

Abstract
Aberrant function of the RNA‐binding protein TDP‐43 has been causally linked to multiple neurodegenerative diseases. Due to its large number of targets, the mechanisms through which TDP‐43 malfunction cause disease are unclear. Here, we report that knockdown, aggregation, or disease‐associated mutation of TDP‐43 all impair intracellular sorting and activity‐dependent secretion of the neurotrophin brain‐derived neurotrophic factor (BDNF) through altered splicing of the trafficking receptor Sortilin. Adult mice lacking TDP‐43 specifically in hippocampal CA1 show memory impairment and synaptic plasticity defects that can be rescued by restoring Sortilin splicing or extracellular BDNF. Human neurons derived from patient iPSCs carrying mutated TDP‐43 also show altered Sortilin splicing and reduced levels of activity‐dependent BDNF secretion, which can be restored by correcting the mutation. We propose that major disease phenotypes caused by aberrant TDP‐43 activity may be explained by the abnormal function of a handful of critical proteins, such as BDNF.
Keywords: brain‐derived neurotrophic factor, dendritic spines, hippocampus, LTP, protein sorting
Subject Categories: Molecular Biology of Disease, Neuroscience
Introduction
RNA‐binding proteins (RBPs) are involved in virtually all steps of post‐transcriptional control, regulating the fate and function of most RNA transcripts in the cell. Post‐transcriptional regulation by RBPs is highly complex, as a typical RBP can have hundreds of RNA targets, many of which overlap with those of other RBPs. TDP‐43 (encoded by the TARDBP gene) is a widely expressed RBP that controls different aspects of RNA metabolism, including mRNA splicing, stability, and trafficking (Lee et al, 2011). TDP‐43 has been identified as a major constituent of the protein inclusions found in most forms of amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD) (Sreedharan et al, 2008), as well as a significant fraction of other neurodegenerative syndromes, such as Alzheimer's disease and other dementias (Baloh, 2011; Lee et al, 2011; Wilson et al, 2011), regardless of whether the disease‐causing mutation is in TARDBP or in another gene, and even in sporadic cases. TDP‐43 is a 414‐aminoacid long protein with two RNA recognition motifs and a carboxy‐terminal glycine‐rich domain involved in protein–protein interactions. Most of the TARDBP mutations associated with disease described to date are within the latter domain (Mackenzie et al, 2010). The unique features of neurodegenerative diseases with TDP‐43 pathology are the mislocalization of TDP‐43 protein aggregates to the cytoplasm and the loss of its normal nuclear localization. It is unclear which of these are mechanistically linked to neurodegeneration, or whether TDP‐43 dysfunction involves gain of a toxic function, loss of a normal function, or both. Wild‐type TDP‐43 is intrinsically aggregation‐prone, but disease‐associated mutations appear to significantly accelerate aggregation and increase toxicity (Johnson et al, 2009). TDP‐43 negatively autoregulates its own expression levels (Ayala et al, 2011), leading to a vicious cycle in which cytoplasmic aggregation causes a drop in nuclear levels, triggering increased production, in turn leading to more cytoplasmic aggregation and nuclear depletion (Buratti & Baralle, 2012). Although there is good consensus that TDP‐43 is mechanistically linked to neurodegeneration, a principal difficulty in establishing direct connections between TDP‐43 malfunction and specific pathophysiological events arises from the large number of processes and mRNA targets that can be affected by TDP‐43 dysfunction in neurons and glial cells.
The neurotrophins are a family of secreted proteins that play vital functions in the developing and adult nervous system. They are essential for neuron survival and differentiation, as well as synapse formation, maintenance, and function; and through these activities affect neurodegeneration, regeneration, learning, and memory (Ibáñez & Simi, 2012; Park & Poo, 2013). Synaptic plasticity is tightly controlled by neurotrophins, particularly brain‐derived neurotrophic factor (BDNF), which elicits diverse and profound effects on central synapses, including enhancement of hippocampal long‐term potentiation (LTP) and formation of new spines (Lu et al, 2013). BDNF activity in the hippocampus is essential for the acquisition, maintenance, and retrieval of episodic memories (Lu et al, 2013). BDNF is produced and secreted in an activity‐dependent manner (Lu et al, 2013), thereby selectively recruiting active synapses, an essential mechanism underlying learning. Key to this activity is the sorting of BDNF to the regulated secretory pathway in neurons, allowing packaging of BDNF in vesicles competent for activity‐mediated release at synapses (Lu et al, 2013). The Vps10p domain protein‐sorting receptor Sortilin (encoded by the SORT1 gene) interacts with the pro‐domain of BDNF in the Golgi and is essential for BDNF sorting to the regulated secretory pathway in neurons (Chen et al, 2005). A common polymorphism in the human BDNF gene introduces a missense Val66Met mutation in the BDNF pro‐domain that impairs Sortilin binding (Chen et al, 2005), and affects its intracellular trafficking and activity‐dependent secretion (Egan et al, 2003; Chen et al, 2004). The Met allele has been associated with poorer episodic memory and abnormal hippocampal activation as measured by functional MRI (Egan et al, 2003; Hariri et al, 2003). These and other observations have provided substantial evidence linking activity‐dependent release of BDNF to long‐lasting synaptic changes and, ultimately, learning and memory in both animal models as well as humans.
Given the pleiotropic nature of TDP‐43 activity and the importance of neurotrophins in nervous system integrity and function, it is sensible to ask whether the two pathways may be connected. Intriguingly, TDP‐43 interacts with the mRNA encoding Sortilin and regulates its splicing (Polymenidou et al, 2011). Loss of TDP‐43 leads to the inclusion of an additional exon (i.e., exon 17b) in the Sort1 mRNA (Polymenidou et al, 2011), resulting in the generation and release of a truncated Sortilin isoform to the extracellular space in both mouse and human cells (Prudencio et al, 2012). Whether TDP‐43 aggregation or mutation also leads to production of soluble Sortilin is still unclear. FTLD patients show elevated levels of this isoform (Prudencio et al, 2012), but its contribution to TDP‐43 pathology remains unknown. We hypothesized that production of soluble Sortilin may represent a link between TDP‐43 dysfunction and aberrant neurotrophin activity, and thus investigated possible alterations in activity‐dependent BDNF secretion and synaptic plasticity as a result of abnormal TDP‐43 function.
Results
Tardbp knockdown induces production of a soluble form of Sortilin that impairs BDNF sorting to the regulated pathway and activity‐dependent secretion in mouse hippocampal neurons
We used a lentivirus vector carrying an shRNA targeting Tardbp mRNA (encoding TDP‐43 protein) to knockdown TDP‐43 expression in primary cultures of embryonic mouse hippocampal neurons. Tardbp mRNA levels were reduced by more than 80% in knockdown neurons compared to control scrambled shRNA, without affecting total SORT1 mRNA expression (Fig 1A). However, TDP‐43 knockdown significantly increased the level of SORT1 mRNA carrying exon 17b (Fig 1A). In mouse cells, it has been proposed that the extra region encoded by exon 17b facilitates the access of proteases to the stalk region of Sortilin, leading to enhanced protein shedding to the extracellular space (Prudencio et al, 2012). In agreement with the mRNA data, TDP‐43 knockdown significantly reduced TDP‐43 protein levels and resulted in the production of a truncated Sortilin protein in the culture supernatant (Fig 1B). In neuronal cells, activity‐dependent BDNF secretion depends upon BDNF sorting to the regulated secretory pathway (Mowla et al, 1999; Lou et al, 2005), a trafficking event that has been shown to require membrane‐bound Sortilin (Chen et al, 2005; Evans et al, 2011). In hippocampal neurons, colocalization of BDNF with secretogranin II (SCG2), a marker of secretory granules of the regulated pathway (Mowla et al, 1999; Lou et al, 2005), was significantly reduced after TDP‐43 knockdown compared to control scrambled shRNA (Fig 1C and D), suggesting impaired BDNF sorting in the absence of TDP‐43. In order to test whether impaired BDNF sorting after TDP‐43 knockdown in hippocampal neurons was indeed caused by altered Sort1 mRNA splicing and loss of membrane‐bound Sortilin, we performed a rescue experiment on the knockdown neurons using a bicistronic lentivirus to express shRNA targeting the 3′ end of Sort1 mRNA (to cancel endogenous Sortilin expression) and full‐length Sort1 cDNA lacking exon 17b (Appendix Fig S1). Hippocampal neurons doubly infected with Tardbp shRNA and Sortilin rescue viruses still lacked TDP‐43 expression, but expressed basal levels of Sort1 mRNA containing exon 17b and undetectable soluble Sortilin in the medium (Appendix Fig S2A and B). Under these conditions, colocalization of BDNF with SCG2 was rescued to normal levels compared to neurons that received only Tardbp shRNA virus (Fig 1E). Next, we assessed activity‐dependent BDNF secretion in hippocampal neuron cultures that had been infected with control or Tardbp shRNA lentiviruses. To facilitate the detection of the small amounts of endogenous BDNF released by these neurons, we used an in situ BDNF ELISA (Balkowiec & Katz, 2000). Basal BDNF secretion was low in the cultures, but 1‐h stimulation with KCl induced robust BDNF release in control cultures which was significantly suppressed by TDP‐43 knockdown (Fig 1F). Interestingly, the Sortilin rescue virus completely restored activity‐dependent BDNF secretion levels in TDP‐43 knockdown cells (Fig 1G). Together, these results indicated that the effects of TDP‐43 knockdown on BDNF sorting to the regulated secretory pathway and activity‐dependent secretion were mediated by altered splicing of Sort1 mRNA and not through another target.
Figure 1. Tardbp knockdown induces production of a soluble form of Sortilin that impairs BDNF sorting to the regulated pathway and activity‐dependent secretion in mouse hippocampal neurons.
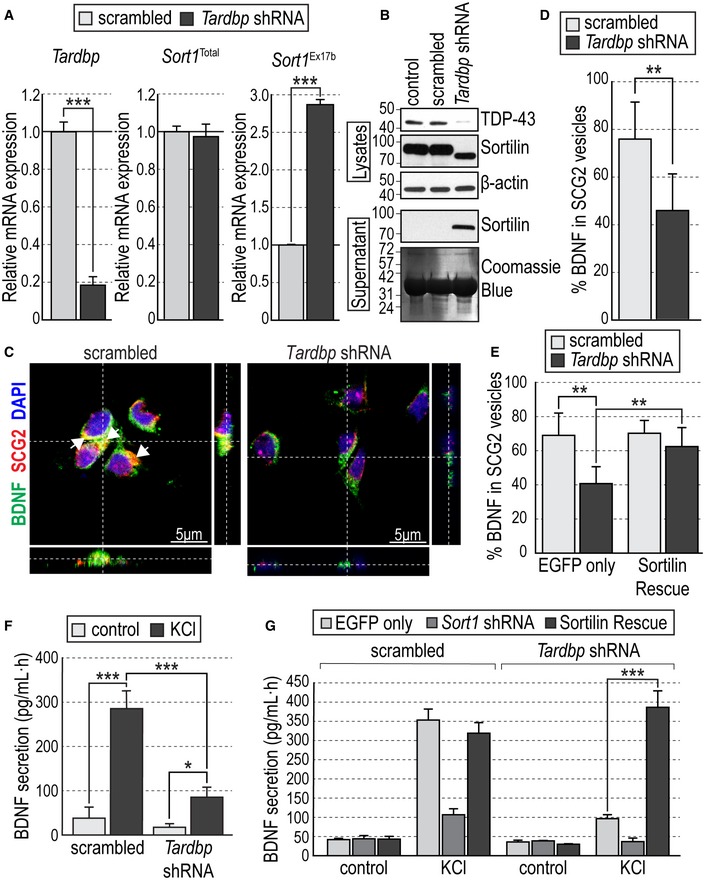
- Expression of Tardbp, total Sort1, and exon 17b Sort1 mRNAs quantified by qPCR in primary hippocampal neurons infected with either scrambled or Tardbp shRNA lentiviruses. Values were first normalized to Gapdh mRNA levels and then plotted as average ± SEM relative to the value of scrambled shRNA (N = 3 independent experiments; n = 3 wells per condition in each experiment). ***P < 0.001.
- Western blots of cell lysates and culture supernatants showing TDP‐43 and Sortilin expression in primary hippocampal neurons infected with scrambled or Tardbp shRNA lentiviruses. Uninfected cultures were used as an additional control. β‐actin immunoblotting and Coomassie Blue staining were used to control equal loading of lysates and supernatants, respectively. Molecular weights are indicated in kDa.
- Representative photomicrographs of cultured hippocampal neurons infected with scrambled or Tardbp shRNA lentiviruses, immunolabeled for BDNF (green), SCG2 (red), and counterstained with DAPI (blue) in orthogonal views (dashed lines) showing colocalization of BDNF and SCG2 in the soma (white arrows).
- Colocalization between BDNF‐ and SCG2‐positive vesicles expressed as percentage of total BDNF immunoreactivity ± SEM in cultured hippocampal neurons infected with scrambled or Tardbp shRNA lentiviruses (N = 3 independent experiments, n > 50 individual cells per condition in each experiment). **P < 0.01.
- Colocalization between BDNF‐ and SCG2‐positive vesicles in hippocampal neurons infected with scrambled or Tardbp shRNA lentiviruses together with either EGFP or Sortilin rescue viruses. Histogram bars show average ± SEM BDNF/SCG2 colocalization expressed as percentage of total BDNF immunoreactivity (N = 3 independent experiments, n > 50 individual cells per condition in each experiment). **P < 0.01.
- BDNF secretion induced by KCl treatment assessed by in situ BDNF ELISA in cultured hippocampal neurons infected with scrambled or Tardbp shRNA lentiviruses. Histogram bars show average ± SEM BDNF levels normalized to a standard curve (N = 3 independent experiments; n = 3 wells per condition in each experiment). *P < 0.05; ***P < 0.001.
- BDNF secretion in cultured hippocampal neurons infected with scrambled (left) or Tardbp (right) shRNA lentiviruses together with either EGFP, Sort1 shRNA, or Sortilin rescue viruses. Histogram bars show average ± SEM BDNF levels normalized to a standard curve (N = 3 independent experiments; n = 3 wells per condition in each experiment). ***P < 0.001.
Specific loss of TDP‐43 in CA1 hippocampal neurons impairs structural and functional plasticity in vivo through altered Sortilin splicing
Structural and functional plasticity, such as spine growth and LTP, are known to be exquisitely dependent on activity‐dependent release of BDNF (Korte et al, 1996; Tanaka et al, 2008; Lu et al, 2013; Edelmann et al, 2014). Synaptic plasticity at the connection between hippocampal CA3 and CA1 neurons (a.k.a. the Schaffer collateral) can be regulated by BDNF released in an activity‐dependent manner from post‐synaptic sites in CA1 neurons (Korte et al, 1996; Tanaka et al, 2008; Edelmann et al, 2015; Harward et al, 2016). BDNF secretion from presynaptic sites has also been proposed, possibly exerting distinct functions (Edelmann et al, 2014). To investigate whether TDP‐43 regulates Sort1 splicing and synaptic plasticity in vivo, we generated conditional mutant mice specifically deficient in TDP‐43 expression in CA1 neurons by crossing Tardbp fx/fx mice (Chiang et al, 2010) with mice carrying a CamKIIa CRE allele that specifically directs expression of CRE recombinase to those neurons (Tsien et al, 1996) (CamKIIa CRE;Tardbp fx/fx mice). We verified recombination driven by the CamKIIa CRE allele using a Rosa26 dTom reporter. At 1 month of age, recombination was predominantly targeted to neurons in the CA1 region of the hippocampus and their dendrites, but it affected other brain regions at later stages (Appendix Fig S3). All subsequent analyses were therefore performed in 1‐month‐old mice. Immunohistochemistry for TDP‐43 revealed fewer cells expressing TDP‐43 in hippocampal CA1 of CamKIIa CRE;Tardbp fx/fx mice compared to Tardbp fx/fx controls (Fig 2A and B). In addition, the remaining CA1 neurons that were still positive for TDP‐43 showed significantly lower levels of expression (Fig 2C), possibly due to hemizygotic deletion or slow turnover of the remaining TDP‐43 protein (as CamKIIa CRE expression begins 1–2 weeks postnatally). Importantly, the rest of the hippocampus (minus CA1) was not as severely affected (Fig 2B and C). Specific reduction in Tardbp mRNA and protein in hippocampal CA1 of CamKIIa CRE;Tardbp fx/fx mice was confirmed by qPCR (Fig 2D) and Western blotting (Fig 2E and F), respectively, in extracts of micro‐dissected CA1 area. Again, no significant differences were detected outside CA1 in the hippocampus of the mutant mice. Analysis of Sort1 mRNA revealed significantly elevated levels of mRNA containing exon 17b in the CA1 region of CamKIIa CRE;Tardbp fx/fx mice compared to Tardbp fx/fx controls, without differences in total levels (Fig 2G). In agreement with this, the level of truncated Sortilin protein, corresponding to the soluble isoform, was increased in CA1 of CamKIIa CRE;Tardbp fx/fx mice compared to controls (Fig 2E and H). Together, these data indicated that loss of TDP‐43 in hippocampal CA1 resulted in altered Sort1 mRNA splicing and increased levels of a truncated Sortilin protein isoform, identical in molecular weight to the soluble fragment recovered from culture supernatants.
Figure 2. CA1‐specific knockout of TDP‐43 increases levels of exon 17b Sort1 mRNA and soluble Sortilin protein.
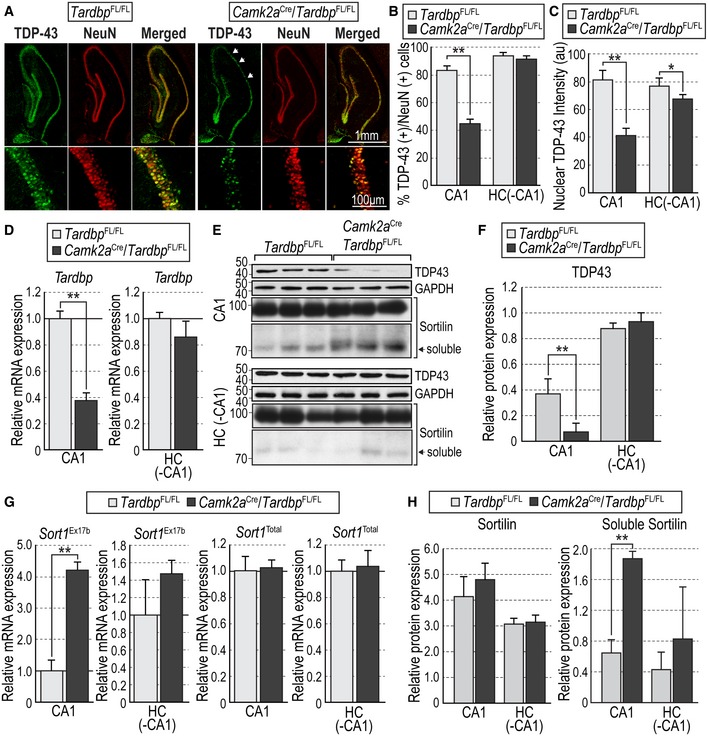
- Representative photomicrographs of TDP‐43 (green) and NeuN (red) immunohistochemistry in hippocampal CA1 of Tardbp fx/fx and CamKIIa CRE;Tardbp fx/fx mice. Arrows denote areas of depleted TDP‐43 immunoreactivity in the mutant.
- Percentage of TDP‐43‐positive cells relative to NeuN cells in CA1 and the remaining hippocampus (HC [‐CA1]) of Tardbp fx/fx and CamKIIa CRE;Tardbp fx/fx mice. Results are presented as average ± SEM (N = 3 mice per condition; 5 sections per mouse). **P < 0.01.
- TDP‐43 intensity in TDP‐43/NeuN double‐positive cells in CA1 and the remaining hippocampus of Tardbp fx/fx and CamKIIa CRE;Tardbp fx/fx mice. Results are presented as average ± SEM (N = 3 mice per condition; 5 sections per mouse). *P < 0.05; **P < 0.01.
- Expression of Tardbp mRNA in micro‐dissected CA1 and the remaining hippocampus of Tardbp fx/fx and CamKIIa CRE;Tardbp fx/fx mice quantified by qPCR. Results are presented as average ± SEM relative to levels in Tardbp fx/fx mice, after normalization to Gapdh mRNA (N = 3 mice per condition; **P < 0.01).
- Western blots of micro‐dissected CA1 and the remaining hippocampus showing expression of TDP‐43, full‐length, and truncated (soluble) Sortilin, and GAPDH in Tardbp fx/fx and CamKIIa CRE;Tardbp fx/fx mice. Bands corresponding to soluble Sortilin are indicated (black arrows). GAPDH immunoblotting was used to control for equal loading. Molecular weights are indicated in kDa.
- TDP‐43 expression in micro‐dissected CA1 and the remaining hippocampus of Tardbp fx/fx and CamKIIa CRE;Tardbp fx/fx mice. Results are presented as average ± SD of densitometry relative to GAPDH (N = 3 mice per condition; **P < 0.01).
- Expression of exon 17b Sort1 and total Sort1 mRNAs in micro‐dissected CA1 and the remaining hippocampus of Tardbp fx/fx and CamKIIa CRE;Tardbp fx/fx mice quantified by qPCR. Results are presented as average ± SD relative to levels in Tardbp fx/fx mice, after normalization to Gapdh mRNA (N = 3 mice per condition; **P < 0.01).
- Expression of full‐length (left) and truncated (soluble, right) Sortilin protein in micro‐dissected CA1 and the remaining hippocampus of Tardbp fx/fx and CamKIIa CRE;Tardbp fx/fx mice. Results are presented as average ± SD of densitometry relative to GAPDH (N = 3 mice per condition; **P < 0.01).
Next, we looked at possible structural and functional alterations in hippocampal CA1 region of CamKIIa CRE;Tardbp fx/fx mice. We found decreased complexity in apical and basal dendritic arbors of CA1 neurons in the mutant mice compared to Tardbp fx/fx controls, as assessed by Golgi staining followed by Sholl analysis (Fig 3A and B). In addition, we also found reduced numbers of spines in dendrites of CA1 neurons of CamKIIa CRE;Tardbp fx/fx mice compared to Tardbp fx/fx controls (Fig 3C and D). In order to assess whether these deficits were caused by altered Sort1 mRNA splicing, we performed stereotactic injections of Sortilin rescue virus in hippocampal CA1 region of these mice (Fig 3E). Analysis of mRNA expression in micro‐dissected CA1 area from CamKIIa CRE;Tardbp fx/fx mice injected with this virus confirmed the reduced levels of Tardbp mRNA in the mutant mice (Fig 3F left) and, importantly, showed normalized levels of Sort1 mRNA containing exon 17b (Fig 3F right), without significant alteration in total Sort1 mRNA levels (Fig 3F center). Under these conditions, spine density in CA1 dendrites was restored to normal levels (Fig 3G). Functional plasticity was assessed as long‐term potentiation induced by theta burst stimulation (TBS‐LTP) in CA1 neurons, a specific form of hippocampal LTP that is critically dependent on activity‐mediated BDNF secretion (Chen et al, 1999). Hippocampal slices from control (Tardbp fx/fx) mice showed pronounced potentiation induced by TBS that was sustained for at least 3 h (Fig 4A). In contrast, synaptic potentiation was not maintained in slices derived from CamKIIa CRE;Tardbp fx/fx mutant mice (Fig 4A and Appendix Fig S4A). Interestingly, the lack of TBS‐LTP in the mutant was completely rescued by stereotactic injection of Sortilin rescue virus in hippocampal CA1 (Fig 4B and Appendix Fig S4B), confirming that it was due to abnormal Sort1 mRNA splicing. Moreover, addition of BDNF to the bath could also rescue the LTP deficit in the mutant slices, without inducing further change in wild‐type slices (Fig 4C and Appendix Fig S4C), suggesting that the deficits were due to low extracellular BDNF in the mutant. Finally, we investigated whether the structural and functional defects observed in CA1 neurons of CamKIIa CRE;Tardbp fx/fx mice affected spatial memory, a form of memory mediated by hippocampal circuitry. In the Barnes maze, control mice always showed significant preference for the target quadrant when tested for either short‐term (20–30 min), long‐term (24 h), or remote (14 days) memory (Fig 4D). In contrast, mutant mice did not show any preference for the target quadrant at any time tested (Fig 4D), indicating impaired spatial memory after Tardbp deletion in hippocampal CA1 neurons.
Figure 3. CA1‐specific knockout of TDP‐43 reduces dendrite complexity and spine number through altered Sortilin splicing.
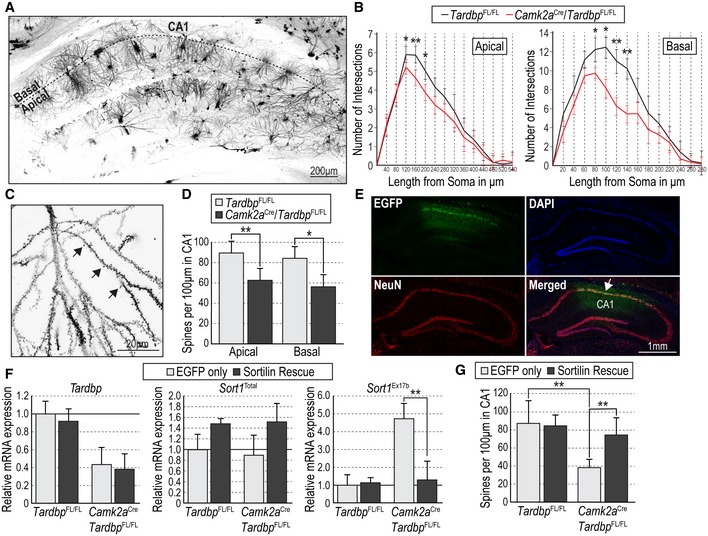
- Representative photomicrograph of Golgi staining in hippocampus of a Tardbp fx/fx mouse.
- Sholl analysis of apical and basal dendritic arbors of CA1 pyramidal neurons from Tardbp fx/fx and CamKIIa CRE;Tardbp fx/fx mice. Results are presented as average ± SEM (N = 3 mice per condition, 20 neurons per mouse). *P < 0.05; **P < 0.01.
- High‐magnification representative photomicrograph of Golgi staining of the dendritic arbor of a CA1 pyramidal neuron in a Tardbp fx/fx mouse. Black arrows denote selected dendritic spines.
- Spine density in apical and basal dendrites of CA1 pyramidal neurons of Tardbp fx/fx and CamKIIa CRE;Tardbp fx/fx mice. Results are presented as average ± SD (N = 3 mice per condition, 15 sections per mouse; *P < 0.05; **P < 0.01).
- Representative photomicrographs of a CamKIIa CRE;Tardbp fx/fx hippocampus after stereotaxic injection of EGFP‐expressing lentivirus immunostained for EGFP (green) and NeuN (red), and counterstained with DAPI (blue). The injected area within CA1 is indicated in the merged panel.
- Expression of Tardbp (left), total Sort1 (center), and exon 17b Sort1 (right) mRNAs quantified by qPCR in micro‐dissected CA1 of Tardbp fx/fx and CamKIIa CRE;Tardbp fx/fx mice after injection of EGFP‐expressing lentivirus or Sortilin rescue virus. Results are presented as average ± SEM relative to levels in Tardbp fx/fx mice injected with EGFP virus, after normalization to Gapdh mRNA. (N = 3 mice per condition). **P < 0.01.
- Spine density in apical dendrites of CA1 pyramidal neurons of Tardbp fx/fx and CamKIIa CRE;Tardbp fx/fx mice after injection of EGFP‐expressing lentivirus or Sortilin rescue virus. Results are presented as average ± SEM (N = 3 mice per condition, 15 sections per mouse). **P < 0.01.
Figure 4. CA1‐specific knockout of TDP‐43 impairs spatial memory and abolishes BDNF‐dependent TBS‐LTP through altered Sortilin splicing.
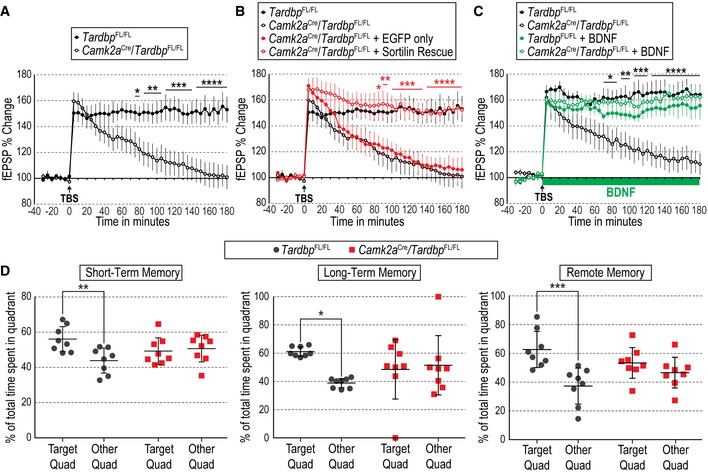
- Percentage of change in field excitatory post‐synaptic potential (fEPSP) recorded from hippocampal area CA1 after theta burst stimulation (TBS) in Schaffer collaterals of hippocampal slices from Tardbp fx/fx and CamKIIa CRE;Tardbp fx/fx mice. Results are presented as average normalized to t = 0 ± SEM (N = 7 slices from three mice per condition). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Representative traces are shown in Appendix Fig S4A.
- Percentage fEPSP change in CA1 after TBS in Schaffer collaterals of hippocampal slices from Tardbp fx/fx and CamKIIa CRE;Tardbp fx/fx mice stereotactically injected with EGFP‐expressing lentivirus or Sortilin rescue virus (red) superimposed to the same data shown in panel (A) (black). Results are presented as average normalized to t = 0 ± SEM (N = 7 slices from three mice per condition). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Representative traces are shown in Appendix Fig S4B.
- Percentage fEPSP change in CA1 after TBS in Schaffer collaterals of hippocampal slices from Tardbp fx/fx and CamKIIa CRE;Tardbp fx/fx mice under control conditions (black) or with BDNF (200 ng/ml) perfused from t = 0 (green). Results are presented as average normalized to t = 0 ± SEM (N = 7 slices from 3 mice per condition). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Representative traces are shown in Appendix Fig S4C.
- Percentage of total time spent in the target quadrant or in the remaining three quadrants of the Barnes maze for Tardbp fx/fx and CamKIIa CRE;Tardbp fx/fx mice at three different probe times: 20–30 min after first training session (short‐term memory) and 24 h or 14 days after last training session (long‐term or remote memory, respectively). Results are presented as average ± SEM (N = 8 mice per condition; *P < 0.05, **P < 0.01, ***P < 0.001).
Aggregation or disease‐associated mutation of TDP‐43 impairs activity‐dependent BDNF secretion through altered Sortilin splicing
While the results presented so far, based on Tardbp knockdown and deletion, reveal a novel physiological role for TDP‐43 in the regulation of activity‐dependent BDNF secretion and hippocampal synaptic plasticity, they do not establish whether these activities have a pathophysiological significance in the context of aberrant TDP‐43 function due to aggregation or mutation. We begun to address this question using a model that allows investigation of effects mediated by TDP‐43 aggregation within cells without affecting the expression of the endogenous gene (Budini et al, 2015). It has been shown that exogenous expression of tandem repeats of the prion‐like Q/N‐rich region of human TDP‐43 fused to additional TDP‐43 protein sequences trigger formation of aggregates that are able to sequester endogenous TDP‐43, depleting its nuclear levels and inducing malfunction at the pre‐mRNA splicing level (Budini et al, 2015). In our experiments, expression of this construct (TARDBP 12xQN) from a lentivirus vector in hippocampal neurons resulted in 60% reduction of TDP‐43 nuclear immunoreactivity (Fig 5A and B). While total levels of endogenous Tardbp or Sort1 mRNAs were not affected in neurons expressing TARDBP 12xQN, Sort1 mRNA containing exon 17b was greatly increased, to an even higher level than that seen after Tardbp knockdown (Fig 5C). In agreement with our previous results, activity‐dependent secretion of BDNF from these neurons was severely impaired (Fig 5D), but could be rescued by restoring expression of full‐length Sort1 mRNA lacking exon 17b (Fig 5E). These data indicated that, similar to Tardbp knockdown and deletion, aggregation of endogenous TDP‐43 also impairs activity‐dependent BDNF secretion through altered Sort1 mRNA splicing.
Figure 5. Aggregation of TDP‐43 impairs activity‐dependent BDNF secretion through altered Sortilin splicing.
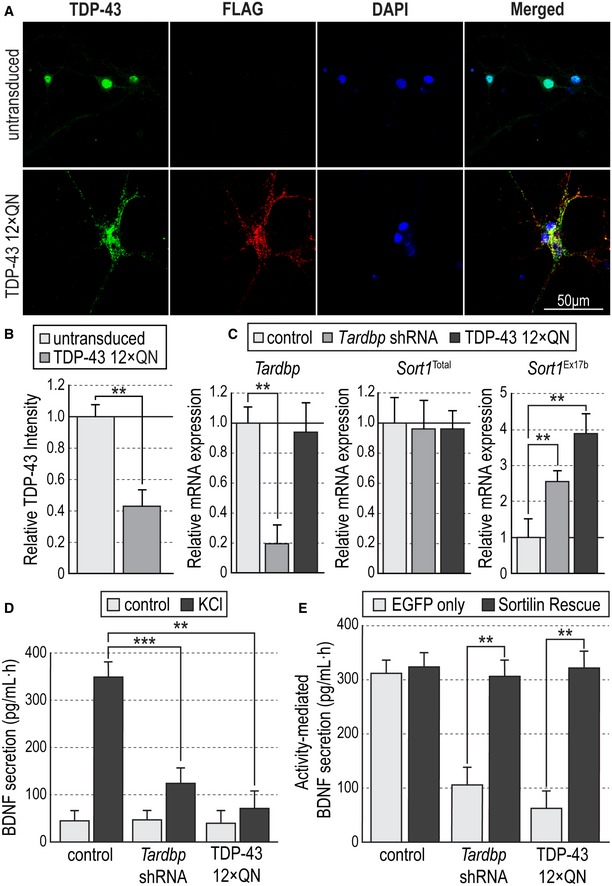
- Representative photomicrographs of cultured hippocampal neurons immunostained for TDP‐43 (green), FLAG (red), and counterstained with DAPI (blue) in control conditions (untransduced) or infected with lentivirus expressing FLAG‐tagged human TDP‐43 12xQN construct.
- TDP‐43 intensity in cell nuclei of untransduced or TDP‐43 12xQN‐infected hippocampal neurons normalized to mean nuclear intensity in untransduced neurons. Results are presented as average ± SEM (N = 3 independent experiments; n = 3 wells per condition in each experiment). Student's t‐test; **P < 0.01.
- Expression of Tardbp (left), total Sort1 (center), and exon 17b Sort1 (right) mRNAs quantified by qPCR in hippocampal neurons untransduced (control) or infected with lentiviruses expressing Tardbp shRNA or human TDP‐43 12xQN constructs. Values were first normalized to Gapdh mRNA levels and then plotted as average ± SEM relative to levels in untransduced neurons (N = 3 independent experiments; n = 3 wells per condition in each experiment). One‐way ANOVA with Tukey's post hoc; **P < 0.01.
- BDNF secretion induced by KCl treatment as assessed by in situ BDNF ELISA in cultured hippocampal neurons untransduced (control) or infected with lentiviruses expressing Tardbp shRNA or human TDP‐43 12xQN constructs. Histogram bars show average ± SEM BDNF levels normalized to a standard curve (N = 5 independent experiments; n = 5 wells per condition in each experiment). **P < 0.01; ***P < 0.001.
- BDNF secretion in cultured hippocampal neurons untransduced (control) or infected with lentiviruses expressing Tardbp shRNA or human TDP‐43 12xQN constructs together with either EGFP or Sortilin rescue viruses. Histogram bars show average ± SEM BDNF levels normalized to a standard curve (N = 5 independent experiments; n = 5 wells per condition in each experiment). **P < 0.01.
We investigated the impact of disease‐associated mutation of TDP‐43 on Sort1 mRNA splicing and activity‐dependent BDNF secretion in mouse hippocampal neurons co‐infected with Tardbp shRNA lentivirus, to knockdown endogenous TDP‐43 expression, and a second lentivirus construct to express mutant variants of human TDP‐43 (Appendix Fig S1). Exogenous expression of human TDP‐43 at physiological levels required the inclusion of the 3′ UTR of the human TARDBP mRNA, which mediates the autoregulation of TDP‐43 expression (Ayala et al, 2011). Expression of wild‐type human TARDBP lacking the 3′ UTR in Tardbp knockdown neurons yielded total levels of Tardbp mRNA and TDP‐43 protein that were significantly higher than the endogenous levels (Fig 6A left, and B). This resulted in 50% reduction in Sort1 mRNA containing exon 17b compared to the uninfected control (Fig 6A right). In contrast, inclusion of the 3′ UTR restored total Tardbp mRNA and TDP‐43 protein expression in knockdown neurons to levels indistinguishable from uninfected controls (Fig 6A left, and B), which remained stable by 3 days post‐transduction (Appendix Fig S5A). Importantly, this also normalized the inclusion of exon 17b in Sort1 mRNA (Fig 6A, right and Appendix Fig S5C), without altering total Sort1 mRNA levels (Fig 6A, center and Appendix Fig S5B). Interestingly, even slight overexpression (≈20%) of human TARDBP mRNA above endogenous levels (Appendix Fig S5A, 2 days post‐transduction) resulted in significant reduction of exon 17b inclusion below uninfected controls (Appendix Fig S5C), highlighting the exquisite sensitivity to TDP‐43 dosage of this splicing event. We used this assay to assess the capacity of different disease‐associated mutant variants of human TDP‐43 to restore normal levels of Sort1 mRNA containing exon 17b in mouse hippocampal neurons lacking endogenous TDP‐43 expression. Many TDP‐43 mutations have been associated with sporadic and familial cases of ALS and FTLD (Lee et al, 2011). We tested the effects of four mutations (i.e., A315T, M337V, Q343R, and N267S) found within the carboxy‐terminal region of the human TDP‐43 protein. Expression levels of mutant variants of TARDBP mRNAs and TDP‐43 proteins were comparable to endogenous levels in the transduced neurons (Fig 6A and B). Interestingly, although wild‐type TARDBP was able to restore Sort1 mRNA containing exon 17b back to control levels in knockdown neurons, three of the mutants tested left significantly elevated levels of exon 17b; one of them, M337V, by as much as 50% over the normal baseline (Fig 6A right). Levels of total Sort1 mRNA were unaffected (Fig 6A center). Importantly, the effects of the TDP‐43 mutants on Sort1 mRNA splicing were sufficient to impair activity‐dependent BDNF secretion in these neurons, to a degree that was directly proportional to the inclusion of Sort1 exon 17b (Fig 6C). Similar to TDP‐43 knockdown and aggregation, co‐infection with the Sortilin rescue virus completely rescued activity‐dependent BDNF secretion to normal levels in the neurons expressing mutant variants of human TDP‐43 (Fig 6D). These results indicated that disease‐associated mutation of TDP‐43 can also impair activity‐dependent BDNF secretion through altered Sort1 mRNA splicing.
Figure 6. Disease‐associated mutations in TDP‐43 increase levels of exon 17b Sort1 mRNA and impair activity‐dependent BDNF secretion in mouse hippocampal neurons and patient‐derived human neurons.
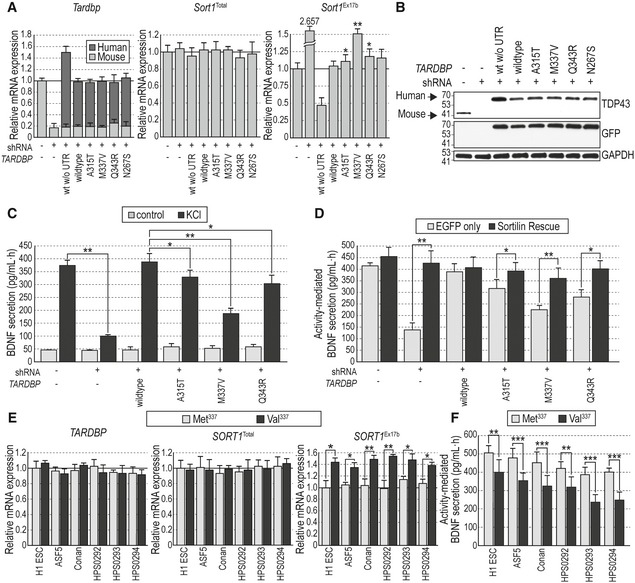
- Expression of Tardbp (left), total Sort1 (center), and exon 17b Sort1 (right) mRNAs quantified by qPCR in hippocampal neurons infected with Tardbp shRNA and mutant human TARDBP lentivirus constructs as indicated. Absolute levels of mouse Tardbp and human TARDBP mRNAs were determined using a standard curve (see Methods) and added together to assess total Tardbp mRNA levels relative to control uninfected cells after normalization to Gapdh mRNA. Results are presented as average ± SEM (N = 3 independent experiments; n = 3 wells per condition in each experiment). *P < 0.05; **P < 0.01.
- Western blots of cell lysates showing total TDP‐43 expression in primary hippocampal neurons infected with Tardbp shRNA and mutant human TARDBP lentivirus constructs as indicated. The antibody used here detects both mouse and human TDP‐43 proteins. GAPDH immunoblotting was used to control for equal loading. Molecular weights are noted in kDa. Bands corresponding to mouse and human TDP‐43 are indicated.
- BDNF secretion induced by KCl treatment assessed by in situ BDNF ELISA in cultured hippocampal neurons infected with Tardbp shRNA and mutant human TARDBP lentivirus constructs as indicated. Histogram bars show average ± SEM BDNF levels normalized to a standard curve (N = 5 independent experiments; n = 5 wells per condition in each experiment). *P < 0.05; ***P < 0.001.
- BDNF secretion induced by KCl treatment in cultured hippocampal neurons infected with Tardbp shRNA and mutant human TARDBP lentivirus constructs together with either EGFP or Sortilin rescue viruses. Histogram bars show average ± SEM BDNF levels normalized to a standard curve (N = 5 independent experiments; n = 5 wells per condition in each experiment). *P < 0.05; **P < 0.01.
- Expression of TARDBP (left), total SORT1 (center), and exon 17b SORT1 (right) mRNAs quantified by qPCR in human neurons derived from the indicated stem cells carrying either wild‐type Met337 (gray bars) or disease‐associated Val337 (black bars) TARDBP alleles. Results are presented as average ± SEM (N = 3 independent experiments; n = 3 wells per condition in each experiment). *P < 0.05; **P < 0.01.
- BDNF secretion in stem‐cell‐derived human neurons carrying wild‐type Met337or Val337 TARDBP alleles, infected with proBDNF lentivirus 5 days prior to depolarization by KCl treatment. Results are presented as average ± SEM (N = 3 independent experiments; n = 5 wells per condition in each experiment). **P < 0.01; ***P < 0.001.
Human neurons derived from patient iPSCs carrying mutated TDP‐43 show altered Sortilin splicing and reduced levels of activity‐dependent BDNF secretion
Having established that mouse hippocampal neurons reconstituted with human M337V TDP‐43 display abnormally elevated levels of Sort1 mRNA containing exon 17b and impaired activity‐dependent BDNF secretion, we sought to determine whether this mutation could have similar effects in human neurons. We used CRISPR/Cas9 gene editing to introduce the M337V mutation in three human pluripotent stem cells, namely H1 embryonic stem cells (ESCs) and ASF5 and Conan induced pluripotent stem cells (iPSCs) (Appendix Fig S6A). Using the same methodology, we corrected the M337V mutation in three iPSC isolates derived from a patient carrying this mutation (Egawa et al, 2012). These two strategies resulted in a set of 6 human pluripotent stem cell lines carrying the M337V mutation in heterozygosity, and corresponding isogenic control lines carrying two normal copies of the wild‐type Met337 allele. We derived neurons from these 12 cell lines using a standard neuronal differentiation protocol designed to preferentially yield excitatory cortico/hippocampal‐like neurons (Egawa et al, 2012) (Appendix Fig S6B–D). Human neurons derived from all lines displayed similar levels of total TARDBP and SORT1 mRNAs regardless of the status of the TDP‐43 codon at position 337 (Fig 6E). However, all carriers of Val337 displayed elevated levels of SORT1 mRNA containing exon 17b, between 40 and 60% increase over that detected in neurons derived from stem cells carrying the wild‐type Met337 allele (Fig 6E). Because of the way in which human neurons were obtained (i.e., 21‐day differentiation from iPSCs and ESCs), we could not use the in situ BDNF ELISA to quantify secretion of endogenous BDNF. To facilitate the detection of BDNF released to the medium, we transduced the neurons with a lentivirus expressing human proBDNF from the human SYN1 promoter and used a conventional ELISA to assess BDNF levels in the culture supernatant. Interestingly, all the neurons carrying Val337 in the TARDBP gene showed a significant decrease in activity‐dependent BDNF secretion (Fig 6F). These results indicate that human neurons carrying a disease‐associated mutation in the gene encoding TDP‐43 display abnormal activity‐dependent secretion of BDNF, an important determinant of synaptic plasticity and learning. Crucially, our gene editing studies demonstrate that this defect is directly caused by the mutation and not due to other factors.
Discussion
A principal difficulty in elucidating specific pathways by which RBPs can cause disease lies in the large number of targets that can be affected by anyone of these proteins. From the point of view of network theory, RBPs can be thought of as highly connected hubs, linked to hundreds or thousands of other proteins. As many other complex systems, cellular networks follow a power‐law distribution: Some nodes have a very high number of connections to other nodes (i.e., hubs), while the majority of nodes have very few (Barabási & Oltvai, 2004). Such networks, also known as scale‐free networks, are very resistant to accidental failures, but vulnerable to coordinated attacks on their principal hubs. At the two extremes, the pathophysiology of diseases caused by RBP malfunction could be explained either by the summation of the effects of all their many targets (as in a random network model) or by failure of only a small subset, those that are also network hubs themselves (as predicted by a scale‐free network model) (Barabási & Oltvai, 2004). Numerous studies have taken advantage of different genome‐wide approaches to identify possible TDP‐43 targets important in ALS/FTLD pathophysiology [e.g., (Polymenidou et al, 2011; Sephton et al, 2011; Xiao et al, 2011; White et al, 2018)]. These studies have identified hundreds, sometimes thousands, of mRNAs bound or somehow affected by TDP‐43, mainly based on correlative evidence. To the best of our knowledge, our study is among the first to perform rescue experiments showing that major pathophysiological consequences of TDP‐43 malfunction can be reverted by restoring the correct expression of only one of its targets. Sortilin controls the production and function of neurotrophins, in turn some of the most powerful and pleiotropic molecules in the nervous system. BDNF, arguably the most important neurotrophin in the brain, is also a highly connected hub, controlling a myriad of processes, from axonal growth and synaptogenesis, to synapse plasticity and function. Based on our findings, we suggest that major disease phenotypes caused by aberrant TDP‐43 activity may be explained by the abnormal function of a handful of critical “hub” proteins, such as BDNF.
A common form of FTLD is caused by mutations in the GRN gene encoding progranulin, a secreted glycoprotein with multiple functions both inside and outside the nervous system (Baker et al, 2006; Cruts et al, 2006). FTLD caused by GRN gene mutations also displays the characteristic TDP‐43 aggregates, suggesting that progranulin may be upstream of TDP‐43 in disease pathogenesis. In neurons, progranulin has been attributed some neurotrophic activities, including an ability to induce neurite outgrowth (Van Damme et al, 2008). Although Sortilin has been described as cellular receptor for progranulin (Hu et al, 2010), studies on Sort1 −/− knockout mice have shown that the neurotrophic effects of progranulin are independent of Sortilin (Gass et al, 2012). Soluble Sortilin, produced as a result of exon 17b inclusion, can still bind progranulin (Prudencio et al, 2012), but the consequence of this interaction is unclear. It has alternatively been proposed to have detrimental effects, by blocking progranulin binding to neuronal receptors (Prudencio et al, 2012), or beneficial effects, by protecting progranulin from endocytosis while preserving its neurotrophic activity (Molgaard et al, 2016). Based on the former line of thought, it has been proposed that Sortilin antagonists and strategies to decrease Sortilin levels may be able to restore normal progranulin levels in FTLD patients (Lee et al, 2014). Our present studies suggest that such approaches may have severe negative effects by compromising activity‐dependent BDNF secretion, a fundamental process required for normal synaptic function and cognition.
The mechanisms by which TDP‐43 dysfunction cause neuropathology remain contentious. The ubiquitous presence of TDP‐43 aggregates in many neurodegenerative diseases propelled the notion of a neurotoxic mode of action, thus supporting a gain‐of‐function model for TDP‐43 pathophysiology. On the other hand, concomitant depletion of TDP‐43 from the cell nucleus has favored a loss‐of‐function mechanism. It has also been shown that TDP‐43 aggregation and TDP‐43 knockdown affect the expression levels of a common set of proteins (Prpar Mihevc et al, 2016), suggesting that TDP‐43 neuropathology involves, at least in part, a loss of normal TDP‐43 function. Several animal models have shown that overexpression of TDP‐43, wild type or mutant, is in itself toxic in a dose‐dependent manner and able to produce neurodegeneration, often without formation of aggregates (Baloh, 2011; Arnold et al, 2013), suggesting that TDP‐43 inclusions are not necessary to promote neurotoxicity. Two recent studies generated knock‐in mice carrying a Q331K substitution, equivalent to a mutation found in human patients, and found mRNA splicing patterns in TDP‐43 target genes consistent with enhanced TDP‐43 activity, suggesting a gain‐of‐function mechanism (Fratta et al, 2018; White et al, 2018). Intriguingly, although Q331K knock‐in mice showed reduced levels of Sort1 mRNA containing exon 17b, they displayed significantly elevated levels of mutant Tardbp mRNA and TDP‐43 protein, by as much as 45% (White et al, 2018). Several studies, including the present one, have shown that even a relatively mild elevation (≈20%) in TDP‐43 levels is sufficient to reduce Sort1 exon 17b inclusion below the endogenous baseline, suggesting that the enhanced exclusion of Sort1 exon 17b observed in Q331K knock‐in mice could have been due to increased TDP‐43 levels, rather than a gain‐of‐function in the TDP‐43 molecule itself. In the present study, we describe a previously unknown consequence of TDP‐43 dysfunction, namely impaired activity‐dependent secretion of BDNF, triggered by either knockdown, aggregation, or disease‐associated mutation of TDP‐43. All three strategies led to elevated levels of Sort1 mRNA containing exon 17b, and in all cases, activity‐dependent BDNF secretion could be rescued by restoring the expression of full‐length Sort1 mRNA lacking exon 17b, regardless of how TDP‐43 dysfunction was achieved. These results reveal a hitherto unidentified neuropathological phenotype that can be attributed to the loss of normal TDP‐43 function.
Activity‐dependent secretion of BDNF is crucial for normal brain function. In humans carrying the Val66Met mutation in the BDNF pro‐domain, disruption of this process leads to abnormal hippocampal and cortical activation, and has been linked to impaired episodic, working, visual, and verbal memories [reviewed in (Dincheva et al, 2012)]. The Val66Met mutation impairs activity‐dependent BDNF secretion by disrupting proBDNF binding to Sortilin (Egan et al, 2003; Chen et al, 2004, 2005). Our results show that a similar phenotypic outcome results from increased inclusion of Sort1 exon 17b caused by abnormal TDP‐43 function, leading to production of a soluble form of Sortilin that diverts trafficking of proBDNF away from the regulated secretory pathway, thereby impairing activity‐dependent BDNF secretion. A disease‐associated mutation in TDP‐43 produced similar effects in human neurons. We have shown that, at least in mice, this resulted in reduced numbers of dendritic spines, a main site of synapse formation, as well as impaired hippocampal TBS‐LTP, both of which could be rescued by restoring membrane‐anchored Sortilin in the affected neurons. These mice showed impaired spatial memory in the Barnes maze. Based on these results, we suggest that a similar mechanism may be present in human subjects with abnormal TDP‐43 function, either due to aggregation or mutation, which could potentially explain cognitive defects present in some ALS/FTLD patients.
In summary, we have uncovered a previously unknown connection between two critical signaling hubs in neurons, TDP‐43 and BDNF, mediated by the alternative splicing of the mRNA encoding the protein‐sorting receptor Sortilin. Using rescue experiments, we have validated this functional relationship, and shown that restoring the expression of only one critical target, among the many hundreds linked to TDP‐43, is sufficient to revert major neuropathological events caused by TDP‐43 dysfunction.
Materials and Methods
Lists of plasmids, antibodies, qPCR primers, CRISPR templates, and human pluripotent stem cell lines used in this study are presented in Appendix Tables S1–S5, respectively.
Plasmids
A summary of all the plasmids used is presented in Appendix Table S1. pLKO.1 vector (Addgene plasmid #10878) was modified to replace the puromycin resistance cassette with an mCherry reporter by restriction digestion. mCherry construct was obtained by high‐fidelity PCR using the Q5® High‐Fidelity Polymerase (NEB) from the pmCherry‐N1 construct (Clontech). Target sequence used for knockdown of mouse Tardbp was GAGTGGAGGTTATGGTCAA and for mouse Sort1 GTTGAAGCTCTTTAAGCCA. Both constructs included a CTCGAG hairpin. We developed a bicistronic lentiviral vector (pESU6L) for concurrent knockdown and cDNA expression in neurons, with shRNA expression under a U6 promoter and cDNA overexpression under a CMV‐enhanced human synapsin I (SYN1) promoter. All subcloning was done using Gibson Assembly (NEB) of cassettes generated by high‐fidelity PCR. The lentiviral expression cassette was obtained from FUGW plasmid (Addgene plasmid #14883); U6 shRNA cassette from pLKO.1‐TRC cloning vector (Addgene plasmid #10878); IRES‐EGFP cassette and multiple cloning site from the pIRES2‐EGFP vector (Clontech). The enhanced human SYN1 promoter was obtained by Gibson Assembly using the CMV enhancer from the pcDNA3.1(+) vector (Invitrogen) and the human SYN1 promoter; the latter was obtained by high‐fidelity PCR from genomic DNA isolated from HEK293T cells (ATCC) using the DNeasy Blood & Tissue Kit (Qiagen). A second vector (pESL) was derived from pESU6L by site‐directed mutagenesis to remove the U6 RNA expression cassette and was used for cDNA expression that did not require concurrent knockdown. Full‐length cDNAs encoding human TDP‐43, mouse Sortilin, and human proBDNF were reverse transcribed using the GoScript Reverse Transcription System (Promega) from RNA obtained from either WiCell HES‐01 human embryonic stem cells (for human constructs) or mouse primary cortical tissue (for mouse constructs). cDNAs were subcloned into the pESL or pESU6L vectors by standard restriction site cloning techniques. Mutations in human TDP‐43 and proBDNF were introduced using the Q5 Site‐directed Mutagenesis Kit (NEB) and verified by Sanger sequencing. The human TDP‐43‐3′UTR construct was made by modifying the pESL‐hTDP‐43 vector. The entire 3′UTR of human TARDBP (2,813 bp) was amplified by high‐fidelity PCR using a forward primer matching the 5′ end of the UTR and a reverse primer targeting the 3′ end of the polyA cassette, and subcloned into the pESL‐hTDP‐43 vector through Gibson Assembly (NEB).
Animals
C57BL/6NTac mice were used for primary culture studies. Embryos of both sexes were used in these studies. B6(SJL)‐Tardbptm1.1Pcw/J (Tardbp fx) mice, B6.Cg‐Tg(Camk2a‐cre)T29‐1Stl/J (CamKIIa CRE), and B6.Cg‐Gt(ROSA)26Sortm9(CAG‐tdTomato)Hze/J (Rosa26 dTom) mice were obtained from The Jackson Laboratory. Tardbp fx/fx mice were crossed with CamKIIa CRE mice to generate CamKIIa CRE;Tardbp fx/+ mice which were then intercrossed to bring the Tardbp fx allele to homozygosity. CamKIIa CRE mice were intercrossed with Rosa26 dTom mice to homozygosity to demonstrate CRE recombinase expression in CamKIIa CRE mice. The data collected in this study were derived from 1‐month‐old offsprings of CamKIIa CRE;Tardbp fx/+ males mated to Tardbp fx/fx non‐littermate females. qPCR, Western blotting, and Golgi studies on CamKIIa CRE;Tardbp fx/fx mice used animals from both sexes. All other studies used only male mice. All animals were housed in the National University of Singapore Comparative Medicine vivarium in a 12‐h light/dark cycle on a standard chow diet. All animal procedures were approved by the National University of Singapore Institutional Animal Care and Use Committee.
Neuron primary culture
Pregnant C57BL/6NTac females were euthanized at the 17th day of gestation by administration of sodium pentobarbital, with euthanasia confirmed with cervical dislocation. The hippocampal structures were aseptically removed from the embryos and digested in Papain (Sigma‐Aldrich) for 30 min at 37°C and rinsed in neuronal maintenance media. Neurons were triturated into a single cell suspension and counted with a hemocytometer. They were then transferred to coverslips or cell culture vessels coated with 0.01% poly‐d‐lysine (Sigma‐Aldrich) and 1 μg/ml mouse Cultrex® Laminin (Trevigen). Cultured hippocampal neurons were maintained in serum‐free defined Neurobasal media supplemented with B27 (Invitrogen), GlutaMAX (Invitrogen), and 50 μg/ml Gentamicin (Invitrogen) at 37°C in 5% CO2 conditions.
BDNF ELISA
We adapted the BDNF Emax® ImmunoAssay Kit (Promega) for in situ detection of endogenous BDNF produced in mouse neuron cultures (in situ BDNF ELISA). Nunc MaxiSorp® flat‐bottom 96‐well plates were coated with anti‐BDNF monoclonal antibody provided in the kit (G700, Promega, 1:1,000) for 16 h at 4°C in 2.5 mM Na bicarbonate buffer pH 9.7. Plates were washed once with sterile PBS and coated with 0.1% poly‐d‐lysine (Sigma‐Aldrich) diluted in PBS at 37°C overnight. Plates were washed twice using sterile PBS and coated with 5 μg/ml Cultrex® Laminin (Trevigen) diluted in PBS. Hippocampal neurons were seeded onto 96‐well plates (1,000 cells per well) in neuron culture medium as described above and simultaneously infected with lentiviral particles at a five multiplicity of infection (MOI). Medium was replaced on the following day. After 5 days in vitro, the medium was refreshed 2 h prior to treatment. Neurons were depolarized with 56 mM KCl for 1 h, placed on ice, washed twice with ice‐cold PBS, and then washed with mild detergent buffer (0.1% Triton X‐100 in PBS) three times of 5 min. The wells were then treated with blocking agent provided in the kit. Detection of bound BDNF followed manufacturer's instructions for all subsequent ELISA detection steps. Detection of human BDNF in culture supernatants of human neurons transduced with proBDNF‐expressing lentivirus was done with the BDNF Emax® ImmunoAssay Kit (Promega) according to manufacturer's instructions.
Lentivirus generation and transduction
HEK293FT cells (Invitrogen) were maintained in OptiMEM with 5% fetal bovine serum and appropriate antibiotics (Invitrogen). Cells were co‐transfected with lentiviral constructs for shRNA or overexpression with the packaging vectors Δ8.9 and Vsv‐g (Addgene) using FuGENE®‐6 (Promega). Supernatants containing viral particles were aseptically collected 3 days post‐transfection, filtered using 0.4‐μm PES filters (Sartorius), concentrated 50–100 times, and dialyzed into sterile Dulbecco's phosphate‐buffered saline (DPBS) by centrifugation in 100 kDa cutoff Amicon Ultra‐15 centrifugal filters (Millipore). Viruses were aliquoted and snap‐frozen in liquid‐nitrogen. Viruses were titrated by serial dilution on HEK293T cells (for shRNA constructs) or serial dilution on primary dissociated cortical neurons and quantified for mCherry or EGFP expression with a Ti‐E inverted fluorescence microscope. Lentiviral transduction of hippocampal neurons was performed after 1 day in vitro (DIV1) at a multiplicity of infection (MOI) = 5 and left for 24 h before the media was changed. The infected cultures were used at 5 days post‐infection.
Stereotaxic injection of lentivirus
Male mice were anesthetized with isoflurane, fixed onto a stereotaxic frame (RWD Life Science) equipped with a digital displacement transducer and a syringe pump (Harvard Apparatus). Mice were kept deeply anesthetized by constant isoflurane infusion and monitored by pinch withdrawal. Animals were injected with 1uL of lentiviral constructs at a titer of approximately 1 × 109 IFU per ml in the CA1 region of the dorsal hippocampus (± 2.1 mm lateral to midline, 2.0 mm anterior to bregma, and 1.4 mm ventral to dura) using a gas‐tight Neuros micro‐injector (Hamilton). The injections were performed at 200 nl/min, with the needle left in place for 5 min post‐injection to minimize backflow. Mice were allowed to recover for 7 days to induce expression of the transgene.
Hippocampus microdissection
For analysis of mRNA and protein expression, mice were anesthetized with isoflurane 7 days post‐injection and perfused intracardially with cold PBS. Brains were removed and placed into cold HEPES buffer saline sucrose (HBSS) (Invitrogen), containing 3% sucrose and 1 mM HEPES buffer (Sigma‐Aldrich). Hippocampi were dissected, and 2‐mm‐thick transverse sections were made using a surgical scalpel. Manual isolation of the CA1 area and the remaining of the hippocampal region was performed under a stereomicroscope (Olympus). Microdissection of EGFP‐positive CA1 area from hippocampi following lentiviral transduction was performed under fluorescence stereomicroscope (Olympus).
RNA isolation and qPCR
For isolation of RNA from cell cultures, cells were washed with ice‐cold PBS and RNA isolated using the ReliaPrep RNA Miniprep System (Promega) as per manufacturer's instructions. For isolation of RNA from micro‐dissected hippocampal tissue, we used the Aurum Total RNA Fatty and Fibrous Tissue Kit (Bio‐Rad) as per manufacturer's instructions. Tissue fragments were homogenized with a bead homogenizer (Bertin) in the buffer of the Aurum kit. RNA samples were digested with DNAse I (Promega) prior to reverse transcription with the GoScript Reverse Transcription Kit (Promega). qPCR was performed on Viia‐7 and ABI QuantStudio five instruments (Applied Biosystems) with primers indicated in Appendix Table S2. To facilitate the quantitative analysis of total transcripts of human and mouse Tardbp mRNA, standard curves for each transcript were obtained using Tardbp cDNA fragments of the corresponding species purified by gel extraction of restriction enzyme digests and quantified by NanoDrop quantification (Thermo Fisher). Standard curves were obtained during each qPCR run to calculate absolute levels of mRNA transcripts for mouse and human Tardbp.
Protein isolation and Western blotting
Cultured cells were first briefly washed in PBS, and cell lysis was performed in 50 mM Tris/HCl pH 7.4 in 1 mM EDTA, 270 mM sucrose, 1% (v/v) Triton X‐100 in the presence of protease and phosphatase inhibitors (Nacalai Tesque) as per manufacturer's instructions. The cellular extracts were then centrifuged at 13,000 ×g at 4°C using a benchtop centrifuge for 5 min whereupon supernatants were collected. For analysis of micro‐dissected hippocampal tissue, the tissue fragments were placed in RIPA buffer (Abcam), homogenized with a bead homogenizer (Bertin), and cleared by centrifugation at 13,000 ×g. Protein concentrations of cellular or tissue extracts were determined by the Bradford Assay (Bio‐Rad). SDS–PAGE was followed by Western blotting. The antibodies used are listed in Appendix Table S3. Immunoblots were developed using the ECL Western Blotting Kit or the SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher) and exposed to Kodak X‐Omat AR films. Densitometric analysis of X‐ray films was done using the ImageJ software (NIH).
Immunocytochemistry and immunohistochemistry, Golgi, and CLARITY
Hippocampal neurons cultured on coverslips were briefly washed in PBS, fixed for 15 min in 4% formaldehyde solution (Sigma‐Aldrich), and blocked in 10% normal donkey serum (Millipore) with 1% bovine serum albumin (Thermo Fisher) in Tris‐buffered saline (TBS). Fixed cells were then incubated for 16 h at 4°C with the appropriate antibodies as listed in Appendix Table S2, followed by incubation in fluorophore‐conjugated secondary antibodies (Molecular Probes). Coverslips were mounted onto microscopy slides using ProLong Diamond anti‐fade mounting media (Invitrogen). Confocal laser scanning microscopy was performed on a Leica SP8 microscope.
For immunohistochemistry, 1‐month‐old mice were deeply anesthetized and intracardially perfused with cold phosphate‐buffered saline (PBS) followed by 4% formaldehyde (Sigma‐Aldrich). Brains were dissected and post‐fixed for 16 h at 4°C. After cryoprotection in 40% sucrose for 16 h, 40‐μm coronal sections were collected on a cryostat. Antigen retrieval was performed by boiling sections in citrate buffer in a pressure cooker prior to immunostaining. Sections were blocked in 5% donkey serum in TBS before incubation with appropriate primary and secondary antibodies as listed in Appendix Table S3.
Golgi staining using superGolgi Kit (Bioenno) was used to visualize dendrite arbors and spines in CA1 hippocampal neurons as per manufacturer's specifications. Briefly, brains from mice that had been intracardially perfused with cold PBS were immersed in Golgi‐Cox solution for 10 days at 4°C, then transferred into post‐impregnation buffer, and sectioned with a vibratome (Campden Instruments) at a thickness of 200 μm. Slices were mounted, stained, and developed as per manufacturer's instructions. Microscopy images of pyramidal layers of the CA1 of the hippocampus were captured using a 63× oil‐immersion objective on a SP8 confocal microscope (Leica) using confocal reflection microscopy for 3D visualization.
The CLARITY staining procedure (Chung & Deisseroth, 2013) was used to visualize EGFP‐positive neuronal processes and spines of neurons infected with Sortilin rescue lentivirus. After intracardial perfusion with cold PBS, mice were reperfused with cold hydrogel solution containing bis‐acrylamide and 4% paraformaldehyde together with VA‐044 as a polymerization initiator. Brains were incubated in hydrogel solution for 3 days to allow further diffusion of hydrogel solution into the tissue, degassed under vacuum, overlaid with mineral oil to prevent ingress of atmospheric oxygen, and incubated at 37°C for additional 3 h. Brains were sectioned to a width of 1 mm on a vibratome (Campden Instruments) and passively cleared in clearing solution containing SDS and boric acid for 1 week. Sections were washed in an excess of PBS and 0.01% Tween‐20 (PBST) for 24 h, and stained in primary antibody against EGFP (Appendix Table S3) for 48 h at 37°C under gentle rocking. Sections were washed for 24 h in PBST and incubated in appropriate secondary antibody for 2 days at 37°C with gentle rocking. Sections were washed for 24 h, incubated in 85% glycerol overnight at 4°C, and mounted on MAS‐coated slides (Matsunami). Samples were imaged using an SP8 confocal microscope (Leica) using a 40× oil‐immersion objective.
Image analysis
For colocalization studies, overlap between BDNF and secretogranin II (SCG2) immunoreactivity was quantified as a percentage of total BDNF staining and averaged across each field. More than 50 neurons were quantified in triplicate wells for each condition. For quantification of TDP‐43 staining in hippocampal CA1, TDP‐43‐positive nuclei were counted as a percentage of total NeuN‐positive nuclei in the hippocampal CA1 region and in the remaining hippocampal formation. Nuclear TDP‐43 intensity was also quantified by averaging TDP‐43 immunoreactivity in TDP‐43‐positive nuclei. In both cases, quantifications were derived from three mice per condition (5 sections per animal). For dendrite length and spine analysis, apical and basal dendrites of CA1 pyramidal neurons were quantified separately using Imaris (Bitplane) on both Golgi‐stained and CLARITY images. For Sholl analysis, the number of intersections at 20 μm (basal) or 40 μm (apical) intervals from the cell soma of Golgi‐stained neurons was quantified. The soma of each neuron was superimposed over the center point of the circle, and dendrite crossings were manually counted and plotted as a number of intersections against distance from soma. Dendritic spines across the entire cross‐section of each dendrite were counted and averaged over a 100 μm distance.
Electrophysiology
Our electrophysiological procedures are described in greater detail in Shetty et al (2015). Briefly, mice were decapitated after anesthetization using CO2 and the brains were quickly removed into cold (4°C) artificial cerebrospinal fluid (ACSF: 124 mM NaCl, 3.7 mM KCl, 1.0 mM MgSO4, 2.5 mM CaCl2, 1.2 mM KH2PO4, 24.6 mM NaHCO3, and 10 mM d‐glucose, equilibrated with 95% O2/5% CO2 (carbogen; total consumption 16 l/h). From each mouse, 6–8 transverse hippocampal slices (400‐μm‐thick) were prepared from the right hippocampus by using a manual tissue chopper. Slices were incubated at 32°C in an interface chamber (Scientific System Design) at an ACSF flow rate of 1 ml/min. One monopolar, lacquer‐coated, stainless steel electrode (5 MΩ; AM Systems) was positioned at an adequate distance within the stratum radiatum of the CA1 region for stimulating synaptic inputs of one neuronal population, thus evoking field excitatory post‐synaptic potential (fEPSP) from Schaffer collateral‐commissural‐CA1 synapses. For recording, another electrode was placed in the CA1 apical dendritic layer. The signals were amplified by a differential amplifier (Model 1700, AM Systems) and were digitized using a CED 1401 analog‐to‐digital converter (Cambridge Electronic Design). After the pre‐incubation period of 2 h, an input–output curve (afferent stimulation versus fEPSP slope) was plotted prior to experiments. To set the test stimulus intensity, a fEPSP of 40% of its maximal amplitude was determined. Biphasic constant current pulses were used for stimulation. Late long‐term potentiation (L‐LTP) was induced using a theta burst stimulation (TBS) protocol which consists of 50 bursts (consisting of 4 stimuli) at an inter‐stimulus interval of 10 ms. The 50 bursts were applied over a period of 30 s at 5 Hz (or at an inter‐burst interval of 200 ms). The slopes of fEPSPs were monitored online. The baseline was recorded for 30 min. For baseline recording and testing at each time point, four 0.2 Hz biphasic constant current pulses (0.1 ms/polarity) were used. fEPSPs were recorded every 5 min from 30 min before stimulation up to 180 min after stimulation across the CA1‐CA3 Schaffer collaterals and normalized against t = 0.
Barnes maze
The Barnes maze spatial memory test was carried out as previously described (Bach et al, 1995). Spatial cues were placed around the maze and these were kept constant throughout the study. On the first day of training, the mouse was placed in the escape box for 1 min. The animal was then placed in the center of the maze inside a black chamber. As in all subsequent sessions, the chamber was removed after 10 s, whereupon a buzzer (80 dB) and a light (400 lux) were turned on, and the mouse was free to explore the maze for 3 min or until the mouse entered the escape tunnel. The tunnel was always located underneath the same hole, which was randomly determined for each mouse. The platform was moved every day by 90° to avoid any odorant cue but the spatial cues and the tunnel position remained the same. Mice were trained using this protocol once daily for 4 days. For the test sessions, the escape tunnel was removed and the mouse was allowed to freely explore the maze for a maximum of 3 min to assess spatial memory. The short‐term memory test was conducted 20–30 min after the first training. Long‐term and remote memory tests were conducted 24 h and 14 days after the last training, respectively. To quantify the preference for the trained target quadrant, time spent in the target quadrant and time spent in the other quadrants were measured.
Human pluripotent stem cells: maintenance, gene editing by CRISPR/Cas9, and neuronal differentiation
All human induced pluripotent stem cells (iPSCs) and embryonic stem cell (ESC) lines used in this study (Appendix Table S4) were approved by the NUS Institutional Review Board (IRB). Human induced pluripotent stem cells HPS0290/0291/0292 were initially cultured on gamma‐irradiated SNL feeders in Primate ES cell media (ReproCELL) containing 4 ng/ml bFGF (Stem Cell Technologies). They were then transitioned to a feeder‐free TeSR‐E8 system (Stem Cell Technologies) on Geltrex (Invitrogen)‐coated plates. Other human pluripotent stem cell lines (H1 embryonic stem cells, ASF5, and Conan) were maintained in TeSR‐E8 media on Geltrex‐coated plates. Feeder‐free human pluripotent stem cells were used for all subsequent experiments.
crRNA guide sequence was designed using the CRISPR Design Tool, identifying TCTTATAGTGGCTCTAATTC as the guide sequence with TGG as PAM. Targeting constructs for genome editing had 50 bp homology arms (Appendix Table S5) and were made as a gBlock® Gene Fragment (IDT). crRNA together with Alt‐R® tracrRNA‐FAM, Alt‐R® S.p. HiFi Cas9 Nuclease 3NLS (IDT), and targeting constructs was transfected using Lipofectamine RNAiMAX (Invitrogen) into pluripotent stem cells and grown for 48 h. FACS sorting using a 488 laser against the FAM‐tag on the tracrRNA was performed using a MoFlo XDP Cell Sorter (Beckman Coulter) into Geltrex‐coated 96‐well plates. Clones were grown in CloneR® hPSC Cloning Supplement (Stem Cell Technologies) and expanded for genomic DNA extraction (Qiagen) and continued culture. To identify positive edited sequences, ARMS‐PCR of gDNA using the following primers was performed: In‐F: GCAGCACTACAGAGCAGTTGGGGTAGGG, In‐R: GTTCTGCTGGCTGG CTAACATGCCAAT, Out‐F: TCCAATGCCGAACCTAAGCACAATAGCA, Out‐R: AAAGCCTCCATTAAAACCACTGCCCGAC; and wild‐type and heterozygous Met337Val clones were identified by agarose gel electrophoresis. The Met337Val mutation was introduced in wild‐type hPSC lines as heterozygote.
A scheme depicting the main steps of neuronal differentiation of pluripotent stem cells is shown in Appendix Fig S6B. Human pluripotent stem cells were passaged on Geltrex (Invitrogen)‐coated plates at 1:10 split ratio 1 day before in TeSR‐E8 media. The protocol for differentiation of human pluripotent stem cells into human neurons was adapted from Li et al (2011). Cells were washed in pre‐warmed DMEM/F12 media and differentiated in neural induction medium (NIM) containing advanced DMEM/F12:Neurobasal media (1:1 ratio), 1× N2 supplement, 1× B27 supplement, 1× GlutaMAX, 5 μg/ml BSA (Invitrogen), 3 μM SB431542 (Sigma‐Aldrich), 4 μM CHIR99021, 0.1 μM Compound E, and 10 ng/ml recombinant human LIF (Stem Cell Technologies). Media was changed every day until 7th day of differentiation, when cells were passaged onto Geltrex‐coated plates in NIM without Compound E, but containing 2 μM ROCK inhibitor Y‐27632 (Wako). The cells (i.e., neural progenitors) were then seeded at 20,000 cells/cm2 on plates coated with 0.01% poly‐d‐ornithine (Sigma‐Aldrich) and 20 μg/ml Cultrex® Laminin (Trevigen) in NIM containing only SB431542, CHIR99021, and hLIF. Two days later, the cells were differentiated toward a neuronal lineage in neuronal differentiation medium containing DMEM/F12, 1× N2 supplement, 1× B27 supplement (Invitrogen), 300 ng/ml dibutyrl‐cAMP (Sigma‐Aldrich), 5 μg/ml l‐ascorbic acid (Sigma‐Aldrich), 10 ng/ml brain‐derived neurotrophic factor (BDNF), and 10 ng/ml glial cell line‐derived neurotrophic factor (GDNF) (R&D Systems) for 7 days with media changes every other day. After 7 days of differentiation, culture medium was changed to a neuronal maturation medium containing Neurobasal media, 1× B27 supplement (Invitrogen), 300 ng/ml dibutyrl‐cAMP (Sigma‐Aldrich), 5 μg/ml l‐ascorbic acid (Sigma‐Aldrich), 10 ng/ml BDNF, and 10 ng/ml GDNF. After 2 days in maintenance culture, the medium was changed and the cells were infected proBDNF lentiviral particles at MOI = 5. Medium was changed every other day thereafter. Assessment of activity‐dependent BDNF secretion was done in cell cultured supernatant harvested 5 days after lentiviral infection. The cells were then lysed for RNA analysis as explained above.
Statistical analysis
Results are presented as average ± standard error of the mean (SEM). Unless otherwise indicated, all statistical tests were performed using 2‐way ANOVA with Sidak post hoc analysis. Statistical significance: *P < 0.05; **P < 0.01; ***P < 0.001, and ****P < 0.0001.
Author contributions
The concept of the study was conceived by JYT and CFI. JYT performed all experiments, except the electrophysiological studies, which were performed by L‐WW. CFI and SS provided supervision. JYT prepared the figures and CFI wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Source Data for Appendix
Review Process File
Source Data for Figure 1B
Source Data for Figure 2E
Source Data for Figure 6B
Acknowledgements
We thank Ling Shuo‐Chien for providing Tardbp fx/fx mice, Edward Koo for CamKIIa CRE mice, Francisco Baralle for 12xQN construct, Chai Chou and Lim Kah Leong for ASF5 cells, Chris Shaw for Conan cells, Wong Peiyan for help with behavioral studies, and Goh Ket Yin and Eunice Sim for technical assistance. Support for this research was provided by grants from the National University of Singapore (Strategic ODPRT Award), Ministry of Education (MOE) of Singapore; the Swedish Research Council and the European Research Council (339237‐p75ntr). We thank Drs. Francis Lee and Shawn Je for advice.
The EMBO Journal (2019) 38: e100989
References
- Arnold ES, Ling S‐C, Huelga SC, Lagier‐Tourenne C, Polymenidou M, Ditsworth D, Kordasiewicz HB, McAlonis‐Downes M, Platoshyn O, Parone PA, Da Cruz S, Clutario KM, Swing D, Tessarollo L, Marsala M, Shaw CE, Yeo GW, Cleveland DW (2013) ALS‐linked TDP‐43 mutations produce aberrant RNA splicing and adult‐onset motor neuron disease without aggregation or loss of nuclear TDP‐43. Proc Natl Acad Sci USA 110: E736–E745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala YM, De Conti L, Avendaño‐Vázquez SE, Dhir A, Romano M, D'Ambrogio A, Tollervey J, Ule J, Baralle M, Buratti E, Baralle FE (2011) TDP‐43 regulates its mRNA levels through a negative feedback loop. EMBO J 30: 277–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach ME, Hawkins RD, Osman M, Kandel ER, Mayford M (1995) Impairment of spatial but not contextual memory in CaMKII mutant mice with a selective loss of hippocampal LTP in the range of the theta frequency. Cell 81: 905–915 [DOI] [PubMed] [Google Scholar]
- Baker M, Mackenzie IR, Pickering‐Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C et al (2006) Mutations in progranulin cause tau‐negative frontotemporal dementia linked to chromosome 17. Nature 442: 916–919 [DOI] [PubMed] [Google Scholar]
- Balkowiec A, Katz DM (2000) Activity‐dependent release of endogenous brain‐derived neurotrophic factor from primary sensory neurons detected by ELISA in situ . J Neurosci 20: 7417–7423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baloh RH (2011) TDP‐43: the relationship between protein aggregation and neurodegeneration in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. FASEB J 278: 3539–3549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barabási A‐L, Oltvai ZN (2004) Network biology: understanding the cell's functional organization. Nat Rev Genet 5: 101–113 [DOI] [PubMed] [Google Scholar]
- Budini M, Romano V, Quadri Z, Buratti E, Baralle FE (2015) TDP‐43 loss of cellular function through aggregation requires additional structural determinants beyond its C‐terminal Q/N prion‐like domain. Hum Mol Genet 24: 9–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E, Baralle FE (2012) TDP‐43: gumming up neurons through protein‐protein and protein‐RNA interactions. Trends Biochem Sci 37: 237–247 [DOI] [PubMed] [Google Scholar]
- Chen G, Kolbeck R, Barde Y‐A, Bonhoeffer T, Kossel A (1999) Relative contribution of endogenous neurotrophins in hippocampal long‐term potentiation. J Neurosci 19: 7983–7990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z‐Y, Patel PD, Sant G, Meng C‐X, Teng KK, Hempstead BL, Lee FS (2004) Variant brain‐derived neurotrophic factor (BDNF) (Met66) alters the intracellular trafficking and activity‐dependent secretion of wild‐type BDNF in neurosecretory cells and cortical neurons. J Neurosci 24: 4401–4411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z‐Y, Ieraci A, Teng H, Dall H, Meng C‐X, Herrera DG, Nykjaer A, Hempstead BL, Lee FS (2005) Sortilin controls intracellular sorting of brain‐derived neurotrophic factor to the regulated secretory pathway. J Neurosci 25: 6156–6166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang P‐M, Ling J, Jeong YH, Price DL, Aja SM, Wong PC (2010) Deletion of TDP‐43 down‐regulates Tbc1d1, a gene linked to obesity, and alters body fat metabolism. Proc Natl Acad Sci USA 107: 16320–16324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung K, Deisseroth K (2013) CLARITY for mapping the nervous system. Nat Methods 10: 508–513 [DOI] [PubMed] [Google Scholar]
- Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin J‐J, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP et al (2006) Null mutations in progranulin cause ubiquitin‐positive frontotemporal dementia linked to chromosome 17q21. Nature 442: 920–924 [DOI] [PubMed] [Google Scholar]
- Dincheva I, Glatt CE, Lee FS (2012) Impact of the BDNF Val66Met polymorphism on cognition. Neuroscientist 18: 439–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelmann E, Leßmann V, Brigadski T (2014) Pre‐ and postsynaptic twists in BDNF secretion and action in synaptic plasticity. Neuropharmacology 76: 610–627 [DOI] [PubMed] [Google Scholar]
- Edelmann E, Cepeda‐Prado E, Franck M, Lichtenecker P, Brigadski T, Leßmann V (2015) Theta burst firing recruits BDNF release and signaling in postsynaptic CA1 neurons in spike‐timing‐dependent LTP. Neuron 86: 1041–1054 [DOI] [PubMed] [Google Scholar]
- Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, Zaitsev E, Gold B, Goldman D, Dean M, Lu B, Weinberger DR (2003) The BDNF val66met polymorphism affects activity‐dependent secretion of BDNF and human memory and hippocampal function. Cell 112: 257–269 [DOI] [PubMed] [Google Scholar]
- Egawa N, Kitaoka S, Tsukita K, Naitoh M, Takahashi K, Yamamoto T, Adachi F, Kondo T, Okita K, Asaka I, Aoi T, Watanabe A, Yamada Y, Morizane A, Takahashi J, Ayaki T, Ito H, Yoshikawa K, Yamawaki S, Suzuki S et al (2012) Drug screening for ALS using patient‐specific induced pluripotent stem cells. Sci Transl Med 4: 145ra104 [DOI] [PubMed] [Google Scholar]
- Evans SF, Irmady K, Ostrow K, Kim T, Nykjaer A, Saftig P, Blobel C, Hempstead BL (2011) Neuronal brain‐derived neurotrophic factor is synthesized in excess, with levels regulated by sortilin‐mediated trafficking and lysosomal degradation. J Biol Chem 286: 29556–29567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fratta P, Sivakumar P, Humphrey J, Lo K, Ricketts T, Oliveira H, Brito Armas JM, Kalmar B, Ule A, Yu Y, Birsa N, Bodo C, Collins T, Conicella AE, Mejia Maza A, Marrero Gagliardi A, Stewart M, Mianne J, Corrochano S, Emmett W et al (2018) Mice with endogenous TDP‐43 mutations exhibit gain of splicing function and characteristics of amyotrophic lateral sclerosis. EMBO J 37: e98684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gass J, Lee WC, Cook C, Finch N, Stetler C, Jansen‐West K, Lewis J, Link CD, Rademakers R, Nykjaer A, Petrucelli L (2012) Progranulin regulates neuronal outgrowth independent of sortilin. Mol Neurodegener 7: 33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariri AR, Goldberg TE, Mattay VS, Kolachana BS, Callicott JH, Egan MF, Weinberger DR (2003) Brain‐derived neurotrophic factor val66met polymorphism affects human memory‐related hippocampal activity and predicts memory performance. J Neurosci 23: 6690–6694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harward SC, Hedrick NG, Hall CE, Parra‐Bueno P, Milner TA, Pan E, Laviv T, Hempstead BL, Yasuda R, McNamara JO (2016) Autocrine BDNF‐TrkB signalling within a single dendritic spine. Nature 538: 99–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu F, Padukkavidana T, Vaegter CB, Brady OA, Zheng Y, Mackenzie IR, Feldman HH, Nykjaer A, Strittmatter SM (2010) Sortilin‐mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron 68: 654–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibáñez CF, Simi A (2012) p75 neurotrophin receptor signaling in nervous system injury and degeneration: paradox and opportunity. Trends Neurosci 35: 431–440 [DOI] [PubMed] [Google Scholar]
- Johnson BS, Snead D, Lee JJ, McCaffery JM, Shorter J, Gitler AD (2009) TDP‐43 is intrinsically aggregation‐prone, and amyotrophic lateral sclerosis‐linked mutations accelerate aggregation and increase toxicity. J Biol Chem 284: 20329–20339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte M, Griesbeck O, Gravel C, Carroll P, Staiger V, Thoenen H, Bonhoeffer T (1996) Virus‐mediated gene transfer into hippocampal CA1 region restores long‐term potentiation in brain‐derived neurotrophic factor mutant mice. Proc Natl Acad Sci USA 93: 12547–12552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EB, Lee VMY, Trojanowski JQ (2011) Gains or losses: molecular mechanisms of TDP43‐mediated neurodegeneration. Nat Rev Neurosci 13: 38–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WC, Almeida S, Prudencio M, Caulfield TR, Zhang Y‐J, Tay WM, Bauer PO, Chew J, Sasaguri H, Jansen‐West KR, Gendron TF, Stetler CT, Finch N, Mackenzie IR, Rademakers R, Gao F‐B, Petrucelli L (2014) Targeted manipulation of the sortilin‐progranulin axis rescues progranulin haploinsufficiency. Hum Mol Genet 23: 1467–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Sun W, Zhang Y, Wei W, Ambasudhan R, Xia P, Talantova M, Lin T, Kim J, Wang X, Kim WR, Lipton SA, Zhang K, Ding S (2011) Rapid induction and long‐term self‐renewal of primitive neural precursors from human embryonic stem cells by small molecule inhibitors. Proc Natl Acad Sci USA 108: 8299–8304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou H, Kim S‐K, Zaitsev E, Snell CR, Lu B, Loh YP (2005) Sorting and activity‐dependent secretion of BDNF require interaction of a specific motif with the sorting receptor carboxypeptidase e. Neuron 45: 245–255 [DOI] [PubMed] [Google Scholar]
- Lu B, Nagappan G, Guan X, Nathan PJ, Wren P (2013) BDNF‐based synaptic repair as a disease‐modifying strategy for neurodegenerative diseases. Nat Rev Neurosci 14: 401–416 [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Rademakers R, Neumann M (2010) TDP‐43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 9: 995–1007 [DOI] [PubMed] [Google Scholar]
- Molgaard S, Demontis D, Nicholson AM, Finch NA, Petersen RC, Petersen CM, Rademakers R, Nykjaer A, Glerup S (2016) Soluble sortilin is present in excess and positively correlates with progranulin in CSF of aging individuals. Exp Gerontol 84: 96–100 [DOI] [PubMed] [Google Scholar]
- Mowla SJ, Pareek S, Farhadi HF, Petrecca K, Fawcett JP, Seidah NG, Morris SJ, Sossin WS, Murphy RA (1999) Differential sorting of nerve growth factor and brain‐derived neurotrophic factor in hippocampal neurons. J Neurosci 19: 2069–2080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Poo M‐M (2013) Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci 14: 7–23 [DOI] [PubMed] [Google Scholar]
- Polymenidou M, Lagier‐Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling S‐C, Sun E, Wancewicz E, Mazur C, Kordasiewicz H, Sedaghat Y, Donohue JP, Shiue L, Bennett CF, Yeo GW, Cleveland DW (2011) Long pre‐mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP‐43. Nat Neurosci 14: 459–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prpar Mihevc S, Baralle M, Buratti E, Rogelj B (2016) TDP‐43 aggregation mirrors TDP‐43 knockdown, affecting the expression levels of a common set of proteins. Sci Rep 6: 33996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudencio M, Jansen‐West KR, Lee WC, Gendron TF, Zhang Y‐J, Xu Y‐F, Gass J, Stuani C, Stetler C, Rademakers R, Dickson DW, Buratti E, Petrucelli L (2012) Misregulation of human sortilin splicing leads to the generation of a nonfunctional progranulin receptor. Proc Natl Acad Sci USA 109: 21510–21515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sephton CF, Cenik C, Kucukural A, Dammer EB, Cenik B, Han Y, Dewey CM, Roth FP, Herz J, Peng J, Moore MJ, Yu G (2011) Identification of neuronal RNA targets of TDP‐43‐containing ribonucleoprotein complexes. J Biol Chem 286: 1204–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty MS, Sharma M, Hui NS, Dasgupta A, Gopinadhan S, Sajikumar S (2015) Investigation of synaptic tagging/capture and cross‐capture using acute hippocampal slices from rodents. J Vis Exp 103: e53008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle FE, de Belleroche J, Mitchell JD, Leigh PN, Al‐Chalabi A, Miller CC, Nicholson G, Shaw CE (2008) TDP‐43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319: 1668–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka J‐I, Horiike Y, Matsuzaki M, Miyazaki T, Ellis‐Davies GCR, Kasai H (2008) Protein synthesis and neurotrophin‐dependent structural plasticity of single dendritic spines. Science 319: 1683–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S (1996) Subregion‐ and cell type‐restricted gene knockout in mouse brain. Cell 87: 1317–1326 [DOI] [PubMed] [Google Scholar]
- Van Damme P, Van Hoecke A, Lambrechts D, Vanacker P, Bogaert E, van Swieten J, Carmeliet P, Van Den Bosch L, Robberecht W (2008) Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J Cell Biol 181: 37–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MA, Kim E, Duffy A, Adalbert R, Phillips BU, Peters OM, Stephenson J, Yang S, Massenzio F, Lin Z, Andrews S, Segonds‐Pichon A, Metterville J, Saksida LM, Mead R, Ribchester RR, Barhomi Y, Serre T, Coleman MP, Fallon JR et al (2018) TDP‐43 gains function due to perturbed autoregulation in a Tardbp knock‐in mouse model of ALS‐FTD. Nat Neurosci 21: 552–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AC, Dugger BN, Dickson DW, Wang D‐S (2011) TDP‐43 in aging and Alzheimer's disease ‐ a review. Int J Clin Exp Pathol 4: 147–155 [PMC free article] [PubMed] [Google Scholar]
- Xiao S, Sanelli T, Dib S, Sheps D, Findlater J, Bilbao J, Keith J, Zinman L , Rogaeva E, Robertson J (2011) RNA targets of TDP‐43 identified by UV‐CLIP are deregulated in ALS. Mol Cell Neurosci 47: 167–180 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Source Data for Appendix
Review Process File
Source Data for Figure 1B
Source Data for Figure 2E
Source Data for Figure 6B