blebbistatin treatment prolonged its duration at the ICB to 80 ± 17 min (n = 13) (Figure 3E, right). Similarly, F-actin, as revealed by GFP-tagged LifeAct, was enriched and prolonged at the ICB (Figure 3F; Video S5). Importantly, regression was always preceded by the removal of actin and myosin filaments from the ICB (Figures 3D–3F). These data indicate that NM-II motor activity is required for the efficient removal of actin and myosin filaments from the ICB, and that these filaments are likely involved in the attachment of the midbody or its associated microtubule (MT) arrays to the plasma membrane.
NM-II Motor Activity Is Required for Midbody Maturation and the Generation of a Site of Constriction
To further explore the mechanisms for the role of NM-IIs at the terminal stage of cytokinesis, we synchronized HeLa-Kyoto cells using the sequential treatments of thymidine, nocodazole, and MG132 (see Transparent Methods for details) and then examined the localizations of the endogenous Cep55 (a midbody marker), NM-IIA, NM-IIB, Sept9, and F-actin in 7.5 μM Blebbistatin- or DMSO-treated cells at the early or late midbody stages, as indicated by tubulin staining, respectively. As expected, Cep55 localized to the midbody regardless Blebbistatin treatment or the midbody stage (Figure 4A). However, the diameters of the midbodies were clearly different in Blebbistatin- versus DMSO-treated cells. During the early midbody stage, the diameters for the drug-treated and untreated cells were 2.6 ± 0.9 μm (n = 169) and 1.8 ± 0.3 μm (n = 117), respectively (Figure 4B). During the late midbody stage, the diameters for the drug-treated and untreated cells were 2.2 ± 0.8 μm (n = 111) and 1.5 ± 0.3 μm (n = 111), respectively (Figure 4B). Strikingly, in DMSO-treated cells, an SOC was formed at both sides of the midbody during the early stage, and this site presumably became the SOA during the late stage. The distance between the SOC and the midbody was 1.0 ± 0.4 μm (n = 89) (Figure 4B). The diameter of the midbody displayed a small change from the early (∼1.8 μm) to late stage (∼1.5 μm), whereas the diameter of the SOC was reduced substantially from 1.1 ± 0.3 μm (n = 117) during the early stage to 0.5 ± 0.3 μm (n = 111) during the late stage (Figure 4B). Remarkably, Blebbistatin treatment abolished the generation of the SOC (Figure 4A). Not only did the midbody diameter fail to shrink but also the MT arrays at the ICB failed to condense, as indicated by the diameter of the ICB at the corresponding position of the SOC (2.5 ± 0.8 μm, n = 111; measured at 1.2 μm away from the midbody) (termed illusionary SOC (iSOC) hereafter for convenience and simplicity), which is significantly larger than the diameter of the midbody (Figure 4B). Taken together, these data indicate that NM-II motor activity is required for midbody and ICB maturation as well as for the generation of a SOC.
Figure 4.

NM-II Motor Activity Is Required for Midbody Maturation and the Generation of a Site of Constriction
(A) The sites of constriction are abolished by treatment with 7.5 μM Blebbistatin. HeLa-Kyoto cells were synchronized sequentially with thymidine, nocodazole, and MG132, and then released into fresh medium for 45 min. Two aliquots of these cells were treated with DMSO or 7.5 μM Blebbistatin for 60 min (early midbody stage) or 120 min (late midbody stage), respectively, before being fixed and stained with anti-Cep55 (green) and anti-α-tubulin (red) antibodies. Arrows, sites of constriction; arrowheads, the site of abscission. Scale bar, 5 μm.
(B) Measurements of the diameter of the midbody, the diameter at the site of constriction, as well as the distance between these cellular structures in DMSO- or Blebbistatin-treated cells during the early and late midbody stages. The samples used for this quantification include the cells synchronized at the early and late midbody stages that were double-stained with an anti-α-tubulin (red) antibody, in combination with an anti-Cep55 (green) (see A), anti-NM-IIA (green) (see Figure 5A), anti-NM-IIB (green) (see Figure 5B), phalloidin (green) (see F-actin in Figure 5C), or anti-Sept9 antibody (green) (see Figure 5D). MD, midbody; SOC, site of constriction; iSOC, illusionary SOC.
(C) Addition of Blebbistatin before the start of furrowing or at the end of furrowing causes furrow regression or a delay in abscission, respectively. HeLa cells stably expressing mCherry-H2B and EGFP-α-tubulin were treated with either DMSO or 7.5 μM Blebbistatin at the indicated times and followed by time-lapse microscopy. Maximum projection of EGFP-α-tubulin (12 z-sections with the step size of 0.7 μm) for a representative cell of each category is shown here. Regression was judged based on both the bright-field and the EGFP-α-tubulin images. Arrowhead indicates the site of constriction that becomes the site of abscission.
(D) NM-II motor activity is required for the thinning of the ICB. The same images as described in (C) were used for measuring the diameter at the midpoint of the spindle as well as the diameter at the thinnest part near the midpoint of the MT array at the ICB (presumably the “site of constriction” in DMSO-treated cells). Individual traces for individual cells of indicated categories are presented here. Time point “0” is the time when furrow ingression was completed (based on bright-field images).
(E) The same data from (D) are presented as mean ± SD.
To further determine the role of NM-II motor activity at the terminal stage of cytokinesis, we performed live imaging on HeLa cells stably transfected with EGFP-α-tubulin and mCherry-H2B in the presence of DMSO or 7.5 μM Blebbistatin. In the presence of DMSO, all cells underwent abscission with duration at the midbody stage of 111 ± 14 min (n = 16) (Figure 4C). The initial SOC became the future SOA (Figure 4C, arrowhead). The diameter at the SOC was progressively reduced over time, going from 1.2 ± 0.2 μm to zero (n = 16) (Figures 4D and 4E). In contrast, when the Blebbistatin was added before furrow ingression, majority of the cells (12 of 16 cells) underwent furrow ingression, followed by furrow regression 176 ± 39 min later (Figures 4C–4E). All cells failed to form an obvious SOC (Figure 4C). The rate of furrow ingression, as reflected by the rate of diameter change for the spindle, was slightly reduced when compared with that of DMSO-treated cells, and the diameter of the thinnest part of the ICB (presumably referring to the “midbody” and then “its adjacent region,” based on the data in Figures 4A and 4B) was reduced from its initial size of 2.2 ± 0.5 μm (n = 15) to 1.3 ± 1.0 μm (n = 12) during the longest time span required for abscission in the DMSO-treated cells (Figures 4C–4E). Thus, consistent with data on the fixed cells described above, these results indicate that NM-II motor activity is required for midbody and ICB maturation as well as for the generation of a SOC, which becomes the future SOA.
When Blebbistatin was added after furrow ingression, 6 of 10 cells showed delayed abscission with duration at the midbody stage of 184 ± 20 min (Figures 4C–4E), 1 cell displayed furrow ingression followed by regression, and the remaining 3 did not display clear abscission. As expected, the rate of furrow ingression was not affected (Figures 4D and 4E) and the diameter of the thinnest part of the ICB (presumably referring to the SOC, which should be formed at the time of Blebbistatin addition based on the data in Figures 4A and 4B) was in between the diameter of the SOC in the DMSO-treated cells and the diameter of the midbody in the Blebbistatin (added before furrowing)-treated cells (Figures 4D and 4E). Importantly, the choice of abscission delay versus furrow regression appeared to strictly correlate with the diameter at the iSOC. These data indicate that NM-II motor activity plays a post-furrow role in cytokinesis, i.e., the generation of a SOC.
NM-IIB, Actin Filaments, and Septin 9 Are Associated with the Site of Constriction
To determine how NM-IIs might be involved in the generation of a SOC, we performed localization studies on NM-IIA and NM-IIB during cytokinesis and abscission in synchronized cells using isoform-specific antibodies. Both IIA and IIB localized to the division site during furrow ingression. IIA did not enrich at any part of the ICB from early to late midbody stage in DMSO-treated cells (Figure 5A) (for the early stage, 22 of 22 cells, and for the late stage, 18 of 18). However, Blebbistatin treatment resulted in its enrichment at the midbody throughout the terminal stage of cytokinesis and IIA was apparently present as a single ring surrounding the midbody during the late stage (Figure 5A) (for the early stage 19 of 19, and for the late stage 17 of 17). In contrast, IIB was enriched at both SOCs during early midbody stage (18 of 18) and then localized to the midbody as well as to a SOC or the SOA during late midbody stage (9 of 16) in DMSO-treated cells (Figure 5B). This was more clearly visualized in a montage of the z-section images (Figure S5). IIB could be detected at the SOC with a diameter as small as ∼0.3 μm. In Blebbistatin-treated cells, IIB was enriched over the entire midbody during the early stage (15 of 15) and was apparently present as a double ring sandwiching the midbody during the late stage (19 of 19) (Figure 5B). Thus, consistent with the time-lapse data on the GFP-IIA and GFP-IIB localization (Figures 3D and 3E), the endogenous IIA and IIB display distinct localization patterns during terminal stage of cytokinesis. Most importantly, these data suggest that IIB is in the right place at the right time to drive the formation of the SOCs, which presumably become the future SOAs.
Figure 5.

NM-IIB, Actin Filaments, and Septin 9 Are Associated with the Site of Constriction
(A) IIA is not associated with the site of constriction. Cells were synchronized at the early or late midbody stage, treated with DMSO or 7.5 μM Blebbistatin, and double-stained for IIA (green) and tubulin (red) as described for Cep55 in Figure 4A. Arrows, sites of constriction; arrowheads, the site of abscission. Scale bar, 5 μm.
(B) IIB is associated with the site of constriction. Cells were processed and double-stained for IIB (green) and tubulin (red) as described for IIA in (A). Arrows, sites of constriction; arrowheads, the site of abscission. Scale bar, 5 μm. See also Figure S5.
(C) F-actin is associated with the site of constriction. Cells were processed and double-stained for F-actin (green) and tubulin (red) as described for IIA in (A). Arrows, sites of constriction; arrowheads, the site of abscission. Scale bar, 5 μm. See also Figure S6.
(D) Septin 9 is associated with the site of constriction. Cells were processed and double-stained for Sept9 (green) and tubulin (red) as described for IIA in (A). Arrows, sites of constriction; arrowheads, the site of abscission. Scale bar, 5 μm. See also Figure S7.
The idea of IIB producing a local constrictive force at the shoulder of the midbody predicts the presence of F-actin at the same location. Indeed, this is the case. F-actin formed a ring-like structure surrounding the SOC during both early and late midbody stages (Figures 5C and S6). As expected, Blebbistatin treatment abolished SOC formation. Under this condition, F-actin was enriched over the midbody region (Figure 5C). This, together with the IIB data, suggests that a secondary AMR is formed to generate the SOC for the future abscission.
Septins are known to scaffold the full activation of NM-II by its kinases (MLCK, ROCK, and citron) during later stages of cytokinesis (Joo et al., 2007). Septin 9 (Sept9) is required for abscission (Estey et al., 2010). Septins are also known to recognize micron-scale membrane curvature (Bridges et al., 2016). These observations raise the possibility that septins might scaffold IIB activation to generate the SOC and stabilize the membrane curvature there. To test this possibility, we examined Sept9 localization in the synchronized cells. We found that Sept9 localized to the SOCs (14 of 22) or to both the SOCs and the MT array (8 of 22) during early midbody stage (Figures 5D and S7). Blebbistatin treatment caused Sept9 accumulation surrounding the midbody (Figure 5D). Sept9 localized to both the SOCs and the MT array (16 of 21) during late midbody stage (Figures 5D and S7). Blebbistatin caused various Sept9 localizations, the majority displaying association with the MT array (14 of 17), some displaying MT association and localization at the iSOC (6 out of 17), some displaying midbody and MT localization (3 of 17) (Figure 5D), some with midbody localization only (3 of 17), and some with MT localization only (5 of 17). These data suggest that Sept9 likely acts together with IIB and F-actin to generate the SOC and stabilizes the membrane curvature.
The ESCRT-III Complex Is Incapable of Generating a Site of Constriction in the Absence of NM-II Motor Activity
To determine how NM-II motor activity might affect abscission, we examined the localization of the endogenous CHMP4B in synchronized cells using immunofluorescence. During early midbody stage, CHMP4B did not localize to the ICB regardless of DMSO or Blebbistatin treatment (Figure 6). However, during late midbody stage, CHMP4B clearly localized to the midbody and “spread” to the SOA in the DMSO-treated cells (6 of 14) (Figure 6). This is consistent with a previous report (Elia et al., 2011). In contrast, CHMP4B localized only to the midbody region in all the Blebbistatin-treated cells (n = 24) (Figure 6). This is not surprising as the formation of SOC was abolished under this condition. Importantly, these data suggest that the ESCRT-III complex is incapable of generating a SOC in the absence of NM-II motor activity.
Figure 6.

The ESCRT-III Complex Is Incapable of Generating a Site of Constriction in the Absence of NM-II Motor Activity
Cells were processed and double-stained for CHMP4B (green) and tubulin (red) as described for Sept9 in Figure 5D. Arrows, sites of constriction; arrowheads, the site of abscission. Scale bar, 5 μm.
Discussion
The prevailing view of cytokinesis is that NM-II is required only for early furrow ingression as a part of the contractile ring. Disassembly of the ring at the midbody stage then allows other factors such as the ESCRT-III complex to take over and drive abscission (Hurley and Hanson, 2010, Guizetti et al., 2011, Elia et al., 2011, Agromayor and Martin-Serrano, 2013). However, our work suggests that NM-II motor activity plays a post-furrow role in cytokinesis, which is to generate a SOC for the ultimate abscission. Although all three isoforms likely share essential roles in both furrow ingression and abscission, our localization data suggest that IIA might be more specialized for furrow ingression and IIB and IIC are more directly involved in abscission. This notion is supported by the following observations: (1) both the endogenous IIA and IIB localize to the division site during furrow ingression, but IIA disappears from the division site at the midbody stage, whereas IIB remains associated with the midbody and the SOC (Figure 5B); (2) knockdown of IIC by isoform-specific siRNA in human lung tumor cells A549 results in a significant delay at the midbody stage, although these cells eventually underwent abscission (Jana et al., 2006); and (3) IIA translocates actin filaments much faster than IIB or IIC, and the half maximal inhibitory concentration (IC50) of Blebbistatin for IIA is significantly higher than that for IIB or IIC (Zhang et al., 2017).
How does NM-II act in abscission mechanistically? The initial diameter of the ICB is similar to that of a midbody, which is about 1.8 ± 0.3 μm (Figure 4B). This is consistent with the previous report of 1.5–2.0 μm for the midbody diameter (Mullins and Biesele, 1973, Mullins and Biesele, 1977). However, ESCRT-III components can only assemble into much smaller spiral filaments (∼40–250 nm in diameter) or rings (50–65 nm in diameter) (Henne et al., 2012, Agromayor and Martin-Serrano, 2013, Chiaruttini et al., 2015, Alonso et al., 2016). Thus, to enable ESCRT filament assembly, the diameter of the ICB must be substantially reduced from its initial size. However, the driving force for ICB thinning has remained obscure. Our study suggests that NM-II, together with actin filaments, produces forces to generate a SOC with a diameter as small as ∼200–300 nm that is stabilized by the curvature-sensing septins, and this SOC allows ESCRT complex to assemble into filaments and carry out its function in membrane fission.
There are at least two actomyosin-based forces, a “global force” and a “local force,” that act in concert to drive ICB thinning. The global force produced at the cortex of the dividing daughter cells during late stage of cytokinesis may drive them apart, which leads to the generation of tension across the ICB that contributes to its general thinning. The existence of such a force is supported by the following observations. (1) NM-IIA and NM-IIB localize to the leading edge and stress fibers, respectively, and are required for cell spreading and migration (Betapudi et al., 2006, Sandquist and Means, 2008). (2) Profilin 1, which promotes formin-nucleated actin filament assembly, is required for abscission during chondrocyte cytokinesis in mice (Bottcher et al., 2009). Cells lacking profilin 1 can undergo normal AMR constriction, but cannot produce strong traction force to drive cell spreading and migration during abscission (Bottcher et al., 2009). (3) Finally, tension rises at the ICB before abscission (Burton and Taylor, 1997).
The local force is produced by a secondary AMR, which, together with the septins, generates and stabilizes the SOC, which becomes the future SOA. This local force is responsible for local thinning of the ICB. The existence of such a force is supported by the following observations: (1) GFP-tagged IIB and IIC0 are clearly localized at or near the midbody region before abscission (Figures 1 and S1); (2) endogenous NM-IIB and NM-IIC, but not NM-IIA, were observed to sandwich the midbody (Maupin et al., 1994, Daniels et al., 2004, Jana et al., 2006) (Figure 5B), and, in addition, the endogenous IIB also localizes to the SOCs (Figure 5B); (3) actin filaments are present at the ICB during the early stage of abscission (Murthy and Wadsworth, 2005, Guizetti et al., 2011) (Figure 3F) and form a ring-like structure surrounding the SOC (Figure 5C); and (4) finally, in Drosophila testis, the germline stem cells undergo AMR-driven furrow ingression that is followed by actin ring disassembly while leaving NM-II in place. Shortly thereafter, a secondary AMR-like structure (∼1.2 μm in diameter) is formed to delay abscission. Only after disassembly of this secondary actin structure can abscission occur (Lenhart and Dinardo, 2015). It remains possible that this secondary AMR might also promote abscission before its disassembly. Collectively, the global and local forces act in concert to sculpt the ICB into a membrane tube with the correct size and shape that allows ESCRT filament assembly and function.
NM-II motor activity could also contribute to abscission by promoting efficient removal of actin filaments at the ICB. Cytoskeletal elements such as MTs and actin filaments play pivotal roles in furrow positioning and ingression, but they must be removed from the ICB before abscission, as they would physically block membrane fission. ESCRT-recruited spastin, an MT-severing enzyme, is required for the removal of MTs at the midbody (Yang et al., 2008), whereas cofilin, an actin-filament-severing protein, as well as Rab35 and its effector MICAL1 are required to clear actin filaments from the ICB in Drosophila germ cells and mammalian cells, respectively (Lenhart and Dinardo, 2015, Fremont et al., 2017a, Fremont et al., 2017b). Clearance of actin and myosin filaments would not only remove a physical block but also release tension, which is essential for abscission (Lafaurie-Janvore et al., 2013). NM-II motor activity might facilitate F-actin removal by sliding actin filaments and making them more accessible to the actin clearance machinery. The prolonged retention of myosin and actin filaments at the ICB in 7.5 μM Blebbistatin-treated cells as well as their decreased turnover in Blebbistatin-treated cells, as revealed by FRAP analysis (Murthy and Wadsworth, 2005), support the proposed role of NM-II in abscission.
Finally, the septins or the AMR at the SOC could function as a scaffold, akin to the role of NM-II in cytokinesis in budding yeast (Wloka et al., 2013), for the localization of the ESCRT complex, which, in turn, directs the deposition of the endosome-carried spastin (Yang et al., 2008, Connell et al., 2009) to depolymerize MT at the SOC for the eventual abscission.
In summary, this study suggests that NM-II motor activity may coordinate multiple cellular events including tension generation across the ICB, MT removal, formation of the SOC, F-actin clearance, and tension release to execute the elaborate process of abscission.
Limitations of the Study
In this study, we have clearly shown that Sept9, NM-IIB, and F-actin are associated with the sites of constriction during the early midbody stage and with the SOA during the late midbody stage. In addition, we have clearly demonstrated that treatment of HeLa cells with a low dose of Blebbistatin abolishes the formation of constriction sites. Our study lays the framework for how NM-II initiates the formation of an abscission site. However, detailed mechanisms concerning the relative contributions of different NM-II isoforms to this process as well as how HeLa and other cell types execute abscission in the absence of specific NM-II isoforms (via CRISPR/Cas9-mediated gene knockouts, instead of siRNA depletion, dominant negative NM-II constructs, or inhibition of NM-II motor activity) are not a focus of this study and warrant further investigation in the future.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Michael Lampson and Francis Barr for cell lines; Kerstin Kutsche for plasmids; Ting Wang for help with cell culture; Andrea Stout for assistance in imaging; Stephen DiNardo, Kari Lenhart, Elias Spiliotis, Joseph Marquardt, and Hiroki Okada for discussion and comments on the manuscript; and Hiroki Okada for the illustration of the concept in the Graphical Abstract. This work was supported by NIH grant GM115420 to E.B. and a Ph.D. fellowship from the Boehringer Ingelheim Fonds to C.W.
Author Contributions
K.W and C.W. designed and performed all experiments, did data analysis, and prepared the manuscript. E.B. supervised the project, did data interpretation, and wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: March 29, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.02.010.
Contributor Information
Carsten Wloka, Email: carstenwloka@gmail.com.
Erfei Bi, Email: ebi@pennmedicine.upenn.edu.
Supplemental Information
References
- Agromayor M., Martin-Serrano J. Knowing when to cut and run: mechanisms that control cytokinetic abscission. Trends Cell Biol. 2013;23:433–441. doi: 10.1016/j.tcb.2013.04.006. [DOI] [PubMed] [Google Scholar]
- Alonso Y.A.M., Migliano S.M., Teis D. ESCRT-III and Vps4: a dynamic multipurpose tool for membrane budding and scission. FEBS J. 2016;283:3288–3302. doi: 10.1111/febs.13688. [DOI] [PubMed] [Google Scholar]
- Bastos R.N., Barr F.A. Plk1 negatively regulates Cep55 recruitment to the midbody to ensure orderly abscission. J. Cell Biol. 2010;191:751–760. doi: 10.1083/jcb.201008108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach J.R., Egelhoff T.T. Myosin II recruitment during cytokinesis independent of centralspindlin-mediated phosphorylation. J. Biol. Chem. 2009;284:27377–27383. doi: 10.1074/jbc.M109.028316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach J.R., Shao L., Remmert K., Li D., Betzig E., Hammer J.A., 3rd Nonmuscle myosin II isoforms coassemble in living cells. Curr. Biol. 2014;24:1160–1166. doi: 10.1016/j.cub.2014.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betapudi V., Licate L.S., Egelhoff T.T. Distinct roles of nonmuscle myosin II isoforms in the regulation of MDA-MB-231 breast cancer cell spreading and migration. Cancer Res. 2006;66:4725–4733. doi: 10.1158/0008-5472.CAN-05-4236. [DOI] [PubMed] [Google Scholar]
- Bhutta M.S., Mcinerny C.J., Gould G.W. ESCRT function in cytokinesis: location, dynamics and regulation by mitotic kinases. Int. J. Mol. Sci. 2014;15:21723–21739. doi: 10.3390/ijms151221723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottcher R.T., Wiesner S., Braun A., Wimmer R., Berna A., Elad N., Medalia O., Pfeifer A., Aszodi A., Costell M., Fassler R. Profilin 1 is required for abscission during late cytokinesis of chondrocytes. EMBO J. 2009;28:1157–1169. doi: 10.1038/emboj.2009.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges A.A., Jentzsch M.S., Oakes P.W., Occhipinti P., Gladfelter A.S. Micron-scale plasma membrane curvature is recognized by the septin cytoskeleton. J. Cell Biol. 2016;213:23–32. doi: 10.1083/jcb.201512029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton K., Taylor D.L. Traction forces of cytokinesis measured with optically modified elastic substrata. Nature. 1997;385:450–454. doi: 10.1038/385450a0. [DOI] [PubMed] [Google Scholar]
- Chiaruttini N., Redondo-Morata L., Colom A., Humbert F., Lenz M., Scheuring S., Roux A. Relaxation of loaded ESCRT-III spiral springs drives membrane deformation. Cell. 2015;163:866–879. doi: 10.1016/j.cell.2015.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell J.W., Lindon C., Luzio J.P., Reid E. Spastin couples microtubule severing to membrane traffic in completion of cytokinesis and secretion. Traffic. 2009;10:42–56. doi: 10.1111/j.1600-0854.2008.00847.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'avino P.P., Giansanti M.G., Petronczki M. Cytokinesis in animal cells. Cold Spring Harb. Perspect. Biol. 2015;7:a015834. doi: 10.1101/cshperspect.a015834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels M.J., Wang Y., Lee M., Venkitaraman A.R. Abnormal cytokinesis in cells deficient in the breast cancer susceptibility protein BRCA2. Science. 2004;306:876–879. doi: 10.1126/science.1102574. [DOI] [PubMed] [Google Scholar]
- Elia N., Sougrat R., Spurlin T.A., Hurley J.H., Lippincott-Schwartz J. Dynamics of endosomal sorting complex required for transport (ESCRT) machinery during cytokinesis and its role in abscission. Proc. Natl. Acad. Sci. U S A. 2011;108:4846–4851. doi: 10.1073/pnas.1102714108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estey M.P., Di Ciano-Oliveira C., Froese C.D., Bejide M.T., Trimble W.S. Distinct roles of septins in cytokinesis: SEPT9 mediates midbody abscission. J. Cell Biol. 2010;191:741–749. doi: 10.1083/jcb.201006031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X., Luo J., Nishihama R., Wloka C., Dravis C., Travaglia M., Iwase M., Vallen E.A., Bi E. Biphasic targeting and cleavage furrow ingression directed by the tail of a myosin-II. J. Cell Biol. 2010;191:1333–1350. doi: 10.1083/jcb.201005134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremont S., Hammich H., Bai J., Wioland H., Klinkert K., Rocancourt M., Kikuti C., Stroebel D., Romet-Lemonne G., Pylypenko O. Oxidation of F-actin controls the terminal steps of cytokinesis. Nat. Commun. 2017;8:14528. doi: 10.1038/ncomms14528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremont S., Romet-Lemonne G., Houdusse A., Echard A. Emerging roles of MICAL family proteins - from actin oxidation to membrane trafficking during cytokinesis. J. Cell Sci. 2017;130:1509–1517. doi: 10.1242/jcs.202028. [DOI] [PubMed] [Google Scholar]
- Guizetti J., Schermelleh L., Mantler J., Maar S., Poser I., Leonhardt H., Muller-Reichert T., Gerlich D.W. Cortical constriction during abscission involves helices of ESCRT-III-dependent filaments. Science. 2011;331:1616–1620. doi: 10.1126/science.1201847. [DOI] [PubMed] [Google Scholar]
- Henne W.M., Buchkovich N.J., Zhao Y., Emr S.D. The endosomal sorting complex ESCRT-II mediates the assembly and architecture of ESCRT-III helices. Cell. 2012;151:356–371. doi: 10.1016/j.cell.2012.08.039. [DOI] [PubMed] [Google Scholar]
- Hurley J.H., Hanson P.I. Membrane budding and scission by the ESCRT machinery: it's all in the neck. Nat. Rev. Mol. Cell Biol. 2010;11:556–566. doi: 10.1038/nrm2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana S.S., Kawamoto S., Adelstein R.S. A specific isoform of nonmuscle myosin II-C is required for cytokinesis in a tumor cell line. J. Biol. Chem. 2006;281:24662–24670. doi: 10.1074/jbc.M604606200. [DOI] [PubMed] [Google Scholar]
- Joo E., Surka M.C., Trimble W.S. Mammalian SEPT2 is required for scaffolding nonmuscle myosin II and its kinases. Dev. Cell. 2007;13:677–690. doi: 10.1016/j.devcel.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Kondo T., Hamao K., Kamijo K., Kimura H., Morita M., Takahashi M., Hosoya H. Enhancement of myosin II/actin turnover at the contractile ring induces slower furrowing in dividing HeLa cells. Biochem. J. 2011;435:569–576. doi: 10.1042/BJ20100837. [DOI] [PubMed] [Google Scholar]
- Kovacs M., Toth J., Hetenyi C., Malnasi-Csizmadia A., Sellers J.R. Mechanism of blebbistatin inhibition of myosin II. J. Biol. Chem. 2004;279:35557–35563. doi: 10.1074/jbc.M405319200. [DOI] [PubMed] [Google Scholar]
- Lafaurie-Janvore J., Maiuri P., Wang I., Pinot M., Manneville J.B., Betz T., Balland M., Piel M. ESCRT-III assembly and cytokinetic abscission are induced by tension release in the intercellular bridge. Science. 2013;339:1625–1629. doi: 10.1126/science.1233866. [DOI] [PubMed] [Google Scholar]
- Lenhart K.F., Dinardo S. Somatic cell encystment promotes abscission in germline stem cells following a regulated block in cytokinesis. Dev. Cell. 2015;34:192–205. doi: 10.1016/j.devcel.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limouze J., Straight A.F., Mitchison T., Sellers J.R. Specificity of blebbistatin, an inhibitor of myosin II. J. Muscle Res. Cell Motil. 2004;25:337–341. doi: 10.1007/s10974-004-6060-7. [DOI] [PubMed] [Google Scholar]
- Maliga Z., Junqueira M., Toyoda Y., Ettinger A., Mora-Bermudez F., Klemm R.W., Vasilj A., Guhr E., Ibarlucea-Benitez I., Poser I. A genomic toolkit to investigate kinesin and myosin motor function in cells. Nat. Cell Biol. 2013;15:325–334. doi: 10.1038/ncb2689. [DOI] [PubMed] [Google Scholar]
- Maupin P., Phillips C.L., Adelstein R.S., Pollard T.D. Differential localization of myosin-II isozymes in human cultured cells and blood cells. J. Cell Sci. 1994;107(Pt 11):3077–3090. doi: 10.1242/jcs.107.11.3077. [DOI] [PubMed] [Google Scholar]
- Mierzwa B., Gerlich D.W. Cytokinetic abscission: molecular mechanisms and temporal control. Dev. Cell. 2014;31:525–538. doi: 10.1016/j.devcel.2014.11.006. [DOI] [PubMed] [Google Scholar]
- Mullins J.M., Biesele J.J. Cytokinetic activities in a human cell line: the midbody and the intercellular bridge. Tissue Cell. 1973;5:47–61. doi: 10.1016/s0040-8166(73)80005-9. [DOI] [PubMed] [Google Scholar]
- Mullins J.M., Biesele J.J. Terminal phase of cytokinesis in D-985 cells. J. Cell Biol. 1977;73:672–684. doi: 10.1083/jcb.73.3.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy K., Wadsworth P. Myosin-II-dependent localization and dynamics of F-actin during cytokinesis. Curr. Biol. 2005;15:724–731. doi: 10.1016/j.cub.2005.02.055. [DOI] [PubMed] [Google Scholar]
- Nagaraj N., Wisniewski J.R., Geiger T., Cox J., Kircher M., Kelso J., Paabo S., Mann M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol. 2011;7:548. doi: 10.1038/msb.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandquist J.C., Means A.R. The C-terminal tail region of nonmuscle myosin II directs isoform-specific distribution in migrating cells. Mol. Biol. Cell. 2008;19:5156–5167. doi: 10.1091/mbc.E08-05-0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straight A.F., Cheung A., Limouze J., Chen I., Westwood N.J., Sellers J.R., Mitchison T.J. Dissecting temporal and spatial control of cytokinesis with a myosin II Inhibitor. Science. 2003;299:1743–1747. doi: 10.1126/science.1081412. [DOI] [PubMed] [Google Scholar]
- Ubukawa K., Guo Y.M., Takahashi M., Hirokawa M., Michishita Y., Nara M., Tagawa H., Takahashi N., Komatsuda A., Nunomura W. Enucleation of human erythroblasts involves non-muscle myosin IIB. Blood. 2012;119:1036–1044. doi: 10.1182/blood-2011-06-361907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wloka C., Vallen E.A., Thé L., Fang X., Oh Y., Bi E. Immobile myosin-II plays a scaffolding role during cytokinesis in budding yeast. J. Cell Biol. 2013;200:271–286. doi: 10.1083/jcb.201208030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D., Rismanchi N., Renvoise B., Lippincott-Schwartz J., Blackstone C., Hurley J.H. Structural basis for midbody targeting of spastin by the ESCRT-III protein CHMP1B. Nat. Struct. Mol. Biol. 2008;15:1278–1286. doi: 10.1038/nsmb.1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.M., Ji H.H., Ni T., Ma R.N., Wang A., Li X.D. Characterization of blebbistatin inhibition of smooth muscle myosin and nonmuscle myosin-2. Biochemistry. 2017;56:4235–4243. doi: 10.1021/acs.biochem.7b00311. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.