

Abstract
The sarcoplasmic/endoplasmic reticulum Ca2+‐ATPase 2a (SERCA2a) performs active reuptake of cytoplasmic Ca2+ and is a major regulator of cardiac muscle contractility. Dysfunction or dysregulation of SERCA2a is associated with heart failure, while restoring its function is considered as a therapeutic strategy to restore cardiac performance. However, its structure has not yet been determined. Based on native, active protein purified from pig ventricular muscle, we present the first crystal structures of SERCA2a, determined in the CPA‐stabilized E2 form (3.3 Å) and the Ca2+‐occluded [Ca2]E1‐AMPPCP form (4.0 Å). The structures are similar to the skeletal muscle isoform SERCA1a pointing to a conserved mechanism. We seek to explain the kinetic differences between SERCA1a and SERCA2a. We find that several isoform‐specific residues are acceptor sites for post‐translational modifications. In addition, molecular dynamics simulations predict that isoform‐specific residues support distinct intramolecular interactions in SERCA2a and SERCA1a. Our experimental observations further indicate that isoform‐specific intramolecular interactions are functionally relevant, and may explain the kinetic differences between SERCA2a and SERCA1a.
Keywords: Ca2+ transport, Ca2+‐ATPase, crystal structure, heart failure, molecular dynamics
Subject Categories: Membrane & Intracellular Transport, Structural Biology
Introduction
The cardiac contractility is tightly regulated by the activity of the sarcoplasmic/endoplasmic reticulum Ca2+‐ATPase 2a (SERCA2a), which is responsible for the reuptake of cytosolic Ca2+ into the sarcoplasmic reticulum (SR) of cardiomyocytes. SERCA2a enables cardiac muscle relaxation and determines how much Ca2+ can be released for contraction, which in turn controls contractile strength (Bers, 2002). In the heart, the activity of SERCA2a is regulated by several transmembrane (TM) micropeptides: Phospholamban (PLB) (MacLennan & Kranias, 2003), Sarcolipin (SLN) (Vangheluwe et al, 2005), Dwarf Open Reading Frame (DWORF) (Nelson et al, 2016), and Another‐Regulin (ALN) (Anderson et al, 2016). These small TM proteins regulate SERCA2a activity mainly via controlling the apparent Ca2+ affinity of the pump within a narrow physiological range, thereby allowing a dynamic regulation of cardiac contractility. For instance, in resting conditions PLB inhibits SERCA2a, which is temporally relieved during β‐adrenergic stimulation, and exerts strong inotropic and lusitropic effects (Arkin et al, 1994).
A reduced SERCA2a activity, at least in part, contributes to the progressive deterioration of cardiac contractility in heart failure (HF). Lower SERCA2a expression (reviewed in Periasamy et al, 2008) and protein sumoylation (Kho et al, 2011), together with reduced levels of DWORF (Nelson et al, 2016) and PLB phosphorylation (reviewed in Periasamy et al, 2008), negatively impact on the uptake of Ca2+ into the SR, impairing the contractile performance of the heart. Also, some familial forms of dilated cardiomyopathy are caused by mutations in PLB, further highlighting the central role of SERCA2a dysregulation in HF (Kimura et al, 1998; Haghighi et al, 2003; Trieber et al, 2005). Conversely, restoring SERCA2a activity by elevating its expression or by PLB interference rescues contractility in isolated cardiomyocytes, and in small and large animal models (MacLennan & Kranias, 2003). Therefore, adeno‐associated viral gene delivery of SERCA2a was explored in clinical trials as a therapeutic strategy for HF (Jessup et al, 2011; Hulot et al, 2017), although inadequate SERCA2a gene delivery has been noted (Greenberg et al, 2016). Alternative to gene delivery, small molecule SERCA2a agonists have been reported (Ferrandi et al, 2013; Kaneko et al, 2017), but molecular details of their working mechanism are lacking.
SERCA2a (encoded by the ATP2A2 gene) is expressed in cardiac, smooth, and slow‐twitch skeletal muscles, while SERCA1a (ATP2A1) is the major isoform in fast‐twitch skeletal muscles. Overall, they are similar and transport two Ca2+ ions per ATPase cycle. Porcine SERCA2a shares 99% sequence identity with the human SERCA2a and 84% identity with rabbit and other mammalian SERCA1a. Due to an amino acid deletion in SERCA2a at position 509, the SERCA2a residue numbering from position 509 is shifted by ‐1 as compared to SERCA1a. Based on available SERCA1a crystal structures capturing most conformational states along the functional cycle, the SERCA Ca2+ transport mechanism is well described at the functional and structural level (Bublitz et al, 2013; Toyoshima et al, 2013; Winther et al, 2013). Three cytoplasmic domains (i.e., the nucleotide binding domain, N; phosphorylation domain, P; and actuator domain, A) regulate ATP binding, autophosphorylation, and dephosphorylation, whereas a TM domain controls Ca2+ binding and translocation. During ion transport across the membrane, SERCA undergoes large conformational changes switching between Ca2+‐bound E1 states and Ca2+‐free E2 states (Albers et al, 1963; Post & Sen, 1965; Moller et al, 2010). In the E1 state, cytosolic Ca2+ enters via the Ca2+ entry gate and binds with high affinity at the TM domain. Mg2+‐ATP binds to the N‐domain, resulting in the phosphorylation of an aspartate residue in the P‐domain (Asp351, conserved in all P‐type ATPases) and the formation of an occluded [Ca2]E1P intermediate. A spontaneous conformational transition to an E2P state, typically rate limiting for SERCA1a transport, opens the Ca2+ exit gate to the luminal compartment and lowers the Ca2+ affinity, which promotes Ca2+ release. Subsequently, negatively charged residues of the ion binding sites are partially protonated, which triggers occlusion and dephosphorylation, catalyzed by a conserved glutamate residue of the A‐domain (E183) via an E2‐Pi intermediate (Olesen et al, 2004) and ending with the phosphate‐released E2 state. The conversion of the protein to an E1 state opens the cytoplasmic Ca2+ entry gate, promoting the release of protons from the TM domain possibly via a C‐terminal pathway (Bublitz et al, 2010) and allowing another cycle to begin (Toyoshima et al, 2013; Winther et al, 2013).
Compared to SERCA1a, heart muscle SERCA2a exhibits a lower maximal turnover rate and higher apparent Ca2+ affinity, which relates to kinetic differences in the catalytic cycle (Dode et al, 2002, 2003; Clausen et al, 2012). SERCA2a is marked by a slower E2 to E1 transition than SERCA1a (Dode et al, 2003; Clausen et al, 2012). Also, SERCA2a is marked by a slower E2P to E2 dephosphorylation reaction (Clausen et al, 2012). The molecular mechanism for these kinetic differences is unclear, but underlying structural differences may be responsible. To explore this possibility, we determined E1 and E2 structures of SERCA2a, which represent the first SERCA2a structures, and we compared them with the corresponding E1 and E2 structures of SERCA1a.
Results
Purified SERCA2a from pig heart is active
To purify native SERCA2a from pig left ventricle, a one‐step affinity chromatography protocol was developed, providing a yield of around 2.5 mg per 100 g of tissue (Fig 1A). In short, cardiac microsomes were solubilized with DDM, and SERCA2a was purified via Reactive Green affinity chromatography, which captures ATP‐binding proteins. SERCA2a was subsequently eluted with AMPPCP. Immunoblotting experiments confirmed that the purified sample consists mainly of the SERCA2a isoform (Fig 1B). Only traces of the housekeeping SERCA2b isoform were found, and no SERCA1a was detected (Fig 1B). No bands corresponding to PLB were detected on SDS–PAGE and immunoblotting (Fig 1B). The purified enzyme displayed robust, Ca2+‐dependent ATPase activity, with a specific activity of 4.3 ± 0.7 μmoles/min/mg at 1 μM free Ca2+, Hill coefficient n of 1.3 ± 0.1, and K m value of 0.22 ± 0.01 μM (Fig 1C). Thus, the purified SERCA2a from pig heart remains functional.
Figure 1. SERCA2a purification and activity.
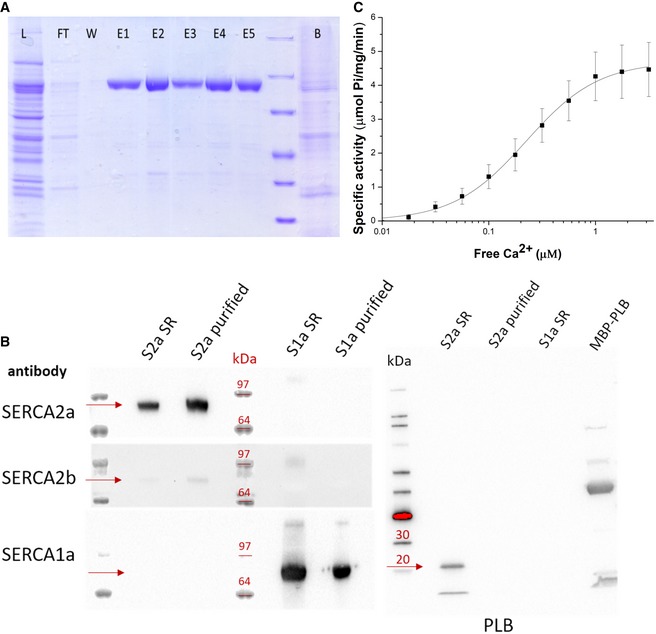
- SDS–PAGE of fractions of the SERCA2a purification procedure (L, total lysate; FT, flow‐through from Reactive Green beads; E, elution fractions; B, beads after elution).
- Immunoblot with sarcoplasmic reticulum (SR) preparations of cardiac (S2a SR) and skeletal (S1a SR) muscles, and purified SERCA2a (S2a purified) and SERCA1a (S1a purified). Detection was performed using antibodies against three SERCA isoforms 1a, 2a, and 2b, or phospholamban (PLB). PLB appears as a monomeric (5 kDa) and pentameric band (25 kDa). A fusion construct of PLB with maltose binding protein (MBP) was used as a positive control for PLB detection. The proteins of interest (e.g., SERCA or PLB) are indicated by the red arrows for each of the Western blots in the figure.
- Ca2+‐dependent ATPase activity of the purified SERCA2a. Error bars represent standard deviation; three biological replicates were analyzed. K m = 0.22 ± 0.01 μM, specific activity is 4.3 ± 0.7 μmoles/min/mg at 1 μM free Ca2+, Hill coefficient n of 1.3.
E2‐AlF4‐CPA and [Ca2]E1‐AMPPCP structures of SERCA2a closely resemble SERCA1a
Next, we determined structures of SERCA2a in a proton‐occluded [H2‐3] form stabilized by the SERCA inhibitor cyclopiazonic acid (CPA), and in a Ca2+‐occluded E1 form using the non‐hydrolyzable ATP analog AMPPCP. The E2‐AlF4‐CPA and [Ca2]E1‐AMPPCP forms were resolved at 3.3 and 4.0 Å resolution, respectively (Fig 2A and B, Table 1). In the [Ca2]E1‐AMPPCP crystals, the TM regions of neighboring proteins interact in an antiparallel packing (i.e., not reflecting physiological contacts). However, the E2‐AlF4‐CPA crystal form is marked by an unusual parallel packing of SERCA2a molecules involving contact points between the A‐ and N‐domains, between the A‐ and P‐domains, as well as N‐domain and L7/8 (Fig EV1). These SERCA2a‐specific regions may play a role in the formation of SERCA2a dimers, which also have been reported in cardiomyocytes (Blackwell et al, 2016). No such parallel packing modes have been observed for SERCA1a crystal forms, and many of the involved residues differ for SERCA2a and SERCA1a, and are conserved within one isoform.
Figure 2. Crystal structures of cardiac SERCA2a in the E2‐CPA and [Ca2]E1‐AMPPCP conformational states.
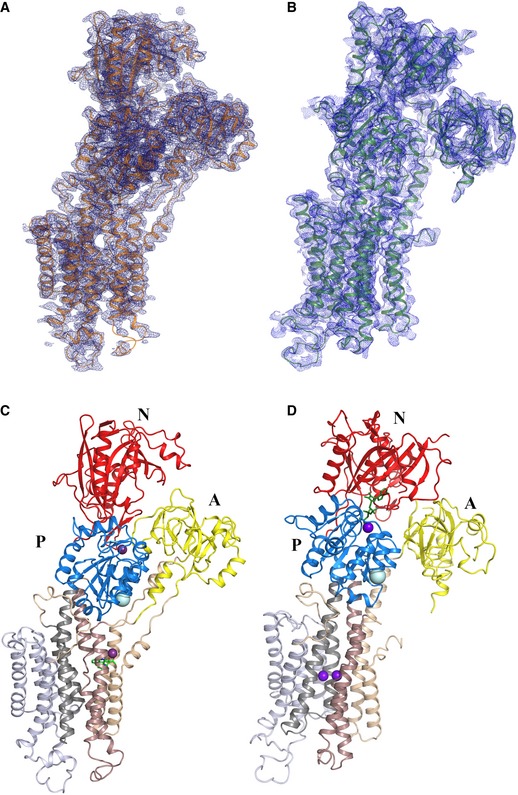
-
A–DElectron density maps of the SERCA2a E2‐CPA (A; PDB 5MPM) and [Ca2]E1‐AMPPCP (B; PDB 6HXB) structures. Domain‐colored SERCA2a in E2‐CPA (C) and [Ca2]E1‐AMPPCP (D) states: The A‐domain (labeled A) is colored in yellow, P‐domain (labeled P) in blue, and N‐domain (labeled N) in red. M1‐M2 are colored in wheat, M3‐M4 in brown, M5‐M6 in dark gray, and M7‐M10 in light gray. Ca2+/Mg2+ ions are represented in purple spheres, and AMPPCP in green sticks. The E2‐CPA structure depicts 992 amino acids of the 997 (no electron density was observed for the last five amino acids). The model of SERCA2a in the E1‐Ca2+‐AMPPCP conformational state contains 993 amino acids (Glu2‐Gly994) of 997 total, with the exception of the residues Leu41‐Leu49, Arg134‐Lys135, Met239‐Gln244, Ala424‐Leu425, Ser504‐Ser509, which were not built due to lack of electron density features.
Table 1.
Data collection and refinement statistics for the SERCA2a structures in the Ca2+‐E1‐AMPPCP and E2‐CPA conformational states
| [Ca2]E1‐AMPPCP | [H2‐3] E2‐CPA | |
|---|---|---|
| Data collection | ||
| Resolution range (Å) | 41.7–4.0 (4.14–4.0) | 63.3–3.3 (3.35–3.3) |
| Space group | P 2 | P 21 |
| Cell dimensions | ||
| a, b, c (Å) | 117.0, 51.8, 125.6 | 53.35 253.3 65.0 |
| α, β, γ (°) | 90, 106.1, 90 | 90.0 100.9 90.0 |
| Mosaicity (°) | 0.38 | 0.17 |
| Total reflections | 45,778 (4,386) | 86,319 (16,193) |
| Unique reflections | 12,439 (1,183) | 25,135 (4,544) |
| Multiplicity | 3.7 (3.7) | 3.4 (3.6) |
| Completeness (%) | 97.9 (99.0) | 99.0 (99.0) |
| Mean I/σ(I) | 3.95 (0.65) | 7.1 (1.0) |
| Copies per ASU | 1 | 1 |
| Rmerge | 0.195 (> 1) | 0.130 (> 1) |
| Rpim | 0.119 (> 1) | 0.115 (> 1) |
| CC1/2 | 0.983 (0.449) | 0.996 (0.598) |
| Refinement statistics | ||
| Resolution (Å) | 41.7–4.0 | 63.3–3.3 |
| Rwork (%) | 31.0 | 20.4 |
| Rfree (%) | 35.4 | 25.8 |
| Average B, all atoms (A2) | 167.0 | 130.0 |
| Favored rotamers (%) | 100 | 99 |
| Ramachandran plot (%) | ||
| Favored | 95.2 | 98 |
| Allowed | 4.5 | 2 |
Figure EV1. Packing of SERCA2a E2‐CPA and [Ca2]E1‐AMPPCP crystals.
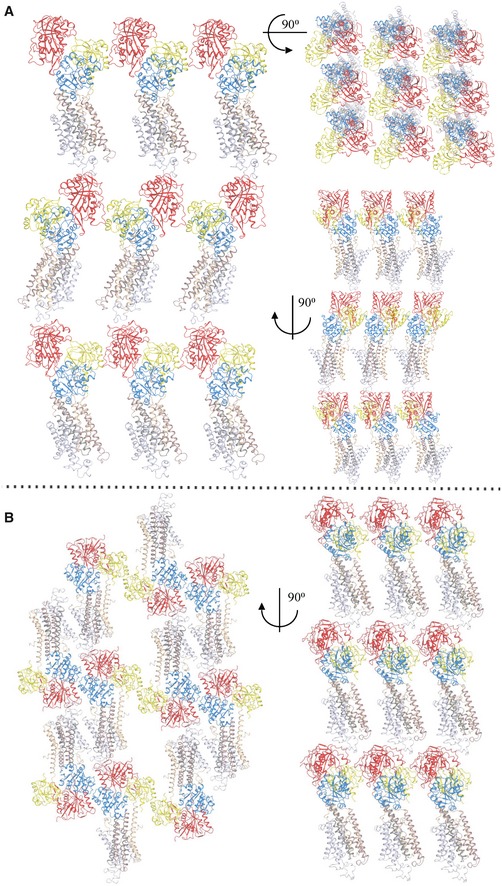
- Parallel packing of the SERCA2a structure in the E2‐CPA conformational state. SERCA2a forms contact points between the A‐domains and N‐domains (Met1‐Asn3, His15 to Glu458‐Lys460) and between the A‐domain (Arg134, Arg139, Lys141) and the P‐domain (Glu659, Glu667, Ser663), as well as N‐domain (Thr506‐Ser509, Asp567‐Arg571) and L7/8 (Asp859‐Arg863, Lys876‐Gly884).
- Parallel packing of the SERCA2a structure in the E2‐CPA conformational state. Contact points are within the A‐domain (Arg35‐Glu40) and P‐domain (Asp658‐Ser663). A‐, P‐, and N‐domains are colored in yellow, blue, and red, respectively. M1‐M2 are colored in wheat, M3‐M4 in brown, M5‐M6 in dark gray, and M7‐M10 in light gray.
SERCA2a has the same overall domain organization as SERCA1a, including three cytoplasmic domains and 10 TM helices (Fig 2C and D). Indeed, the [H2‐3]‐CPA and [Ca2]E1‐AMPPCP structures of SERCA2a are similar to the corresponding structures of SERCA1a (PDB 3FGO and 3N8G, respectively) (Fig 3A and B) with root‐mean‐square deviation (RMSD) for Cα atoms of 0.89 Å (895 Cα atoms) and 1.73 Å (918 Cα atoms), respectively. Even the regions of highest sequence diversity, such as luminal loops L7/8 or L9/10, are similarly positioned in SERCA1a and SERCA2a. In both the E1 and the E2 structures, the SERCA1a headpiece is tilted more toward the membrane as compared to SERCA2a. The [Ca2]E1‐AMPPCP structure contains clear densities for the AMPPCP molecule as well as for bound Ca2+ and K+ ions. Ca2+ and AMPPCP binding in SERCA2a is very similar as in the SERCA1a structure (Fig EV2). The CPA and MgFx molecules as well as K+ ions are easily recognized in the ‐CPA structure. However, AMPPCP is not observed despite being present at high concentrations in the crystallization conditions. Similar as in SERCA1a, CPA binds in a groove between helices M2, M3, and M4, coordinated by a Mg2+ ion and residues Gln56, Asp59, and Asn101, and blocks the proposed Ca2+ entry pathway (Moncoq et al, 2007; Laursen et al, 2009).
Figure 3. Comparison of the SERCA2a structures with the SERCA1a structures in the corresponding conformational states.
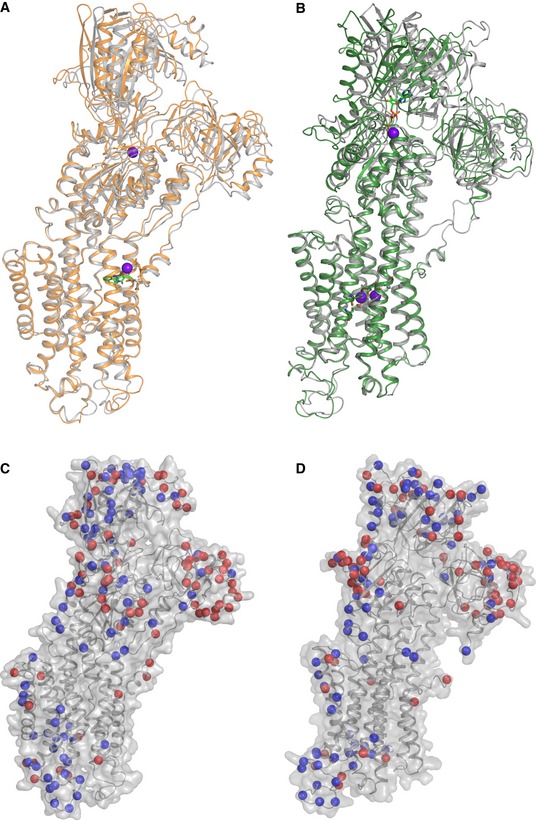
-
AAlignment of the SERCA2a E2‐CPA (orange, PDB 5MPM) and SERCA1a (gray, PDB 3FGO) structures.
-
BAlignment of the SERCA2a [Ca2]E1‐AMPPCP (green, PDB 6HXB) and SERCA1a (gray, PDB 3N8G) structures.
-
C, DIsoform‐specific residues are represented by both red and blue spheres mapped on the SERCA2a E2‐CPA (C) and [Ca2]E1‐AMPPCP (D) structures. Blue spheres depict residues that are identical in more than 90% of a selection of 83 vertebrate orthologues, while red spheres are less conserved.
Figure EV2. Coordination of ions and molecules in the SERCA2a structures.
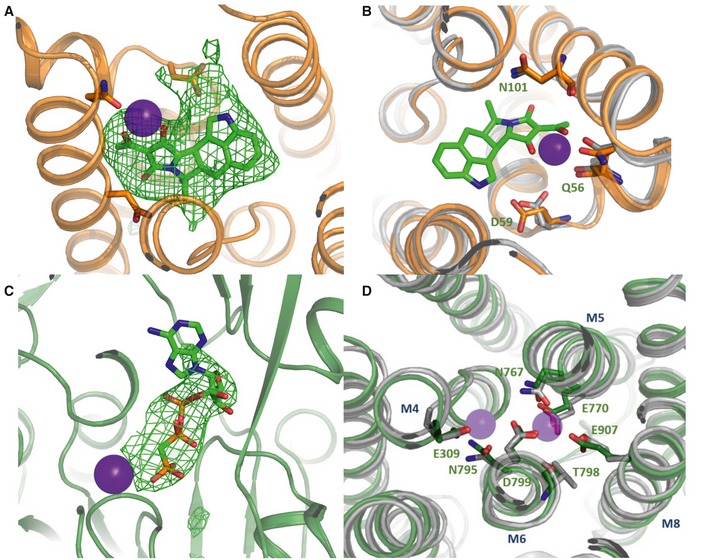
-
A–CCa2+ ions are shown in purple spheres. Omit maps for the CPA (A) and the AMPPCP (C) molecules contoured at 3σ. (B) CPA coordination in [H2‐3]E2‐CPA state of SERCA. SERCA1a is depicted in gray and SERCA2a in orange.
-
DCoordination of Ca2+ ions in the [Ca2]E1‐AMPPCP state. SERCA1a is shown in gray and SERCA2a in green. Residues are marked according to the SERCA2a numbering.
SERCA2a‐specific amino acids are highly conserved, pointing to a functional role
The catalytic core of the Ca2+ pump appears highly conserved (Fig EV3), indicating that the Ca2+ transport mechanism is preserved between SERCA1a and SERCA2a. Despite the 160 amino acid sequence differences between rabbit SERCA1a and pig SERCA2a, their structures do not readily reveal why the isoforms display distinct functional properties (Dode et al, 2003; Clausen et al, 2012). As can be seen from Fig 3C and D, the isoform‐specific sequences are predominantly localized in exposed regions of the N‐, P‐ and A‐domains, near the membrane interface of the TM helices M7 and M10, and in the luminal loops L7/8 and L9/10. Surprisingly, many of the sequence differences between SERCA1a and SERCA2a are highly conserved within each isoform (Dataset EV1), indicating that they may exert isoform‐specific functions. To examine their functional role, we explored whether the isoform‐specific amino acid substitutions (i) modulate protein interactions, (ii) insert/remove sites for post‐translational modifications (PTMs), or (iii) alter the intramolecular dynamics of the Ca2+ pump.
Figure EV3. Sequence alignment of SERCA1a and SERCA2a.
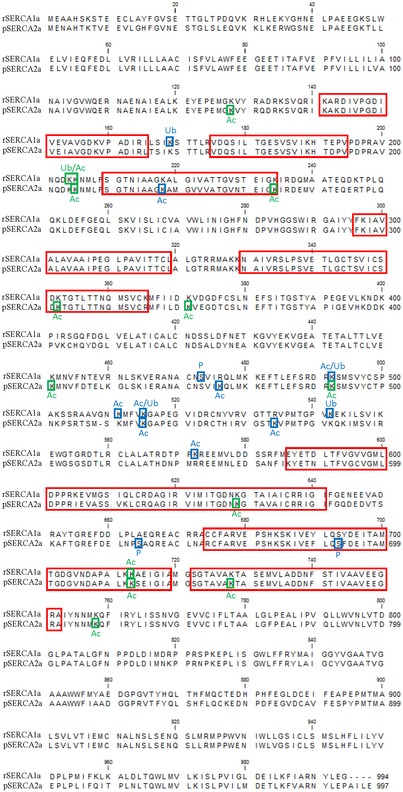
Alignment of rabbit SERCA1a and pig SERCA2a protein sequences indicating the eight most conserved core sequence regions in P‐type ATPases (red squares) (Axelsen & Palmgren, 1998). The post‐translational modifications found by MS in this study are marked by blue (reported before) and green (identified in this study) squares.
The PLB site is highly conserved between SERCA2a and SERCA1a
Isoform‐specific substitutions may introduce, disrupt, or alter the interaction sites of regulatory proteins. Here, we focused on PLB, the major regulator of SERCA2a in the heart, which is also able to functionally interact with SERCA1a. PLB is not present in the purified SERCA2a fraction and is therefore not visible in the structure (Fig 1), but we examined the PLB binding site that is located between the M2, M4, M6, and M9 helices in the SERCA2a [H2‐3]‐CPA structure, and compared it with the available SERCA1a‐PLB (Akin et al, 2013). Due to the different conformational states, the M2 is shifted away by 9.4 Å in SERCA2a, but the relevant side chains in the PLB binding groove are placed in a similar way (Leu802, Ala806, Phe809, Trp932, Leu939, which interact with Leu31 of PLB; and Gly801, Thr805, and Gln108, which interact with Asn34 of PLB), i.e., not pointing to major differences in the PLB acceptor site in the TM domain of SERCA2a and SERCA1a. However, we cannot exclude that isoform‐specific residues are important for the interaction with other regulatory proteins (Vandecaetsbeek et al, 2009a, 2011; Anderson et al, 2015; Nelson et al, 2016), but their interaction sites are less well characterized.
Isoform‐specific sites of post‐translational modifications
Since many of the isoform‐specific residues are exposed and highly conserved, we hypothesized that they may serve as PTM acceptor sites, which may provide isoform‐specific regulatory control. SERCA isoforms are regulated by various PTMs, including sumoylation, phosphorylation, acetylation, glutathionylation, ubiquitination, and nitration (Stammers et al, 2015). A structural map of all previously reported PTMs (Foster et al, 2013; Hornbeck et al, 2015) of SERCA2a and SERCA1a shows that the majority of PTMs (79/87 in SERCA2a and 27/29 in SERCA1a) are located in the cytoplasmic domains, accessible for enzyme interactions (Fig 4A and B; Dataset EV2). Interestingly, 26 previously reported PTMs are located on SERCA2a‐specific residues (i.e., 26/160). This supports the view that isoform‐specific sequence differences provide regulatory control by PTMs. Also, a higher number of unique PTMs has so far been reported for SERCA2a than for SERCA1a (87 versus 29 reported PTMs), suggesting that SERCA2a may be more regulated by PTMs than SERCA1a.
Figure 4. Post‐translational modifications mapped on SERCA2a and SERCA1a structures.
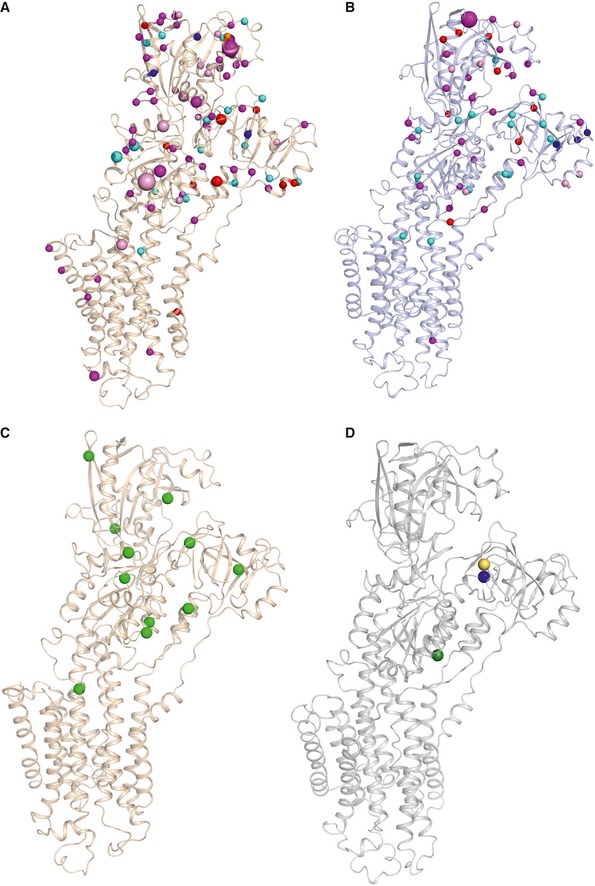
-
A, BPost‐translational modifications (PTMs) that were reported previously in humans, mouse, and rat (Foster et al, 2013; Hornbeck et al, 2015), as well as found in this study (in rabbit and pig), are shown in spheres for SERCA2a E2‐CPA (PDB 5MPM) (A) and SERCA1a (PDB 3FGO) (B). Phosphorylation is shown in magenta spheres, ubiquitination in cyan, sumoylation in orange, acetylation in red, and methylation in dark blue spheres. The residues, which are both acetylated and ubiquitinated, are colored in pink. The size of the sphere correlates with the number of records obtained via a proteomic discovery‐mode mass spectrometry approach (the smallest sized spheres correspond to PTMs that were identified one to five times, the medium‐sized sphere represents 6–25 hits, and the largest sized spheres indicate > 26 records).
-
CNewly identified acetylation sites found in purified SERCA2a in this study are shown in green spheres.
-
DNewly identified PTMs found in purified SERCA1a by MS in this study: Lys204 (blue sphere) or Lys205 (yellow sphere) was found ubiquitinated, as well as Lys205 and Lys713 (green sphere) were found to be acetylated. The list of PTMs depicted in (A and B) is provided in Dataset EV2.
While some PTM sites are found in both SERCA isoforms, each isoform presents a distinct PTM fingerprint (Fig 4). We experimentally confirmed this by comparing the PTMs in the available purified SERCA1a and SERCA2a samples (Dataset EV3). With an average coverage of 58.6% for SERCA1a and 60.3% for SERCA2a, we found a combination of previously reported and newly identified PTMs in both isoforms. We discovered multiple new SERCA2a acetylation sites (Lys128, 205, 234, 352, 371, 451, 492, 628, 712, 727, 757) (Fig 4C, Dataset EV3). In SERCA1a, we identified one new ubiquitination (either Lys204 or 205) and two new acetylation sites (Lys205 and 713) (Fig 4D, Dataset EV3). While most of the experimentally confirmed PTMs take place on lysine and serine residues that are conserved between SERCA1a and SERCA2a, four PTMs are found on SERCA1a‐specific (K572) or SERCA2a‐specific (K476, K533, and S663) residues (Dataset EV2). This experimentally confirms that both isoforms are differently regulated by PTMs due to changes in the primary structure.
Isoform‐specific sequence differences alter molecular dynamics and functional properties
While the PTM analysis supports the view that SERCA1a and SERCA2a are differently regulated, this provides no mechanistic explanation as to why the isoforms present different E2 to E1 conversion rates (Dode et al, 2002, 2003; Clausen et al, 2012). In the absence of clear structural differences, we hypothesized that isoform‐specific amino acids may alter the intramolecular network of hydrogen bond and salt bridge interactions, which may have an impact on the rate of domain rearrangements during the catalytic cycle. To explore this possibility, we performed three 50‐ns MD simulations of the highest resolution structure of SERCA2a ([H2‐3]E2‐AlF4—CPA) and its corresponding SERCA1a structure (3FGO) inserted in a phosphatidylcholine membrane environment. While short 50‐ns MD simulations do not simulate the conformational transitions in the catalytic cycle, they may provide insight into the dynamics and stability of intramolecular hydrogen bond and salt bridge interactions, which likely have an impact on larger domain rearrangements during conformational transitions.
We first compared the dynamics of both isoforms by assessing RMSD values of several domains over the course of the MD trajectory (Fig 5A and B). While the average RMSD values are identical for SERCA1a and SERCA2a for the N‐domain and M6, we observed that the A‐ and P‐domains are more flexible in SERCA1a than in SERCA2a (Fig 5A and B, Table 2). Moreover, our MD analysis supports the view that isoform‐specific residues influence the network of intramolecular interactions. We identified intramolecular hydrogen bonds and salt bridges based on energetic criteria (Dataset EV4), and observed that the following percentage of isoform‐specific residues participate in such interactions: 15.6% of SERCA1a and 12.5% of SERCA2a for salt bridges, and 36.9% of SERCA1a and 53.1% of SERCA2a for hydrogen bonds (Dataset EV4). While we cannot reliably assess the functional contribution of each interaction, the many concurrent observations support our view that intramolecular interactions are functionally relevant.
Figure 5. RMSD values of selected SERCA regions during the MD trajectory.
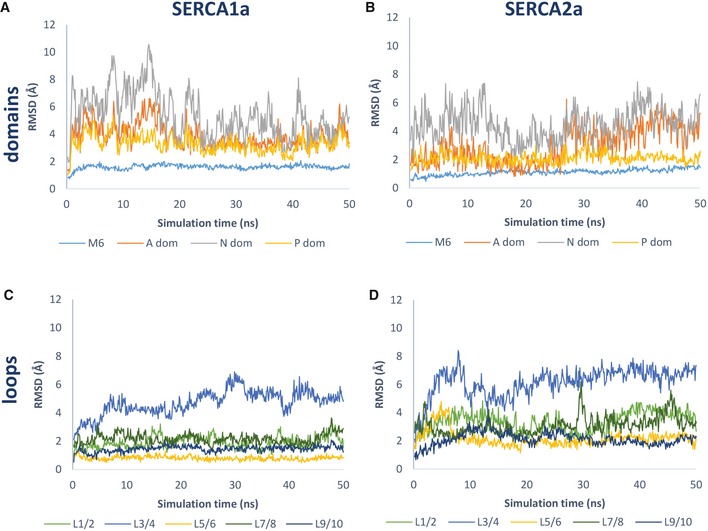
-
A, BRepresentative plot of one MD simulation depicting main chain RMSD values of M6 and A‐, N‐ and P‐domains in SERCA1a (A) and SERCA2a (B) over the course of the MD trajectory.
-
C, DRepresentative plot of one MD simulation depicting RMSD values of SERCA1a (C) and SERCA2a (D) luminal loops L1/2, L3/4, L5/6, L7/8, and L9/10 over the MD trajectory. Average RMSD values and statistical analysis are provided in Table 2. RMSD, root‐mean‐square deviation.
Table 2.
Average RMSD values (Å) of selected SERCA regions from three MD simulations
| RMSD | SERCA1a | SERCA2a |
|---|---|---|
| Average (Å) ± SD | Average (Å) ± SD | |
| M6 | 1.3 ± 0.3 | 1.3 ± 0.3 |
| A‐domain | 3.8 ± 1.0 | 3.0 ± 1.5a |
| N‐domain | 4.6 ± 1.4 | 4.6 ± 1.4 |
| P‐domain | 2.9 ± 0.6 | 2.1 ± 0.6a |
| L1/2 | 2.1 ± 0.5 | 3.0 ± 0.7a |
| L3/4 | 5.2 ± 1.4 | 7.2 ± 2.7a |
| L5/6 | 0.9 ± 0.2 | 3.0 ± 1.2a |
| L7/8 | 2.5 ± 0.6 | 3.4 ± 0.9a |
| L9/10 | 1.3 ± 0.3 | 1.9 ± 0.6a |
P < 0.05.
First, 96% of all identified Darier disease (DD) mutations take place on residues that according to our MD simulations are predicted to form hydrogen bonds or salt bridges. Since DD mutations generally lead to loss of function, this shows that the predicted intramolecular interactions involve functionally relevant residues. Moreover, several DD point mutations occur on SERCA2a‐specific residues and therefore participate in isoform‐specific intramolecular interactions (e.g., DD mutations Leu32Pro/Phe and Thr982Met, which in SERCA1a are replaced by His32 and Ile983) (Dataset EV4). This indicates that predicted isoform‐specific intramolecular interactions also involve functionally relevant residues.
Second, we explored the luminal region nearby luminal loops L7/8 and L9/10, two of the loops that differ the most between both isoforms (Fig 3). The MDs predict several salt bridges and hydrogen bonds between the luminal loops, but the type of these interactions and their duration time differ between the two isoforms (Dataset EV4). For instance, Lys958 in L9/10 of SERCA1a forms salt bridges with Glu83 in L1/2 and Glu892 in L7/8 (55 and 36% of the MD simulation time, respectively), which is absent in SERCA2a due to a substitution by Gln957. Lys958 is functionally relevant, since its mutation reduces the ATPase and Ca2+ transport activities of SERCA1a by 20 and 40%, respectively (Xu et al, 2008). The functional impact of Lys958 may be explained by the predicted hydrogen bond interactions with M6 (Gln791) and M9 (Pro954), connecting L9/10 with upstream elements in the membrane region of the pump (Fig 6).
Figure 6. Isoform‐specific interactions between luminal loops.
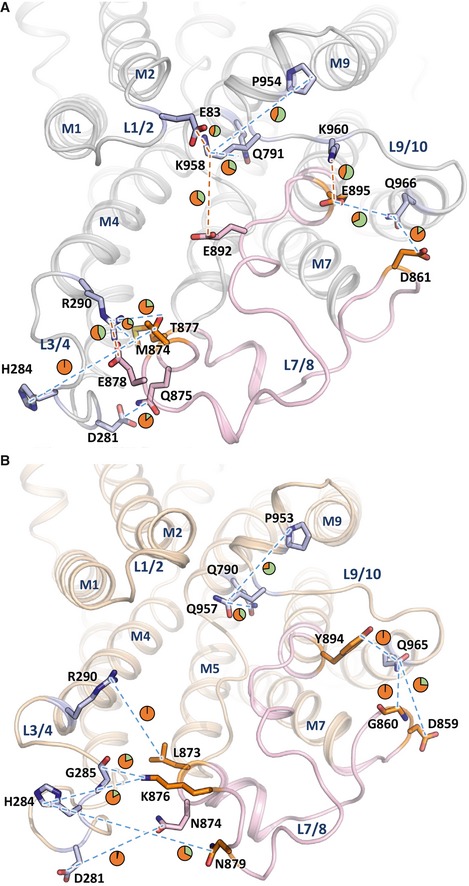
-
A, BSERCA1a (A, PDB 3FGO) and SERCA2a E2‐CPA (B, PDB 5MPM) depicting interactions between residues of L7/8 (colored in pink) and the residues of the other loops. In addition, all predicted interactions of K958 (SERCA1a) and Q957 (SERCA2a) were shown. The interacting residues of L7/8 are depicted in pink (conserved residues between SERCA1a and SERCA2a) or orange (isoform‐specific residues), while the interaction partners on other luminal loops are depicted in blue sticks. Predicted interactions in the MD simulations are highlighted as dashed lines in red (for salt bridges) and blue (for hydrogen bonds). Pie charts indicate the interaction time between the residues as percentage of the total MD simulation time (green, fraction of time when the interaction occurs; orange, fraction of time that no interaction takes place). Note that only those interactions are depicted that were observed in three independent MD simulations. Residues interacting within luminal loops or interacting < 5% of the simulation time are not displayed.
Third, we also assessed the dynamics of each individual luminal loop by plotting their RMSD values over the MD trajectory (Fig 5C and D). All luminal loops appear more flexible in SERCA2a than in SERCA1a, as demonstrated by the statistically higher average RMSD values for each luminal loop (Table 2). The different mobility of L3/4 is surprising because this loop is identical in both isoforms (Fig 5C and D, Table 2). This can only be rationalized by interactions of L3/4 with isoform‐specific neighboring loops, i.e., L7/8, indicating that isoform‐specific intramolecular interactions may affect the dynamic behavior of more remote and conserved protein regions. Of interest, three SERCA1a‐specific residues of L7/8 are predicted to participate in salt bridges (Glu892‐Lys958, and Glu895‐Lys960 and Arg290‐Glu878) that may hold L7/8 more stably together with L3/4 and L9/10 (Fig 6). In addition, a SERCA1a salt bridge can be formed between Lys958 in L9/10 with Glu83 in L1/2. In SERCA2a, these four salt bridges are absent or replaced by weaker hydrogen bonds (Fig 6).
Finally, we hypothesized that an exchange of L7/8 between SERCA1a and SERCA2a may alter the interactions of L7/8 with the neighboring loops or even cause a mismatch. If such interactions are functionally important, this may lead to a loss of function. To test this, we introduced the SERCA1a‐specific L7/8 in SERCA2a (SERCA2a‐L7/8‐1a chimera) and the SERCA2a‐specific L7/8 in SERCA1a (SERCA1a‐L7/8‐2a chimera; Clausen et al, 2012). We determined the functional properties of these chimeras in microsomes of COS cells that equally express all constructs (Fig EV4). The substitution of L7/8 significantly reduced the V max by 20–30% in both isoforms (Table 3), showing that L7/8 is functionally important, but also depends on other elements of the pump to integrate its function properly. These experimental observations are therefore in good agreement with the predictions of the MD simulations and are in line with a functional role of the interactions between luminal loops.
Figure EV4. Expression levels of SERCA1a and SERCA2a constructs.
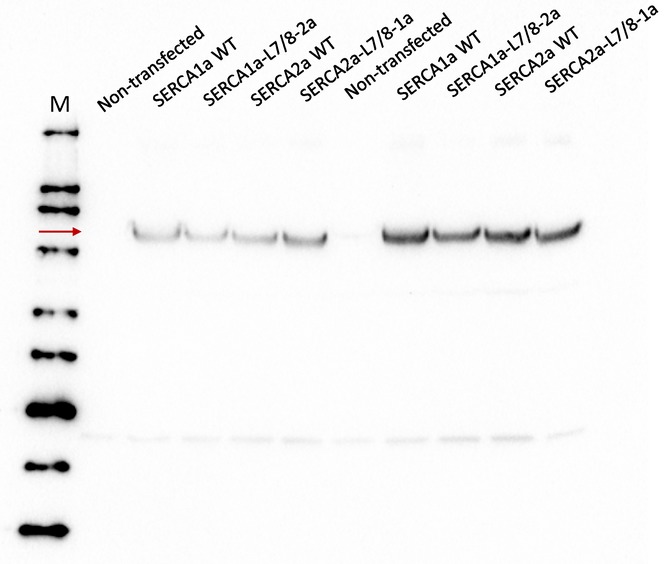
Immunoblot with a non‐isoform‐specific SERCA TRY2 antibody (Mountian et al, 2001) depicting similar protein levels of the WT and chimera constructs of SERCA2a and SERCA1a following transient expression in COS cells.
Table 3.
Non‐normalized (nmol Pi/μg microsomes/min) and normalized (%) activity for protein expression levels as well as the apparent Ca2+ affinity of the SERCA isoforms and chimeras
| K m (μM) | V max (nmol Pi/μg/min) | V max (%) | n | |
|---|---|---|---|---|
| SERCA1a WT | 0.19 ± 0.04 | 0.51 ± 0.17 | 100.9 ± 2.1 | 1.5 ± 0.1 |
| SERCA1a‐L7/8‐2a | 0.16 ± 0.04 | 0.28 ± 0.08 | 80.1 ± 2.5 | 1.5 ± 0.2 |
| SERCA2a WT | 0.14 ± 0.02 | 0.18 ± 0.06 | 100.0 ± 2.8 | 1.5 ± 0.1 |
| SERCA2a‐L7/8‐1a | 0.10 ± 0.02 | 0.13 ± 0.07 | 71.6 ± 4.9 | 1.5 ± 0.1 |
Mean values are given with standard deviation. N = 3 for each measurement.
Discussion
In this study, we developed a high‐yield purification protocol of native SERCA2a from pig heart and determined the first crystal structures of SERCA2a in two conformational states. We combined structural information, MD simulations, mass spectrometry, and a biochemical analysis to compare SERCA2a with SERCA1a. The strong resemblance between the SERCA2a and SERCA1a structures is consistent with the 84% sequence identity between the proteins, and suggests that the Ca2+ transport mechanism may be highly conserved. However, the isoform‐specific amino acids in SERCA2a and SERCA1a may also be functionally relevant. They provide regulatory control via PTMs and/or change the intramolecular network of salt bridge and hydrogen bond interactions, which most likely impact the dynamic behavior and functional properties of the pump.
SERCA2a‐ and SERCA1a‐specific sequences provide regulatory control
Based on the analysis of online PTM repositories and the determination of PTMs in the purified proteins, we concluded that SERCA1a and SERCA2a are marked by distinct PTM fingerprints. Indeed, many PTMs are located in exposed regions of the pumps and take place on isoform‐specific residues. The different PTMs in SERCA1a and SERCA2a may provide isoform‐specific regulatory control. With our protocols, it now becomes possible to determine the PTM fingerprint of purified SERCA2a from normal and disease tissue, which will be important to establish the molecular underpinnings of SERCA2a dysfunction in disease. However, the growing number of PTMs represents a major challenge to decipher the functional impact of each individual modification. To assess the functional role of multiple PTMs, it may become important to correlate PTM patterns identified in purified SERCA2a with its functional properties.
It is well established that SERCA2a is regulated by an increasing number of PTMs and that this regulatory control may be disturbed in pathological conditions such as HF (Kho et al, 2011). For instance, the progressive accumulation of SERCA2a tyrosine nitration has been described during aging (Viner et al, 1999) and SERCA2a nitration levels are clearly correlated with HF (Lokuta et al, 2005). Moreover, nitration of SERCA2a‐containing microsomes has a strong negative effect on Ca2+ uptake (Lokuta et al, 2005), and sumoylation regulates SERCA2a activity and stability and is decreased in HF, while restoration of SERCA2a sumoylation provides cardioprotection (Kho et al, 2011). An increased SERCA2a acetylation is associated with higher activity and altered intracellular Ca2+ dynamics influencing performance of cardiomyocytes (Meraviglia et al, 2018).
While we identified via mass spectrometry several PTMs in the purified protein, the SERCA2a structures did not reveal additional electron densities that may be ascribed to PTMs. The absence of PTMs in the electron density maps may be explained by their low occupancy in the crystal, a high level of disorder, and/or a limited resolution of the structures, as was also discussed for the palmitoylation of SERCA1a‐SLN (Montigny et al, 2014). Additionally, unmodified SERCA2a may have been preferentially incorporated into the crystals during the crystallization process.
Isoform‐specific residues alter the network of intramolecular interactions
The unique SERCA1a and SERCA2a properties are attributed to kinetic differences in various steps of the catalytic cycle, such as slower E2P to E2 dephosphorylation and E2 to E1 conversion rates in SERCA2a (Dode et al, 2002, 2003; Clausen et al, 2012). In this study, we observed that the E2 and E1 structures are surprisingly similar between both isoforms, indicating that the slower E2 to E1 conversion rate of SERCA2a cannot be explained by clear structural differences. Instead, the kinetic variation most likely reflects differences in the dynamic behavior of the two enzymes, which are not captured in the static structures. To compare the intramolecular dynamics of both isoforms, we calculated the intramolecular interactions in MD simulations over a short 50 ns time span, and indeed observed clear differences between SERCA1a and SERCA2a that depend on isoform‐specific residues. Moreover, both isoforms display differences in the dynamics, but this is clearly region‐dependent. The mobility of the A‐ and P‐domains is more pronounced in SERCA1a, while all luminal loops are more flexible in SERCA2a. We propose that the changes in flexibility of these protein regions may alter the rate of conformational transitions and contribute to the different kinetic behavior of both isoforms.
Note that our MDs do not simulate large protein motions or transitions of state, but depict only the dynamics and intramolecular networks within one state that may be at the origin of larger scale dynamic differences determining the kinetic properties. More advanced MD simulations of conformational transitions over longer timescales (μs to ms) will be required to further explore the impact of the intramolecular interactions on large domain rearrangements. Moreover, the presented list of intramolecular interactions was obtained with a fixed number of MD parameters for SERCA2a and SERCA1a. Albeit a limitation of the study, this facilitates a direct comparison of the interaction network of SERCA2a and SERCA1a in the same conditions, which revealed isoform‐specific differences in the dynamic behavior of a subset of regions and in the network of intramolecular interactions. However, the type and duration of intramolecular salt bridges and hydrogen bonds most likely depend on the local environment, which may differ for SERCA1a and SERCA2a in a cellular context where also interacting proteins, ions, PTMs, and lipids are present.
According to the MD simulations, many residues that differ between SERCA1a and SERCA2a participate in isoform‐specific salt bridges and hydrogen bonds. An important question is whether these intramolecular interactions may be functionally relevant. Indeed, several disease‐causing mutations affecting SERCA2a‐specific residues are predicted to form salt bridge or hydrogen bond interactions, highlighting their critical functional role. Although some interactions may impose a limited functional effect, each intramolecular interaction may contribute to the overall properties of the protein. To further assess the functional relevance of intramolecular interactions, we exchanged luminal loop L7/8 between SERCA1a and SERCA2a, which contains many isoform‐specific residues. According to the MD simulations, L7/8 may dynamically interact with other luminal loops, like L3/4 and L9/10. Indeed, L3/4 is identical in both isoforms, but appears more flexible in SERCA2a than in SERCA1a, which most likely depends on interactions with L7/8. In addition, both the type and duration of the luminal loop interactions seem isoform‐specific. Together, the isoform‐specific interactions between luminal loops predict that a chimeric replacement of L7/8 may result in a mismatch, which explains well why the L7/8 replacement impairs the maximal turnover rate in either isoform. Therefore, our results indicate that L7/8 operates together with other luminal loops to fine‐tune the functional properties of the Ca2+ pump.
As one of the most varying regions between SERCA1a and SERCA2a, L7/8 emerges as an important regulatory element of the Ca2+ pump. SERCA isoforms are regulated by a disulfide bridge formed between two conserved Cys residues in L7/8 (Daiho et al, 2001; Ushioda et al, 2016). In SERCA1a, the disulfide bridge inactivates the pump, while in SERCA2b, the reversible formation of a disulfide bridge between C875 and C887 serves as a luminal redox sensor that is controlled by an ER disulfide reductase (Ushioda et al, 2016). Binding of calumenin to L7/8 leads to a reduction in the apparent Ca2+ affinity of the ATPase (Sahoo et al, 2009). In addition, L7/8 serves as the major interaction site for the luminal extension of the SERCA2b C‐terminus that follows an additional transmembrane helix TM11 (Vandecaetsbeek et al, 2009b; Clausen et al, 2012). However, also other luminal loops are important for the SERCA2b‐specific properties of the luminal extension (Vandecaetsbeek et al, 2009b; Clausen et al, 2012) and the SERCA2b C‐terminus may also interact with the other luminal loops (Vandecaetsbeek et al, 2009b). The presence of the SERCA2b luminal extension may therefore alter the interactions between luminal loops, which may change the dynamic behavior and kinetic properties.
Mapping Darier disease mutations on the SERCA2a structure
Several mutations in the ATP2A2 gene are associated with DD, an autosomal dominant skin disorder, which is characterized by keratosis, skin, and nail defects (Sakuntabhai et al, 1999b). Here, we used the SERCA2a structures to map all known DD mutations of various disease impact (Stenson et al, 2017). The map demonstrates that DD mutations are scattered all over the protein, without specific clusters that correlate with disease severity (Fig 7). As highlighted above, only a few DD mutations are amino acid substitutions of SERCA2‐specific residues that are different in SERCA1a. DD mutations affect SERCA2 activity by a reduced protein stability and/or impaired functionality. Thus, haploinsufficiency of SERCA2 may explain the dominant inheritance pattern. Although DD mutations affect all three SERCA2 splice variants (SERCA2a, b, c), clinical manifestations only appear in the skin, where the ubiquitous SERCA2b isoform is expressed, while cardiac functionality is surprisingly well preserved (Tavadia et al, 2001; Mayosi et al, 2006).
Figure 7. Darier disease mutations.
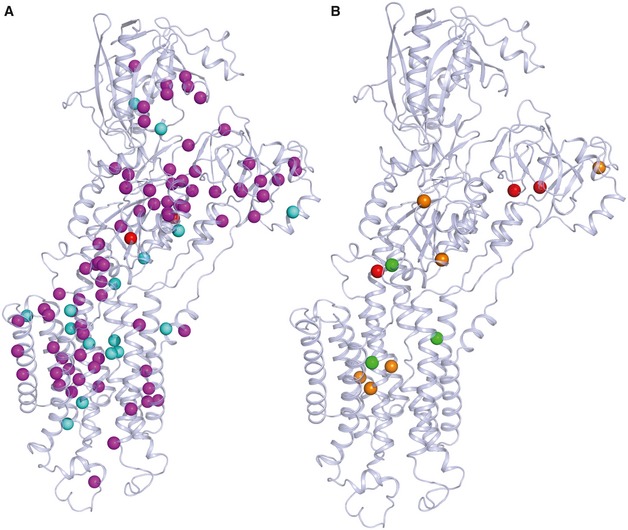
- Darier disease (DD)‐associated mutations shown in magenta spheres are missense/nonsense mutations, in cyan are most likely DD mutations, and in red are associated with acrokeratosis verruciformis/hypertension.
- The review of Nellen et al correlates DD mutations with the severity of the phenotype (Nellen et al, 2017). Mutations leading to a mild phenotype are represented by green spheres, moderate by orange, and severe by red. Some of the mutations with the described severe (Pro160, Gly211, Gln691, Arg750, His943) and moderate phenotypes (His943, Asn795, Gly769, Gln691, Ala672, Arg131) are associated with lower expression and activity levels of the protein (e.g., Arg750, His943, and Arg131) (Ruiz‐Perez et al, 1999; Sakuntabhai et al, 1999a; Sorensen & Andersen, 2000; Ringpfeil et al, 2001; Chao et al, 2002; Miyauchi et al, 2006). Also, Gly211 is related to the slower turnover rate of the protein and Gly769 with a higher affinity and Ca2+ transport (Ruiz‐Perez et al, 1999; Miyauchi et al, 2006; Ren et al, 2006).
Conclusion
The first structures of the SERCA2a isoform presented here represent important steps to facilitate further structural and functional studies of SERCA2a in normal and diseased context. SERCA2a is a recognized target for HF therapy. This study will aid SERCA2a‐based drug discovery efforts and opens the door for the future crystallization of SERCA2a complexes with small molecules or TM regulators like PLB. The SERCA2a and SERCA1a structures are very similar, which indicates a conserved mechanism of Ca2+ transport. The isoform‐specific residues emerge as sites for post‐translational control and/or modulators of the intramolecular dynamics, which may modify the kinetic behavior of the pump. Our findings are of interest for ongoing drug discovery efforts that aim to develop SERCA2a‐specific modulators.
Materials and Methods
Isolation and purification of SERCA2a from pig heart
Pig hearts were collected from a slaughterhouse (Danish Crown in Horsens, Denmark) or obtained from the Department of Cardiovascular Sciences (KU Leuven, Belgium), and were immediately rinsed with an ice‐cold 0.9% (w/v) NaCl solution. All the following procedures were performed at 4°C. Ventricle tissue from the pig heart was homogenized with 10 mM NaHCO3 buffer (1:3 w/w) and centrifuged for 20 min at 12,200 × g. The supernatant was filtered through eight layers of gauze and centrifuged as above. The filtering step was then repeated, and the supernatant was centrifuged at 140,000 × g for 45 min. The pellet was re‐suspended in buffer (0.6 M KCl, 10 mM histidine, pH 7.0), magnetically stirred for 30 min, and then centrifuged again at 140,000 × g for 45 min. The final pellet containing crude SR microsomes was re‐suspended in a stabilizing buffer (8 mM CaCl2, 50 mM MOPS‐KOH, pH 7.0, 20% (vol/vol) added glycerol, 5 mM MgCl2) and flash‐frozen in liquid nitrogen. A similar protocol preparing for purification by Reactive Red 120 beads was published earlier (Yao et al, 1998). The protein was solubilized in n‐dodecyl β‐D‐maltopyranoside (DDM) at a ratio of 1:3 (w/w). Once solubilized, the samples were centrifuged for 45 min at 200,000 × g at 4°C. The supernatant was loaded onto Reactive beads (Green Separopore 4B‐CL, Bio‐world) and incubated for 1 h. Beads were washed with five column volumes of wash buffer (20% (vol/vol) glycerol, 20 mM MOPS‐KOH pH 7.0, 1 mM CaCl2, 1 mg/ml C12E8; 0.35 mg/ml egg‐yolk phosphatidylcholine lipids (EYPC)). Finally, protein was eluted with 2 column volumes elution buffer with 50 mM NaCl and 4 mM Adenosine‐5′‐[β,γ‐methylene]triphosphate (AMPPCP). The protein concentration was measured using the Bradford protein assay (Bio‐Rad). The presence of other SERCA isoforms was tested by Western blot using the following antibodies: S2b AB (SERCA2b pAB serum, Badrilla); S2a AB (SERCA2a pAB serum, Badrilla) 1:100,000; S2 AB (IID8) 1:2,000.
Activity measurements of purified SERCA2a and COS‐1 microsomes
For the activity measurements, the wash and elution buffers for the protein purification contained 0.25 mg/ml C12E8 and no EYPC was added. The Ca2+‐dependent activity of the protein was measured using the Baginski assay probing inorganic phosphate released from ATPase activity (Baginski et al, 1967). Briefly, the reaction mixture containing 50 mM TES/TRIS, pH 6.9, 100 mM KCl, 7 mM MgCl2, 1 mM ethylene glycol‐bis(β‐aminoethyl ether)‐N,N,N′,N′‐tetraacetic acid (EGTA), 5 mM NaN3, 0.2 mM NaMoO4, 50 mM KNO3, and 150 ng (purified enzyme) or 5 μg (COS‐1 microsomes) of the protein, as well as CaCl2 in different concentrations, was initiated with 5 mM ATP (final concentration) and incubated at 37°C for 20 min. The reaction was stopped with the double reaction volume of ascorbic acid solution (170 mM ascorbic acid dissolved 0.5 N HCl and mixed with 4 mM NH4MoO4, solutions kept on ice). The samples treated with ascorbic acid solutions were incubated for 5 min on ice, and then, triple reaction volume of colorimetric solution (210 mM Na3AsO3, 155 mM sodium citrate and 58 mM acetic acid) was added to stabilize the color. The absorbance was measured at 850 nm wavelength after an additional 5‐min incubation at 37°C. The results were analyzed in Excel and Origin (OriginLab Corporation).
Crystallization of SERCA2a
Purified SERCA2a was concentrated to 3.5–6 mg/ml, and trace amounts of aggregated proteins were pelleted by centrifugation at 100,000 × g for 10 min. Crystals were grown using the vapor‐diffusion–hanging drop method, in which 1 μl of protein was mixed with 1 μl reservoir solution and equilibrated against 450 μl reservoir solution. For the CPA‐stabilized form, an egg‐yolk phosphatidylcholine (EYPC)‐to‐protein ratio of 1.7:1 (w/v) was used, and 200 μM CPA, 80 mM KCl, 15 mM MgCl2, 2 mM EGTA, and 10 mM NaF were added. The ‐CPA crystals were grown at 19°C for 4 days using reservoir solutions containing 14–20% PEG4000, 0–5% glycerol, 4–6% 2‐methyl‐2,4‐pentanediol, and 100 mM Na+ malonate in the reservoir and then stored at 4°C. For the E1 conformation, the protein was treated with a final concentration of 10 mM CaCl2, 80 mM KCl, and 3 mM MgCl2. [Ca2]E1‐AMPPCP crystals appeared with reservoir solutions containing 21% PEG2000 monomethyl ether, 20% glycerol, 100 mM NaCl, 5% tert‐BuOH, and 2.4% Zwittergent‐3‐08 and grew within 7–10 days at 19°C. Crystals were fished with nylon loops. The E2‐AlF4‐CPA crystals were swiftly dipped in a mixture of 1 μl reservoir solution and 1 μl of 50% glycerol for cryoprotection. The mounted crystals were flash‐cooled and stored in liquid nitrogen.
Data collection, processing, and structural analysis
Diffraction data were collected at the Swiss Light Source (Villigen, Switzerland), at beamline PXI or PXII using a Pilatus 6M detector, with X‐ray wavelengths at around 1 Å. Data were processed with XDS (Kabsch, 2010) to 3.3 Å maximum resolution in space group P21 for the E2‐AlF4‐CPA form and to 4.0 Å in space group P2 for the [Ca2]E1‐AMPPCP form. The resolution cutoff was determined by CC1/2. The structures were determined by molecular replacement using PHASER‐MR (McCoy et al, 2007) and SERCA1a as a search model (PDB entries 3FGO and 3N8G) and revealing in both cases one SERCA2a molecule in the asymmetric unit. Model building was performed in COOT (Emsley & Cowtan, 2004) using electron density maps calculated using PHENIX (Adams et al, 2010). Refinement of the structural model was done in PHENIX (Adams et al, 2010). Both structures were built using the iMDff technique (Croll & Andersen, 2016; Focht et al, 2017). Structures were analyzed with Molprobity, indicating for the E2‐AlF4‐CPA structure 98% residues in favored, 2% in further allowed, and 0 in non‐favored region, with < 1% rotamer outliers, and for the [Ca2]E1‐AMPPCP structure 96% residues in favored, 4% in further allowed, and 0 in non‐favored regions of the Ramachandran plot, with no outlier rotamers. Figures were prepared, and further, model analysis was performed with PyMOL. The root‐mean‐square deviation (RMSD) for Cα atoms between the SERCA2a and the corresponding SERCA1a structures was determined by the “super” command in PyMOL.
Molecular dynamics
Molecular dynamics (MD) simulations were performed using the Gromacs package (version 4.5.3) (Hess et al, 2008). Each structure was placed in a dodecahedral box (x: 9.483 nm, y: 9.542 nm, z: 17.07 nm), and the system was solvated with TIP3P water molecules (Jorgensen et al, 1983). Lengths of x‐axis and y‐axis were fitted to the membrane dimensions. The energy of the system was minimized with 50,000 steps of steepest descent. Pre‐equilibrated 1,2‐dioleoyl‐sn‐glycero‐3‐phosphocholine (DOPC) membrane structure and parameters were obtained from the Lipidbook website. The membrane system was energy‐minimized with 50,000 steps of steepest descent. Subsequently, all water molecules in the protein and membrane system were removed before merging them and inserting the protein into the membrane using g_membed tool in Gromacs. Next, the protein–membrane system was solvated with TIP3P and counterions were added to neutralize the intrinsic negative charge of the SERCA2a structure. The energy of the protein–membrane system was minimized once more with 50,000 steps of steepest descent. The system was equilibrated using 500 ps of NVT ensemble with the V‐rescale thermostat followed by 2 ns of NPT ensemble with the V‐rescale thermostat and the Parrinello/Rahman barostat. Protein atoms and phosphorous atoms of lipid molecules were position‐restrained during equilibration with a force of 1,000 kJ/mol/nm2. After equilibrium was reached, a full production MD without position restraint was performed for a timescale of 50 ns. In all simulations, an all‐atom AMBER99SB force field supplemented with AMBER‐GAFF force field parameters for DOPC membrane (available on Lipidbook website) was employed. Interactions between residues were identified by energetic criteria. Using CHARMM force field in Brugel, we calculated the electrostatic interactions.
Plasmids, mutagenesis, and cell culture
SERCA1a in pcDNA3.1 and SERCA2a in pMT2 were used for all expression experiments (Dode et al, 2003). Mutants were generated using the QuikChange site‐directed mutagenesis kit (StrataGene) or the Q5 mutagenesis kit (New England Biolabs). A SERCA1a chimera with the luminal loop connecting transmembrane segments 7 and 8 (L7/8) of SERCA2a was previously described (Clausen et al, 2012). The reverse SERCA2a chimera with L7/8 of SERCA1a was generated using BBVC1 and BMGB1 restriction sites. Transfection into COS‐1 cells was performed with GeneJuice transfection reagent (Invitrogen). 72 h after transfection, the microsomal fraction was isolated by differential centrifugation as described previously (Maruyama & MacLennan, 1988).
Mass spectrometry
SERCA1a was purified according to a previously published protocol (Young et al, 1997). 10 μg of purified SERCA2a and SERCA1a was reduced by 5 mM DTT and alkylated with 25 mM iodoacetamide, followed by precipitation (Wessel & Flügge, 1984). The proteins were digested overnight using 0.5 μg trypsin at 37°C in 200 mM ammonium bicarbonate, 5% acetonitrile, 0.01% ProteaseMax (Promega). The resulting peptides were desalted with C18 ZipTip pipette tips (Millipore), and high‐resolution LC‐MS/MS on a 15‐cm EASY‐spray C18 column (Thermo Fisher Scientific) was performed using an Ultimate 3000 nano UPLC system interfaced with a Q Exactive hybrid quadrupole‐orbitrap mass spectrometer. Peptides were identified by MASCOT (Matrix Science) using the SwissProt mammalian database via the Proteome Discoverer 2.2 software. Percolator was incorporated for peptide validation and ptmRS for PTM localization. Carbamidomethylation (C) was used as fixed modification, and acetylation (K), phosphorylation (STY), and ubiquitination (K) were each used in combination with oxidation (M) as variable modifications in the search parameter fields. Other search parameters included two allowed missed cleavages for trypsin digestion, peptide tolerance at 10 ppm and MS/MS tolerance at 20 mmu. Only peptides with a high identification confidence (PEP < 0.01) and a localization confidence of > 99% were taken into account.
Statistics
Results of the Ca2+‐dependent ATPase measurements were fitted with the Hill function using Origin 8.0. One‐way ANOVA with a Bonferroni post hoc test was used for establishing significance.
Author contributions
Conceptualization: CO, PV, PN; Methodology: AS, ND, IV, MDM, EW, CO, PV, PN; Formal analysis: AS, NDD, JLA, RD, SS; Investigation: AS, NDD, RD, SS, JDR, IV, JC; Writing—original draft preparation: AS, PV; Writing—review and editing: AS, JLA, PN, PV; Visualization: AS; Supervision: CO, PV, PN; Funding acquisition: AS, CO, PV, PN.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Review Process File
Acknowledgements
We would like to thank Maike Bublitz and Jesper Lykkegaard Karlsen for their help with data processing and iMDff experiments. We are grateful to Marleen Schuermans, Ingrid Puusta, Tugce Arslan, Anne Lindeman, Anna Marie Nielsen, Lotte Thue Pedersen, and Tetyana Klymchuk for their excellent technical assistance. We would like to thank Oleg Sitsel and Caroline Neumann for help with data analysis and valuable discussions; Howard Young for the initial input to establish a purification protocol; Karin Sipido for supplying us with pig hearts; Chris Ulens for access to crystallization facilities and valuable advice during data processing; the Swiss Light Source (Paul Scherrer Institute) for providing data collection facilities; and the EMBL beamlines at ESRF and DESY for crystal screening, and the Dansync program of the Danish Research Council for support of synchrotron research. This work was funded by the Flanders Research Foundation FWO (G044212N and G0B1115N) and the Inter‐University Attraction Poles program (P7/13) assigned to PV. The work was supported by an ERC grant (BIOMEMOS) to PN. The DANDRITE center is funded by the Lundbeck Foundation (grant no. R248‐2016‐2518). AS was supported by the doctoral scholarship provided by Agentschap Innoveren & Ondernemen (VLAIO).
The EMBO Journal (2019) 38: e100020
Contributor Information
Claus Olesen, Email: ceo@biomed.au.dk.
Peter Vangheluwe, Email: peter.vangheluwe@kuleuven.be.
Poul Nissen, Email: pn@mbg.au.dk.
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L‐W, Kapral GJ, Grosse‐Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akin BL, Hurley TD, Chen Z, Jones LR (2013) The structural basis for phospholamban inhibition of the calcium pump in sarcoplasmic reticulum. J Biol Chem 288: 30181–30191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albers RW, Fahn S, Koval GJ (1963) The role of sodium ions in the activation of electrophorus electric organ adenosine triphosphatase. Proc Natl Acad Sci USA 50: 474–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DM, Anderson KM, Chang CL, Makarewich CA, Nelson BR, McAnally JR, Kasaragod P, Shelton JM, Liou J, Bassel‐Duby R, Olson EN (2015) A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell 160: 595–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DM, Makarewich CA, Anderson KM, Shelton JM, Bezprozvannaya S, Bassel‐Duby R, Olson EN (2016) Widespread control of calcium signaling by a family of SERCA‐inhibiting micropeptides. Sci Signal 9: ra119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkin IT, Adams PD, MacKenzie KR, Lemmon MA, Brünger AT, Engelman DM (1994) Structural organization of the pentameric transmembrane alpha‐helices of phospholamban, a cardiac ion channel. EMBO J 13: 4757–4764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelsen KB, Palmgren MG (1998) Evolution of substrate specificities in the P‐type ATPase superfamily. J Mol Evol 46: 84–101 [DOI] [PubMed] [Google Scholar]
- Baginski ES, Foà PP, Zak B (1967) Microdetermination of inorganic phosphate, phospholipids, and total phosphate in biologic materials. Clin Chem 13: 326–332 [PubMed] [Google Scholar]
- Bers DM (2002) Cardiac excitation contraction coupling. Nature 415: 198–215 [DOI] [PubMed] [Google Scholar]
- Blackwell DJ, Zak TJ, Robia SL (2016) Cardiac calcium ATPase dimerization measured by cross‐linking and fluorescence energy transfer. Biophys J 111: 1192–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bublitz M, Poulsen H, Morth JP, Nissen P (2010) In and out of the cation pumps: P‐type ATPase structure revisited. Curr Opin Struct Biol 20: 431–439 [DOI] [PubMed] [Google Scholar]
- Bublitz M, Musgaard M, Poulsen H, Thøgersen L, Olesen C, Schiøtt B, Morth JP, Møller JV, Nissen P (2013) Ion pathways in the sarcoplasmic reticulum Ca2+‐ATPase. J Biol Chem 288: 10759–10765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao SC, Yang MH, Lee JYY (2002) Mutation analysis of the ATP2A2 gene in Taiwanese patients with Darier's disease. Br J Dermatol 146: 958–963 [DOI] [PubMed] [Google Scholar]
- Clausen JD, Vandecaetsbeek I, Wuytack F, Vangheluwe P, Andersen JP (2012) Distinct roles of the C‐terminal 11th transmembrane helix and luminal extension in the partial reactions determining the high Ca2+ affinity of sarco(endo)plasmic reticulum Ca2+‐ATPase isoform 2b (SERCA2b). J Biol Chem 287: 39460–39469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croll TI, Andersen GR (2016) Re‐evaluation of low‐resolution crystal structures via interactive molecular‐dynamics flexible fitting (iMDFF): a case study in complement C4. Acta Crystallogr Sect D Struct Biol 72: 1006–1016 [DOI] [PubMed] [Google Scholar]
- Daiho T, Yamasaki K, Saino T, Kamidochi M, Satoh K, Iizuka H, Suzuki H (2001) Mutations of either or both Cys876 and Cys888 residues of sarcoplasmic reticulum Ca2+‐ATPase result in a complete loss of Ca2+ transport activity without a loss of Ca2+‐dependent ATPase activity. Role of the CYS876‐CYS888 disulfide bond. J Biol Chem 276: 32771–32778 [DOI] [PubMed] [Google Scholar]
- Dode L, Vilsen B, Van Baelen K, Wuytack F, Clausen JD, Andersen JP (2002) Dissection of the functional differences between sarco(endo)plasmic reticulum Ca2+‐ATPase (SERCA) 1 and 3 isoforms by steady‐state and transient kinetic analyses. J Biol Chem 277: 45579–45591 [DOI] [PubMed] [Google Scholar]
- Dode L, Andersen JP, Leslie N, Dhitavat J, Vilsen B, Hovnanian A (2003) Dissection of the functional differences between sarco(endo)plasmic reticulum Ca2+‐ATPase (SERCA) 1 and 2 isoforms and characterization of Darier disease (SERCA2) mutants by steady‐state and transient kinetic analyses. J Biol Chem 278: 47877–47889 [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Crystallogr Sect D Biol Crystallogr 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Ferrandi M, Barassi P, Tadini‐Buoninsegni F, Bartolommei G, Molinari I, Tripodi MG, Reina C, Moncelli MR, Bianchi G, Ferrari P (2013) Istaroxime stimulates SERCA2a and accelerates calcium cycling in heart failure by relieving phospholamban inhibition. Br J Pharmacol 169: 1849–1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Focht D, Croll TI, Pedersen BP, Nissen P (2017) Improved model of proton pump crystal structure obtained by interactive molecular dynamics flexible fitting expands the mechanistic model for proton translocation in P‐Type ATPases. Front Physiol 8: 202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster DB, Liu T, Rucker J, O'Meally RN, Devine LR, Cole RN, O'Rourke B (2013) The cardiac acetyl‐lysine proteome. PLoS ONE 8: e67513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg B, Butler J, Felker GM, Ponikowski P, Voors AA, Desai AS, Barnard D, Bouchard A, Jaski B, Lyon AR, Pogoda JM, Rudy JJ, Zsebo KM (2016) Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double‐blind, placebo‐controlled, phase 2b trial. Lancet 387: 1178–1186 [DOI] [PubMed] [Google Scholar]
- Haghighi K, Kolokathis F, Pater L, Lynch RA, Asahi M, Gramolini AO, Fan G‐C, Tsiapras D, Hahn HS, Adamopoulos S, Liggett SB, Dorn GW, MacLennan DH, Kremastinos DT, Kranias EG (2003) Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest 111: 869–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess B, Kutzner C, Van Der Spoel D, Lindahl E (2008) GRGMACS 4: algorithms for highly efficient, load‐balanced, and scalable molecular simulation. J Chem Theory Comput 4: 435–447 [DOI] [PubMed] [Google Scholar]
- Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E (2015) PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43: D512–D520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulot JS, Salem JE, Redheuil A, Collet JP, Varnous S, Jourdain P, Logeart D, Gandjbakhch E, Bernard C, Hatem SN, Isnard R, Cluzel P, Le Feuvre C, Leprince P, Hammoudi N, Lemoine FM, Klatzmann D, Vicaut E, Komajda M, Montalescot G et al (2017) Effect of intracoronary administration of AAV1/SERCA2a on ventricular remodelling in patients with advanced systolic heart failure: results from the AGENT‐HF randomized phase 2 trial. Eur J Heart Fail 19: 1534–1541. [DOI] [PubMed] [Google Scholar]
- Jessup M, Greenberg B, Mancini D, Cappola T, Pauly DF, Jaski B, Yaroshinsky A, Zsebo KM, Dittrich H, Hajjar RJ, Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID) Investigators (2011) Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+‐ATPase in patients with advanced heart failure. Circulation 124: 304–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79: 926–935 [Google Scholar]
- Kabsch W (2010) XDS. Acta Crystallogr D Biol Crystallogr 66: 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko M, Yamamoto H, Sakai H, Kamada Y, Tanaka T, Fujiwara S, Yamamoto S, Takahagi H, Igawa H, Kasai S, Noda M, Inui M, Nishimoto T (2017) A pyridone derivative activates SERCA2a by attenuating the inhibitory effect of phospholamban. Eur J Pharmacol 814: 1–8 [DOI] [PubMed] [Google Scholar]
- Kho C, Lee A, Jeong D, Oh JG, Chaanine AH, Kizana E, Park WJ, Hajjar RJ (2011) SUMO1‐dependent modulation of SERCA2a in heart failure. Nature 477: 601–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y, Asahi M, Kurzydlowski K, Tada M, MacLennan DH (1998) Phospholamban domain Ib mutations influence functional interactions with the Ca2+‐ATPase isoform of cardiac sarcoplasmic reticulum. J Biol Chem 273: 14238–14241 [DOI] [PubMed] [Google Scholar]
- Laursen M, Bublitz M, Moncoq K, Olesen C, Møller JV, Young HS, Nissen P, Morth JP (2009) Cyclopiazonic acid is complexed to a divalent metal ion when bound to the sarcoplasmic reticulum Ca2+‐ATPase. J Biol Chem 284: 13513–13518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokuta AJ, Maertz NA, Meethal SV, Potter KT, Kamp TJ, Valdivia HH, Haworth RA (2005) Increased nitration of sarcoplasmic reticulum Ca2+‐ATPase in human heart failure. Circulation 111: 988–995 [DOI] [PubMed] [Google Scholar]
- MacLennan DH, Kranias EG (2003) Phospholamban: a crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol 4: 566–577 [DOI] [PubMed] [Google Scholar]
- Maruyama K, MacLennan DH (1988) Mutation of aspartic acid‐351, lysine‐352, and lysine‐515 alters the Ca2+ transport activity of the Ca2+‐ATPase expressed in COS‐1 cells. Proc Natl Acad Sci USA 85: 3314–3318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayosi BM, Kardos A, Davies CH, Gumedze F, Hovnanian A, Burge S, Watkins H, Watkins H (2006) Heterozygous disruption of SERCA2a is not associated with impairment of cardiac performance in humans: implications for SERCA2a as a therapeutic target in heart failure. Heart 92: 105–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40: 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraviglia V, Bocchi L, Sacchetto R, Florio M, Motta B, Corti C, Weichenberger C, Savi M, D'Elia Y, Rosato‐Siri M, Suffredini S, Piubelli C, Pompilio G, Pramstaller P, Domingues F, Stilli D, Rossini A (2018) HDAC inhibition improves the sarcoendoplasmic reticulum Ca2+‐ATPase activity in cardiac myocytes. Int J Mol Sci 19: 419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyauchi Y, Daiho T, Yamasaki K, Takahashi H, Ishida‐Yamamoto A, Danko S, Suzuki H, Iizuka H (2006) Comprehensive analysis of expression and function of 51 sarco(endo)plasmic reticulum Ca2+‐ATPase mutants associated with darier disease. J Biol Chem 281: 22882–22895 [DOI] [PubMed] [Google Scholar]
- Moller JV, Olesen C, Winther A‐ML, Nissen P (2010) The sarcoplasmic Ca(2+)‐ATPase: design of a perfect chemi‐osmotic pump. Q Rev Biophys 43: 501–566 [DOI] [PubMed] [Google Scholar]
- Moncoq K, Trieber CA, Young HS (2007) The molecular basis for cyclopiazonic acid inhibition of the sarcoplasmic reticulum calcium pump. J Biol Chem 282: 9748–9757 [DOI] [PubMed] [Google Scholar]
- Montigny C, Decottignies P, Le Maréchal P, Capy P, Bublitz M, Olesen C, Møller JV, Nissen P, Le Maire M (2014) S‐Palmitoylation and S‐Oleoylation of rabbit and pig Sarcolipin. J Biol Chem 289: 33850–33861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mountian I, Baba‐Aïssa F, Jonas JC, De Smedt H, Wuytack F, Parys JB (2001) Expression of Ca2+ transport genes in platelets and endothelial cells in hypertension. Hypertension 37: 135–141 [DOI] [PubMed] [Google Scholar]
- Nellen RGL, Steijlen PM, van Steensel MAM, Vreeburg M, Frank J, van Geel M, van Geel M (2017) Mendelian disorders of cornification caused by defects in intracellular calcium pumps: mutation update and database for variants in ATP2A2 and ATP2C1 associated with darier disease and hailey‐hailey disease. Hum Mutat 38: 343–356 [DOI] [PubMed] [Google Scholar]
- Nelson BR, Makarewich CA, Anderson DM, Winders BR, Troupes CD, Wu F, Reese AL, McAnally JR, Chen X, Kavalali ET, Cannon SC, Houser SR, Bassel‐Duby R, Olson EN (2016) A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Sci AAAS 351: 271–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen C, Sørensen TL‐M, Nielsen RC, Møller JV, Nissen P, Sorensen TL, Nielsen RC, Moller JV, Nissen P (2004) Dephosphorylation of the calcium pump coupled to counterion occlusion. Science 306: 2251–2255 [DOI] [PubMed] [Google Scholar]
- Periasamy M, Bhupathy P, Babu GJ (2008) Regulation of sarcoplasmic reticulum Ca2+ ATPase pump expression and its relevance to cardiac muscle physiology and pathology. Cardiovasc Res 77: 265–273 [DOI] [PubMed] [Google Scholar]
- Post RL, Sen AK (1965) An enzymatic mechanism of active sodium and potassium transport. J Histochem Cytochem 13: 105–112 [DOI] [PubMed] [Google Scholar]
- Ren YQ, Gao M, Liang YH, Hou YX, Wang PG, Sun LD, Xu SX, Li W, Du WH, Zhou FS, Shen YJ, Yang S, Zhang XJ (2006) Five mutations of ATP2A2 gene in Chinese patients with Darier's disease and a literature review of 86 cases reported in China. Arch Dermatol Res 298: 58–63 [DOI] [PubMed] [Google Scholar]
- Ringpfeil F, Raus A, DiGiovanna JJ, Korge B, Harth W, Mazzanti C, Uitto J, Bale SJ, Richard G (2001) Darier disease–novel mutations in ATP2A2 and genotype‐phenotype correlation. Exp Dermatol 10: 19–27 [DOI] [PubMed] [Google Scholar]
- Ruiz‐Perez VL, Carter SA, Healy E, Todd C, Rees JL, Steijlen PM, Carmichael AJ, Lewis HM, Hohl D, Itin P, Vahlquist A, Gobello T, Mazzanti C, Reggazini R, Nagy G, Munro CS, Strachan T (1999) ATP2A2 mutations in Darier's disease: variant cutaneous phenotypes are associated with missense mutations, but neuropsychiatric features are independent of mutation class. Hum Mol Genet 8: 1621–1630 [DOI] [PubMed] [Google Scholar]
- Sahoo SK, Kim T, Kang GB, Lee JG, Eom SH, Kim DH (2009) Characterization of calumenin‐SERCA2 interaction in mouse cardiac sarcoplasmic reticulum. J Biol Chem 284: 31109–31121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuntabhai A, Burge S, Monk S, Hovnanian A (1999a) Spectrum of novel ATP2A2 mutations in patients with Darier's disease. Hum Mol Genet 8: 1611–1619 [DOI] [PubMed] [Google Scholar]
- Sakuntabhai A, Ruiz‐Perez V, Carter S, Jacobsen N, Burge S, Monk S, Smith M, Munro CS, O'Donovan M, Craddock N, Kucherlapati R, Rees JL, Owen M, Lathrop GM, Monaco AP, Strachan T, Hovnanian A (1999b) Mutations in ATP2A2, encoding a Ca2+ pump, cause Darier disease. Nat Genet 21: 271–277 [DOI] [PubMed] [Google Scholar]
- Sorensen TL, Andersen JP (2000) Importance of stalk segment S5 for intramolecular communication in the sarcoplasmic reticulum Ca2+‐ATPase. J Biol Chem 275: 28954–28961 [DOI] [PubMed] [Google Scholar]
- Stammers AN, Susser SE, Hamm NC, Hlynsky MW, Kimber DE, Kehler DS, Duhamel TA (2015) The regulation of sarco(endo)plasmic reticulum calcium‐ATPases (SERCA). Can J Physiol Pharmacol 12: 1–12 [DOI] [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, Hussain M, Phillips AD, Cooper DN (2017) The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next‐generation sequencing studies. Hum Genet 136: 665–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavadia S, Tait RC, McDonagh TA, Munro CS (2001) Platelet and cardiac function in Darier's disease. Clin Exp Dermatol 26: 696–699 [DOI] [PubMed] [Google Scholar]
- Toyoshima C, Iwasawa S, Ogawa H, Hirata A, Tsueda J, Inesi G (2013) Crystal structures of the calcium pump and sarcolipin in the Mg2+‐bound E1 state. Nature 495: 260–264 [DOI] [PubMed] [Google Scholar]
- Trieber CA, Douglas JL, Afara M, Young HS (2005) The effects of mutation on the regulatory properties of phospholamban in co‐reconstituted membranes. Biochemistry 44: 3289–3297 [DOI] [PubMed] [Google Scholar]
- Ushioda R, Miyamoto A, Inoue M, Watanabe S, Okumura M, Maegawa K, Uegaki K, Fujii S, Fukuda Y, Umitsu M, Takagi J, Inaba K, Mikoshiba K, Nagata K (2016) Redox‐assisted regulation of Ca2+ homeostasis in the endoplasmic reticulum by disulfide reductase ERdj5. Proc Natl Acad Sci USA 113: E6055–E6063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandecaetsbeek I, Raeymaekers L, Wuytack F, Vangheluwe P (2009a) Factors controlling the activity of the SERCA2a pump in the normal and failing heart. BioFactors 35: 484–499 [DOI] [PubMed] [Google Scholar]
- Vandecaetsbeek I, Trekels M, De Maeyer M, Ceulemans H, Lescrinier E, Raeymaekers L, Wuytack F, Vangheluwe P (2009b) Structural basis for the high Ca2+ affinity of the ubiquitous SERCA2b Ca2+ pump. Proc Natl Acad Sci USA 106: 18533–18538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandecaetsbeek I, Vangheluwe P, Raeymaekers L, Wuytack F, Vanoevelen J (2011) The Ca2+ pumps of the endoplasmic reticulum and Golgi apparatus. Cold Spring Harb Perspect Biol 3: 1–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vangheluwe P, Schuermans M, Zádor E, Waelkens E, Raeymaekers L, Wuytack F (2005) Sarcolipin and phospholamban mRNA and protein expression in cardiac and skeletal muscle of different species. Biochem J 389: 151–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viner RI, Ferrington DA, Williams TD, Bigelow DJ, Scho C (1999) Protein modification during biological aging : selective tyrosine nitration of the SERCA2a isoform of the sarcoplasmic reticulum Ca2+‐ATPase in skeletal muscle. Biochem J 340: 657–669 [PMC free article] [PubMed] [Google Scholar]
- Wessel D, Flügge UI (1984) A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem 138: 141–143 [DOI] [PubMed] [Google Scholar]
- Winther A‐ML, Bublitz M, Karlsen JL, Møller JV, Hansen JB, Nissen P, Buch‐Pedersen MJ (2013) The sarcolipin‐bound calcium pump stabilizes calcium sites exposed to the cytoplasm. Nature 495: 265–269 [DOI] [PubMed] [Google Scholar]
- Xu C, Prasad AM, Inesi G, Toyoshima C (2008) Critical role of Val‐304 in conformational transitions that allow Ca2+ occlusion and phosphoenzyme turnover in the Ca2+ transport ATPase. J Biol Chem 283: 3297–3304 [DOI] [PubMed] [Google Scholar]
- Yao Q, Chen LT, Bigelow DJ (1998) Affinity purification of the Ca‐ATPase from cardiac sarcoplasmic reticulum membranes. Protein Expr Purif 13: 191–197 [DOI] [PubMed] [Google Scholar]
- Young HS, Rigaud JL, Lacapère JJ, Reddy LG, Stokes DL (1997) How to make tubular crystals by reconstitution of detergent‐solubilized Ca2+‐ATPase. Biophys J 72: 2545–2558 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Review Process File