

Abstract
Telomere shortening induces cellular senescence in proliferative cells. Yet, it is presently unclear how it is triggered in post‐mitotic cells such as cardiac myocytes. A new study by Anderson et al (2019) reports that during ageing of the heart, cellular senescence develops independently of telomere length, but is evoked by DNA damage, which preferentially accumulates at the telomere. Removal of senescent cells using senolytic drugs ameliorated cardiac hypertrophy and fibrosis and may inform novel approaches to improve the conditions for the ageing heart.
Subject Categories: Ageing; DNA Replication, Repair & Recombination; Metabolism
Ageing is the single strongest risk factor for heart disease. The senescent heart develops many alterations including structural changes such as vascular stiffening, increased left ventricular wall thickness and myocardial fibrosis, leading to diastolic dysfunction, increased afterload and a condition known as heart failure with preserved ejection fraction (HFpEF; Strait & Lakatta, 2012). At the functional level, there is a reduction of the maximal heart rate, changes in contractility (prolonged contraction and relaxation) and impaired sympathetic signalling. Presently, it is unclear what triggers ageing of the heart.
At the end of each eukaryotic chromosome, telomeres are found, which are special chromatin structures protecting chromosomal ends. Telomeric DNA is composed of non‐coding double‐stranded repeats of the DNA sequence TTAGGG, which is extended several thousand base pairs in length (5–12 kb in man and 25–40 kb in mice). The telomere terminates in a single‐stranded 3′ overhang that recoils into the double‐stranded telomere DNA, forming a complex loop structure (Fig 1). Telomeric DNA is bound by a protein complex termed shelterin, which is essential for the maintenance of the telomere structure. Telomeres are synthesised by an RNA–protein complex that extends telomeric DNA at the 3′ ends of chromosomes through telomerase reverse transcriptase (TERT) and the integral template‐containing telomerase RNA (TER). Both are expressed in the early embryo, embryonic stem cells and gametes but are absent from somatic cells. In the absence of TERT, each time a somatic cell will go through the cell cycle will lead to a reduction in telomere length. Telomere shortening below a critical length will destabilise the shelterin complex and the three‐dimensional structure of telomeric DNA and being recognised as DNA damage leading to the association of the formation of a telomere‐associated foci (TAF) containing proteins of the DNA damage response (DDR), which triggers a cellular senescence programme and cell cycle arrest.
Figure 1. Cellular senescence in cardiac myocytes is trigged by persistent DNA damage at telomeres.
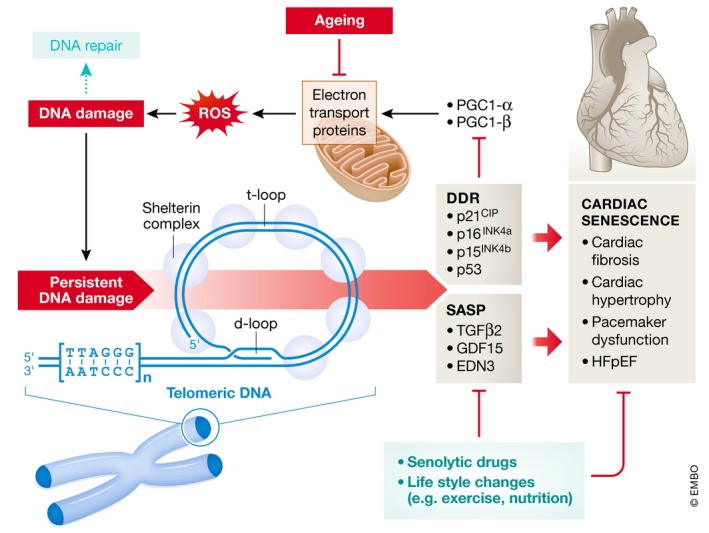
In the ageing heart, decreased expression of mitochondrial electron transport proteins leads to increased production of reactive oxygen species (ROS) and is causing persistent DNA damage at the telomere. A DNA damage response (DDR) is triggered, leading to increased expression of senescence genes and a senescence‐associated secretory phenotype (SASP). These changes are responsible for the development of cardiac senescence. Senolytic drugs are able to improve the conditions for the ageing heart. However, also lifestyle changes impacting on mitochondrial function and ROS production may have an effect.
Presently, it is unclear how cellular senescence is triggered in post‐mitotic cells such as cardiac myocytes, which divide at a very low frequency of < 1% (Bergmann et al, 2015). A study in The EMBO Journal (Anderson et al, 2019) provides novel insight into the process of cellular senescence in rarely dividing cells. Previously, the authors identified a novel mechanism of cellular senescence, which was caused by telomere damage and was found to be independent of telomere shortening (Hewitt et al, 2012). They reasoned that this mechanism might also be active in cardiac myocytes. The number of cardiac myocytes containing TAF complexes as well as the number of TAF per cell increased significantly with age. Interestingly, in old animals (> 30 month of age) cardiac myocytes was the cell type, which contained the highest number of TAF suggesting that age‐dependent accumulation of TAF is more likely to occur in cardiac myocytes than in any other cell type present in the heart. Similar results were obtained by chromatin immunoprecipitation demonstrating an enrichment of TAF marker proteins at telomeres in cardiac myocytes of ageing mice. STED microscopy allowed to measure the volume of individual telomeres, leading to the conclusion that TAF presence was not determined by a reduction in telomere length.
In order to link TAF formation to DNA damage, the authors were irradiating adult cardiac myocytes in vivo. While initially both telomeric DNA and non‐telomeric DNA were found to be damaged, only telomeric DNA damage persisted and triggered TAF formation. Similar results were obtained in cultured cardiac myocytes subjected to free radicals with the help of hydrogen peroxide, suggesting that oxidative stress may represent a trigger to induce DNA damage. DNA damage was also induced by a TRF1‐FokI fusion protein (Dilley et al, 2016), which targets the endonuclease FokI specifically to telomeres. Applying this tool to cultured neonatal cardiac myocytes induced telomere damage and cellular senescence characterised by the expression of senescence‐associated β‐galactosidase (SA‐βGal), the cyclin‐dependent kinase inhibitor p21CIP and increased cardiac hypertrophy. Significantly, when the same experiment was repeated and DNA damage was caused by a non‐telomere‐targeted endonuclease, DNA damage was repaired and did not result in the upregulation of a senescence phenotype.
In proliferative cells, the senescent state is associated with an expression of a number of pro‐inflammatory genes such as interleukins and chemokines collectively termed the senescence‐associated secretory phenotype (SASP; Coppé et al, 2010). However, SASP genes were not present in ageing adult cardiac myocytes. Instead, ageing cardiac myocytes secreted endothelin 3 (Edn3), transforming growth factor beta 2 (TGFβ2) and growth/differentiation factor 15 (Gdf15). In proliferative cells, secretion of SASP‐associated factors will lead to a spreading of the senescence phenotype. Could this also hold true in cardiac myocytes? To address this question, conditioned medium from old cardiac myocytes was applied, which induced smooth muscle α‐actin expression, SA‐β‐GAL and cessation of cell proliferation in cultured fibroblasts. Both, purified Edn3 and TGFβ2 had the same biological activity as conditioned medium, suggesting that these factors probably constitute the SASP in adult cardiac myocytes. Unfortunately, the authors did not characterise the effects of conditioned medium or of Edn3 and TGFβ2 on adult cardiac myocytes. Other investigators previously have shown that both GDF15 and TGFβ2 are induced in the infarcted heart (Frangogiannis, 2017), suggesting that production of these cytokines is not unique to the senescent heart.
Mitochondria have been identified as a driver of cellular senescence (Correia‐Melo et al, 2016). Removing mitochondria from cells was found by Anderson et al (2019) to be sufficient to rescue cells from cellular senescence. Transcriptome analysis of ageing hearts revealed a reduction in mitochondrial genes and in particular of those genes involved in electron transport. In order to study the impact of reactive oxygen species (ROS) on telomeres, a transgenic mouse line overexpressing the pro‐oxidant enzyme monoamine oxidase A (Mao‐A) in the heart was utilised. As was expected, the number of TAF‐positive adult cardiac myocytes was increased in the transgenic animals and reverted to normal levels after antioxidant treatment. The involvement of mitochondria was best documented through the use of the Polg null mutant strain, a model of accelerated ageing and mitochondrial dysfunction. Cardiac myocytes from these mice displayed increased expression of p21CIP and developed cardiac hypertrophy. Thus, overall a strong link is made between telomere erosion and ageing‐induced induction of cellular senescence in cardiac myocytes. Interestingly, there is a direct connection between mitochondrial dysfunction and telomere erosion by p53, which is part of the DDR and binds and represses PGC‐1α and PGC‐1β, key mitochondrial transcription factors, leading to impaired mitochondrial physiology and metabolism (Sahin et al, 2011).
Finally, the question arises, whether cellular senescence of cardiac myocytes can be stopped and therefore the condition of the ageing heart be improved? The authors addressed this by inducing clearance of senescent myocytes from the ageing heart. For this purpose, a mouse line (INK‐ATTAC) was utilised, which allows the ablation of senescent cells expressing p16Ink4A after small molecule treatment (Baker et al, 2016). Clearing senescent cells in ageing INK‐ATTAC mice resulted in a reduced number of TAFs in cardiac myocytes and, furthermore, caused a reduction in cardiac hypertrophy and fibrosis, while heart function was unaltered. Similar beneficial effects were obtained with the senolytic drug Navitoclax, which triggers apoptotic clearance of senescent cells. In this regard, it is noteworthy that novel senolytic compounds are currently developed (Yousefzadeh et al, 2018). A question remaining open is how the heart after senolytic treatment compensates for the loss of senescent myocytes? Fibrosis as well as myocyte size is reduced post‐treatment, while EDU incorporation, as well as the number of Ki67‐positive myocytes, is increasing. These data would suggest that cardiac myocytes in the ageing mouse heart are able to proliferate in response to senolytic treatment. In support of this, hypothetical mechanisms are the observation that some cardiac myocytes were labelled by Aurora B, a protein of the contractile ring, representing strong evidence in favour of the presence of mitotically active cardiac myocytes. Further research is of course required to substantiate this view. The report therefore establishes DNA damage specifically accumulating at telomeres due to impaired mitochondrial function and the accumulation of ROS to induce cellular senescence in the ageing heart and probably causing structural and functional impairment in this organ. Apart from novel treatment options such as senolytic drug treatment to remove senescent cardiac myocytes, this study may also lead to a better understanding of the molecular processes involved in cellular senescence and inform science‐based advice for lifestyle changes to improve the health of the ageing heart.
The EMBO Journal (2019) 38: e101571
See also: R Anderson et al (March 2019)
References
- Anderson R, Lagnado A, Maggiorani D, Walaszczyk A, Dookun E, Chapman J, Birch J, Salmonowicz H, Ogrodnik M, Jurk D, Proctor C, Correia‐Melo C, Victorelli S, Fielder E, Berlinguer‐Palmini R, Owens A, Greaves L, Kolsky KL, Parini A, Douin‐Echinard V et al (2019) Length‐independent telomere damage drives post‐mitotic cardiomyocyte senescence. EMBO J 38: e100492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, Khazaie K, Miller JD, van Deursen JM (2016) Naturally occurring p16 (Ink4a)‐positive cells shorten healthy lifespan. Nature 530: 184–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann O, Zdunek S, Felker A, Salehpour M, Alkass K, Bernard S, Sjostrom SL, Szewczykowska M, Jackowska T, Dos Remedios C, Malm T, Andrä M, Jashari R, Nyengaard JR, Possnert G, Jovinge S, Druid H, Frisén J (2015) Dynamics of cell generation and turnover in the human heart. Cell 161: 1566–1575 [DOI] [PubMed] [Google Scholar]
- Coppé JP, Desprez PY, Krtolica A, Campisi J (2010) The senescence‐associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5: 99–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia‐Melo C, Marques FD, Anderson R, Hewitt G, Hewitt R, Cole J, Carroll BM, Miwa S, Birch J, Merz A, Rushton MD, Charles M, Jurk D, Tait SW, Czapiewski R, Greaves L, Nelson G, Bohlooly‐Y M, Rodriguez‐Cuenca S, Vidal‐Puig A et al (2016) Mitochondria are required for pro‐ageing features of the senescent phenotype. EMBO J 35: 724–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilley RL, Verma P, Cho NW, Winters HD, Wondisford AR, Greenberg RA (2016) Break‐induced telomere synthesis underlies alternative telomere maintenance. Nature 539: 54–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangogiannis NK (2017) The role of transforming growth factor (TGF)‐β in the infarcted myocardium. J Thorac Dis 9(Suppl. 1): S52–S63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt G, Jurk D, Marques FD, Correia‐Melo C, Hardy T, Gackowska A, Anderson R, Taschuk M, Mann J, Passos JF (2012) Telomeres are favoured targets of a persistent DNA damage response in ageing and stress‐induced senescence. Nat Commun 3: 708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin E, Colla S, Liesa M, Moslehi J, Muller FL, Guo M, Cooper M, Kotton D, Fabian AJ, Walkey C, Maser RS, Tonon G, Foerster F, Xiong R, Wang YA, Shukla SA, Jaskelioff M, Martin ES, Heffernan TP, Protopopov A et al (2011) Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 470: 359–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strait JB, Lakatta EG (2012) Aging‐associated cardiovascular changes and their relationship to heart failure. Heart Fail Clin 8: 143–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefzadeh MJ, Zhu Y, McGowan SJ, Angelini L, Fuhrmann‐Stroissnigg H, Xu M, Ling YY, Melos KI, Pirtskhalava T, Inman CL, McGuckian C, Wade EA, Kato JI, Grassi D, Wentworth M, Burd CE, Arriaga EA, Ladiges WL, Tchkonia T, Kirkland JL et al (2018) Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 36: 18–28 [DOI] [PMC free article] [PubMed] [Google Scholar]