

Abstract
Approximately 15% of all human tumors harbor mutant KRAS, a membrane-associated small GTPase and notorious oncogene. Mutations that render KRAS constitutively active will lead to uncontrolled cell growth and cancer. However, despite aggressive efforts in recent years, there are no drugs on the market that directly target KRAS and inhibit its aberrant functions. In the current work, we combined structure-based design with a battery of cell and biophysical assays to discover a novel pyrazolopyrimidine-based allosteric KRAS inhibitor that binds to activated KRAS with sub-micromolar affinity and disrupts effector binding, thereby inhibiting KRAS signaling and cancer cell growth. These results show that pyrazolopyrimidine-based compounds may represent a first-in-class allosteric noncovalent inhibitors of KRAS. Moreover, by studying two of its analogues, we identified key chemical features of the compound that interact with a set of specific residues at the switch regions of KRAS and play critical roles for its high-affinity binding and unique mode of action, thus providing a blueprint for future optimization efforts.
Introduction
Somatic mutations in RAS proteins are associated with about 16% of all human cancers.1,2 KRAS is the most frequently mutated RAS isoform, accounting for 85% of all RAS-related cancers.1,2 Cellular KRAS is tethered to the inner surface of the plasma membrane by a farnesylated polybasic lipid anchor3 and cycles between active guanosine triphosphate (GTP)- and inactive guanosine diphosphate (GDP)-bound conformational states.4 GTPase activating proteins (GAPs) facilitate hydrolysis of GTP by KRAS, whereas guanine nucleotide exchange factors (GEFs) catalyze GDP dissociation.4−6 Upon activation by receptor tyrosine kinases such as epidermal growth factor receptors, GEFs are recruited to KRAS and initiate exchange of GDP for GTP. Active KRAS interacts with effectors such as Raf in the MAPK pathway and PI3K in the AKT pathway,7 driving cell growth and proliferation.8,9 In a regulated RAS cycle, signaling is turned off upon GTP hydrolysis. Oncogenic mutations that impair its GAP-mediated or intrinsic GTPase activity render KRAS constitutively active and thereby cause uncontrolled cell growth/proliferation, leading to cancer.1,2 Mutant KRAS is therefore a highly sought-after anticancer drug target.10,11
Despite decades of efforts, however, drugging KRAS (and RAS proteins in general) remains an unrealized goal.12 Among the many challenges, conservation of the nucleotide-binding site among a diverse group of small GTPases4,13 and the high (picomolar) affinity of RAS for its endogenous ligands, GDP or GTP, are arguably the most significant. These issues made competitive inhibition impractical and avoiding off-target effects difficult. Thus, along with efforts at indirect RAS inhibition by targeting its interaction partner proteins14,15 or membrane localization,16,17 development of direct allosteric KRAS inhibitors is currently a major focus of many laboratories.18 Proof-of-principle studies have established the allosteric nature of RAS11,19,20 and discovered several allosteric small-molecule KRAS binders.21−25 Moreover, a number of recent reports described molecular fragments,23 small molecules,18,24−26 peptidomimetics,27,28 and monobodies29 that bind KRAS and modulate its functions in various ways. Although this paints an optimistic picture of the prospects of allosteric KRAS inhibition, to the best of our knowledge, none of these compounds has made it to clinical trial. Recent efforts toward developing covalent GDP analogues30 or other small-molecule ligands31 targeting G12C mutant KRAS may have a better chance of eventually treating specific tumor types.18 However, their application is likely limited to a few cancer cases such as small-cell lung cancer.10 We believe noncovalent allosteric inhibition will be needed to target some of the most important mutations in KRAS including G12D, G12V, G13D, and Q61H found in biliary tract, small intestine, colorectal, lung, and pancreatic cancers.2,10 Together, these four mutations appear to account for greater than 78% of all KRAS-associated cancers.10
In previous reports, we described four allosteric ligand-binding sites on KRAS using a range of computational approaches,32,33 including molecular dynamics (MD) simulations to sample transient conformations with open allosteric pockets.34−36 Among these, pocket p1 was the best characterized and is well-established as a suitable target with many crystal structures of p1-bound ligand–KRAS complexes available in the protein data bank (PDB). In the current work, we combined MD simulation with a range of biophysical and cell assays to discover and characterize a novel class of inhibitors that bind to the p1 pocket with sub-micromolar affinity and abrogate signaling primarily by directly inhibiting the interaction of KRAS with effector proteins.
Materials and Methods
MD Simulation and Allosteric Pocket Analysis
Most oncogenic RAS mutants are constitutively active because their ability to hydrolyze GTP is compromised.37,38 An inhibitor that selectively targets GTP-bound mutant RAS would therefore be desirable. However, there was no ligand-free high-resolution experimental structure of GTP-bound KRAS (GTPKRAS) when we started this project in 2014, and our target pocket p1 (see below) was closed or was too small in the available GDP-bound KRAS (GDPKRAS) structures. Therefore, we used MD simulation to generate an ensemble of GTPKRAS structures with open p1. The initial structure for the simulation was a 5′-guanosinediphosohate-monothiophosphate (GSP)-bound KRASG12D X-ray structure from the PDB (ID 4DSO) with benzamidine bound at p1 and glycerol between helices 2 and 3.23 After converting GSP to GTP, removing all other molecules except crystal waters and the bound Mg2+, adding hydrogen atoms and solvent, minimization, and restrained simulation, we conducted a 300 ns production run using a protocol identical to that described in a recent report.39 The trajectory was analyzed in terms of volume and other features (such as number of hydrogen bond donors and acceptors) of our target pocket p1, and the conformation with the most open p1 was selected for the virtual screening of ligand libraries.
High-Throughput Virtual Screening
Six million compounds from the Drugs Now subset of the ZINC40 database were docked into pocket p1 of our MD-derived KRASG12D structure (Figure 1A, see also Figure S1). Gasteiger charges and atomic radii were assigned using AutoDock tools, and a first round of docking was conducted with AutoDock,41 as implemented in the parallelization routine DOVIS.42 We used the flexible ligand option with 1.0 Å spacing, along with a Lamarckian search with 150 generations and 1 000 000 energy evaluations. The top ∼4000 compounds with energy score ≤−6.8 kcal/mol were rescreened with VINA v1.1.243 with exhaustiveness set to 12 and energy range set to 4. The top 500 hits in each screen were then evaluated in terms of their ability to form close contact, salt bridge, hydrogen bonding, hydrophobic, cation−π, π–π, and π–stacking interactions with the protein, using distance and angle cutoffs recommended by Durant and McCammon.44 We found 58 ligands that score well in the majority of these metrics, procured ∼30 that were available for purchase, and experimentally tested the 11 compounds listed in Figure S2A using a low-throughput cell signaling assay.
Figure 1.
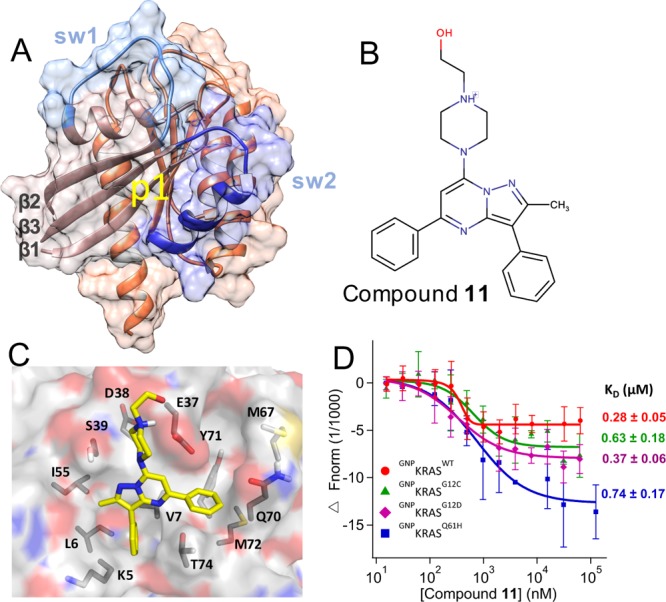
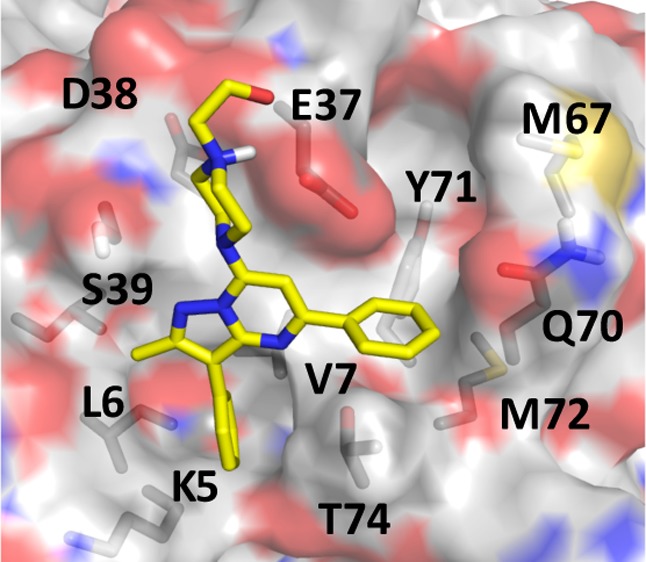
Predicted binding mode and measured affinity of compound 11 to KRAS. (A) Structure of the catalytic domain of KRAS used for the virtual screening. Lobe1 (residues 1–86) and lobe2 (residues 87–166) are highlighted in different colors, as are switches 1 (residues 30–40) and 2 (residues 60–75). The location of our target allosteric pocket p1 is indicated. (B) Chemical structure of compound 11. (C) Predicted binding pose of compound 11, with the key residues that make polar or vdW contacts with the ligand labeled. (D) MST experiments indicating the direct binding of compound 11 to KRAS, along with dissociation constants (KD) derived from the curves. Changes in fluorescence upon titration of 50 nM KRAS with increasing concentration of compound are shown: KRASWT (red), KRASG12C (green), KRASG12D (purple), and KRASQ61H (blue), each bound to the nonhydrolyzable GTP analogue, guanylyl imidodiphosphate (GNP). No or very weak binding was detected toward GDP-bound KRAS, GNP- or GDP-bound NRAS and HRAS, and Rap1B that was used as control (Figure S3).
Cell Signaling
The inhibitory potential of compounds was tested in monoclonal baby hamster kidney (BHK) cell lines stably expressing monomeric green fluorescence protein (mGFP)-tagged KRASG12D, KRAS,G12V and HRASG12V. Cells were cultured in Dulbecco’s modified Eagle’s medium (Hyclone) supplemented with 10% v/v bovine calf serum and incubated with compound or vehicle dimethyl sulfoxide (DMSO) for 3 h without serum. Cells were then harvested in lysis buffer [50 mM Tris (pH 7.5), 75 mM NaCl, 25 mM NaF, 5 mM MgCl2, 5 mM egtazic acid (EGTA), 1 mM dithiothreitol, 100 μM NaVO4, and 1% Nonidet P40 plus protease inhibitors] and subjected to western analysis, controlling protein loading by bicinchoninic acid assay. Lysates were resolved with Bio-Rad polyacrylamide TGX 10% gel, transferred to polyvinylidene fluoride membrane, and immunoblotted using pan-AKT (2920S), GFP (2956S), p-AKTS473 (4060L), p-cRafS338 (9427S), p-ERKT202/Y204 (4370L), ERK1/2 (4695S), or β-actin antibodies (Cell Signaling Technology). IC50 values were calculated with Prism 4-parameter fit.
Pull-Down
We pulled down GFP–RAS with the GST-tagged RAS binding domain (RBD) of cRafA85K (hereafter GST–RafRBD) to monitor RAS–Raf interaction. To prepare GST–RafRBD bound to agarose beads, bacteria (BL21) transfected with a previously cloned GEX plasmid were grown in selection media to optical density levels of 0.5–1.0 before protein expression was initiated with isopropyl β-D-1-thiogalactopyranoside (1:1000). After 4 h, the sample was centrifuged at 6000 rpm for 5 min at 4 °C, the pellet was resuspended with phosphate-buffered saline (PBS) containing 5 mM EGTA, 1% Triton X-100, PIC 1:50, and phenylmethylsulfonyl fluoride 1:100, and the cells were lysed with cycles of freezing and thawing. The lysate was sonicated to break up the DNA and pelleted. The supernatant was incubated with glutathione agarose (Pierce) beads that bind to GST–RafRBD. For all pull-down experiments, equal volumes of lysates from BHK cells expressing GFP–RAS were incubated for 2 h at 4 °C with GST–RBD beads plus control DMSO or compound. Then, the samples were washed with Tris buffer (50 mM Tris, pH 7.4, 1 mM ethylenediaminetetraacetic acid (EDTA), 1 mM EGTA, 150 mM NaCl, 0.1% Trition X-100, and protease inhibitors) and immunoblotted with anti-GFP (Cell Signaling) and anti-GST (Santa Cruz Biotechnology) antibodies.
Fluorescence Lifetime Imaging–Fluorescence Resonance Energy Transfer
Fluorescence lifetime imaging (FLIM)–Fluorescence resonance energy transfer (FRET) experiments were carried out using a lifetime fluorescence imaging attachment (Lambert Instruments, The Netherlands) on an inverted microscope.45 BHK cells transiently expressing mGFP-tagged KRASG12D (donor) alone or with mRFP-tagged cRafWT (acceptor) (1:5 ratio) were prepared and treated with compound for 2 h, washed with PBS, fixed with 4% paraformaldehyde, and quenched with 50 mM NH4Cl. The samples were excited using a sinusoidally modulated 3 W 470 nm light-emitting diode (LED) at 40 MHz under epi-illumination. Fluorescein (lifetime = 4 ns) was used as a lifetime reference standard. Cells were imaged with a Plan APO 60× 1.40 oil objective using an appropriate GFP filter set. The phase and modulation were determined from 12 phase settings using the manufacturer’s software. Resolution of two lifetimes in the frequency domain was performed using a graphical method46 mathematically identical to global analysis algorithms.47,48 The analysis yields the mGFP lifetime of the free mGFP donor (τ1) and the mGFP lifetime in donor/acceptor complexes (τ2). FLIM data were averaged on a per-cell basis. In a separate set of experiments, BHK cells coexpressing GFP–KRASG12D or GFP–HRASG12V with the empty vector pC1 or mCherry-RBD were treated with vehicle DMSO or 1 μM, and GFP fluorescence was measured as described above.
Cell Proliferation
Potential effect of the ligands on cancer cell proliferation was tested in four lung cancer cells, SKLU-1 (KRASWT), H1975 (KRASWT), H441 (KRASG12V), and H522 (KRASG12D), and four oral cancer cell lines, UM-SCC-22A (HRASWT), UM-SCC-22A (HRASG12V), HN31 (HRASG12D), and HN31 (HRASknockdown). One thousand cells were seeded per well in a 96-well plate. After 24 h of seeding, fresh growth medium supplemented with vehicle (DMSO) or varying concentrations of the drug was added. Cells were treated with the drug for 72 h, with the addition of fresh medium containing the drug every 24 h. Then, the cells were washed with PBS and frozen at −80 °C for a minimum of 24 h. The plates were thawed, and CyQUANT dye (in lysis buffer provided in the CyQUANT cell proliferation assay kit, Invitrogen) was added. After 5-minute incubation, fluorescence (excitation: 480 nm emission: 520 nm) was measured with a Tecan Infinite M200 plate reader for the lung cancer cells, and the number of oral cancer cells were quantified using the CyQUANT Proliferation Assay (ThermoFisher), according to manufacturer’s protocol.
Microscale Thermophoresis
Determination of dissociation constants using microscale thermophoresis (MST) was performed following vendor protocols. Purified RAS was labeled with the Monolith MT Protein Labeling Kit RED–NHS (NanoTemper Tech) through buffer-exchange in the labeling buffer [40 mM N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (HEPES), pH 7.5, 5 mM MgCl2, and 500 mM NaCl]. The concentration of the eluted protein was adjusted to 2–20 μM, the dye was added at a 2–3-fold concentration to a final volume of 200 μL, and the mixture was incubated for 30 min at room temperature in the dark. Labeled KRAS was purified using the column provided in the kit. For MST measurements, 16-point serial dilution of the ligand was prepared in an MST assay buffer (40 mM HEPES, pH 7.5, 5 mM MgCl2, 100 mM NaCl, plus 0.05% TWEEN-20, and 2–4% DMSO) and added to an equal volume of 100 nM KRAS solution. The solutions were loaded in capillaries, and measurements were done at room temperature using 20% LED and 40% MST power. The data were fit in Igor Pro using the Hill equation.
Nucleotide Exchange and Release Assays
Loading of fluorescent-labeled GDP (BODIPY–GDP; BGDP from hereon) to KRAS was conducted following previous reports,23,49 with minor modifications. Purified KRAS was buffer-exchanged in an NAP-5 column (GE Life Sciences) in a low Mg2+ buffer (25 mM Tris pH 7.5, 50 mM NaCl, and 0.5 mM MgCl2). The eluate was incubated with 10-fold molar excess of BGDP (Life Technologies) in the presence of 5 mM EDTA and 1 mM dithiothreitol (DTT) for 1.5 h at 20 °C in the dark. Then, 10 mM MgCl2 was added, and the solution was incubated for 30 min at 20 °C. Free nucleotide was removed by gel filtration using a PD-10 column (GE Life Sciences) that had been equilibrated with the reaction buffer (25 mM Tris–HCL, pH 7.5, 50 mM NaCl, 1 mM MgCl2, and 1 mM DTT). The concentration of BGDPKRAS was determined using the Bradford assay and a BGDP standard curve. Then, the effect of ligands on the intrinsic rate of nucleotide release was monitored using the decrease in fluorescence with time as BGDP dissociates from KRAS in a 100 μL reaction mixture (96-well plate) of 0.5 μM BGDPKRAS, 100 μM GTP, and varying concentrations of the ligand (0–25 μM); GTP was added just before the measurements. To measure the rate of SOS-mediated nucleotide release, 0.5 μM SOS (residues 564–1049, Cytoskeleton Inc) was added after adding GTP, and the fluorescence was immediately read (excitation: 485 nm, emission: 510 nm) using a Tecan Infinite M200 plate reader. Intrinsic and SOS-mediated nucleotide exchange rates were monitored with the fluorescence intensity increase of BGTP as it displaces GDP from KRAS. We used a 100 μL reaction mixture containing 0.5 μM each of GDPKRAS and BGTP (and SOS) plus varying concentrations of the ligand (0–5 μM); BGTP was added just before the measurements. Experiments were conducted with minimal light, and the reaction was monitored for 2 h at room temperature. Fluorescence intensities were normalized at 120 s, and the traces were fit with linear or single exponential functions (Igor Pro, Wavemetrics).
Fluorescence Polarization
Fluorescence polarization assay was conducted following previous reports.50,51 KRAS was preloaded with the nonhydrolyzable fluorescent GTP analogue BODIPY-GTP-γ-S (BGTP-γ-S; Life Technologies) using buffer exchange in NAP-5 (GE Life Sciences), as described in the previous section. Then, 0.5 μM (50 μL) BGTP-γ-SKRAS was incubated with an equal volume but varying concentrations (0–2.5 μM) of GST–RafRBD (Raf RBD residues 1–149; Life Technologies) for 30 min in the dark. To determine the effect of ligand on RAS–Raf binding, KRAS was first incubated with a fixed concentration of the ligand for 30 min and then with GST–RafRBD. Fluorescence polarization was measured using a POLARStar OPTIMA plate reader (excitation: 485 nm, emission: 520 nm) at room temperature. GST-tag was used to increase the weight of RafRBD for greater polarization. The dissociation constant for KRAS–Raf binding was determined using a quadratic ligand-binding equation.50
Results and Discussion
Initial Hits from Molecular Modeling and High-Throughput Virtual Screening
We conducted in silico screening of compounds from the ZINC database,40 targeting pocket p1 on an MD-derived structure of GTPKRASG12D. This pocket is located between the functionally critical switches 1 (residues 25–40) and 2 (residues 60–75) and encompasses residues 5–7, 37–39, 50–56, 67, and 70–75 (Figures 1A and S1). Many of these residues, including residues 37–39 on the effector binding loop and residue 71 on switch 2, participate in interactions with effectors and/or GEFs. Therefore, we reasoned that a p1-targeted ligand could disrupt either or both of these interactions. However, p1 was fully or partially closed in the available KRAS structures including the holo forms, which were generally bound to small (<160 Da) ligands (Figure S1). We wanted to have a more open conformation to dock a wide range of “druglike” molecules spanning the ∼150–500 Da molecular weight range common in marketed drugs. We therefore conducted MD simulation to generate an ensemble of GTPKRASG12D structures with open p1. Analysis of the trajectory yielded 119 and 219 Å3 as the mean and maximum volumes of pocket p1, respectively. We performed retrospective comparison of the MD conformer with the most open p1, which we used for molecular docking, with currently available GTP (or analogue)- and GDP-bound crystallographic KRAS structures (Figure S1). We observed three distinct groups of conformers that differ mainly in the orientation of helix 2. In one group, the orientation of helix 2 is such that pocket p1 is nearly or completely closed (orange). All of these structures are GDP-bound and are dominated by structures in complex with covalent ligands. In the second, sampled by both GDP- and GTP-bound KRAS, movement of helix 2 toward helix 3 opens up the pocket to some extent. In group 3, helix 2 moved even farther away from the core β-sheet, allowing for a more open p1. Our MD-derived conformer belongs to the third group and exhibits the largest displacement of helix 2, which, together with side chain reorientations, allowed for a wider pocket p1 (Figure S1). We used this snapshot to conduct an initial screen of 6 000 000 compounds, followed by a secondary screen of the top ∼4000 (see Methods). Analysis of the top 500 ligands in each screen yielded a consensus prediction of 58 initial hits. Eleven of these were purchased and tested in cells (Figure S2A).
Cell Signaling Assays Identify Compound 11 as a Promising Initial Hit
Western analysis was used to quickly assess the potential impact of our predicted hits on MAPK signaling, a major pathway mediated by KRAS. Specifically, we monitored ERK1/2 phosphorylation levels (p-ERK) in BHK cells stably expressing KRASG12D treated with vehicle (DMSO), the MEK inhibitor U0125 (U), or the compound at four different concentrations (1–100 μM). The results showed that the majority of the predicted hits have no effect, whereas few (e.g., 4) increase rather than decrease the p-ERK levels (Figure S2B). Compounds 9 and 11, on the other hand, decreased the p-ERK levels at concentrations ≥50 and ≥1 μM, respectively. To verify the latter observation, we repeated the experiments in an expanded concentration range starting from 0.1 μM. As in the first screen, compound 11 dose-dependently decreased the p-ERK levels, leading to a ∼50% reduction at 5 μM (Figure S2C). However, compound 9 increased the p-ERK levels at 25 and 38 μM in contrast to the decrease observed at higher concentrations (Figure S2B). Although a similar increase and then decrease of KRAS signaling upon increasing of ligand concentration has been observed before,49,52 we selected the more potent and monotonously dose-dependent compound 11 for further analysis.
Compound 11 Binds to WT and Oncogenic KRAS Mutants with High Affinity
Figure 1B,C shows the chemical structure and the predicted complex of compound 11 with KRAS, suggesting that the ligand potentially forms multiple favorable interactions with residues in the p1 pocket. Figure 1D shows that the compound binds to the isolated catalytic domain (residues 1–166) GTPKRASWT with a KD = ∼0.3 μM, suggesting a very tight binding rarely seen in primary screens. The compound has a very similar affinity (KD = ∼0.4–0.7 μM) for oncogenic mutants KRASG12D, KRASG12C, and KRASQ61H in the GTP state (Figure 1D). However, very weak or no binding was detected for KRASWT and KRASG12D in the GDP state, HRASWT and NRASWT in both their GDP and GTP-bound forms, or to our control Rap1b (Figure S3), a RAS-related small GTPase with a homologous structure. Few weak-affinity noncovalent binders that exhibit some selectivity toward GDP- or GTP-KRAS have been reported.23−25 Although further scrutiny is required to establish its true selectivity profile, our initial observations suggest that compound 11 may represent the first small molecule to selectively bind GTP-bound KRAS with high affinity.
In the docked pose (Figure 1C), the 1-piperazineethanol moiety occupies an electronegative cleft near D54 and D38, potentially donating hydrogen bonds to the side chain and backbone atoms of E37. The methylated pyrazolopyrimidine core sits in a trench on top of V7 and L56 with its methyl group pointing toward I55, whereas the pyrimidine-bound benzene ring occupies the space between the central β-sheet (β1−β3) and helix 2 and makes π-stacking interaction with Y71. The pyrazol-attached benzene is buried deep in a tight pocket, stabilized primarily by van der Waals interactions with the side chain carbon atoms of V7, L6, and K5. These interactions are common in the majority of our predicted hits listed in Figure S2A, and redocking of compound 11 after in silico mutation of each of these residues to Ala reduced the AutoDock free energy score by up to 2 kcal/mol. Therefore, we propose that, in addition to potential induced-fit effects, the preference of compound 11 for GTP-bound KRAS may be due to conformational differences of these residues in GTPRAS versus GDPRAS.4 Comparison of available GDP- and GTP-bound RAS structures supports this conclusion. For example, pocket p1 is partially occluded by helix 2 in a large number of GDP-bound KRAS (Figure S1) and HRAS (Figure S4) crystallographic structures. Similarly, the apparent preference of compound 11 for KRAS over HRAS or NRAS may arise from subtle conformational differences. For example, Mattos and colleagues have recently shown that the active site of activated KRAS is more open and dynamic than that of HRAS.53
Compound 11 Disrupts Interaction of KRAS with Raf
We used three different assays to check if our compound inhibits RAS signaling by interfering with effector binding. These included fluorescence polarization and pull-down assays, which directly measure the interaction of KRAS with the RBD of Raf in purified or cell lysate systems, respectively, and FLIM–FRET, which measures the interaction of KRAS with full-length or truncated Raf in the cellular milieu. We used fluorescence polarization of BGTP-γ-S to monitor the binding of the KRAS catalytic domain to GST–RafRBD with and without preincubation with 1 μM compound 11. Figure 2A shows a dramatic decrease in polarization in the entire concentration range of GST–RafRBD. For example, at 2 μM GST–RafRBD, compound treatment reduced the polarization and therefore RAS–RafRBD interaction by >80%. That we observed such a large reduction despite the weaker affinity of the RBD used in this assay (residues 1–149) than the commonly used shorter RBD (residues 51–131) further highlights the major impact of 11 on KRAS/Raf complex formation. The dissociation constant derived from the polarization curves indicate that 11 reduced the affinity of KRAS to RafRBD by ∼13-fold. Consistent with this observation, pull-down of GFP–KRASG12D by GST–RafRBD in compound-treated cell lysates show a significant (e.g., >50% at 1 μM of 11) decrease in GFP–KRASG12D levels (Figure 2B).
Figure 2.
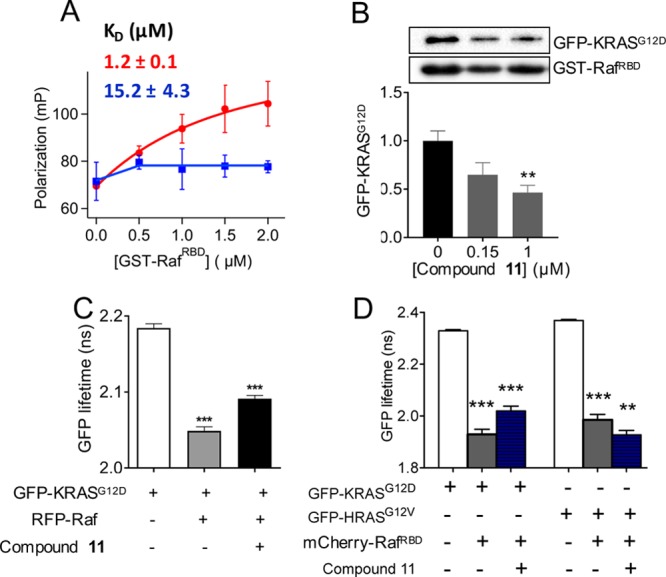
Compound 11 disrupts KRAS–Raf interaction.
(A) Fluorescence polarization of BGTP-γ-SKRAS (0.5 μM) as a function of varying concentration of GST–RafRBD in the absence (red) and presence (blue) of 1 μM
compound 11. Shown above the curves is the KD for KRAS–RafRBD binding obtained by
fitting the data to 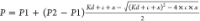 , where P1 is
the polarization
of free KRAS, P2 is the polarization of Raf-bound
KRAS, c is the total concentration of KRAS, and x is the total concentration of RafRBD. (B) Amount
of GFP–KRASG12D pulled down by GST–RafRBD after treatment of cell lysates with compound at the indicated
concentrations (representative westerns shown at the top). An equal
volume of lysates was used, and the data were normalized to GST–RBD
and DMSO control, which also serves as the loading control. The RBD
sequence length was 1–149 and 51–131 in the fluorescence
polarization and pull-down assays, respectively. Whereas the shorter
RBD is sufficient for biochemical assays, the extra amino acids in
the longer RBD increases the size and thereby enhances the signal-to-noise
ratio in the fluorescence polarization assay. (C) GFP fluorescence
lifetime from FLIM–FRET using cells expressing GFP–KRASG12D alone or with RFP–Raf (full-length cRaf: residues
1–648), with or without treatment by 1 μM compound 11. (D) GFP fluorescence lifetime from FLIM–FRET using
cells expressing GFP–KRASG12D or HRASG12V with an empty vector pC1 or mCherry-RafRBD (RBD: residues
51–131), with or without treatment with 1 μM compound 11. In (B–D), data are shown as mean ± standard
error (SE) from three separate experiments; significance was estimated
by one-way analysis of variance (ANOVA) relative to the control for
each bar in B, second bar in C, and second and fourth bars in D, or
relative to the bar immediately to the left of bar 3 in C and bars
3 and 6 in D.
, where P1 is
the polarization
of free KRAS, P2 is the polarization of Raf-bound
KRAS, c is the total concentration of KRAS, and x is the total concentration of RafRBD. (B) Amount
of GFP–KRASG12D pulled down by GST–RafRBD after treatment of cell lysates with compound at the indicated
concentrations (representative westerns shown at the top). An equal
volume of lysates was used, and the data were normalized to GST–RBD
and DMSO control, which also serves as the loading control. The RBD
sequence length was 1–149 and 51–131 in the fluorescence
polarization and pull-down assays, respectively. Whereas the shorter
RBD is sufficient for biochemical assays, the extra amino acids in
the longer RBD increases the size and thereby enhances the signal-to-noise
ratio in the fluorescence polarization assay. (C) GFP fluorescence
lifetime from FLIM–FRET using cells expressing GFP–KRASG12D alone or with RFP–Raf (full-length cRaf: residues
1–648), with or without treatment by 1 μM compound 11. (D) GFP fluorescence lifetime from FLIM–FRET using
cells expressing GFP–KRASG12D or HRASG12V with an empty vector pC1 or mCherry-RafRBD (RBD: residues
51–131), with or without treatment with 1 μM compound 11. In (B–D), data are shown as mean ± standard
error (SE) from three separate experiments; significance was estimated
by one-way analysis of variance (ANOVA) relative to the control for
each bar in B, second bar in C, and second and fourth bars in D, or
relative to the bar immediately to the left of bar 3 in C and bars
3 and 6 in D.
We observed a similar effect in FLIM–FRET experiments in cells. In this experiment, quenching of GFP fluorescence lifetime indicates RAS–cRaf interaction in cells cotransfected with GFP–RAS and RFP–cRaf. In cells coexpressing KRASG12D and wild-type full-length cRaf, quenching of GFP fluorescence lifetime and hence KRASG12D–cRaf interaction is significantly reduced upon compound treatment (Figure 2C). FLIM–FRET was also used to examine the interaction of GFP-tagged RAS mutants and mCherry-tagged RafRBD. As shown in Figure 2D, GFP fluorescence lifetime in cells expressing GFP–KRASG12D with empty vector pC1 was ∼2.3 ns, which decreased to ∼1.93 ns in cells coexpressing GFP–KRASG12D and mCherry–RBD, indicating significant FRET and thus an interaction between the two constructs. Treatment with 1 μM compound 11 for 2 h increased the GFP lifetime to ∼2.02 ns, suggesting reduction of the interaction between KRASG12D and RBD. The same experiments with GFP–HRASG12V and mCherry-RBD show that compound 11 has inexplicably the opposite albeit small effect on the interaction of HRASG12V with RafRBD. These results in cells confirm our observations from pull-downs in lysates and fluorescence polarization in purified systems and support the potential KRAS-selectivity of compound 11 suggested by MST.
Compound 11 Inhibits KRAS Signaling
Figure 3 shows that compound 11 dose-dependently decreases both p-ERK and p-cRaf levels in BHK cells expressing KRASG12D and KRASG12V, suggesting inhibition of RAS signaling via the MAPK pathway. The data also indicate that the ligand has a slightly lower IC50 for its direct effector cRaf (e.g., 0.7 μM in the case of KRASG12D) than the two-steps removed ERK (1.3 μM). Note also that the IC50 for cRaf is very close to the KD of the ligand for GTPKRAS. Changes in phosphorylated AKT (p-AKT) levels show that the compound also inhibits signaling through the AKT pathway but to a lesser extent than the MAPK pathway. Together, these results suggest that the ligand disrupts MAPK signaling by acting on RAS or its upstream modulators. We have also measured p-ERK and p-cRaf levels in BHK cells expressing the constitutively active HRASG12V (Figure 3, right). In these cells and the ligand concentration range that we have tested, compound 11 has less effect on p-ERK and p-cRaf levels and hence on signaling via the MAPK pathway. Similarly, no statistically significant effect on p-AKT levels was observed even though H-Ras is a major driver of the AKT pathway. As a control, treatment of the HRASG12V-expressing BHK cells with 10 μM of U (the MEK inhibitor U0126) almost completely abolished MAPK signaling (Figure 3).
Figure 3.

Compound 11 inhibits mutant KRAS signaling. Representative western blots and their quantification showing levels of phosphorylated cRaf (p-cRaf), ERK (p-ERK), and AKT (p-AKT) in cells expressing KRASG12D (top left), KRASG12V (bottom left), and HRASG12V (right) treated with the indicated concentrations of compound 11, DMSO, or where indicated 10 μM MEK inhibitor U0125 (U). Data are shown as mean ± SE; significance was estimated by one-way ANOVA: * = p < 0.02; ** = p < 0.005; *** = p < 0.0001. t-cRaf, t-ERK, and t-AKT represent total cRaf, ERK and AKT.
We tested the effect of compound 11 on the proliferation of four lung and four oral cancer cell lines and found that the KRAS-expressing lung cancer cells, particularly those with mutant KRAS, are more sensitive to the compound than the HRAS-expressing oral cancer cells (Figure 4A). Also, there is no major difference between HRASWT and HRASG12V/HRASG12D cancer cells or between HN31 cancer cells with and without HRAS knockdown. In Figure 4B, the relative growth of the eight cell lines in the presence of 5 μM compound 11 is shown. Relative to DMSO control, growth of the oral cancer cells with or without mutant HRAS as well as the lung cancer cells with wild-type KRAS is 60–80%, whereas the corresponding number for the lung cancer cells harboring KRASG12D or KRASG12V is 30–35%. In summary, data from the eight cell lines that we have tested suggest that compound 11 more efficiently inhibits signaling through KRAS than HRAS, consistent with its tight binding to activated KRAS (Figure 1) and effect on KRAS–Raf interaction (Figure 2) and its weaker binding (if any) to HRAS and NRAS (Figure S3).
Figure 4.

Cell proliferation assays suggest that cancer cells expressing mutant KRAS are more sensitive to compound 11. (A) Proliferation profile of KRAS-expressing lung cancer cells and HRAS-expressing oral cancer cells upon treatment by increasing concentration of compound 11 and monitored by CyQUANT assay. (B) Relative growth of the KRAS and HRAS cancer cells after treatment with 5 μM compound 11. The lung cancer cells include H1975 and H522 that express KRASWT, SKLU-1 that expresses KRASG12D, and H441 harboring KRASG12V. The oral cancer cells include UM-SCC-22A lines harboring HRASWT and HRASG12V, HN31 cells expressing HRASG12D, and HN31 cells with HRAS knockdown. Data are shown as mean ± SE; significance was estimated by one-way ANOVA with respect to the data for SKLU-1: * = p < 0.02; ** = p < 0.005; *** = p < 0.0001.
Proposed Mechanism of Action and Optimization Route for Pyrazolopyrimidine-Based Kras Inhibitors.
In addition to its effect on effector binding, compound 11 also slightly reduced the rates of both intrinsic and SOS-mediated GDP/GTP exchange reactions of KRAS as well as SOS-mediated GDP release (Supporting Information text and Figure S5). To identify the chemical fingerprints of compound 11 potentially responsible for its high-affinity binding and effect on KRAS function, we studied compounds 12 and 13. Obtained from similarity searches based on 11, these analogues provided valuable insights into the likely mechanism of action of our pyrazolopyrimidine-based ligand. In compound 12, the 1-piperazineethanol functional group of 11 is replaced by 1-methylpiperazine (Figure 5A,B), making it more hydrophobic and less soluble in DMSO. This compound slightly reduced the p-ERK levels at a higher concentration of 2 μM (Figure 5C), but it is nearly as effective as 11 in inhibiting proliferation of lung cancer cells (Figure 5D). However, it has no effect on p-cRaf levels (Figure 5C) or on KRAS–Raf interaction as assessed by FLIM–FRET (Figure 5E), suggesting a potentially different mechanism of inhibition than compound 11 or an off-target effect. The predicted binding mode of 12 is similar to that of 11, but it lacks the capacity for hydrogen-bonding interactions with residues at the effector-binding loop (Figure 5B). Together, these results suggest that the hydroxymethyl group on the piperazine ring, which in compound 11 is predicted to interact with residues in the effector-binding loop (Figure 1C), plays a crucial role in disrupting KRAS–Raf interaction and/or in modulating binding to KRAS.
Figure 5.
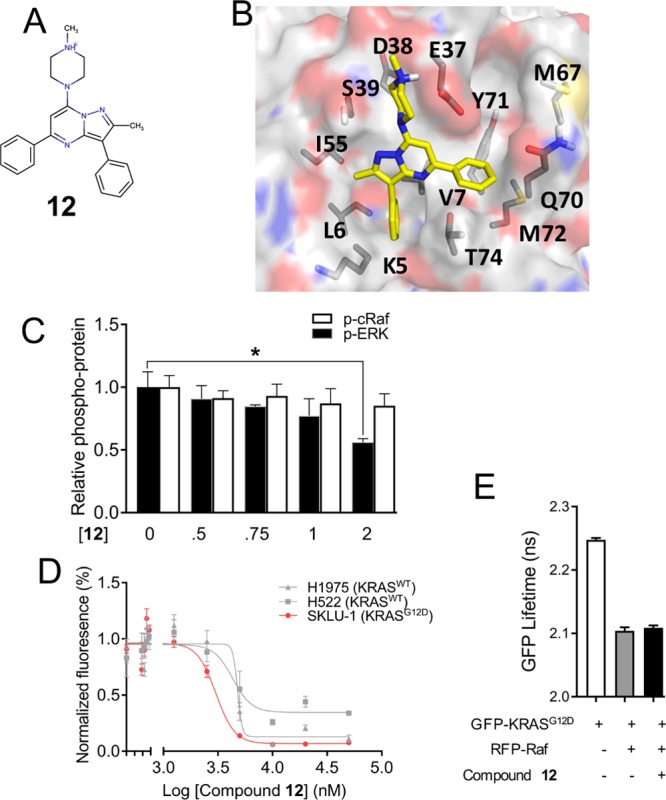
Potential role of the piperazineethanol moiety on compound 11 for abrogating effector binding. (A) Chemical structure of compound 12, an analogue of 11 lacking the terminal hydroxymethyl functional group. (B) Predicted binding pose of compound 12. (C) p-ERK and p-cRaf levels in BHK cells expressing KRASG12D treated with indicated concentrations of 12 or vehicle. (D) Proliferation profile of lung cancer cells upon treatment with increasing concentration of compound 12, monitored by the CyQUANT assay. Data are averages over three independent experiments, and error bars represent SE. (E) GFP fluorescence lifetime from FLIM–FRET using cells expressing GFP–KRASG12D alone or together with RFP–cRaf and with or without treatment with 2 μM compound 12.
However, a derivative with a better solubility profile than 12 and one that preserves the ability of 11 to inhibit effector binding would be more desirable. The less hydrophobic compound 13 (Figure 6A,B), which has a methyl group attached to the pyrimidine in place of the benzene ring found in 11, is readily soluble in DMSO and other common solvents. This allowed us to measure its KD with G12D and other KRAS mutants using MST. The results summarized in Figure 6C show that compound 13 has a 6.5–7.1-fold weaker affinity for KRAS than compound 11. Similar to compound 11, however, 13 does not appear to bind to GDPKRASWT or GDPKRASG12D. Comparison of the docked poses of 11 (Figure 1C) and 13 (Figure 6B) suggests a potential rationale for the observed differences in binding affinity. The benzene ring of compound 11 is involved in a T-shaped π-stacking interaction with the side chain of Y71, which is replaced by the much smaller methyl in 13. This suggests a critical role for the phenyl ring on the pyrimidine core for potency, providing a useful clue for future optimization efforts.
Figure 6.
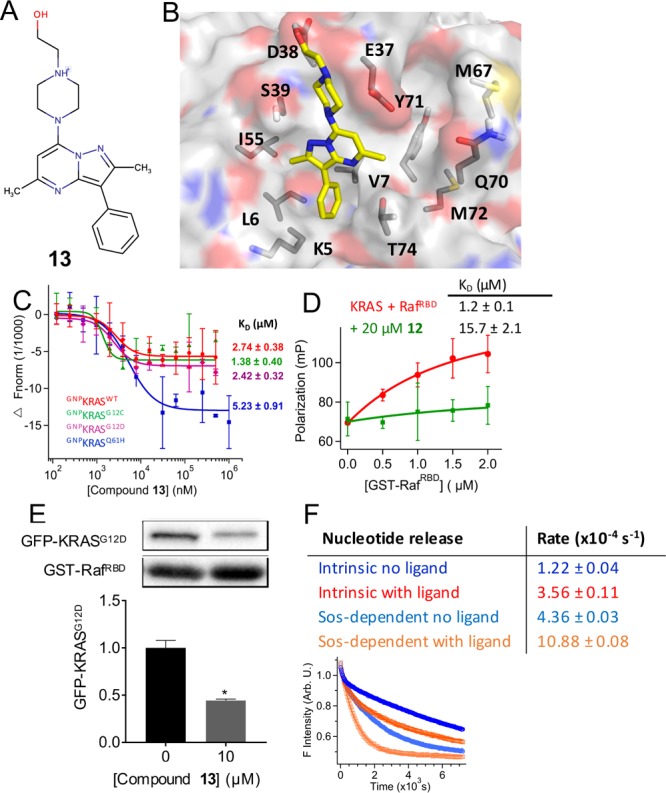
Interaction with switch 2 residues modulate the exchange factor activity. (A) Chemical structure of compound 13, an analogue of 11 without a benzene on the pyrimidine core. (B) Predicted binding pose of 13. (C) Fluorescence intensity and KD from MST experiments on KRAS mutants (see the legend of Figure 2 for details). (D) Fluorescence polarization of BGTP-γ-SKRAS (0.5 μM) with increasing concentration of GST–RafRBD in the absence (red) and presence (green) of 20 μM compound 13. (E) Amount of GFP-KRASG12D pulled down by GST–RafRBD after treatment of whole cell lysates with 10 μM compound 13 (representative western blots shown at the top). An equal volume of lysates was used, and the data are normalized to GST–RBD and DMSO control. (F) Intrinsic and SOS-mediated nucleotide release rates in a mixture of 0.5 μM KRAS (and SOS), 100 μM GTP, and 0 or 50 μM compound 13 (top) derived from changes in the fluorescence intensity during the reaction KRASBGDP + GTP → KRASGTP + BGDP (bottom). Rates were calculated using single exponential fits starting at 120 s.
We then used fluorescence polarization and pull-down assays to test the functional implication of the modification in 13 relative to the parent compound 11. Figure 6D shows that 20 μM compound 13 disrupts the interaction of KRAS with GST–RafRBD as effectively as the parent compound. Our pull-down assay led to the same conclusion: 13 disrupts KRASG12D–RafRBD interaction (Figure 6E). These results demonstrate that modifications can be made on the pyrazolopyrimidine core to optimize for potency without compromising the effect on effector binding. This conclusion is supported by the predicted ligand/KRAS complex structures (Figures 1C and 6B), which show that 11 and 13 are likely to make identical contacts with residues at the effector-binding region via their piperazine ring and especially the piperazineethanol group. This is important because, as we have shown using 12, modification in this part of the ligand may cause loss of effect on Raf binding. We then wondered if interaction with switch 2 residues or lack thereof may play a role in nucleotide release, because the conformation of many switch 2 residues, such as Y71 and Y64, differs between free and GEF-bound RAS.54,55 To test this, we measured the intrinsic and SOS-dependent rates of labeled-GDP release in the absence and presence of 13. We found that, indeed, replacing the benzene ring on the pyrimidine core by methyl dramatically altered the effect on nucleotide release. Whereas 11 had no effect on intrinsic and only modestly decreased the rate of SOS-mediated nucleotide release (Figure S5), 13 dramatically increased both rates (Figure 6F). This result suggests that interaction with switch 2 residues including Y71 may determine how a p1-bound ligand affects GEF activity. The results also provide a strong support for the reliability of the predicted ligand-KRAS complex structures and offer a viable route for additional modifications in future optimization efforts.
Concluding Discussion
Finding a direct inhibitor of KRAS remains a major challenge in the search for cancer therapy. Previous attempts at preventing membrane binding of KRAS by farnesyl transferase inhibitors failed in clinical trials. More recent efforts focused on the dynamics of RAS revealed allosteric pockets suitable for binding of small molecules.32,35 Several small-molecule ligands that bind to some of these pockets and disrupt interaction with GEFs or effectors have been discovered.21−25 However, thus far, none of these ligands have led to a viable lead compound. In the current work, we combined MD simulation to generate a KRAS conformation with open pocket p1 and virtual screening to identify potential hits, followed by biophysical and cell biological experiments for validation. We have discovered a novel high-affinity KRAS inhibitor, compound 11, that has unique structural features. Compound 11 (2-[4-(8-methyl-3,9-diphenyl-2,6,7-triazabicyclo[4.3.0]nona-2,4,7,9-tetraen-5-yl)piperazin-1-yl]ethanol) is druglike (drug likeness = 4.1) and somewhat polar with six hydrogen bond donors and two acceptors (clogP = 0.87). It has a pyrazolopyrimidine core rather than an indole or imidazole ring typical in published ligands. Also, 11 is relatively large (415 Da) with its pyrazol ring methylated and benzylated and its pyrimidine ring β-modified by benzene and 1-piperazineethanol. This allowed it to make more extensive predicted contacts with KRAS p1 residues than is common in most of the published ligands (Figure 1C). Although more work is required to fully establish its selectivity profile, our data suggest that compound 11 binds to GTPKRAS with submicromolar affinity (Figures 1 and S3), inhibits MAPK signaling (Figure 3), and reduces the growth of cancer cells expressing mutant KRAS more efficiently than those expressing HRAS (Figure 4). Moreover, we used fluorescence polarization, pull-down, and FLIM–FRET assays to demonstrate that compound 11 inhibits MAPK signaling primarily by abrogating interaction with effector proteins (Figure 2), in contrast to many published KRAS ligands that mainly affect GEF activity.22−24 At a high concentration, 11 exhibits a small effect on intrinsic and GEF-catalyzed guanine nucleotide exchange rates (Figure S5), but this effect is too small at concentrations used in the cell-based assays to explain the significant inhibitory activity of the compound. For example, there is a maximum of ∼5% reduction in the rates of both intrinsic and SOS-mediated nucleotide release or exchange reactions at 1 μM compound 11. In contrast, the p-ERK levels dropped by about 50% after a 3 h treatment using the same concentration of the compound (Figure 3).
The above conclusions are also supported by data from comparative analyses of compound 11 and its analogues 12 and 13. Compound 13 retains the effect of the parent compound on Raf binding even though it has a weaker (low μM) affinity for KRAS. Intriguingly, 13 accelerates both intrinsic and SOS-mediated rates of nucleotide release, in contrast to 11 which has no effect on the intrinsic and only modestly decreases the SOS-mediated reaction rate. Compound 12 has no effect on KRAS/Raf interaction and displays some inhibitory activities via an unknown mechanism. The distinct behavior of the derivatives and the parent compound, especially 11 and 13 for which we have data for direct KRAS binding, suggest altered protein–ligand interactions. We propose that the piperazineethanol group interacts with switch 1 of KRAS and plays a critical role in abrogating effector binding, whereas the potentially switch 2-interacting nonpolar moieties attached to the pyrazolopyrimidine core modulate GEF activity and contribute to high-affinity binding. These insights provide ideal starting points for further optimization of our highly promising lead compounds.
Acknowledgments
We thank current and former members of the Gorfe and Hancock laboratories for illuminating discussions, Prof. X. Cheng for help with MST and for providing fluorescent-labeled Rap1b, and Prof. S. Cunha for encouragements and help with pull-down assays. We gratefully acknowledge Prof. C. Mattos (Northeastern University) for purified GNP-bound KRAS samples, Prof. Sharon Campbell for HRAS and NRAS samples, and Prof. J. Putkey and Prof X. Wang (University of Texas Health Science Center) for purified GDP-bound KRAS samples and technical advice. We thank Dr. Jeffrey Myers of The University of Texas MD Anderson Cancer Center for providing oral cancer cell lines. This work was supported by the Cancer Prevention and Research Institute of Texas (CPRIT grant no DP150093). K.J.C. was supported in part by NIH (grant no. R00CA188593). A.K.G. was supported in part by a postdoctoral training fellowship from the Keck Center Computational Cancer Biology Training Program (CCBTP) of the Gulf Coast Consortia (CPRIT grant No. RP170593).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b03308.
Comparison of MD-derived and experimental KRAS structures; list of predicted hits and initial experimental tests; additional binding data; overlay of an MD-derived KRAS structure and experimental HRAS structures; effect of compound 11 on intrinsic and GEF-dependent nucleotide release and exchange reactions (PDF)
Author Contributions
A.A.G. designed and oversaw the project; M.J.M. performed high-throughput virtual screening and cell signaling and proliferation assays; C.V.P. performed biophysical assays; P.P. performed MD simulations and structure analysis; M.J.M., K.J.C., and Y.Z. performed FLIM–FRET assays; L.H., A.K.G., and A.N. performed additional cell-based assays; K.J.C., D. vdH., J.F.H., and Y.Z contributed to the design and interpretation of cell assays; M.J.M., C.V.P., P.P., K.J.C., and A.A.G. wrote the paper. All authors contributed toward reagents/analysis tools, interpretation, and critically reviewed the manuscript.
The authors declare no competing financial interest.
Notes
The data that support the findings of this study, including MD-derived coordinates of KRAS used for docking, are available from the corresponding author upon reasonable request.
Supplementary Material
References
- Cox A. D.; Der C. J. Ras history: The saga continues. Small GTPases 2014, 1, 2–27. 10.4161/sgtp.1.1.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior I. A.; Lewis P. D.; Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. 10.1158/0008-5472.can-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock J. F.; Magee A. I.; Childs J. E.; Marshall C. J. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell 1989, 57, 1167–1177. 10.1016/0092-8674(89)90054-8. [DOI] [PubMed] [Google Scholar]
- Vetter I. R.; Wittinghofer A. Signal transduction - The guanine nucleotide-binding switch in three dimensions. Science 2001, 294, 1299–1304. 10.1126/science.1062023. [DOI] [PubMed] [Google Scholar]
- Prakash P.; Gorfe A. A. Overview of simulation studies on the enzymatic activity and conformational dynamics of the GTPase Ras. Mol. Simul. 2014, 40, 839–847. 10.1080/08927022.2014.895000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash P.; Gorfe A. A. Lessons from computer simulations of Ras proteins in solution and in membrane. Biochim. Biophys. Acta 2013, 1830, 5211–5218. 10.1016/j.bbagen.2013.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick F.; Wittinghofer A. Interactions between Ras proteins and their effectors. Curr. Opin. Biotechnol. 1996, 7, 449–456. 10.1016/s0958-1669(96)80123-6. [DOI] [PubMed] [Google Scholar]
- Schubbert S.; Shannon K.; Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 2007, 7, 295–308. 10.1038/nrc2109. [DOI] [PubMed] [Google Scholar]
- Hancock J. F. Ras proteins: different signals from different locations. Nat. Rev. Mol. Cell Biol. 2003, 4, 373–385. 10.1038/nrm1105. [DOI] [PubMed] [Google Scholar]
- Stephen A. G.; Esposito D.; Bagni R. K.; McCormick F. Dragging ras back in the ring. Cancer Cell 2014, 25, 272–281. 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- Gorfe A. Mechanisms of allostery and membrane attachment in Ras GTPases: implications for anti-cancer drug discovery. Curr. Med. Chem. 2010, 17, 1–9. 10.2174/092986710789957832. [DOI] [PubMed] [Google Scholar]
- Singh H.; Longo D. L.; Chabner B. A. Improving prospects for targeting Ras. J. Clin. Oncol. 2015, 33, 3650–3659. 10.1200/jco.2015.62.1052. [DOI] [PubMed] [Google Scholar]
- Rojas A. M.; Fuentes G.; Rausell A.; Valencia A. The Ras protein superfamily: evolutionary tree and role of conserved amino acids. J. Cell Biol. 2012, 196, 189–201. 10.1083/jcb.201103008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauschild A.; Grob J.-J.; Demidov L. V.; Jouary T.; Gutzmer R.; Millward M.; Rutkowski P.; Blank C. U.; Miller W. H.; Kaempgen E.; Martín-Algarra S.; Karaszewska B.; Mauch C.; Chiarion-Sileni V.; Martin A.-M.; Swann S.; Haney P.; Mirakhur B.; Guckert M. E.; Goodman V.; Chapman P. B. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. 10.1016/s0140-6736(12)60868-x. [DOI] [PubMed] [Google Scholar]
- Zimmermann G.; Papke B.; Ismail S.; Vartak N.; Chandra A.; Hoffmann M.; Hahn S. A.; Triola G.; Wittinghofer A.; Bastiaens P. I. H.; Waldmann H. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature 2013, 497, 638–642. 10.1038/nature12205. [DOI] [PubMed] [Google Scholar]
- van der Hoeven D.; Cho K.-j.; Ma X.; Chigurupati S.; Parton R. G.; Hancock J. F. Fendiline inhibits K-Ras plasma membrane localization and blocks K-Ras signal transmission. Mol. Cell. Biol. 2012, 33, 237–251. 10.1128/mcb.00884-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho K.-j.; Park J.-H.; Piggott A. M.; Salim A. A.; Gorfe A. A.; Parton R. G.; Capon R. J.; Lacey E.; Hancock J. F. Staurosporines disrupt phosphatidylserine trafficking and mislocalize Ras proteins. J. Biol. Chem. 2012, 287, 43573–43584. 10.1074/jbc.m112.424457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph J.; Stokoe D. Selective inhibition of mutant Ras protein through covalent binding. Angew. Chem., Int. Ed. 2014, 53, 3777–3779. 10.1002/anie.201400233. [DOI] [PubMed] [Google Scholar]
- Grant B. J.; Lukman S.; Hocker H. J.; Sayyah J.; Brown J. H.; McCammon J. A.; Gorfe A. A. Novel Allosteric Sites on Ras for Lead Generation. PLoS One 2011, 6, e25711. 10.1371/journal.pone.0025711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant B. J.; Gorfe A. A.; McCammon J. A. Large conformational changes in proteins: signaling and other functions. Curr. Opin. Struct. Biol. 2010, 20, 142–147. 10.1016/j.sbi.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel J.; Cromm P. M.; Zimmermann G.; Grossmann T. N.; Waldmann H. Small-molecule modulation of Ras signaling. Nat. Chem. Biol. 2014, 10, 613–622. 10.1038/nchembio.1560. [DOI] [PubMed] [Google Scholar]
- Hocker H. J.; Cho K.-J.; Chen C.-Y. K.; Rambahal N.; Sagineedu S. R.; Shaari K.; Stanslas J.; Hancock J. F.; Gorfe A. A. Andrographolide derivatives inhibit guanine nucleotide exchange and abrogate oncogenic Ras function. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 10201–10206. 10.1073/pnas.1300016110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer T.; Garrenton L. S.; Oh A.; Pitts K.; Skelton N. J.; Fauber B. P.; Pan B.; Malek S.; Stokoe D.; Bowman K. K.; Wu J.; Giannetti A. M.; Starovasnik M. A.; Mellman I.; Jackson P. K.; Rudolph J.; Wang W.; Fang G. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 5299–5304. 10.1073/pnas.1116510109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q.; Burke J. P.; Phan J.; Burns M. C.; Olejniczak E. T.; Waterson A. G.; Lee T.; Rossanese O. W.; Fesik S. W. Discovery of Small Molecules that Bind to K-Ras and Inhibit Sos-Mediated Activation. Angew. Chem., Int. Ed. 2012, 51, 6140–6143. 10.1002/anie.201201358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima F.; Yoshikawa Y.; Ye M.; Araki M.; Matsumoto S.; Liao J.; Hu L.; Sugimoto T.; Ijiri Y.; Takeda A.; Nishiyama Y.; Sato C.; Muraoka S.; Tamura A.; Osoda T.; Tsuda K.-i.; Miyakawa T.; Fukunishi H.; Shimada J.; Kumasaka T.; Yamamoto M.; Kataoka T. In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 8182–8187. 10.1073/pnas.1217730110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C.; Li Y.; Li L.-L.; Fan X.-X.; Wang Y.-W.; Wei C.-L.; Liu L.; Leung E.-L.; Yao X.-J. Identification of a New Potent Inhibitor Targeting KRAS in Non-small Cell Lung Cancer Cells. Front. Pharmacol. 2017, 8, 823. 10.3389/fphar.2017.00823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh T. B.; Upadhyaya P.; Qian Z.; Pei D. Discovery of a Direct Ras Inhibitor by Screening a Combinatorial Library of Cell-Permeable Bicyclic Peptides. ACS Comb. Sci. 2015, 18, 75–85. 10.1021/acscombsci.5b00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhyaya P.; Qian Z.; Habir N. A.; Pei D. Direct Ras Inhibitors Identified from a Structurally Rigidified Bicyclic Peptide Library. Tetrahedron 2014, 70, 7714–7720. 10.1016/j.tet.2014.05.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer-Smith R.; Koide A.; Zhou Y.; Eguchi R. R.; Sha F.; Gajwani P.; Santana D.; Gupta A.; Jacobs M.; Herrero-Garcia E.; Cobbert J.; Lavoie H.; Smith M.; Rajakulendran T.; Dowdell E.; Okur M. N.; Dementieva I.; Sicheri F.; Therrien M.; Hancock J. F.; Ikura M.; Koide S.; O’Bryan J. P. Inhibition of RAS function through targeting an allosteric regulatory site. Nat. Chem. Biol. 2016, 13, 62–68. 10.1038/nchembio.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y.; Lu J.; Hunter J.; Li L.; Scott D.; Choi H. G.; Lim S. M.; Manandhar A.; Gondi S.; Sim T.; Westover K. D.; Gray N. S. Covalent Guanosine Mimetic Inhibitors of G12C KRAS. ACS Med. Chem. Lett. 2016, 8, 61–66. 10.1021/acsmedchemlett.6b00373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrem J. M.; Peters U.; Sos M. L.; Wells J. A.; Shokat K. M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548. 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant B. J.; Lukman S.; Hocker H. J.; Sayyah J.; Brown J. H.; McCammon J. A.; Gorfe A. A. Novel allosteric sites on Ras for lead generation. PLoS One 2011, 6, e25711. 10.1371/journal.pone.0025711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhrman G.; O′Connor C.; Zerbe B.; Kearney B. M.; Napoleon R.; Kovrigina E. A.; Vajda S.; Kozakov D.; Kovrigin E. L.; Mattos C. Analysis of Binding Site Hot Spots on the Surface of Ras GTPase. J. Mol. Biol. 2011, 413, 773–789. 10.1016/j.jmb.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy M.; Prakash P.; Gorfe AA Computational allosteric ligand binding site identification on Ras proteins. Acta Biochim Biophys Sin (Shanghai) 2016, 48, 3–10. 10.1093/abbs/gmv100. [DOI] [PubMed] [Google Scholar]
- Prakash P.; Hancock J. F.; Gorfe A. A. Binding hotspots on K-ras: consensus ligand binding sites and other reactive regions from probe-based molecular dynamics analysis. Proteins 2015, 83, 898–909. 10.1002/prot.24786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash P.; Sayyed-Ahmad A.; Gorfe A. A. pMD-Membrane: A Method for Ligand Binding Site Identification in Membrane-Bound Proteins. PLoS Comput. Biol. 2015, 11, e1004469. 10.1371/journal.pcbi.1004469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal S. E.; Eccleston J. F.; Hall A.; Webb M. R. Kinetic Analysis of the Hydrolysis of GTP by p21 N-Ras. J. Biol. Chem. 1988, 263, 19717–19722. [PubMed] [Google Scholar]
- Hunter J. C.; Manandhar A.; Carrasco M. A.; Gurbani D.; Gondi S.; Westover K. D. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res. 2015, 13, 1325–1335. 10.1158/1541-7786.mcr-15-0203. [DOI] [PubMed] [Google Scholar]
- Sayyed-Ahmad A.; Prakash P.; Gorfe A. A. Distinct dynamics and interaction patterns in H- and K-Ras oncogenic P-loop mutants. Proteins 2017, 85, 1618–1632. 10.1002/prot.25317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin J. J.; Sterling T.; Mysinger M. M.; Bolstad E. S.; Coleman R. G. ZINC: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M.; Goodsell D. S.; Halliday R. S.; Huey R.; Hart W. E.; Belew R. K.; Olson A. J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. 10.1002/(sici)1096-987x(19981115)19:14<1639::aid-jcc10>3.0.co;2-b. [DOI] [Google Scholar]
- Jiang X.; Kumar K.; Hu X.; Wallqvist A.; Reifman J. DOVIS 2.0: an efficient and easy to use parallel virtual screening tool based on AutoDock 4.0. Chem. Cent. J. 2008, 2, 18. 10.1186/1752-153x-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant J. D.; McCammon J. A. BINANA: a novel algorithm for ligand-binding characterization. J. Mol. Graphics Modell. 2011, 29, 888–893. 10.1016/j.jmgm.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plowman S. J.; Ariotti N.; Goodall A.; Parton R. G.; Hancock J. F. Electrostatic interactions positively regulate K-Ras nanocluster formation and function. Mol. Cell. Biol. 2008, 28, 4377–4385. 10.1128/mcb.00050-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton A. H. A.; Hanley Q. S.; Verveer P. J. Graphical representation and multicomponent analysis of single-frequency fluorescence lifetime imaging microscopy data. J. Microsc. 2004, 213, 1–5. 10.1111/j.1365-2818.2004.01265.x. [DOI] [PubMed] [Google Scholar]
- Verveer P. J.; Bastiaens P. I. H. Evaluation of global analysis algorithms for single frequency fluorescence lifetime imaging microscopy data. J. Microsc. 2003, 209, 1–7. 10.1046/j.1365-2818.2003.01093.x. [DOI] [PubMed] [Google Scholar]
- Esposito A.; Gerritsen H. C.; Wouters F. S. Fluorescence lifetime heterogeneity resolution in the frequency domain by lifetime moments analysis. Biophys. J. 2005, 89, 4286–4299. 10.1529/biophysj.104.053397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns M. C.; Sun Q.; Daniels R. N.; Camper D.; Kennedy J. P.; Phan J.; Olejniczak E. T.; Lee T.; Waterson A. G.; Rossanese O. W.; Fesik S. W. Approach for targeting Ras with small molecules that activate SOS-mediated nucleotide exchange. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 3401–3406. 10.1073/pnas.1315798111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gremer L.; Merbitz-Zahradnik T.; Dvorsky R.; Cirstea I. C.; Kratz C. P.; Zenker M.; Wittinghofer A.; Ahmadian M. R. Germline KRAS mutations cause aberrant biochemical and physical properties leading to developmental disorders. Hum. Mutat. 2010, 32, 33–43. 10.1002/humu.21377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakhaeizadeh H.; Amin E.; Nakhaei-Rad S.; Dvorsky R.; Ahmadian M. R. The RAS-Effector Interface: Isoform-Specific Differences in the Effector Binding Regions. PLoS One 2016, 11, e0167145. 10.1371/journal.pone.0167145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos P. I.; Zhang C.; Bollag G.; Shokat K. M.; Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010, 464, 427–430. 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J. A.; Volmar A. Y.; Pavlopoulos S.; Mattos C. K-Ras Populates Conformational States Differently from Its Isoform H-Ras and Oncogenic Mutant K-RasG12D. Structure 2018, 26, 810–820 e4. 10.1016/j.str.2018.03.018. [DOI] [PubMed] [Google Scholar]
- Boriack-Sjodin P. A.; Margarit S. M.; Bar-Sagi D.; Kuriyan J. The structural basis of the activation of Ras by Sos. Nature 1998, 394, 337–343. 10.1038/28548. [DOI] [PubMed] [Google Scholar]
- Hall B. E.; Bar-Sagi D.; Nassar N. The structural basis for the transition from Ras-GTP to Ras-GDP. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 12138–12142. 10.1073/pnas.192453199. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.