

Abstract
In light of the increasing number of identified cancer‐driven gain‐of‐function (GOF) mutants of p53, it is important to define a common mechanism to systematically target several mutants, rather than developing strategies tailored to inhibit each mutant individually. Here, using RNA immunoprecipitation‐sequencing (RIP‐seq), we identified the Polycomb‐group histone methyltransferase EZH2 as a p53 mRNA‐binding protein. EZH2 bound to an internal ribosome entry site (IRES) in the 5′UTR of p53 mRNA and enhanced p53 protein translation in a methyltransferase‐independent manner. EZH2 augmented p53 GOF mutant‐mediated cancer growth and metastasis by increasing protein levels of mutant p53. EZH2 overexpression was associated with worsened outcome selectively in patients with p53‐mutated cancer. Depletion of EZH2 by antisense oligonucleotides inhibited p53 GOF mutant‐mediated cancer growth. Our findings reveal a non‐methyltransferase function of EZH2 that controls protein translation of p53 GOF mutants, inhibition of which causes synthetic lethality in cancer cells expressing p53 GOF mutants.
Keywords: EZH2, gain‐of‐function mutation, metastasis, non‐methyltransferase activity, p53
Subject Categories: Cancer, Molecular Biology of Disease, Protein Biosynthesis & Quality Control
Introduction
EZH2 is a SET domain‐containing protein that belongs to the Polycomb‐group (PcG) family (Margueron & Reinberg, 2011). Working coordinately with other PcG proteins in the Polycomb repressive complex 2 (PRC2), EZH2 primarily functions as an methyltransferase by catalyzing histone H3 lysine 27 trimethylation (H3K27me3; Cao et al, 2002; Czermin et al, 2002; Kuzmichev et al, 2002; Muller et al, 2002). The Polycomb‐dependent (PcD) function of EZH2 is not only important for developmental patterning, X chromosome inactivation, stem cell maintenance, and cell‐fate decision (Plath et al, 2003; Boyer et al, 2005; Ezhkova et al, 2009; Margueron & Reinberg, 2011), but also implicated in cancer (Varambally et al, 2002; Cha et al, 2005; Chen et al, 2010). Overexpression of EZH2 often correlates with aggressive, metastatic solid tumors such as prostate and breast cancers (Varambally et al, 2002; Kleer et al, 2003; Karanikolas et al, 2009). Active mutations in the SET domain of EZH2 frequently occur in human lymphomas (Morin et al, 2010; Sneeringer et al, 2010; McCabe et al, 2012a), resulting in aberrant activation of PcD and elevation of H3K27me3.
In a Polycomb‐independent (PcI) manner, EZH2 acts in “solo” to regulate actin polymerization and the oncogenic activities of transcription factors such as the androgen receptor (AR), but the PcI function remains methyltransferase‐dependent (Su et al, 2005; Xu et al, 2012). Because of the importance of the methyltransferase‐dependent PcD and PcI functions in cancer, targeting the enzymatic activity of EZH2 is the current focus to develop small molecule inhibitors of EZH2 for cancer treatment (McCabe et al, 2012b). A few such inhibitors have been developed and exhibit apparent anti‐cancer activities by decreasing cell proliferation and tumor growth in various cancer models (McCabe et al, 2012b; Wu et al, 2016). While the methyltransferase activities of EZH2 are well studied, it remains unexplored whether or not EZH2 also possesses non‐methyltransferase function(s) that might also be important for oncogenesis.
TP53 is a well‐studied tumor suppressor gene (Levine, 1997; Li et al, 2012). It is commonly mutated in advanced tumors. While losing the tumor suppressor activity, some mutants of p53 acquire a dominant negative function to inhibit the activity of the remaining wild‐type p53 or gain completely new functions (GOF) to drive cancer progression, which include the functions to promote cell proliferation, migration, metastasis, and metabolism in various types of cancer (Dittmer et al, 1993; Olive et al, 2004; Freed‐Pastor et al, 2012; Weissmueller et al, 2014). Due to the presence of many different types of p53 GOF mutations with distinctive roles in driving cancer progression, various strategies have been explored to target mutant p53 for cancer therapy, including the degradation of p53 mutant proteins, conversion back to the wild‐type p53 or targeting downstream signaling pathways of p53 mutants (Adorno et al, 2009; Muller & Vousden, 2014; Weissmueller et al, 2014; Zhu et al, 2015). However, such approaches largely depend on the types of p53 mutations a tumor carries, which potentially limits the broad use of each strategy in clinic. Thus, it becomes very critical to identify common regulators of different p53 GOF mutants for effective treatment of cancers.
Protein translation can be carried out by both cap‐dependent and cap‐independent pathways. When cap‐dependent translation is globally inhibited under conditions such as cellular stresses, cells can continuously synthesize full‐length p53 protein and ΔNp53 isoform by utilizing two IRES elements residing in the 5′UTR (hereafter termed IRES1) and the coding region (hereafter termed IRES2) of p53 mRNA, respectively (Candeias et al, 2006; Ray et al, 2006; Yang et al, 2006). The importance of IRES‐dependent p53 protein production is further manifested in unstressed cells (Weingarten‐Gabbay et al, 2014). A few proteins, including polypyrimidine tract binding protein (PTB), translation initiation factor DAP5, HDMX, and HDM2, have been identified as putative IRES trans‐acting factors (ITAFs) that preferentially bind to IRES2 of p53 mRNA (Grover et al, 2008; Malbert‐Colas et al, 2014; Weingarten‐Gabbay et al, 2014). To date, however, proteins that preferentially bind to p53 IRES1 remain unidentified. In the present study, we identified EZH2 as a p53 mRNA‐binding protein. We demonstrated that EZH2 bound to IRES1 of p53 mRNA and enhanced p53 protein translation in a methyltransferase‐independent manner. We further showed that inhibition of such function of EZH2 induced synthetic lethality in p53 GOF‐driven cancer cells.
Results
RIP‐seq analysis identifies EZH2 as a p53 mRNA‐binding protein
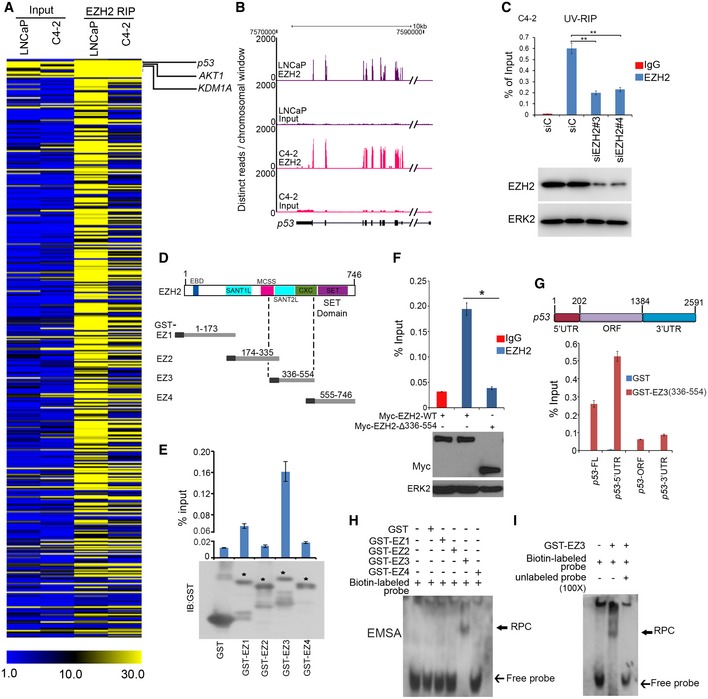
Previous studies suggest that EZH2 regulates gene expression through interaction with long non‐coding RNAs (lncRNAs; Wang et al, 2015; Chen et al, 2018). To define previously unrecognized oncogenic functions of EZH2, we sought to identify new EZH2‐interacting RNAs in cancer cells. A previous study determined that EZH2 nonselectively binds to RNAs, at least under in vitro conditions while findings from other studies suggest that the PRC2 complex as a whole may not do the same in live cells (Davidovich et al, 2013; Cifuentes‐Rojas et al, 2014). Since the crosslink‐based RIP may be susceptible to contamination with non‐specific RNAs (Kaneko et al, 2014), we performed native EZH2 RIP‐seq in two prostate cancer cell lines without crosslink. In addition to binding with lncRNAs, EZH2 selectively bound to a subset of messenger RNA (mRNA) encoding proteins highly relevant to cancer such as p53 (Fig 1A and B, and Table EV1). Using ultraviolet crosslinked RNA immunoprecipitation (UV‐RIP) and quantitative polymerase chain reaction (qPCR), we confirmed EZH2 selective association with p53 mRNA (Fig 1C). EZH2 specific binding with mRNA of other genes such as AKT1 and KDM1A, but not the unbound target SKP2 was also confirmed by RIP‐qPCR (Figs 1A and EV1A–D). These data indicate that EZH2 protein selectively binds to mRNA of a subset of cancer‐relevant genes including TP53 in cells.
Figure 1. EZH2 binds to 5′UTR of p53 mRNA .
-
AHeat map showing a subset of mRNAs of genes immunoprecipitated by anti‐EZH2 antibody in LNCaP and C4‐2 prostate cancer cell lines, which was generated based on the distinct RIP‐seq reads on specific gene exons. The units of the heatmap values were reads per kilobase million (RPKM).
-
BScreen shots from the UCSC genome browser showing signal profiles of p53 mRNA immunoprecipitated by anti‐EZH2 antibody in LNCaP and C4‐2 cells.
-
CC4‐2 cells were subjected to UV‐RIP assay. RT–qPCR measurement of p53 mRNA immunoprecipitated by IgG or anti‐EZH2 antibody. Data shown as means ± SD (n = 3). Statistical significance was determined by two‐tailed Student's t‐test. **P < 0.01.
-
DSchematic diagram of four GST‐EZH2 recombinant proteins (EZ1–EZ4). EBD, EED binding domain; SANT1(/2)L, “Swi3, Ada2, N‐Cor, TFIIIB”1(/2) like; MCSS, motif connecting SANT1L and SANT2L; CXC, cysteine‐rich domain; SET, catalytic domain.
-
ETop, RT–qPCR analysis of p53 mRNA in C4‐2 cell lysate pulled down by GST or GST‐EZH2 recombinant proteins EZ1‐EZ4. Bottom, Western blotting analysis of GST or GST‐EZH2 proteins used for GST pull‐down assay. Asterisks indicate the protein bands at expected molecular weight. Data shown as means ± SD (n = 3). Statistical significance was determined by two‐tailed Student's t‐test.**P < 0.01.
-
FC4‐2 cells were transfected with Myc‐tagged EZH2‐WT or EZH2‐∆336–554 for 24 h, and cells were harvested for RIP with IgG or anti‐Myc‐tag antibody. Transfected proteins and pull‐down p53 mRNAs were analyzed by Western blot and RT–qPCR, respectively. Data shown are means ± SD (n = 3). *P < 0.01. ERK2, a loading control.
-
GGST pull‐down assay using in vitro transcribed different fragments of p53 mRNA and indicated GST proteins followed by RT–qPCR analysis of pull‐down p53 mRNA. FL, full length; ORF, open reading frame; UTR, untranslated region.
-
H, IRNA EMSA evaluation of EZh2 binding of p53 mRNA. GST‐EZH2 recombinant proteins (EZ1–EZ4) were incubated with biotin‐labeled in vitro transcribed p53 5′UTR (biotin‐labeled probe) in the presence or absence of 100‐fold unlabeled p53 5′UTR (unlabeled probe), followed by PAGE and immune blotting with HRP‐conjugated streptavidin.
Source data are available online for this figure.
Figure EV1. Assessment of binding and regulation of mRNAs by EZH2. Related to Fig 1 .
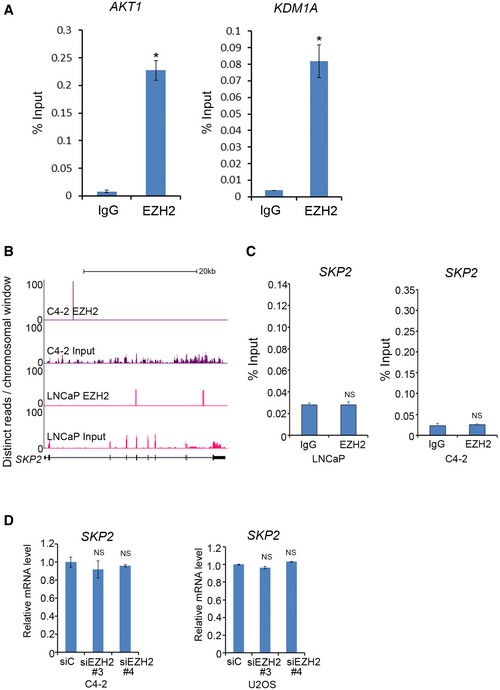
- RT–qPCR analysis of mRNAs of AKT1 and KDM1A immunoprecipitated by IgG or EZH2 antibody in C4‐2 cells. These genes are on the top of the list of mRNAs highly enriched by anti‐EZH2 antibody. Data shown are mean values ± SD from three replicates. *P < 0.01.
- Screen shots from UCSC genome browser showing signal profiles of SKP2 mRNA immunoprecipitated by anti‐EZH2 antibody in LNCaP and C4‐2 cells. Total RNA was used as the input control of RIP‐seq.
- RIP‐qPCR analysis of SKP2 mRNA immunoprecipitated by IgG and anti‐EZH2 antibody in LNCaP and C4‐2 cells. Data shown are mean values ± SD from three replicates.
- Effect of EZH2 on SKP2 mRNA expression. C4‐2 prostate cancer and U2OS osteosarcoma cell lines were transfected with non‐specific control (siC) or two independent EZH2‐specific siRNAs. 48‐h post‐transfection expression of SKP2 mRNA was measured by RT–qPCR. Error bar showing SD from three replicates. NS, no significance.
EZH2 binds to IRES1 of p53 mRNA
The p53 protein is a cellular gatekeeper that plays essential roles in maintaining genomic integrity and regulating cell growth, survival, and energy metabolism (Levine, 1997; Li et al, 2012). We chose to further characterize the molecular basis of the interaction between EZH2 protein and p53 mRNA and the biological consequences. We first examined which region of EZH2 protein is responsible for p53 mRNA binding. Glutathione S‐transferase (GST)‐EZH2 recombinant proteins were purified from bacteria as described (Chen et al, 2010; Fig 1D) and incubated with lysate of LNCaP cells which express endogenous wild‐type p53. GST pull‐down products were subjected to RNA purification and reverse transcription quantitative PCR (RT–qPCR) analysis. The p53 mRNA‐binding domain is the central region (amino acids 336–554, aa336–554; Fig 1D and E). RIP assays demonstrated that deletion of aa336–554 abolished EZH2 binding of p53 mRNA (Fig 1F). In vitro RNA binding assay showed that the aa336–554 region in EZH2 bound primarily to the 5′UTR, but not the coding region and the 3′UTR of p53 mRNA (Fig 1G). These data suggest that EZH2 binds directly to p53 mRNA 5′UTR. To further validate this observation, we performed in vitro RNA electrophoretic mobility shift assay (EMSA) using purified human EZH2 and biotin‐labeled p53 5′UTR as a probe. Consistent with GST pull‐down results (Fig 1E and F), GST‐EZ3 (aa336–554), but not GST alone or other GST‐EZH2 recombinant proteins formed an RNA–protein complex (RPC) with p53 5′UTR (Fig 1H). The binding was dose‐dependent and blocked by excessive amount of unlabeled p53 5′UTR (Figs 1I and EV2A), confirming that the interaction between EZH2 and p53 mRNA 5′UTR is specific. Together, these data suggest that EZH2 protein directly binds to the 5′UTR of p53 mRNA.
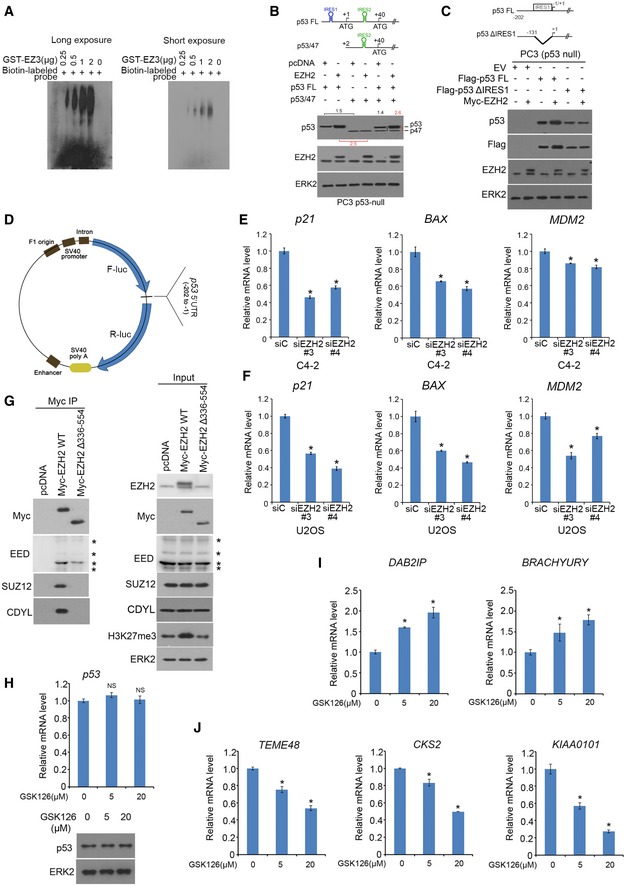
Figure EV2. EZH2 regulation of expression of p53 downstream target genes. Related to Fig 1 .
-
AEZH2 fragment binding to p53 5′UTR determined by RNA EMSA. Different doses of GST‐EZH2 recombinant proteins (GST‐EZ3) were incubated with 1 μg of biotin‐labeled p53 mRNA 5′UTR probe for 1 h on ice. The RNA–protein complex (RPC) was detected by PAGE followed by immune blotting with HRP‐conjugated streptavidin.
-
BpcDNA3.1‐based expression vectors for Flag‐p53 FL and/or Flag‐p53/47 in combination with empty vector or Myc‐EZH2 were transfected into PC3 cells. Forty‐eight hours after transfection cells was lysed in RIPA buffer for Western blots with indicated antibodies. ERK2, a loading control.
-
CPC3 cells were transfected with indicated plasmids. Forty‐eight hours after transfection cells was lysed for Western blot.
-
DDiagram of the map for pp53‐5′UTR‐F/Rluc vector. F means firefly luciferase gene. R means Renilla luciferase gene.
-
E, FExpression of mRNAs for p21 CIP1 , BAX, and MDM2 genes was measured by RT–qPCR in C4‐2 (E) and U2OS (F) cells 48 h after transfection with non‐specific control (siC) or two independent EZH2‐specific siRNAs. GAPDH was used as internal control. Data shown are mean values ± SD (error bar) from three replicates. *P < 0.01 comparing EZH2 siRNA‐transfected with siC‐transfected cells.
-
GC4‐2 cells were transfected with indicated plasmids for 24 h, and cells were harvested for co‐IP and Western blot analysis. Asterisks indicate different EED isoforms.
-
H–JC4‐2 cells treated with vehicle (DMSO) or different concentrations (5 and 20 μM) of GSK126 for 24 h were harvested for analysis of expression of p53 mRNA and protein using RT–qPCR and Western blot, respectively (H), mRNA expression of EZH2 repressed genes DAB2IP and BRACHYURY (I), and EZH2‐activated genes TEME48, CKS2, and KIAA0101 (J). The GAPDH was used as internal control. Data shown are mean values ± SD (error bar) from three replicates. *P < 0.01 comparing GSK126‐treated with mock‐treated cells.
Source data are available online for this figure.
EZH2 enhances IRES1‐mediated p53 protein translation
Consistent with the results of in vitro RNA binding assay (Fig 1G), reciprocal biotin‐labeled RNA pull‐down assays showed that endogenous EZH2 protein from LNCaP cell lysate were bound strongly by p53 mRNA 5′UTR, but very weakly by the 3′UTR and ORF (Fig 2A). As a positive control, EZH2 was readily pulled down by the HOTAIR lncRNA (Fig 2A). We further demonstrated that a 122‐nucleotide (nt) region (−122 to −1 nt) immediately adjacent to the translation start in the 5′UTR of p53 mRNA is critical for EZH2 binding (Fig 2B). Notably, this region almost completely overlaps with IRES1 (−130 to −1 nt) reported previously (Ray et al, 2006; Yang et al, 2006). To determine the IRES1 effect on p53 protein level, p53‐null PC3 cells were co‐transfected with EZH2 in combination with pcDNA‐p53‐FL (5′UTR + CDS + 3′UTR) and/or pcNDA‐p53/47 (ΔNp53), an IRES1‐deletion mutant encoding a protein translated from the start codon at +40. While EZH2 overexpression increased protein level of p53‐FL, its expression had no overt effect on p53/47 in PC cells (Fig EV2B). Similar results were obtained for p53ΔIRES1, an IRES1 deletion (deletion of −130 to −1 nt) mutant in PC3 cells, although p53ΔIRES1 encodes a full‐length p53 (Fig EV2C). These data suggest that EZH2 regulates p53 protein level through IRES1.
Figure 2. EZH2 upregulates p53 expression via binding to the IRES motif in 5′UTR .
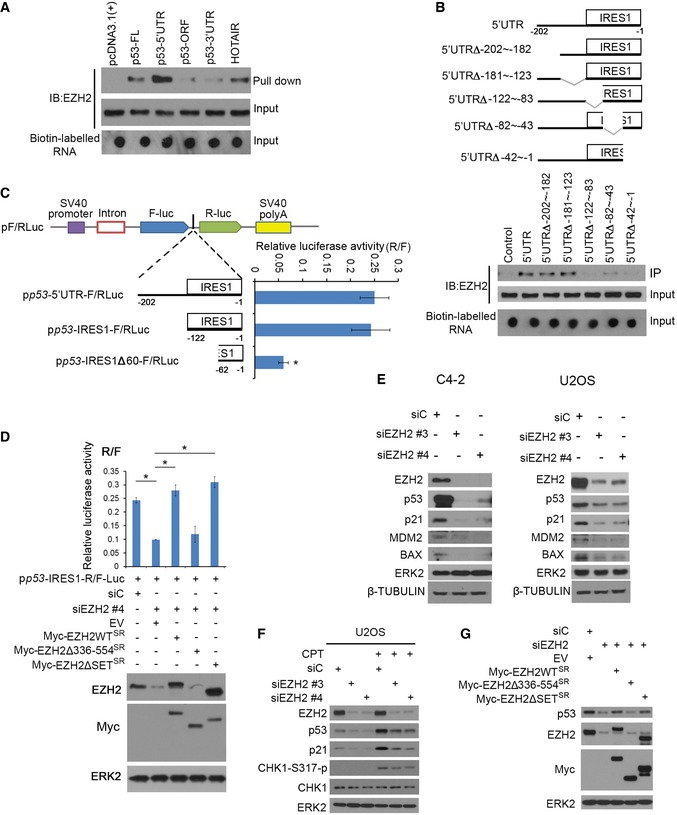
- Biotin pull‐down assay by incubating biotin‐labeled different fragments of p53 mRNA and HOTAIR (positive control) with C4‐2 cell lysate followed by Western blot with EZH2 antibody.
- Biotin pull‐down assay as in (A) using unmutated and various internally deleted mutants of 5′UTR of p53 mRNA. Top, diagram of different p53 5′UTR deletion mutants.
- Upper, the linear map of the pRF bicistronic report plasmid. The SV40 promoter (purple box) was used to drive firefly luciferase (Fluc) and Renilla luciferase (Rluc) gene transcription. Different p53 5′UTR fragments were inserted between the Fluc and Rluc genes. Lower, at 24 h after transfection, cells were lysed and luciferase activities were measured using a dual‐luciferase kit and the ratio of Fluc/Rluc was calculated. Data shown as means ± SD (n = 3). Statistical significance was determined by two‐tailed Student's t‐test. *P < 0.01.
- EZH2 knockdown C4‐2 cells were transfected with the bicistronic reporter vector in combination with empty vector, Myc‐tagged EZH2 WT, or deletion mutants followed by Western blot analysis with indicated antibodies (bottom) and luciferase assay (top). Data shown as means ± SD (n = 3). *P < 0.01.
- C4‐2 and U2OS cell lines were transfected with non‐specific control (siC) or two independent EZH2‐specific siRNAs and harvested for Western blot analysis with indicated antibodies. ERK2 and β‐TUBULIN, loading controls.
- U2OS cells were transfected with control (siC) or EZH2‐specific siRNAs and treated with 200 nM of CPT followed by Western blot analysis for indicated proteins.
- C4‐2 cells were transfected with control (siC) or EZH2‐specific siRNAs and plasmids for empty vector, EZH2 WT, or deletion mutants followed by Western blot analysis for indicated proteins.
Source data are available online for this figure.
We further employed a dual reporter assay to determine whether EZH2 regulates p53 protein translation by binding to IRES1. We generated a series of luciferase reporter constructs by cloning different portions of p53 5′UTR into a bicistronic plasmid (Figs 2C and EV2D). Similar to the previous reports in MCF7 and HeLa cells (Ray et al, 2006; Yang et al, 2006), both the entire 5′UTR and the IRES1 region of p53 mRNA exhibited translation‐promoting activity in comparison with the empty vector (Fig 2C). However, deletion of 60 nt in the 5′ end of IRES1 (IRES1∆60) largely diminished the activity of IRES1 (Fig 2C). Knockdown of endogenous EZH2 by small interference RNAs (siRNAs) significantly reduced the luciferase activity of the IRES‐F/RLuc construct, and this effect was completely reversed by restored expression of siRNA‐resistant wild‐type EZH2 (EZH2‐WTSR), SET domain deletion (methyltransferase‐deficient) mutant (EZH2∆SETSR), but not the aa336–554‐deletion mutant (EZH2∆336–554SR; Fig 2D). These data indicate that EZH2 enhances IRES1‐dependent translation of p53 mRNA and this effect is mediated through the RNA‐binding function, but not the methyltransferase activity of EZH2.
We also determined the effect of EZH2 on the steady‐state level of p53 protein under physiological conditions. We knocked down EZH2 using two independent siRNAs in C4‐2 prostate cancer cell line and U2OS osteosarcoma cell line, both of which express wild‐type p53. Knockdown of endogenous EZH2 decreased expression of endogenous p53 proteins in both cell lines and p53 downstream targets p21CIP1, MDM2, and BAX at both protein and mRNA levels (Figs 2E, and EV2E and F). Thus, EZH2 regulates expression of p53 protein and its downstream genes in unstressed cells.
p53 remains at low activity under normal conditions and becomes highly activated in response to genotoxic stresses. We treated U2OS cells with camptothecin (CPT), a chemotherapeutic drug that inhibits the re‐ligation activity of topoisomerase‐1 and therefore causes DNA breaks. CPT treatment increased expression of p53 protein and the downstream target p21CIP1 in control siRNA‐treated cells (Fig 2F). The effectiveness of CPT was evident by increased phosphorylation of CHK1, a DNA damage checkpoint kinase (Fig 2F). Most importantly, CPT‐induced upregulation of p53 protein expression was largely diminished by EZH2 knockdown (Fig 2F). These results suggest that EZH2 also regulates p53 protein expression in cells under genotoxic stress.
To determine whether EZH2 augments p53 protein expression via aa336–554, we performed knockdown and rescue experiments. EZH2 knockdown‐induced downregulation of p53 protein was almost completely reversed by restored expression of siRNA‐resistant EZH2 (EZH2‐WTSR) and EZH2∆SETSR, but not EZH2∆336–554SR mutant in C4‐2 cells (Fig 2G). To rule out the potential impact of EZH2Δ336–554 on PRC2 complex formation, we examined the ability of EZH2Δ336–554 to bind to other PRC2‐related proteins. EZH2Δ336–554 was able to bind to EED, but not SUZ12 and CDYL (Fig EV2G), indicating that EZH2Δ336–554 cannot form a functional PRC2 complex. These data suggest that EZH2 enhances p53 protein expression in cells in a manner dependent on the p53 mRNA‐binding domain.
The effect of EZH2∆336–554 shown in Fig 2G also indicates that EZH2 regulates p53 protein level in a manner independent of the methyltransferase activity. To further confirm this observation, we treated C4‐2 cells with the EZH2 enzymatic inhibitor GSK126 (McCabe et al, 2012b). As expected, GSK126 treatment effectively increased expression of EZH2‐repressed PcD genes [e.g., DAB2IP and BRACHYURY (Wang et al, 2013)] and decreased expression of EZH2‐activated PcI genes [e.g., TEME48, CKS2, and KIAA0101 (Xu et al, 2012)], but it had no overt effect on p53 mRNA and protein expression (Figs EV2H–J). Together, these data suggest that the function to bind to p53 mRNA, but not the methyltransferase activity is important for EZH2 to increase p53 protein expression in cells.
EZH2 interacts with eIF4G and eIF4A and regulates p53 mRNA binding with polysomes
Without the requirement of eIF4E, both viral and cellular IRESs can mediate cap‐independent protein translation through non‐canonical binding with the eIF4G‐eIF4A complex (Pestova et al, 2001; Jackson et al, 2010; Komar & Hatzoglou, 2011). We performed unbiased tandem affinity purification and mass spectrometry (TAP‐MS) in 293T cells transfected with an empty vector containing S, Flag, and Biotin‐binding‐protein‐(streptavidin)‐binding‐peptide (SFB) tags or SFB‐tagged EZH2. The key components of PRC2, including SUZ12, EED, and JARID2, were effectively pulled down by SFB‐EZH2, but not the SFB empty vector (Fig 3A and Table EV2), indicating that the purification was successful. Importantly, eIF factors involved in cap‐independent translation including eIF4G, eIF4A, and eIF3 as well as poly(A)‐binding protein‐1 (PABP1) were also specifically immunoprecipitated by SFB‐EZH2, but the cap‐binding protein eIF4E was not present in immunoprecipitants (Fig 3A and Table EV2). Reciprocal co‐immunoprecipitation assays further confirmed the interaction of endogenous or Myc‐tagged EZH2 with eIF4G2 and PABP1 in C4‐2 cells (Figs 3B and EV3A). eIF4Gs serve as ITAFs and form a complex with RNA and other proteins (Lopez‐Lastra et al, 2005). To determine whether EZH2 binding with eIF4G or PABP1 is mediated indirectly through RNA or DNA, we treated cell lysate with RNaseA or DNaseI prior to IP. We demonstrated that EZH2 was no longer able to bind to eIF4G2 and PABP1 following RNaseA, but not DNaseI treatment (Figs 3C and EV3B), suggesting that EZH2 bound to eIF4G2 and PABP1 in an RNA‐dependent manner. To further validate the role of RNA in mediating EZH2 interaction with PABP1 and eIF4G2, we performed in vitro protein binding assay in the presence or absence of p53 FL mRNA using GST‐PABP1 and GST‐eIF4G2 purified from bacteria and in vitro transcribed and translated Myc‐EZH2. GST‐PABP1 or GST‐eIF4G2 bound to EZH2 in vitro only in the presence of p53 FL mRNA (Fig EV3C). UV‐RIP assays showed that p53 mRNA was specifically pulled down by EZH2, but not EED and SUZ12 antibodies (Fig EV3D). Moreover, different from the finding that EZH2 knockdown markedly decreased p53 level, knockdown of endogenous SUZ12 and EED only slightly decreased p53 protein level (Fig EV3E). These effects were likely mediated by slightly decreased EZH2 level in SUZ12‐ and EED‐knockdown cells (Fig EV3E), a finding similar to the previous report that the EZH2 level is dependent on the intact PRC2 complex (Pasini et al, 2004; Xu et al, 2015). These data suggest that intact PRC2, but independent of its methyltransferase activity, is required for RNA binding and for p53 upregulation via IRES1. Together, our data indicate that EZH2 association with eIF4G2 and PABP1 is mediated through RNAs such as p53 mRNA.
Figure 3. EZH2 binds with cap‐independent translation complex and increases p53 protein translation.
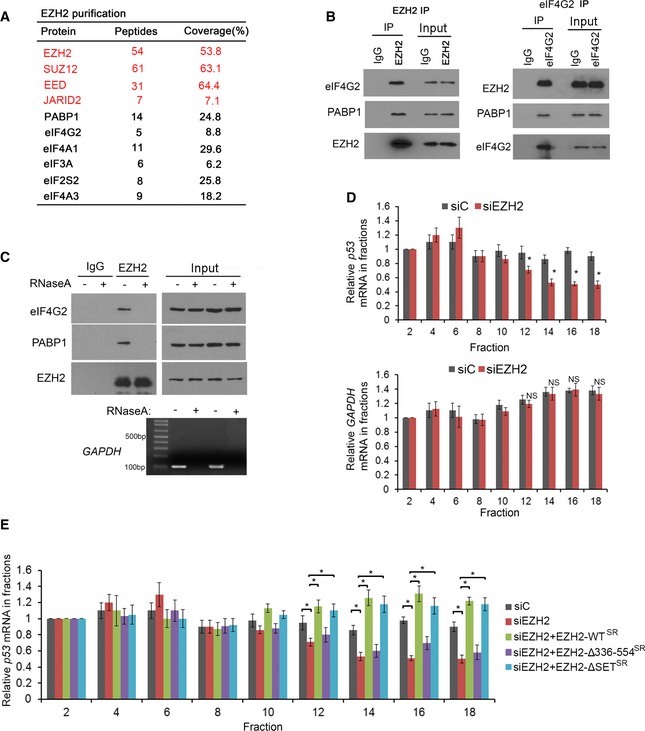
- List of top hits of EZH2‐bound proteins identified by TAP‐MS. The number of peptides and coverage of each protein are indicated.
- Reciprocal co‐IP of endogenous EZH2 and eIF4G2 or PABP1 in C4‐2 cells.
- Co‐IP of endogenous EZH2 with eIF4G2 or PABP1 from C4‐2 cell lysate pre‐treated with RNaseA in 37°C for 30 min. The effectiveness of RNaseA treatment was monitored by RT–PCR analysis of the presence of GAPDH mRNA.
- C4‐2 cells were transfected control (siC) or EZH2‐specific siRNA and lysed for polysome fractionation. RNA was extracted from even‐number fractions followed by RT–qPCR analysis for p53 and GAPDH mRNA. The β‐ACTIN mRNA was used as an internal control. Data shown as means ± SD (n = 3). Statistical significance was determined by two‐tailed Student's t‐test. *P < 0.01; NS, no significance.
- C4‐2 cells were transfected with control (siC) or EZH2‐specific siRNAs in combination with empty vector, EZH2 WT, or different deletion mutants followed by polysome fractionation and RT–qPCR. β‐ACTIN mRNA was used as an internal control. Data shown as means ± SD (n = 3). *P < 0.01.
Source data are available online for this figure.
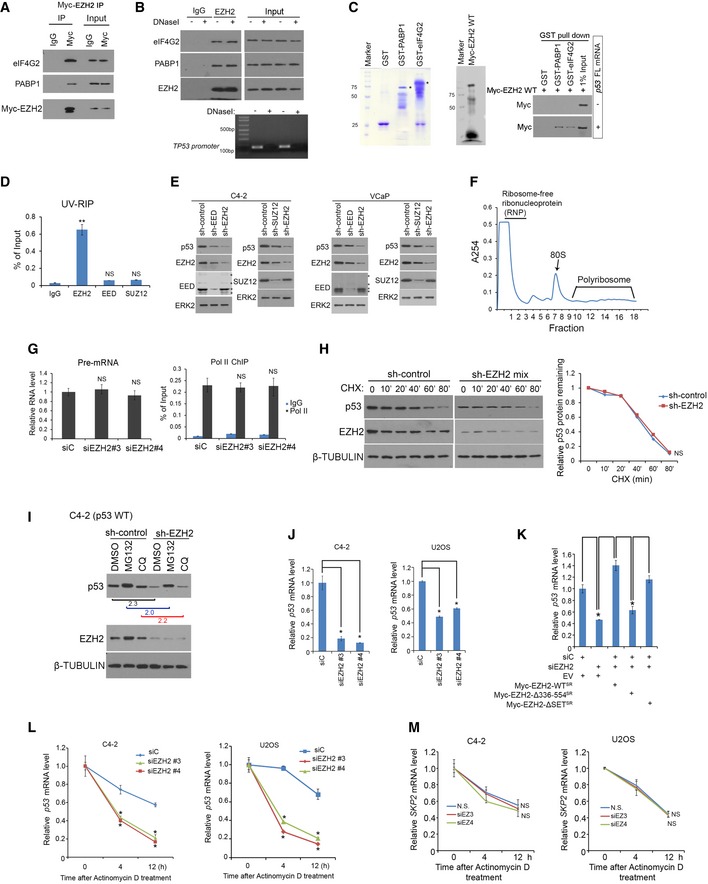
Figure EV3. The effect of the EZH2‐specific enzymatic inhibitor GSK126 on p53 mRNA expression and stability. Related to Fig 3 .
-
AC4‐2 cells transfected with Myc‐EZH2 WT for 24 h were lysed for co‐IP and Western blot analysis.
-
BC4‐2 cells were lysed in IP buffer and treated with DNaseI in 37°C for 15 min followed by co‐IP and Western blot analysis. Genomic DNA in input samples was assessed by PCR using primers for TP53 promoter.
-
CGST, GST‐eIF4G2, and GST‐PABP1 proteins were purified from BL21 bacteria. Myc‐EZH2 was produced using Quick coupled transcription/translation kit through T7 promoter in vitro. p53 full‐length (FL) mRNA was transcribed by T7 enzyme in vitro. These purified proteins were used for GST pull‐down assay in the presence or absence of p53 FL mRNA, followed by Western blot analysis.
-
DUV‐RIP‐qPCR analysis of p53 mRNA immunoprecipitated by IgG, anti‐EED, anti‐SUZ12, or anti‐EZH2 antibody from lysate of C4‐2 cells treated with 4SU for 8 h. Data shown are mean values ± SD from three replicates. **P < 0.01 comparing EZH2 IP with IgG IP. NS, no significance.
-
EC4‐2 (Left) and VCaP (Right) cells were infected with lentivirus for non‐specific control (sh‐Control) or two independent EED (or SUZ12)‐specific shRNAs for 48 h followed by Western blot analysis. ERK2, a loading control.
-
FRNA was extracted from sucrose gradient fractions of C4‐2 cell lysate. 40S, 60S, 80S, and polyribosomes were indicated above fraction numbers. A254, absorbance at 254 nm.
-
GC4‐2 cells were infected with lentivirus for si‐control or siEZH2#3 or siEZH2#4 for 48 h followed by RT–PCR analysis of p53 pre‐mRNA expression (Left) and ChIP‐PCR analysis of Pol II (N20) occupancy (Right).
-
HControl or EZH2 knockdown C4‐2 cells were treated with cycloheximide (CHX, 50 mg/ml) for different periods of time and harvested for Western blot analysis. β‐TUBULIN, a loading control.
-
IControl or EZH2 knockdown C4‐2 cells were treated with MG132 (20 mg/ml) or chloroquine (CQ 25 mg/ml) for 8 h followed by Western blot analysis. β‐TUBULIN, a loading control.
-
JExpression of p53 mRNA was measured by RT–qPCR in C4‐2 cells 48 h after transfection with non‐specific control (siC) or two independent EZH2‐specific siRNAs. GAPDH was used as internal control. Data shown are mean values ± SD from three replicates. *P < 0.01.
-
KExpression of p53 mRNA was measured by RT–qPCR in C4‐2 cells 48 h after transfection with non‐specific control (siC) or EZH2‐specific siRNAs in combination with or without empty vector (EV) or siRNA‐resistant Myc‐EZH2 wild‐type (WT) or mutants. GAPDH was used as an internal control. Data shown are mean values ± SD from three replicates. *P < 0.01.
-
L, Mp53 (L) and SKP2 (M) mRNA stability was assessed in C4‐2 and U2OS cells transfected with non‐specific siRNA (siC) or EZH2‐specific siRNA followed by actinomycin D treatment. GAPDH was used as internal control. Mean values ± SD from three replicates. *P < 0.01.
Source data are available online for this figure.
To further assess EZH2 regulation of p53 protein translation, we performed polysome profiling in C4‐2 cells with or without EZH2 knockdown. EZH2 knockdown resulted in approximately 50% reduction of p53 mRNA presented in polysomes (Figs 3D and EV3F). To determine whether this effect was specifically mediated by EZH2, we performed rescue experiments similar to the one shown in Fig 2G. Restored expression of EZH2 WTSR and EZH2∆SETSR, but not EZH2∆336–554SR mutant was able to restore the level of p53 mRNA in polysomes (Fig 3E). We also determined whether EZH2 affects p53 mRNA transcription and p53 protein degradation. EZH2 knockdown did not affect either p53 pre‐mRNA level or Pol II Ser‐2p occupancy in the TP53 promoter (Fig EV3G). Further analyses showed that EZH2 did not affect the half‐life and the steady‐state level of p53 protein in the presence or absence of the proteasome inhibitor MG132 and the lysosome inhibitor chloroquine (CQ) in C4‐2 cells (Fig EV3H and I). We provide evidence that EZH2 regulates p53 mRNA association with polysomes in a methyltransferase‐independent fashion.
EZH2 increases p53 mRNA stability
Increasing evidence suggests that eIF4G‐PABP1 interaction promotes formation of a “closed” mRNA loop that not only enhances ribosomal recruitment but also prevents mRNA decay. We sought to determine whether EZH2 regulates p53 mRNA stability. We first examined the effect of EZH2 on the steady‐state level of p53 mRNA. Knockdown of endogenous EZH2 by two independent siRNAs invariably decreased mRNA level of endogenous wild‐type p53 in both C4‐2 and U2OS cell lines (Fig EV3J). This effect was completely reversed by restored expression of siRNA‐resistant EZH2‐WTSR and EZH2ΔSETSR, but not the EZH2Δ336–554SR mutant (Figs 2D and EV3K). By measuring the rate of p53 mRNA decay, we demonstrated EZH2 knockdown shortened the half‐life of p53 mRNA in both C4‐2 and U2OS cell lines (Fig EV3L). Consistent with the finding that EZH2 did not bind to SKP2 mRNA (Fig EV1C), EZH2 knockdown had no overt effect on the half‐life of SKP2 mRNA in these two cell lines (Fig EV3M). These results suggest that in addition to regulating p53 protein translation, EZH2 also enhances p53 expression by increasing mRNA stability.
Ezh2 knockout decreases p53 mRNA and protein level in a prostate cancer mouse model
Nucleotide sequence comparison showed that there is approximately 70% sequence similarity between human p53 mRNA 5′UTR and mouse and rat counterparts (Fig EV4A). Similar to the human homolog, the predicted secondary structure of rodent p53 mRNA 5′UTRs also exhibits three major potential stem‐loop moieties (Fig EV4B). In agreement with these observations, RIP‐qPCR assay showed that Ezh2 associated with p53 mRNA in murine cell lines (Fig EV4C).
Figure EV4. Ezh2 binds to p53 mRNA in murine cells. Related to Fig 4 .
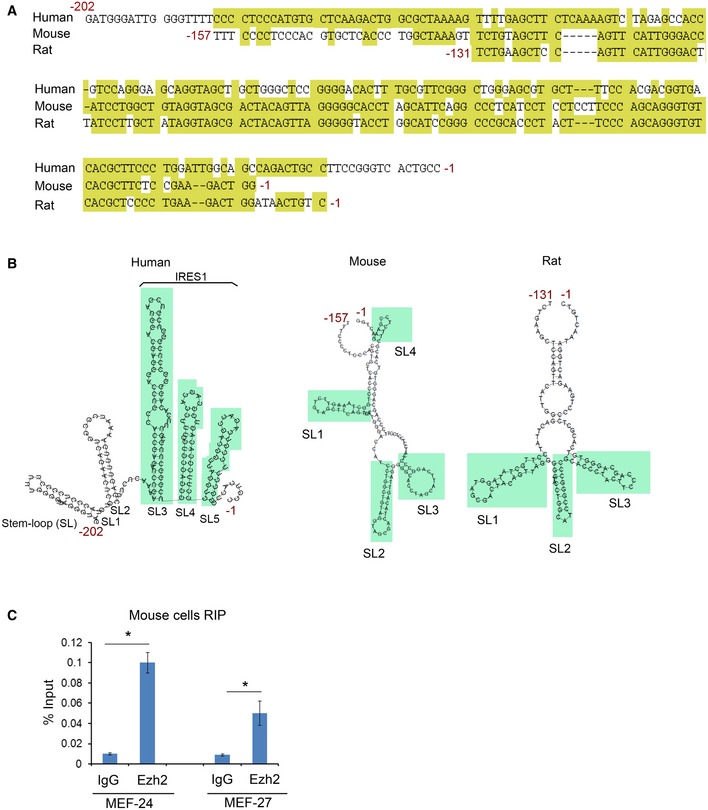
- Alignment of nucleotide sequence of the 5′UTR of p53 mRNA of human, mouse, and rat. The conserved nucleotides are highlighted in yellow.
- The predicted secondary structure of 5′UTR of p53 mRNA of human, mouse, and rat. SL, stem loop.
- RIP assay showing p53 mRNA immunoprecipitated by anti‐EZH2 antibody in MEF‐27 and MEF‐24 cells as measured by RT–qPCR. Data shown as means ± SD (n = 3). *P < 0.01.
PTEN is a tumor suppressor gene that is frequently mutated or deleted in human prostate cancers, and homozygous deletion of the Pten gene invariably promotes tumorigenesis in the mouse prostate. Homozygous deletion of Pten induces upregulation of p53 in prostate cancers in mice (Chen et al, 2005), but the underlying mechanism remains poorly understood. Several studies independently show that Pten‐knockout upregulates Ezh2 mRNA and proteins in mouse prostate tumors (Kuzmichev et al, 2005; Mulholland et al, 2011; Ding et al, 2014). Thus, the Pten‐knockout mice represent an ideal model to determine whether EZH2 plays a causal role in regulation of p53 expression in vivo. We examined whether increased expression of Ezh2 correlates with p53 expression in prostate tumors in this model. As expected, homozygous Pten deletion resulted in upregulation of both p53 and Ezh2 proteins in prostate tumors in mice (Fig 4A and B). Importantly, Ezh2 level highly correlated with p53 protein expression in a cohort of Pten‐knockout mouse prostate tumors (Fig 4C). To determine the causal role of Ezh2 in p53 upregulation in Pten‐null mouse prostate tumors, we generated prostate‐specific Pten;Ezh2 double knockout mice. Ezh2 deletion decreased expression of p53 protein and mRNA as well as its downstream gene Bax in the background of Pten deletion (Fig 4A, D, and E). Thus, similar to the effect in human cancer cell lines cultured in vitro, Ezh2 also plays a causal role in the regulation of p53 mRNA and protein expression under in vivo conditions.
Figure 4. Ezh2 knockout decreases p53 expression in the Pten‐null prostate cancer mouse model.
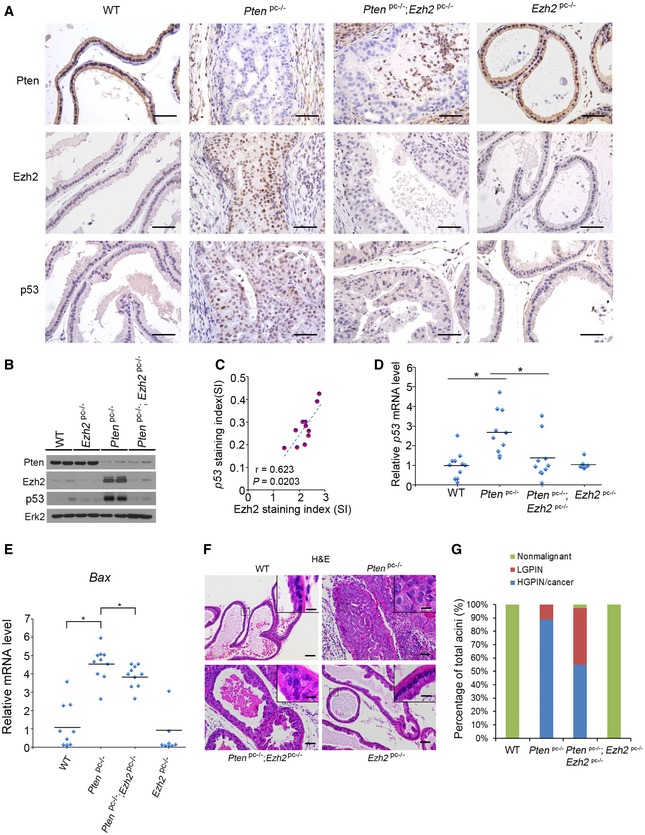
- IHC analysis of Pten, Ezh2, and p53 proteins in prostatic tissues of 4‐month‐old mice with different genotypes: Pten pc−/− (n = 11); Ezh2 pc−/− (n = 8); Pten pc−/−;Ezh2 pc−/− (n = 10) and “wild‐type” littermate controls (n = 10). Scale bars, 50 μm.
- Western blot analysis of expression of indicated proteins in four different groups of mice as described in (A) (n = 2 mice/group).
- Analysis of correlation of Ezh2 and p53 protein levels determined by IHC in Pten‐knockout (Pten pc−/−) mice (n = 11). See IHC scoring details in Materials and Methods section.
- RT–qPCR analysis of p53 mRNA expression in four different genotypes of mice as described in (A). Statistical significance was determined by two‐tailed Student's t‐test. *P < 0.01.
- RT–qPCR analysis of mRNA expression of p53 downstream target gene Bax in four different genotypes of mice as indicated. Statistical significance was determined by two‐tailed Student's t‐test. *P < 0.01.
- H&E analysis of ventral prostate (VP) of 4‐month‐old mice with indicated genotypes. The inset shows a high magnification image of the representative (framed) area in each panel. Scale bars, 50 μm. Scale bars in insets, 10 μm.
- Quantification of nonmalignant, low‐grade PIN (LGPIN), and high‐grade PIN (HGPIN) or cancerous acini in the prostates [including anterior prostate (AP), ventral prostate (VP), and dorsolateral prostate (DLP)] of mice with the indicated genotypes and number as in (A).
Source data are available online for this figure.
Consistent with the previous report that EZH2 level is extremely low in normal human prostatic tissues (Varambally et al, 2002), we demonstrated that Ezh2 protein was barely detectable in the prostate of wild‐type mice (Fig 4A and B). It is not surprising that homozygous deletion of Ezh2 in prostatic epithelium had little or no effect on p53 mRNA and protein expression and prostate epithelium morphogenesis (Fig 4A, B, D, F, and G). In Pten‐deleted tumors, Ezh2 protein level was substantially elevated (Fig 4A and B), suggesting that increased EZH2 expression may play a causal role in mediating PTEN deletion‐induced prostate tumorigenesis. To our surprise, there were a significant portion of acini remained at the high‐grade prostatic intraepithelial neoplasia (HGPIN)/cancer stage after co‐deletion of Ezh2 in the Pten‐deleted tumors (Fig 4F and G). These results are consistent with the finding that the level of p53 mRNA and protein and its downstream target gene Bax were significantly downregulated in Pten;Ezh2 double knockout tumors compared with Pten single knockout tumors (Fig 4D and E). Thus, depletion of EZH2 in malignant tissues that express high levels of EZH2 results in undesirable downregulation of wild‐type p53 tumor suppressor, which may therefore contribute to tumorigenesis. These findings also provide a plausible explanation for the phenomenon that homozygous deletion of Ezh2 failed to completely block Pten deletion‐induced tumorigenesis in the p53 wild‐type prostate.
Expression of EZH2 and p53 positively correlates in human cancers
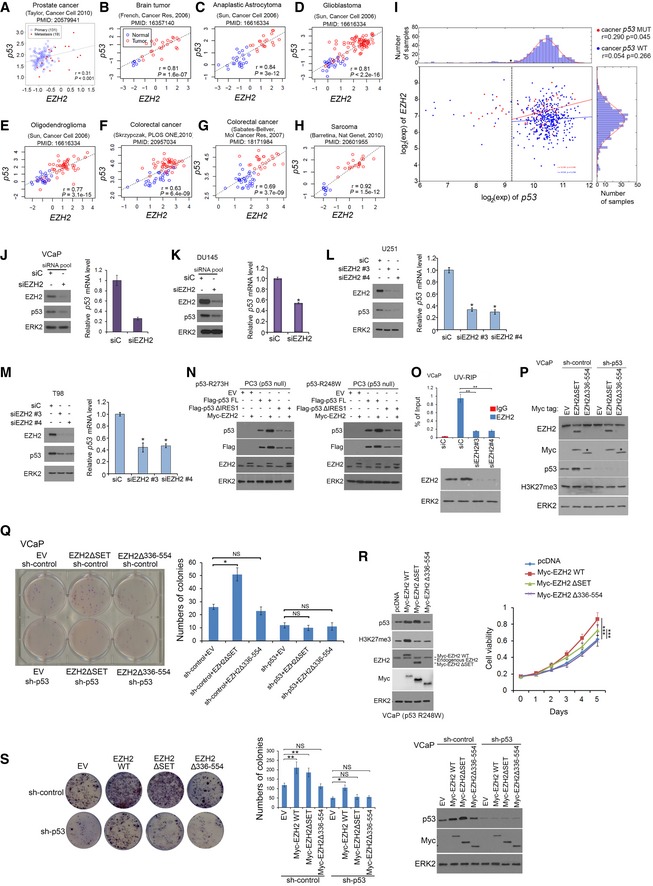
The finding that EZH2 increases p53 protein levels by enhancing mRNA stability and protein translation in cultured human cancer cells and mouse tumors prompted us to determine the correlation of these two proteins in cancer patient specimens. Meta‐analyses of previously published gene expression profiling data revealed a positive correlation between EZH2 and p53 mRNA in a variety of human cancer types examined, including prostate, brain, colorectal cancer, and sarcoma (Fig EV5A–I). Notably, the correlation between p53 and EZH2 RNA levels in the TCGA cohort was stronger in tumors that carry TP53 mutations as compared to those that retain wild‐type p53 (Fig EV5I), supporting the notion that the selection pressures may favor the correlation in p53 mutant tumors. The correlation between EZH2 and p53 was further confirmed at both mRNA and protein levels by RT–qPCR and IHC, respectively, in prostate cancer specimens from two independent cohorts of patients (Fig 5A–C). IHC analysis also showed that EZH2 protein expression correlated with p53 protein levels in various other cancer types, including brain, colorectal, pancreatic cancer, and sarcoma (Fig 5D and E). These results indicate that EZH2 and p53 expression are correlated positively at both mRNA and protein levels in many types of human cancer.
Figure EV5. Positive correlation between EZH2 and p53 mRNA expression in different types of human cancer. Related to Figs 5 and 6 .
-
A–HAnalysis of correlation between EZH2 and p53 mRNA expression in published datasets obtained from different types of human cancer. The correlation coefficient (Pearson's r) and P value were shown in each figure.
-
ICorrelation analysis of EZH2 and p53 (WT or mutant) mRNA level in primary prostate cancer specimens of the TCGA cohort.
-
J–MWestern blot and RT–qPCR analysis of p53 protein and mRNA expression in VCaP (J), DU145 (K), glioblastoma U251 (L), and T98 (M) cells at 48 h after transfected with non‐specific control (siC) or EZH2‐specific siRNAs. GAPDH was used as internal control. Data shown are mean values ± SD from three replicates. *P < 0.01 comparing EZH2 siRNA‐transfected with siC siRNA‐transfected cells.
-
NPC3 cells were transfected with plasmids for R273H (Left)‐ or R248W (Right)‐mutated Flag‐tagged p53 full‐length or ΔIRES1 mutant for 48 h followed by Western blots.
-
OControl or EZH2 knockdown VCaP cells were treated with 100 μM 4‐thiouridine (4SU) for 8 h for Western blot analysis and UV‐RIP assay followed by RT–qPCR measurement of p53 mRNA immunoprecipitated by IgG or anti‐EZH2 antibody. Data shown as means ± SD (n = 3). **P < 0.01.
-
PExpressions of p53, Myc‐tagged EZH2, H3K27me3, and total EZH2 protein were measured by Western blot in VCaP cells infected lentivirus for sh‐control or p53‐specific shRNAs in combination with Myc‐tagged empty vector (EV) or EZH2 truncation mutants. ERK2 was used as the loading control. Asterisks indicate that Myc‐tagged EZH2Δ336–554 protein cannot be recognized by the anti‐EZH2 antibody.
-
QVCaP cells were infected lentivirus as in (P) and plated into 6‐well plate for colony formation assay. At 14 days after plating, colonies were fixed, stained, photographed (Left), and quantified (Right). *P < 0.01; NS, no significance.
-
RVCaP cells expressing p53 R248W mutant were transfected indicated plasmids followed by Western blot analysis (Left) and MTS assay (Right). Data are means ± SD from experiments with three replicates. ***P < 0.001. ERK2 was used as a loading control.
-
SVCaP cells were infected with lentivirus for indicated shRNAs or WT EZH2 or mutants for 48 h followed by colony formation assay (Left and Middle) and Western blot (Right). Data are means ± SD from experiments with three replicates. **P < 0.01, NS, no significance. ERK2 was used as a loading control.
Source data are available online for this figure.
Figure 5. EZH2 correlates with p53 expression in various types of human cancer.
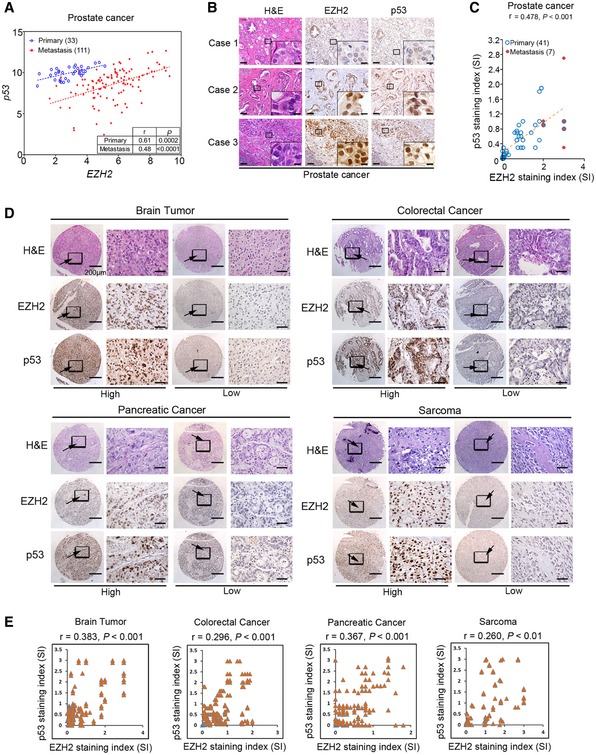
- Correlation analysis of EZH2 and p53 mRNA expression measured by RT–qPCR in primary and metastatic prostate cancer specimens of a cohort of patients from the University of Washington.
- IHC analysis of EZH2 and p53 protein expression in human prostate cancer tissues of a cohort of patients from Mayo Clinic. Cases 1 and 2 show low and high expression in primary tissues, respectively; case 3 shows high expression in lymph node metastasis. Scale bar, 50 μm. Scale bars in insets, 10 μm.
- Correlation analysis of EZH2 and p53 protein expression in primary and metastatic prostate cancer specimens of the Mayo Clinic cohort.
- IHC analysis of EZH2 and p53 protein expression in commercially purchased TMA with patient specimens of brain, colorectal, pancreatic cancer, and sarcoma. Scale bars in low magnification images, 200 μm; scale bars in high magnification images, 50 μm. Arrows, indicated the zoom out‐box.
- IHC data as shown in (D) were quantified and used for correlation analysis of EZH2 and p53 protein expression in patient specimens of brain, colorectal, pancreatic cancer, and sarcoma. Pearson correlation r and P‐values in each cancer type are indicated.
EZH2 increases the level of mutated p53 mRNA and protein in cancer cells
It is generally accepted that mutated p53 protein is much easier to be detected by IHC in cancer specimens than the unmutated counterpart, and therefore, detection of high‐level p53 protein expression is often utilized as a proxy for the presence of p53 mutations (Muller & Vousden, 2014). A previous association study reveals that EZH2 overexpression positively correlates with the high‐level expression of p53 protein in esophagus squamous cell carcinomas, many of which were shown to express mutated p53, although the molecular mechanism underlying the correlation was not explored (Yamada et al, 2011). Since both WT and mutated p53 mRNAs share the same IRES1 in the 5′UTR, we sought to determine whether EZH2 modulates the expression of mutated p53 in a manner similar to WT p53. Knockdown of EZH2 invariably decreased the steady‐state level of different p53 mutants at both protein and mRNA level in various cancer cell lines with different tissue origins, including VCaP (R248W) and DU145 (P223L and V274F) prostate cancer cell lines and U251 (R273H) and T98 (M237I) glioblastoma cell lines (Fig EV5J–M). EZH2 overexpression upregulated protein level of full‐length mutant p53 (R273H and R248W), but not the p53ΔIRES1 mutant in PC3 cells (Fig EV5N). These data indicate that EZH2 also regulates p53 mutant expression in cancer cells.
EZH2 cooperates with p53 GOF mutants to promote cancer growth and metastasis
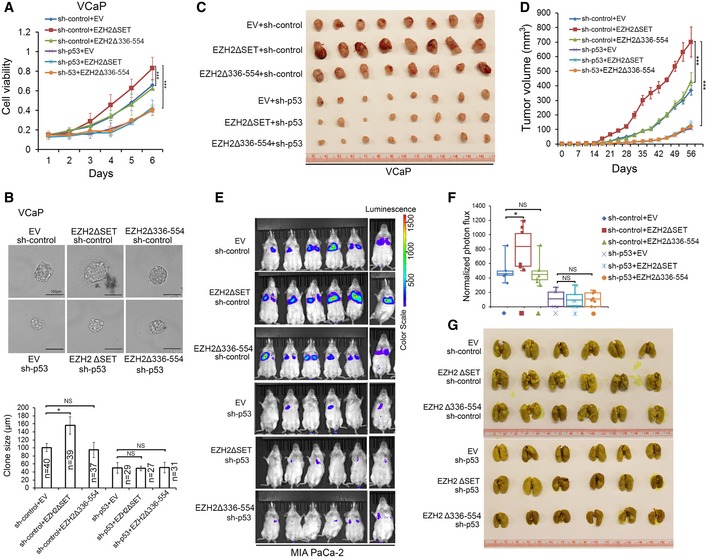
Most p53 mutants in human cancer are missense mutations and often clustered at a few “hotspots” (Muller & Vousden, 2014). Previous studies in cell culture, xenograft, and genetically engineered mouse models suggest that some of the “hotspot” mutations, such as R175H, R248W, and R273H, are GOF mutants that gain new functions in promoting cancer invasion, metastasis, and progression (Dittmer et al, 1993; Olive et al, 2004; Freed‐Pastor et al, 2012; Weissmueller et al, 2014). We sought to determine whether the cancer‐promoting functions of p53 GOF mutants are regulated by EZH2. VCaP cells express R248W GOF mutant of p53. We confirmed EZH2 binding with R248W p53 in VCaP cells by UV‐RIP assay (Fig EV5O). Knockdown of R248W mutant by shRNAs largely decreased VCaP cell growth in 2D and 3D culture conditions and in mice (Figs 6A–D, and EV5P and Q). Importantly, ectopic expression of EZH2ΔSET, but not the p53 mRNA‐binding‐deficient mutant EZH2Δ336–554 significantly increased growth of VCaP cells in culture and in mice, and no such effect was detected in R248W mutant knockdown cells (Figs 6A–D, and EV5P and Q). The effect of EZH2Δ336–554 and EZH2ΔSET on cell growth was unlikely mediated by H3K27me3‐dependent functions since their expression did not affect H3K27me3 level (Fig EV5P and Q). Different from WT EZH2 and EZH2ΔSET, EZH2Δ336–554 expression failed to increase R248W mutant level and cell growth (Fig EV5R). We observed similar results in colony formation assays, but such effects of WT EZH2 and EZH2ΔSET were largely diminished by knockdown of GOF mutant p53 in VCaP cells (Fig EV5S). Thus, EZH2 augments p53 GOF mutant‐mediated cancer growth in a methyltransferase‐independent manner.
Figure 6. EZH2 enhances p53 GOF mutant‐mediated cancer growth and metastasis independently of its methyltransferase activity.
-
A, BVCaP cells stably expressing control (sh‐control) or p53‐specific shRNA were infected with lentivirus for empty vector (EV) or deletion mutants of EZH2. Cell growth in 2D (A) and 3D (B) conditions were determined by MTS assay and measurement of clone size, respectively. Statistical significance was determined by two‐tailed Student's t‐test. *P < 0.05, ***P < 0.001.
-
C, DVCaP cells (1 × 107) infected with lentivirus as in (A) were injected subcutaneously into NSG mice (n = 8/group). Tumors were measured by caliper twice a week. Tumors at the end point of measurement were isolated and photographed (C), and data are shown as means ± SD (D). Statistical significance was determined by two‐tailed Student's t‐test for tumors at day 56. ***P < 0.001.
-
E–GLuciferase‐expressing MIA PaCa‐2 cells (2 × 106) infected with lentivirus as in (A) were injected via tail vein into NSG mice (n = 6/group). At 12 weeks after injection, mice were subjected to bioluminescent imaging, and images were recorded (E) and bioluminescent signals were quantified (F). Bioluminescent flux (photons/s/sr/cm2) was determined for lesions in lung, the ends of the box are the upper and lower quartiles and the box spans the interquartile range; the median is marked by a vertical line inside the box; the whiskers are the two lines outside the box that extend to the highest and lowest observations. Lungs were isolated from mice, stained with Bouin's solution, and photographed (G). The white spots on lungs (stained in yellow) are metastatic tumors. *P < 0.05; NS, no significance.
p53 GOF mutants can also cause cancer metastasis (Yue et al, 2015). We sought to determine to what extent EZH2 regulation of p53 GOF mutant expression contributes to cancer cell invasion and metastasis. Ectopic expression of full‐length p53 R273H and R248W mutants largely enhanced invasion of p53‐null PC3 prostate cancer cells (Appendix Fig S1A–F). EZH2 knockdown not only decreased protein level of these mutants, but also substantially inhibited cell invasion augmented by R248W and R273H mutant (Appendix Fig S1A–F). MIA PaCa‐2 is a highly metastatic pancreatic cancer cell line which expresses an endogenous p53 GOF mutant R248W. Expression of EZH2ΔSET mutant not only elevated R248W protein level, but also largely increased cell invasion, but no such effects were observed for the p53 mRNA‐binding‐deficient mutant EZH2Δ336–554 (Appendix Fig S1G and H). EZH2ΔSET‐induced cell invasion was completely abolished by knockdown of endogenous p53 R248W mutant (Appendix Fig S1G and H). While expression of Y641F EZH2, a point mutation with enhanced H3K27me3 function (Yap et al, 2011) expectedly increased H3K27me3 level, this mutant‐enhanced cell growth and invasion was also largely diminished by knockdown of endogenous p53 R248W mutant in Mia‐PaCa2 cells (Appendix Fig S1I). The results from both loss‐ and gain‐of‐function studies indicate that EZH2 regulates the level and activity of GOF mutant p53 protein in a manner independent of its methyltransferase activity.
We further examined whether EZH2 promotes cancer metastasis through interaction with GOF mutant p53 mRNA. We generated MIA PaCa‐2 cells expressing a luciferase gene. Consistent with the results of cell invasion assay (Appendix Fig S1G and H), ectopic expression of EZH2ΔSET mutant, but not the p53 mRNA‐binding‐deficient mutant EZH2Δ336–554 largely increased MIA PaCa‐2 cell metastasis to lung (Fig 6E–G and Appendix Fig S1J). Most importantly, knockdown of endogenous p53 R248W mutant almost completely abolished EZH2ΔSET mutant‐induced lung metastasis of MIA PaCa‐2 cells in mice (Fig 6E–G and Appendix Fig S1J).
SHARP1 and CCNG2 are two cancer metastasis‐associated genes reported as the targets of mutated p53 (Adorno et al, 2009). Expression of these two genes was upregulated by the methyltransferase‐deficient mutant EZH2ΔSET, but not EZH2Δ336–554 in control knockdown MIA PaCa‐2 cells (Appendix Fig S1K). However, this effect was completely abolished by knockdown of endogenous p53 R248W mutant (Appendix Fig S1K). These data suggest that EZH2 binding of p53 mRNA is important for p53 GOF mutant‐mediated cancer cell invasion and metastasis, implying that EZH2 is a viable target for treatment of tumors harboring p53 GOF mutations.
EZH2 depletion induces synthetic vulnerability in p53 GOF mutant‐expressing cancer cells
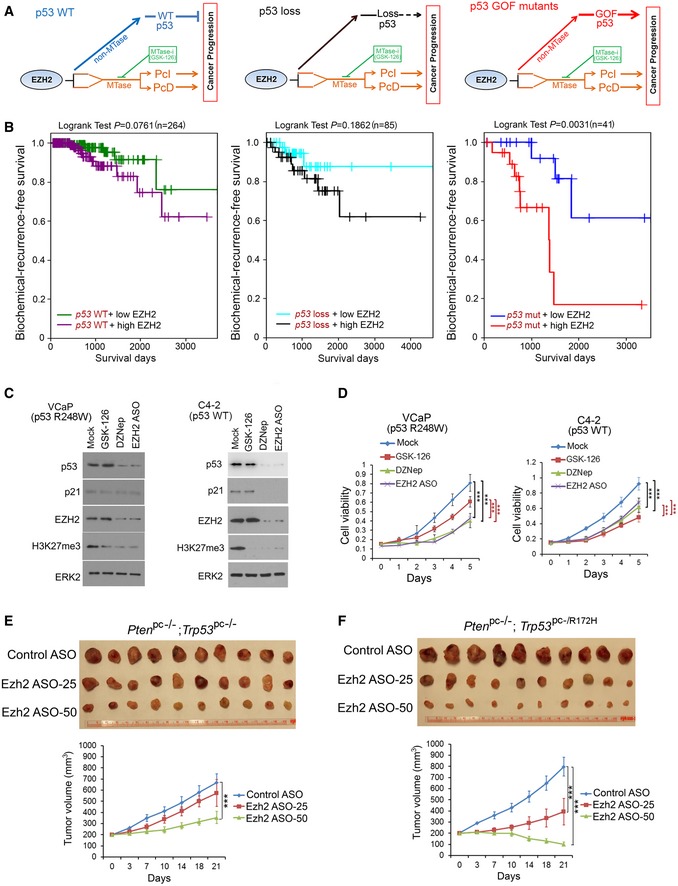
Based on the data described above, we hypothesized that in cancer cells expressing WT p53, EZH2‐enhanced expression of p53 acts against EZH2‐mediated oncogenesis (Fig 7A, left). In contrast, in cancer cells expressing p53 GOF mutants, EZH2 and mutated p53 work cooperatively in favor of cancer progression (Fig 7A, right). In support of this hypothesis, analysis of TCGA prostate cancer data showed that high levels of EZH2 proteins significantly correlated with the worse biochemical recurrence‐free survival only in patients with tumors expressing mutated p53, but not when p53 is WT or lost (Fig 7B). These data suggest that EZH2 represents a promising therapeutic target for p53‐mutated cancer cells, especially those with GOF mutants. To test this notion, we compared the effect of EZh2 inhibition on cell viability and growth of cancer cells expressing R248W GOF mutant versus having p53 WT. We treated VCaP (p53 GOF mutant R248W) cells with different EZH2 inhibitory agents, including GSK126 (inhibits EZH2 methyltransferase enzymatic activity; McCabe et al, 2012b), DZNep (inhibits EZH2 protein expression and the methyltransferase enzymatic activity; Tan et al, 2007), and EZH2 antisense oligonucleotides (ASOs; inhibits EZH2 protein expression). As expected, treatment of these agents invariably decreased H3K27me3 levels, increased expression of PcD gene DAP2IP, and downregulated expression of PcI genes CEP76, RAD51C, and TEME48 in VCaP cells (Fig 7C and Appendix Fig S1L). However, we found a higher concentration of these inhibitors was needed to achieve similar effects on H3K27me3 inhibition and EZH2 target gene expression in p53 WT C4‐2 cells (Fig 7C and Appendix Fig S1L). Moreover, different from the effect in R248W‐expressing VCaP cells, treatment with DZNep and ASO, but not GSK126 also decreased expression of p53 downstream target p21CIP1, a growth‐inhibitory protein in C4‐2 cells (Fig 7C). Accordingly, treatment with DZNep and ASO resulted in much greater inhibition of VCaP cell viability assessed in 2D, 3D culture and colony formation compared to C4‐2 cells (Fig 7D and Appendix Fig S2A and B). Importantly, similar results were obtained in a panel of cell lines of different cancer types, including breast cancer cell lines MCF7 (p53 WT), MDA‐MB‐231 (R280K), MDA‐MB‐435 (G266E), prostate cancer cell lines (LNCaP (p53 WT), 22RV1 (Q331R), PC3 (p53 loss), and PC3 cells transfected with R248W (Appendix Fig S2C and D). The effect of DZNep on p53 GOF mutant was not mediated by EZH1 (Appendix Fig S2E). These data show that depletion of EZH2 protein expression, but not the inhibition of its enzymatic activity induces synthetic vulnerability in p53 GOF mutant‐expressing cancer cells.
Figure 7. Depletion of EZH2 expression inhibits growth of p53 GOF‐mutated cancer cells.
-
ASchematic diagram depicting the distinctive impacts of the functional interplay between EZH2 protein and p53 mRNA on progression of cancers expressing WT p53, no p53, or GOF‐mutated p53. MTase, methyltransferase.
-
BKaplan–Meier plots showing the association of EZH2 overexpression with biochemical recurrence of prostate cancer in patients from the TCGA cohort. Tumors were separated into two groups according to EZH2 expression levels (high or low) and three groups based on the mutation status of p53 (WT, loss or mutated).
-
C, DVCaP and C4‐2 cells were treated with GSK‐126, DZNep (5 μM for VCaP and 10 μM for C4‐2), and EZH2 ASO, followed by Western blots with indicated antibodies (C) and MTT assay (D). Data shown as means ± SD (n = 6). Statistical significance was determined by two‐tailed Student's t‐test. ***P < 0.001.
-
E, FEffect of Ezh2 ASOs on growth of murine prostate cancer allografts. Mice (n = 10) bearing the Pten pc−/−;Trp53 pc−/− (E) and Pten pc−/−;Trp53 pc−/R172H allografts (F) were treated according the scheme shown in (Appendix Fig S2G) with control ASOs (50 mg/kg) or Ezh2 ASOs at different doses (25 and 50 mg/kg). Tumor growth was measured twice a week for 3 weeks, and the data are shown in the bottom panels. Each allograft was isolated by the end of ASO treatment and photographed (top panels). Data shown as means ± SD (n = 10). Statistical significance was determined by two‐tailed Student's t‐test. ***P < 0.001.
Source data are available online for this figure.
EZH2 depletion inhibits growth of p53 GOF mutant‐expressing tumors in vivo
To determine the inhibitory effect of EZH2 depletion on growth of p53 GOF mutant‐expressing tumors in mice, we generated murine prostate cancer cell lines from prostate‐specific Pten pc−/−;Trp53 pc−/− and Pten pc−/−;Trp53 pc−/R172H mutant mice (Blee et al, 2018; Appendix Fig S2F). These tumor cell lines were inoculated subcutaneously into NSG mice. When the size of allografts reached up to 200 mm3, mice were randomly grouped and treated intraperitoneally (i.p.) with control or Ezh2‐specific ASOs twice a week for 3 weeks (Appendix Fig S2G). Volumes of Pten pc−/−;Trp53 pc−/− tumors were slightly decreased in mice treated with 25 mg of Ezh2 ASO (Ezh2 ASO‐25) but significantly decreased after treated with 50 mg of Ezh2 ASO (Ezh2 ASO‐50) in comparison with control ASO treatment (Fig 7E). Most importantly, p53 GOF mutant‐expressing Pten pc−/−;Trp53 pc−/R172H tumors were much more sensitive to Ezh2 ASO treatment compared to p53‐deficient Pten pc−/−;Trp53 pc−/− tumors (Fig 7E and F). Notably, little or no effect of Ezh2 ASO treatment on mouse weight loss was detected (Appendix Fig S2H). Furthermore, ectopic expression of p53 R248W increased PC3 xenograft tumor growth in mice, but p53 R248 tumors were more sensitive to EZH2 ASO treatment compared to p53‐null PC3 tumors (Appendix Fig S2I and J). RT–qPCR analysis indicated that expression of Ezh2 and p53 mRNA expression was effectively depleted by Ezh2 ASO treatment, especially at the high dose in Ptenpc−/−;Trp53pc−/R172H tumors (Appendix Fig S2K and L). In contrast, only modest reduction in Ezh2 mRNA levels was observed in a few normal tissues such as brain and testis, but not in the majority of tissues examined including heart, intestine, kidney, liver, lung, and muscle, and the alterations were not statistically significant except two occasions (Appendix Fig S2K). Importantly, no significant changes were observed in p53 mRNA levels in all the normal tissues examined and this was presumably due to the fact that Ezh2 levels were much lower in normal tissues than that in tumors (Appendix Fig S2K and L). These data are consistent with the effect of Ezh2 on p53 expression in Ezh2 knockout prostates (Fig 4A, B, and D), implying the negligible level of EZH2 may have limited leverage in regulation of p53 expression in normal tissues. Together, these data suggest that depletion of EZH2 protein with strategies such as ASOs can be a viable therapeutic arsenal for treatment of advanced cancer, especially those expressing p53 GOF mutants.
Discussion
Non‐methyltransferase function of EZH2 in cancer
PcD and PcI are two major functions of EZH2 identified thus far. Since both roles are dependent on the methyltransferase activity and implicated in cancer development and progression (Varambally et al, 2002; Xu et al, 2012), a significant amount of effort has been put into developing EZH2 enzymatic inhibitors. A few such inhibitors including GSK126 and EPZ‐6438 have been developed and are currently being tested in phase I clinical trials for treatment of B‐cell lymphomas and advanced solid tumors (McCabe et al, 2012b; Knutson et al, 2014; Vaswani et al, 2016). To our knowledge, however, no favorable report has been documented yet (Italiano et al, 2018). In the present study, we provide evidence that EZH2 binds to IRES1 in the 5′UTR of both wild‐type and mutated p53 mRNA and increases mRNA stability and cap‐independent protein translation. We further show that this function of EZH2 is independent of its methyltransferase activity. Thus, we identify a previously unrecognized non‐methyltransferase function of EZH2 in cancer. Our findings also suggest that targeting EZH2 expression rather than targeting its enzymatic activity could be a more effective therapeutic option, particularly in tumors expressing GOF‐mutated p53.
Mechanistic explanation of the dichotomous role of EZH2 in cancer
Paradoxical roles of EZH2 and PRC2 have been seen in different types of human cancer. It is well established that EZH2 plays an oncogenic role and correlates with the progressiveness or stages in most types of solid cancer. However, deletions of EZH2 and other PcG genes such as SUZ12 occur in a subset of hematopoietic malignancies such as T‐cell acute lymphoblastic leukemia (T‐ALL; Ntziachristos et al, 2012). Our discovery of the positive regulation of p53 mRNA and protein by EZH2 supports the model wherein EZH2 may exert dichotomous roles in oncogenesis of cells expressing a wild‐type p53 (Fig 7A, left). We also provide evidence that in tumors harboring p53 GOF mutant, EZH2 upregulates and works cooperatively with mutated p53, thereby favoring cancer progression (Fig 7A, right). Moreover, similar to our findings in solid tumors (Fig 7B), the prognosis of leukemia patients with a mutation in the TP53 gene is worse than those expressing a wild‐type p53 (Peller & Rotter, 2003). This model is further supported by a previous report that EZH2 overexpression and p53 mutations frequently occur in late‐stage cancers (Varambally et al, 2002). Thus, our findings not only provide a plausible explanation for the dichotomous roles of EZH2 occurring in certain cancer types, such as a subset of hematopoietic malignancies, but also reveal a biological and mechanistic basis for the co‐occurrence of EZH2 overexpression and p53 mutations in many advanced cancer types.
EZH2 binding of mRNA of cancer‐relevant genes
EZH2 is a known RNA‐binding protein. By binding to non‐coding RNAs (ncRNAs) such as XIST, RepA and HOTAIR, EZH2 has been shown to work together with other PcG proteins to promote X chromosome inactivation, developmental patterning, and maintenance of stem cell pluripotency (Plath et al, 2003; Rinn et al, 2007; Zhao et al, 2008; Wang et al, 2013). Binding of EZH2 with ncRNAs such as MALATA1 and NEAT1 also promotes cancer growth and metastasis (Wang et al, 2015; Chen et al, 2018). Therefore, the biological and clinical significance of EZH2 interaction with ncRNAs have been extensively studied. However, the specificity of EZH2 binding of so many RNAs remains a topic for study (Long et al, 2017; Wang et al, 2017). Using the unbiased RIP‐seq approach, in the present study we demonstrated that EZH2 binds to a group of mRNAs which encode functionally important proteins such as p53. Most importantly, we provide evidence that EZH2 binding of p53 mRNA is functional. Specifically, we showed that EZH2 increases p53 mRNA stability and promotes cap‐independent protein translation of p53 mRNA by binding to the IRES element in the 5′UTR. Thus, our finding of EZH2 binding of mRNA largely broadens our understanding of the functional significance of RNA binding of EZH2, thereby representing a framework for the discovery of new roles of EZH2 in developmental and cancer biology.
A new strategy to inhibit p53 GOF mutants in cancer
Given that mutated p53 proteins are generally considered less druggable (Lehmann & Pietenpol, 2012), endeavor has been largely focused on targeting the downstream effectors of each individual GOF mutant of p53 (Adorno et al, 2009; Weissmueller et al, 2014; Zhu et al, 2015). We demonstrated that different from the inhibition of EZH2 enzymatic activity, depletion of EZH2 protein expression with ASOs selectively suppresses growth of cancer cells expressing GOF‐mutated p53, highlighting that targeting EZH2 induces synthetic vulnerability in p53 GOF‐mutated cancer cells. While we found that EZH2 regulates mRNA and protein levels in cultured cancer cells, we provided evidence that homozygous deletion of Ezh2 in normal tissues (e.g., normal prostate gland) or treatment with Ezh2 ASOs had no drastic impact on Trp53 mRNA and protein expression in a large spectrum of mouse tissues. In agreement with these observations, we also show that EZH2 expression is much lower in normal tissues than that in tumors, and therefore, it is not surprising that negligible EZH2 expression level may have limited leverage in regulation of p53 expression in normal tissues. These findings stress that targeting the upstream regulator such as EZH2 offers a new opportunity to specifically inhibit various p53 GOF mutants with diversified functions in cancer, and such treatment appears to have little or no harm on normal tissues, which is fully supported by our observation that Ezh2 ASO treatment almost had no effect on body weight of mice. Thus, our findings not only indicate EZH2 is a viable therapeutic target in p53‐mutated cancer, but also suggest that inhibition of functionally diversified p53 GOF mutants is achievable by single targeting of EZH2 as a common upstream regulator, which therefore represents a new paradigm of targeted therapy of p53‐mutated cancer.
Materials and Methods
Cell lines, cell culture, reagents, and antibodies
LNCaP, PC3, VCaP, 22RV1, MCF7, MDA‐MB‐231, MDA‐MB‐435, MIA PaCa‐2, and U2OS cell lines were purchased from ATCC. C4‐2 cell line was purchased from UroCorporation. MEF‐24 and MEF‐27 cell lines were kindly provided by Dr. Zhenbang Chen at Meharry Medical College (Chen et al, 2005). LNCaP, PC3, 22RV1, and C4‐2 cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific) and 500 μg/ml penicillin‐streptomycin glutamine (Thermo Fisher Scientific). VCaP, MCF7, MDA‐MB‐231, MDA‐MB‐435, MIA PaCa‐2 cells were cultured in high glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific) and 500 μg/ml penicillin‐streptomycin glutamine (Thermo Fisher Scientific). U2OS cells were cultured in Mccoy's 5A with 10% FBS. Human glioblastoma cell lines T98 and U251 were kindly provided by Dr. Jann N. Sarkaria (Mayo Clinic) and cultured in DMEM medium supplemented with 10% FBS. Cells were incubated in 37°C with 5% CO2. Actinomycin D (A1410) and Bouin's solution (HT10132‐1l) were purchased from Sigma‐Aldrich. DZNep (CAS 120964‐45‐6) was purchased from Calbiochem. GSK126 (S7061) was purchased from Selleckchem. EZH2‐specific generation‐2.5 antisense oligonucleotides (ASOs) for human EZH2 (633365) and mouse Ezh2 (633418 and 640638) and control ASOs were kindly provided by Ionis Pharmaceuticals Inc (Carlsbad, CA). VCaP cells were treated with 5 μM GSK‐126, DZNep, and EZH2 ASO. MCF7, LNCaP, C4‐2, MDA‐MB‐435, MDA‐MB‐231, 22RV1, and PC3 cells were treated with 10 μM GSK‐126, DZNep, and EZH2 ASO, followed by Western blots with indicated antibodies and MTT assay. Antibodies used are as follows: anti‐p21CIP1 (F‐5), anti‐p53 (DO‐1), anti‐p53 (FL‐393), anti‐CHK1 (FL‐467), EZH1 (H‐4), CDYL (E‐8), total Pol II (N‐20), anti‐SUZ12 (D‐10), and ERK2 (Santa Cruz Biotechnology); anti‐EED (09‐727; Millipore); anti‐Flag (M2) and anti‐β‐TUBULIN (Sigma); anti‐eIF4G2 (D88B6), anti‐PABP1 (#4992), anti‐phospho‐CHK1 (Ser317; D12H3), anti‐PTEN, anti‐MDM2, anti‐BAX, Myc‐tag (71D10), and anti‐EZH2 (D2C9; Cell Signaling Technology); and anti‐H3K27me3 (ab6002; Abcam).
In vitro invasion assay
Cell invasion was performed with Corning matrigel invasion chamber assay according to manufacturer's instructions (Corning). Briefly, cells were diluted to a density of 6 × 104 cells per well in serum‐free medium and plated to the inside of matrigel chamber in 24‐well plates, outside the chamber was added the medium with 10% FBS. After 24 h, cells were fixed in methanol for 15 min and then stained with 1 mg/ml crystal violet staining for 20 min. The membranes of chamber were covered by coverslip and observed using microscope after multiple‐time washing. Wells were repeated in triplicate, and the invaded cells were quantified per field of view. Six fields of three independent replicates were recorded and analyzed.
Mouse xenograft generation and tumor growth measurement
The mouse study was approved by Mayo Clinic IACUC. Six‐week‐old NSG male mice were injected with 1 × 107 of cancer cells infected with lentivirus or shRNAs and/or expression vectors in 100 μl PBS with 100 μl of Matrigel matrix (BD Bioscience) in both flanks. After injection of tumor cells into mice, tumors were monitored until they reach maximum tumor volumes of 1,000 mm3 and tumor growth was measured with caliper twice a week.
Plasmids and mutagenesis
The mammalian expression vector for Myc‐tagged EZH2‐WT and bacterial expression vectors for GST‐EZH2 recombinant proteins are described previously (Chen et al, 2010; Wang et al, 2013). GST‐eIF4G2 and GST‐PABP1 were cloned into pGEX‐4T1 plasmid using the primers as described in Table EV3. p53/47 (ΔNp53), p53ΔIRES1 (WT, R248W or R273H), Myc‐tagged EZH2 mutants EZH2ΔSET and EZH2Δ336–554, and all siRNA‐resistant mutants were generated by KOD Plus Mutagenesis Kit (TOYOBO) following the manufacturer's instructions. All shRNA plasmids were purchased from Sigma‐Aldrich. R5 EMCV (#51733) was purchased from Addgene (USA). LentiCrisperV2 was purchased from Addgene (#52961). EZH1 knockout plasmids were cloned into LentiCrisperV2 plasmid using the primers as described in Table EV3.
RNA immunoprecipitation and high‐throughput sequencing (RIP‐seq)
LNCaP and C4‐2 cells grown to ~ 70–80% confluence were washed twice with 1 × PBS and trypsinized and washed twice with ice‐cold 1 × PBS containing 1 × protease inhibitor cocktail (PIC). Cells were lysed with RIPA buffer [50 mM Tris–HCl (pH 7.9), 0.25 M NaCl, 1% Nonidet P‐40 (NP‐40), 10 mM EDTA, and RNase inhibitor (Promega)], incubated for 30 min at 4°C. Lysates obtained by centrifugation (13,400 g for 10 min at 4°C) and precleared with 30 μl protein G agarose beads for 2 h at 4°C. Add 100 μl antibody/beads to samples and incubated for 18 h at 4°C. Protein–RNA complexes bound to beads were washed three times in NT2 buffer [50 mM Tris–HCl (pH 7.4), 300 mM NaCl, 1 mM MgCl2, 0.05% Nonidet P‐40 (NP‐40), 1 × PIC, RNase inhibitor], followed by incubation with DNase I for 15 min at 37°C and further washed twice with NT2 buffer. Resuspend the beads in 100 μl of NT2 Buffer. Co‐purified RNA was extracted by RNA purification kit (RNAeasy MiniElute kit, QIAGEN), and cDNA was synthesized using the SuperScript kit from Thermo Fisher Scientific and the quality of cDNA was analyzed by real‐time (quantitative) polymerase chain reaction (PCR). For high‐throughput sequencing, double‐strained cDNA was synthesized and fragmented by sonication. Library was prepared with the adapter mix from the Illumina. Samples were sequenced using the Illumina HiSeq2000 platform at the Mayo Genome Core Facility after size selection (150–300 bp). RIP‐seq raw reads were mapped to human reference genome (hg19/GRCh37) using Tophat (v1.4.0; Trapnell et al, 2009). Raw count mapped to each Refseq exon was calculated using RSeQC package (Wang et al, 2012). Hypergeometric test was applied to evaluate the significance of enrichment of RIP‐seq reads at each exon. P‐values were then corrected using Benjamini‐Hochberg procedure to control false discovery rate.
Pten and Ezh2 conditional knockout mice
The mouse study was approved by IACUC at Mayo Clinic. Pten loxP/loxP mice (Wang et al, 2003) were purchased from Jackson Laboratory, and Ezh2 loxP/loxP mice (Su et al, 2003) were kindly provided by Dr. Alexander Tarakhovsky at the Rockefeller University. Prostate‐specific Pten and/or Ezh2 deletion mice (Pten pc−/−; Ezh2 pc−/− and Pten pc−/−;Ezh2 pc−/−) and “wild‐type” control mice were obtained by initial cross breeding between PB‐Cre4;Pten loxP/loxP males (Zhong et al, 2014) and Ezh2 loxP/loxP females. Mouse tail DNA was isolated for PCR analysis using primers for Cre, loxped alleles of Pten as described previously (Zhong et al, 2014) and for loxped alleles of Ezh2 (Table EV3). As we reported recently, Ptenpc−/−;p53pc−/R172H and Ptenpc−/−;p53pc−/− mice were littermates generated by crossing PB‐Cre4;Pten loxP/loxP mice with p53loxP/LSL‐R172H mice (via multiple rounds of breeding). Thus, p53 loss and p53 mutant mice had similar genetic background.
RNA isolation from cultured cells and mouse tissues and quantitative reverse transcription PCR (RT–qPCR)
Total RNA was isolated from cultured cell lines and mouse prostate tissues using TRIzol reagent (Thermo Fisher Scientific), and cDNA was synthesized using the SuperScript kit from Thermo Fisher Scientific. RT–qPCR was performed using the SYBR Green Mix (Bio‐Rad) and an iCycler iQTM system (Bio‐Rad). For normalization, ΔC t values were calculated relative to the levels of GAPDH transcripts. The PCR primer sequences are provided in Table EV3.
GST recombinant protein purification and pull‐down of in vitro transcribed RNA
GST‐EZH2 recombinant proteins covering different portions of EZH2 were expressed in Escherichia coli (BL21) and purified with glutathione Sepharose 4B beads (GE Healthcare) as described previously (Chen et al, 2010). Expression plasmids for full‐length p53 and p53‐ORF (Zhang et al, 2013) were kindly provided by Dr. Xinbin Chen from the University of California at Davis. Expression plasmids for p53 5′UTR and 3′UTR alone were cloned into pcDNA3 vector. Plasmids were linearized by digestion with Xho I followed by purification with Gel Extraction kit (QIAGEN). 200 ng linearized plasmid DNAs were transcribed in vitro using T7 RNA polymerase and treated with DNase I to digest the template DNA. In vitro transcribed RNA was incubated at 90°C for 2 min and cooled down to form second structure. Purified GST recombinant proteins were incubated with in vitro transcribed RNA in NT2 buffer [50 mM Tris (pH 7.4), 150 mM NaCl, 1 mM MgCl2, 0.05% NP‐40, 1 × PIC, and RNase inhibitor]. After five‐time washing, RNAs were eluted by RNeasy MinElute Cleanup Kit (QIAGEN). Precipitated RNA was detected by RT–qPCR.
Biotin‐labeled RNA pull‐down and Western blot analysis
In vitro biotin‐labeling of RNAs was performed using Biotin RNA Labeling Mix (Roche) and T7 polymerase (New England Biolabs). C4‐2 cells were lysed in modified RIPA buffer [50 mM Tris–HCl (pH 7.9), 0.25 M NaCl, 1% NP‐40, 10 mM EDTA, 1 × PIC, and RNase inhibitor] and incubated for 30 min at 4°C. Cell lysates were incubated with biotin‐labeled RNAs and streptavidin beads at 4°C overnight. The beads were washed in NT2 buffer [50 mM Tris (pH 7.4), 300 mM NaCl, 0.05% NP‐40, 1 mM MgCl2, 1 × PIC, and RNase inhibitor] at 4°C for five times. The samples were subjected to Western blot analysis.
RNA interference
Smart pools and individual siRNAs for human EZH2 (M‐004218‐03), p53 (L‐003329), and non‐specific control siRNAs were purchased from GE Healthcare Dharmacon. The targeting sequences of EZH2 individual siRNAs siEZ3 and siEZ4 were shown in Table EV3. Transfection of cells with siRNAs was performed following the manufacturer's instruction.
Prostate cancer patient samples for RT–qPCR analysis
RT–qPCR analyses of p53 and EZH2 mRNA expression in prostate cancer patient samples were approved by the Institutional Review Board (IRB) of the University of Washington Medical Center. Primary prostate tumor tissues were obtained from 25 untreated patients undergoing radical prostatectomy (RP) for localized prostate cancer, and 111 metastatic tumors from 46 men with CRPC were obtained through the University of Washington Prostate Cancer Donor Autopsy Program (Roudier et al, 2003). A total of 16 Gleason pattern 3 and 17 Gleason pattern 4 tumor foci from primary tumors as well as 111 metastases were laser‐capture microdissected as previously described (True et al, 2006). Total RNA was isolated using the Arcturus PicoPure RNA Isolation kit (Molecular Devices, Sunnyvale, CA) with DNase treatment and amplified two rounds using the Ambion MessageAmp RNA Kit (Ambion Inc, Austin, TX).
EZH2 and p53 transcript levels were measured using RT–qPCR in triplicate reactions using 0.5 ng cDNA, 0.2 μmol/l of each primer pair, and SYBR Green PCR master mix (Applied Biosystems). We normalized mean cycle threshold (C t) for each gene to a housekeeping gene, RPL13A, in the same sample using the ΔC T method (primer sequences in Table EV3). Pearson correlations of the mean‐centered ΔΔC T ratios and associated P‐values were computed in Prism (GraphPad Software, La Jolla, CA).
Prostate cancer patient samples for histology analysis and immunohistochemistry (IHC)
Forty‐one prostate cancer tissues [Gleason score (GS10), one case; GS9, five cases; GS8, five cases; GS7, 19 cases; GS6, 10 cases and GS5, one case] and seven prostate cancer lymph node metastases [GS9, four cases; GS8, two cases; GS7, one case] were selected randomly from patients who have been treated at the Mayo Clinic between January 1995 and January 2014. The age of the patients ranged from 47 to 78 years. The study was approved by the Mayo Clinic IRB.
Tissue microarray slides (H&E stained or unstained) for brain tumors, colorectal cancer, pancreatic cancer, and osteosarcoma were purchased from US Biomax (Cat. # BC000110, CNS2081, PA1921, and OS804a). Human tissue slides were subjected for IHC staining of EZH2 (dilution 1:100) and p53 (dilution 1:100). Different lobes of the mouse prostate were dissected and fixed in 10% formalin overnight and transferred to 1 × PBS prior to paraffin embedding. Sections (3 μm) were cut from formalin‐fixed paraffin‐embedded tissues and stained with hematoxylin and eosin (H&E) for histology analysis or subjected to IHC staining with anti‐Ezh2, anti‐p53, and anti‐Pten antibodies as previously described (Ding et al, 2014).
The scoring method of IHC staining
The scoring of IHC staining was performed by three investigators including a GU pathologist. A staining index (SI) was calculated as follows: Staining intensity and staining percentage for any given TMA spot was graded accordingly (intensity: 0 = no staining, 1 = low staining, 2 = media staining, and 3 = strong staining); a final SI score for each TMA element was obtained by multiplying values obtained from staining percentage and intensity and used for correlation analysis.
Measurement of mRNA stability
The stability of p53 mRNA was measured by blocking transcription with actinomycin D. Briefly, cells were transfected with non‐specific (N.S.) or EZH2‐specific siRNAs for 36 h, and then, cells were treated with 3 μM actinomycin D. Cells were harvested at various time points, and mRNA was quantified by RT–qPCR. The mRNA decay was determined as the percentage of mRNA remaining over time compared with the level by the time of adding actinomycin D.
Lentiviral constructs and stable cell lines
Lentivirus was produced by transfection of 293T cells with pTSiN‐based lentiviral constructs and the packaging plasmids pVSV‐G and pCMV‐HR. Forty‐eight hours after transfection supernatant was collected and filtered through a 0.45‐μm filter. Cell lines were infected with virus medium (DMEM, 10% FBS, P/S, 12 μg/ml polybrene) at 1:1 ratio, and the medium was changed 24 h later and puromycin was added at concentration of 1.2 μg/ml for selection of infected cells.
RNA electrophoretic mobility shift assay (RNA EMSA)
Biotin‐labeled RNA probes were generated by in vitro transcription using cDNA containing T7 promoter and the IRES1 region in the 5′ UTR of p53 mRNA. For the RNA EMSA assay, recombinant GST or different GST‐EZH2 recombinant protein, 100 ng/ml tRNA, and 1 μg of biotin‐labeled RNA probe were mixed in binding buffer (10 mM Tris–Cl, pH 7.5, 25 mM KCl, 10 mM MgCl2, 1 mM DTT) for 20 min at 25°C. RNA–protein complexes were digested by adding 100 units of RNase‐T1 for 15 min at 37°C and then separated in 6% of native poly acrylamide gel. RNA–protein complexes were blotted with HRP‐conjugated streptavidin and visualized by autoradiography.
Co‐immunoprecipitation (Co‐IP)
To immunoprecipitate the ectopically expressed Myc‐tagged proteins, transfected cells were lysed 24 h post‐transfection in BC100 buffer. The whole‐cell lysates were immunoprecipitated with the monoclonal anti‐Myc antibody‐conjugated agarose beads (Sigma‐Aldrich) at 4°C overnight. After three washes with lysis buffer, followed by two washes with BC100 buffer, the bound proteins were eluted using Myc‐Peptide (Sigma‐Aldrich) prepared in BC100 for 3 h at 4°C. The eluted protein sample was resolved by SDS–PAGE. To immunoprecipitate the endogenous proteins, cells were lysed with 1 × 106 cell lysis buffer (Cell Signaling Technology), and the lysate was centrifuged. The supernatant was precleared with protein A/G beads (Sigma‐Aldrich) and incubated with indicated antibody and protein A/G beads at 4°C overnight. Beads were washed six times with lysis buffer and resuspended in sample buffer and analyzed by SDS–PAGE.
Western blot
Cell lysates or immunoprecipitates were subjected to SDS–PAGE, and proteins were transferred to nitrocellulose membranes (GE Healthcare Sciences). The membranes were blocked in Tris‐buffered saline (TBS, pH 7.4) containing 5% non‐fat milk and 0.1% Tween‐20, washed twice in TBS containing 0.1% Tween‐20, and incubated with primary antibody overnight at 4°C, followed by secondary antibody for 1 h at room temperature. The proteins of interest were visualized using ECL chemiluminescence system (Santa Cruz Biotechnology).
3D culture of cells
For three‐dimensional (3D) cultures, approximately 2 × 103 cells were resuspended in 250 μl plain medium and seeded on the top of a thin layer of Matrigel in a 24‐well plate. After 30 min, when the cells were settled down, they were covered with another layer of 10% Matrigel diluted with DMEM/F12 medium. The medium was changed with 500 μl of fresh and warm DMEM/F12 plus 5% FBS medium every 3 days.
Clonogenic formation assay
The procedure was carried out by following a published study (Franken et al, 2006). Briefly, 5 × 102 cells were plated onto each well of 6‐well plate. Approximately 16 days later, the colonies were fixed with acetic acid:methanol (1:7) for 30 min and stained with (0.5% w/v) crystal violet for 1 h. The colonies were gently washed with running tape water. The colonies with more than 50 cells were counted.
Generation and treatment of murine prostate cancer allografts with no expression of p53 or expression of a GOF mutant p53
Pten loxp/loxp conditional mice were acquired from Jackson Laboratory (004597) and originally generated in the laboratory of Dr. Hong Wu at University of California, Los Angeles, CA (Wang et al, 2003); Trp53 loxp/loxp conditional mice were acquired from the NCI Mouse Repository and originally generated in the laboratory of Dr. Tyler Jacks at Massachusetts Institute of Technology at Cambridge, MA (Olive et al, 2004); and Trp53 loxp‐STOP‐loxp‐R172H (LSL) conditional mice were acquired from the NCI Mouse Repository and originally generated in the laboratory of Dr. Tyler Jacks at Massachusetts Institute of Technology at Cambridge, MA (Olive et al, 2004). All animal study was approved by the Mayo Clinic Institutional Animal Care and Use Committee (IACUC). All mice were housed in standard conditions with a 12‐h light/12‐h dark cycle and access to food and water ad libitum. NOD‐SCID IL‐2 receptor γ‐null (NSG) mice were generated in house and, at 4 weeks of age, were randomly divided into different experimental treatment groups as indicated (ten mice per group). Ptenpc−/−;p53pc−/R172H and Ptenpc−/−;p53pc−/− prostate cancer cells were generated from corresponding mice, and 5 × 106 cells of each genotype were suspended in 0.1 ml of 50% PBS and 50% Corning Matrigel Matrix and implanted by subcutaneous injection into the left flank of each NSG mouse (one implantation per mouse). Once the implanted cells grew to reach a size of ~ 200 mm3 measured externally with calipers (approximately one and half week post transplantation), mice were randomly grouped and treated with control or Ezh2 ASOs at given doses (control ASO at 50 mg/kg in 100 μl PBS solution, Ezh2 ASO at 25 mg/kg or 50 mg/kg in 100 μl PBS solution) by i.p. injection, twice per week for 3 weeks. Mouse weight and tumor size was determined twice a week by measuring tumor length (L) and width (W) using a caliper, and tumor volume (TV) was calculated with the following formula: TV = (L × W2)/2. At the end of treatment, tumors and normal tissues of different organs were harvested from mice (n = 4 per group) and collected for subsequent studies.
Generation and culture of murine prostate cancer cell lines
Prostate cancer tissues dissected from a 5‐month‐old Ptenpc−/−;p53pc−/R172H and Ptenpc−/−;p53pc−/− mice were minced, digested with 0.5% collagenase type I, and tissue fragments were filtered through a 40‐μm mesh. Fragments trapped by the mesh were plated in tissue culture dishes coated with type I collagen. Epithelial cells growing out of the tissue fragments were collected and single cells plated into a 96‐well plate. Ptenpc−/−;p53pc−/R172H and Ptenpc−/−;p53pc−/− prostate cancer cell lines were established as a spontaneously immortalized line from one well. The line was maintained in DMEM supplemented with 10% fetal bovine serum, 25 μg/ml bovine pituitary extract, 5 μg/ml bovine insulin, and 6 ng/ml recombinant human epidermal growth factor (EGF; Watson et al, 2005).
Mouse tail‐vein injection of tumor cells and bioluminescent imaging
The mouse study was approved by Mayo Clinic IACUC. Male NSG mice were used for the experiment. Mia PaCa2 cells stably expressing the luciferase gene were infected with lentivirus for empty vector or EZH2 mutants (EZH2ΔSET or EZH2Δ336–554) in combination with or without control shRNA or p53‐specific shRNA. 2 × 106 infected cells in 0.2 ml PBS were injected via tail vein into individual mice (six mice each group). Mice were monitored weekly by bioluminescent imaging. Mice were injected with luciferin (300 mg/kg) 10 min before imaging. Mice were anaesthetized (3% isoflurane) and imaged using the IVIS spectrum imaging system (Xenogen, Life Sciences). Images were analyzed with Living Image software (Xenogen, Life Sciences). Bioluminescent flux (photons/s/sr/cm2) was determined for lesions in lung. At 12 weeks, lung tissues were collected and stained with Bouin's solution. Half of lung was fixed in formalin solution overnight and embedded in paraffin, from which sections were made and stained with hematoxylin and eosin. The other half was fast‐frozen into RNA‐ladder buffer (NEB) and stored at −80°C.
Analysis of p53 and EZH2 correlation in the TCGA dataset
The mutation status and copy‐number variation (CNV) of TP53 gene in the prostate cancer TCGA dataset were downloaded from cBioPortal (see attached TP53_mutation.txt and TP53_GISTIC_CNV.txt), and we separated those patients into three groups based on the mutation and CNV status of TP53: (i) wild‐type group—in which all patients without TP53 mutant and excluding patients with GIST value equals −1 (hemi‐deletion) and −2 (homo‐deletion) of CNV status; (ii) mutant group—in which all patients with TP53 mutant; (iii) lost group—in which all patients with −1 (hemi‐deletion) or −2 (homo‐deletion) CNV deletion status but excluding those patients with TP53 mutant. We further required the availability of RNA expression and relapse‐free survival (RFS) information for the above categorized patients and this led to the final three sets: 41 patients in the mutant group, 264 patients in the wild‐type group, and 85 patients in the lost group. In each group, we split the samples into two subgroups (low and high) by the median value of EZH2 RNA‐expression value (log2(RPKM)) and applied the survival analysis package (ref: http://lifelines.readthedocs.io) on these two subgroups for comparison, to measure the disease‐free survival of individuals under the two different conditions, and the log rank test was used to compare different Kaplan–Meier curves.
Ultraviolet crosslinked RNA immunoprecipitation (UV‐RIP)
5 × 106 C4‐2 or VCaP cells were washed with cold PBS one time, and cells were irradiated once with 150 mJ/cm2 at 254 nm using a Stratalinker. Cells were lysed in lysis buffer (50 mM Tris–HCL, pH 7.4, 100 mM NaCl, 1% NP‐40, 0.1% SDS, 0.5% sodium deoxycholate, protease inhibitor cocktail, and RNase inhibitors) with protease inhibitors (1 ml) and transferred to 1.5‐ml microtubes. Lysate was partially digested by 1 U/μl RNaseT1 for 15 min at 22°C. RNA was immunoprecipitated with EZH2, EED, or SUZ12 antibodies and protein A/G beads for 4 h at 4°C. After washed for four times, RNA was phosphorylated by T4 PNK and ligated RNA between 3′ and 5′ ends by RNA T4 ligase. SDS–PAGE loading buffer was added, and the mixture was incubated at 70°C for 10 min. After running the SDS–PAGE gel, the RNA–protein complexes were transferred from gel to a nitrocellulose membrane using a wet transfer apparatus (30 V for 1 h). The membrane with target protein was cut up, and the targeted membrane piece was incubated with ProteinaseK for de‐crosslink. After de‐crosslink, RNA was reverse transcribed into cDNA and subjected to real‐time qPCR analysis.
Polyribosome analysis
Polyribosome analysis was performed as previously described (Hu et al, 2009). The gradient station was kindly provided by Dr. Wenqian Hu at the Mayo Clinic. Specifically, cells grown to middle log phase were treated with cycloheximide to a final concentration of 100 μg/ml and collected by centrifugation. Cell pellets were lysed in the buffer (10 nM Tris, pH 7.4, 100 mM NaCl, 1 mM DTT, 20 mM MgCl2, 500 μg/ml heparin, and 100 μg/ml cycloheximide) by bead bashing, and Triton X‐100 was added to a final concentration of 1%. All gradients were made on a BioComp gradient maker and were 5–50% weight/weight (sucrose) with buffer (50 nM TrisAcetate pH 7.0, 50 mM NH4Cl, 10 mM MgCl2, 1 mM DTT). 20 units (OD260) of cell lysate were loaded onto each gradient. Gradients were centrifuged at 23,460 g for 3 h at 4°C in a Beckman SW‐41ti rotor. The gradient was fractionated using a gradient station (BioComp) with an ultraviolet detector (Bio‐Rad EM‐1). RNA from each collected fraction was isolated using the method previously described (Zhang et al, 2017) and subjected to RT–qPCR analysis.
Statistical analysis
Experiments were carried out with three or more replicates unless otherwise stated. Statistical analyses were performed by Student's t‐test for most studies. Pearson's product moment correlation coefficient was used to measure the association between EZH2 and p53 expression. Association P‐value was calculated using paired t‐test. Kaplan–Meier curves were used logrank test. P < 0.05 is considered statistically significant.
Author contributions
HH conceived the study. YZ, LD, DW, YH, LM, YP, QW, KP, XH, SJW, CH, RZ, RC, IC, RJK, PSN conducted experiments, data collection and analysis, generated mouse models, cell lines, and/or providing other reagents. JZ supervised IHC staining and JZ, YZ, LD scored the IHC data. ZY, LW performed bioinformatics analysis. YZ, HH wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 7
Acknowledgements
We thank the patients and their families for their altruism in participating in research studies. We also thank Xinbin Chen, Qiang Yu, Zhenbang Chen, and Da‐Qing Yang for reagents and suggestions; Wenqian Hu from Mayo Clinic for providing facilities for polysome fractionation; Youngsoo Kim and Robert MacLeod from Ionis Pharmaceuticals Inc for providing EZH2 ASOs; members of Huang laboratory for their constructive comments for the study; Colm Morrissey, Robert Vessella, Larry True, Xiaotun Zhang, and all other members of the University of Washington rapid autopsy team for their tremendous efforts. This work was supported in part by grants from the National Institutes of Health (CA134514, CA130908, CA193239, and CA203849 to H.H.), the Mayo Clinic Center for Individualized Medicine (to H.H.), the Kendall Fellowship in Biochemistry Award (to Y.Z.), the Department of Defense (W81XWH‐09‐1‐622 and W81XWH‐14‐1‐0486 to H.H. and W81XWH‐17‐1‐0415 to P.S.N.), the Pacific Northwest SPORE in Prostate Cancer P50CA097186 (to P.S.N.), and the Prostate Cancer Foundation (to P.S.N.).
The EMBO Journal (2019) 38: e99599
Data accessibility
High‐throughput EZH2 RIP‐seq data have been deposited to the GEO database with the GEO number GSE63230.
References
- Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, Solari A, Bobisse S, Rondina MB, Guzzardo V, Parenti AR, Rosato A, Bicciato S, Balmain A, Piccolo S (2009) A Mutant‐p53/Smad complex opposes p63 to empower TGFbeta‐induced metastasis. Cell 137: 87–98 [DOI] [PubMed] [Google Scholar]
- Blee AM, He Y, Yang Y, Ye Z, Yan Y, Pan Y, Ma T, Dugdale J, Kuehn E, Kohli M, Jimenez R, Chen Y, Xu W, Wang L, Huang H (2018) TMPRSS2‐ERG controls luminal epithelial lineage and antiandrogen sensitivity in PTEN and TP53‐mutated prostate cancer. Clin Cancer Res 24: 4551–4565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG, Gifford DK, Melton DA, Jaenisch R, Young RA (2005) Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122: 947–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candeias MM, Powell DJ, Roubalova E, Apcher S, Bourougaa K, Vojtesek B, Bruzzoni‐Giovanelli H, Fahraeus R (2006) Expression of p53 and p53/47 are controlled by alternative mechanisms of messenger RNA translation initiation. Oncogene 25: 6936–6947 [DOI] [PubMed] [Google Scholar]
- Cao R, Wang L, Wang H, Xia L, Erdjument‐Bromage H, Tempst P, Jones RS, Zhang Y (2002) Role of histone H3 lysine 27 methylation in Polycomb‐group silencing. Science 298: 1039–1043 [DOI] [PubMed] [Google Scholar]
- Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT, Ping B, Otte AP, Hung MC (2005) Akt‐mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science 310: 306–310 [DOI] [PubMed] [Google Scholar]
- Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, Cordon‐Cardo C, Pandolfi PP (2005) Crucial role of p53‐dependent cellular senescence in suppression of Pten‐deficient tumorigenesis. Nature 436: 725–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Bohrer LR, Rai AN, Pan Y, Gan L, Zhou X, Bagchi A, Simon JA, Huang H (2010) Cyclin‐dependent kinases regulate epigenetic gene silencing through phosphorylation of EZH2. Nat Cell Biol 12: 1108–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Cai J, Wang Q, Wang Y, Liu M, Yang J, Zhou J, Kang C, Li M, Jiang C (2018) Long noncoding RNA NEAT1, regulated by the EGFR pathway, contributes to glioblastoma progression through the WNT/beta‐catenin pathway by scaffolding EZH2. Clin Cancer Res 24: 684–695 [DOI] [PubMed] [Google Scholar]
- Cifuentes‐Rojas C, Hernandez AJ, Sarma K, Lee JT (2014) Regulatory interactions between RNA and polycomb repressive complex 2. Mol Cell 55: 171–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V (2002) Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell 111: 185–196 [DOI] [PubMed] [Google Scholar]
- Davidovich C, Zheng L, Goodrich KJ, Cech TR (2013) Promiscuous RNA binding by Polycomb repressive complex 2. Nat Struct Mol Biol 20: 1250–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Chen S, Liu P, Pan Y, Zhong J, Regan KM, Wang L, Yu C, Rizzardi A, Cheng L, Zhang J, Schmechel SC, Cheville JC, Van Deursen J, Tindall DJ, Huang H (2014) CBP loss cooperates with PTEN haploinsufficiency to drive prostate cancer: implications for epigenetic therapy. Cancer Res 74: 2050–2061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmer D, Pati S, Zambetti G, Chu S, Teresky AK, Moore M, Finlay C, Levine AJ (1993) Gain of function mutations in p53. Nat Genet 4: 42–46 [DOI] [PubMed] [Google Scholar]
- Ezhkova E, Pasolli HA, Parker JS, Stokes N, Su IH, Hannon G, Tarakhovsky A, Fuchs E (2009) Ezh2 orchestrates gene expression for the stepwise differentiation of tissue‐specific stem cells. Cell 136: 1122–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C (2006) Clonogenic assay of cells in vitro . Nat Protoc 1: 2315–2319 [DOI] [PubMed] [Google Scholar]
- Freed‐Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez‐Barrueco R, Barsotti A, Chicas A, Li W, Polotskaia A, Bissell MJ, Osborne TF, Tian B, Lowe SW, Silva JM, Borresen‐Dale AL, Levine AJ, Bargonetti J, Prives C (2012) Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 148: 244–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover R, Ray PS, Das S (2008) Polypyrimidine tract binding protein regulates IRES‐mediated translation of p53 isoforms. Cell Cycle 7: 2189–2198 [DOI] [PubMed] [Google Scholar]
- Hu W, Sweet TJ, Chamnongpol S, Baker KE, Coller J (2009) Co‐translational mRNA decay in Saccharomyces cerevisiae . Nature 461: 225–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Italiano A, Soria JC, Toulmonde M, Michot JM, Lucchesi C, Varga A, Coindre JM, Blakemore SJ, Clawson A, Suttle B, McDonald AA, Woodruff M, Ribich S, Hedrick E, Keilhack H, Thomson B, Owa T, Copeland RA, Ho PTC, Ribrag V (2018) Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B‐cell non‐Hodgkin lymphoma and advanced solid tumours: a first‐in‐human, open‐label, phase 1 study. Lancet Oncol 19: 649–659 [DOI] [PubMed] [Google Scholar]
- Jackson RJ, Hellen CU, Pestova TV (2010) The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol 11: 113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Bonasio R, Saldana‐Meyer R, Yoshida T, Son J, Nishino K, Umezawa A, Reinberg D (2014) Interactions between JARID2 and noncoding RNAs regulate PRC2 recruitment to chromatin. Mol Cell 53: 290–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karanikolas BD, Figueiredo ML, Wu L (2009) Polycomb group protein enhancer of zeste 2 is an oncogene that promotes the neoplastic transformation of a benign prostatic epithelial cell line. Mol Cancer Res 7: 1456–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF, Sabel MS, Livant D, Weiss SJ, Rubin MA, Chinnaiyan AM (2003) EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA 100: 11606–11611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang KC, Xiao Y, Kadowaki T, Uesugi M, Kuznetsov G, Kumar N, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Waters NJ, Smith JJ, Porter‐Scott M, Chesworth R, Moyer MP, Copeland RA et al (2014) Selective inhibition of EZH2 by EPZ‐6438 leads to potent antitumor activity in EZH2‐mutant non‐Hodgkin lymphoma. Mol Cancer Ther 13: 842–854 [DOI] [PubMed] [Google Scholar]
- Komar AA, Hatzoglou M (2011) Cellular IRES‐mediated translation: the war of ITAFs in pathophysiological states. Cell Cycle 10: 229–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmichev A, Nishioka K, Erdjument‐Bromage H, Tempst P, Reinberg D (2002) Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev 16: 2893–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmichev A, Margueron R, Vaquero A, Preissner TS, Scher M, Kirmizis A, Ouyang X, Brockdorff N, Abate‐Shen C, Farnham P, Reinberg D (2005) Composition and histone substrates of polycomb repressive group complexes change during cellular differentiation. Proc Natl Acad Sci USA 102: 1859–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann BD, Pietenpol JA (2012) Targeting mutant p53 in human tumors. J Clin Oncol 30: 3648–3650 [DOI] [PubMed] [Google Scholar]
- Levine AJ (1997) p53, the cellular gatekeeper for growth and division. Cell 88: 323–331 [DOI] [PubMed] [Google Scholar]
- Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, Gu W (2012) Tumor suppression in the absence of p53‐mediated cell‐cycle arrest, apoptosis, and senescence. Cell 149: 1269–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long Y, Bolanos B, Gong L, Liu W, Goodrich KJ, Yang X, Chen S, Gooding AR, Maegley KA, Gajiwala KS, Brooun A, Cech TR, Liu X (2017) Conserved RNA‐binding specificity of polycomb repressive complex 2 is achieved by dispersed amino acid patches in EZH2. Elife 6: e31558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Lastra M, Rivas A, Barria MI (2005) Protein synthesis in eukaryotes: the growing biological relevance of cap‐independent translation initiation. Biol Res 38: 121–146 [DOI] [PubMed] [Google Scholar]
- Malbert‐Colas L, Ponnuswamy A, Olivares‐Illana V, Tournillon AS, Naski N, Fahraeus R (2014) HDMX folds the nascent p53 mRNA following activation by the ATM kinase. Mol Cell 54: 500–511 [DOI] [PubMed] [Google Scholar]
- Margueron R, Reinberg D (2011) The Polycomb complex PRC2 and its mark in life. Nature 469: 343–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe MT, Graves AP, Ganji G, Diaz E, Halsey WS, Jiang Y, Smitheman KN, Ott HM, Pappalardi MB, Allen KE, Chen SB, Della Pietra A III, Dul E, Hughes AM, Gilbert SA, Thrall SH, Tummino PJ, Kruger RG, Brandt M, Schwartz B et al (2012a) Mutation of A677 in histone methyltransferase EZH2 in human B‐cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc Natl Acad Sci USA 109: 2989–2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A III, Diaz E, LaFrance LV, Mellinger M, Duquenne C, Tian X, Kruger RG, McHugh CF, Brandt M, Miller WH, Dhanak D, Verma SK et al (2012b) EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2‐activating mutations. Nature 492: 108–112 [DOI] [PubMed] [Google Scholar]
- Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, Yap D, Humphries RK, Griffith OL, Shah S, Zhu H, Kimbara M, Shashkin P, Charlot JF, Tcherpakov M, Corbett R et al (2010) Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B‐cell lymphomas of germinal‐center origin. Nat Genet 42: 181–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, Wang S, Plaisier S, Garraway IP, Huang J, Graeber TG, Wu H (2011) Cell autonomous role of PTEN in regulating castration‐resistant prostate cancer growth. Cancer Cell 19: 792–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller J, Hart CM, Francis NJ, Vargas ML, Sengupta A, Wild B, Miller EL, O'Connor MB, Kingston RE, Simon JA (2002) Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 111: 197–208 [DOI] [PubMed] [Google Scholar]
- Muller PA, Vousden KH (2014) Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell 25: 304–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntziachristos P, Tsirigos A, Van Vlierberghe P, Nedjic J, Trimarchi T, Flaherty MS, Ferres‐Marco D, da Ros V, Tang Z, Siegle J, Asp P, Hadler M, Rigo I, De Keersmaecker K, Patel J, Huynh T, Utro F, Poglio S, Samon JB, Paietta E et al (2012) Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat Med 18: 298–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, Jacks T (2004) Mutant p53 gain of function in two mouse models of Li‐Fraumeni syndrome. Cell 119: 847–860 [DOI] [PubMed] [Google Scholar]
- Pasini D, Bracken AP, Jensen MR, Lazzerini Denchi E, Helin K (2004) Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J 23: 4061–4071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peller S, Rotter V (2003) TP53 in hematological cancer: low incidence of mutations with significant clinical relevance. Hum Mutat 21: 277–284 [DOI] [PubMed] [Google Scholar]
- Pestova TV, Kolupaeva VG, Lomakin IB, Pilipenko EV, Shatsky IN, Agol VI, Hellen CU (2001) Molecular mechanisms of translation initiation in eukaryotes. Proc Natl Acad Sci USA 98: 7029–7036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plath K, Fang J, Mlynarczyk‐Evans SK, Cao R, Worringer KA, Wang H, de la Cruz CC, Otte AP, Panning B, Zhang Y (2003) Role of histone H3 lysine 27 methylation in X inactivation. Science 300: 131–135 [DOI] [PubMed] [Google Scholar]
- Ray PS, Grover R, Das S (2006) Two internal ribosome entry sites mediate the translation of p53 isoforms. EMBO Rep 7: 404–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, Chang HY (2007) Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129: 1311–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roudier MP, True LD, Higano CS, Vesselle H, Ellis W, Lange P, Vessella RL (2003) Phenotypic heterogeneity of end‐stage prostate carcinoma metastatic to bone. Hum Pathol 34: 646–653 [DOI] [PubMed] [Google Scholar]
- Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM, Copeland RA (2010) Coordinated activities of wild‐type plus mutant EZH2 drive tumor‐associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B‐cell lymphomas. Proc Natl Acad Sci USA 107: 20980–20985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su IH, Basavaraj A, Krutchinsky AN, Hobert O, Ullrich A, Chait BT, Tarakhovsky A (2003) Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat Immunol 4: 124–131 [DOI] [PubMed] [Google Scholar]
- Su IH, Dobenecker MW, Dickinson E, Oser M, Basavaraj A, Marqueron R, Viale A, Reinberg D, Wulfing C, Tarakhovsky A (2005) Polycomb group protein ezh2 controls actin polymerization and cell signaling. Cell 121: 425–436 [DOI] [PubMed] [Google Scholar]
- Tan J, Yang X, Zhuang L, Jiang X, Chen W, Lee PL, Karuturi RK, Tan PB, Liu ET, Yu Q (2007) Pharmacologic disruption of Polycomb‐repressive complex 2‐mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev 21: 1050–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA‐Seq. Bioinformatics 25: 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- True L, Coleman I, Hawley S, Huang CY, Gifford D, Coleman R, Beer TM, Gelmann E, Datta M, Mostaghel E, Knudsen B, Lange P, Vessella R, Lin D, Hood L, Nelson PS (2006) A molecular correlate to the Gleason grading system for prostate adenocarcinoma. Proc Natl Acad Sci USA 103: 10991–10996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar‐Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP, Rubin MA, Chinnaiyan AM (2002) The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419: 624–629 [DOI] [PubMed] [Google Scholar]
- Vaswani RG, Gehling VS, Dakin LA, Cook AS, Nasveschuk CG, Duplessis M, Iyer P, Balasubramanian S, Zhao F, Good AC, Campbell R, Lee C, Cantone N, Cummings RT, Normant E, Bellon SF, Albrecht BK, Harmange JC, Trojer P, Audia JE et al (2016) Identification of (R)‐N‐((4‐Methoxy‐6‐methyl‐2‐oxo‐1,2‐dihydropyridin‐3‐yl)methyl)‐2‐methyl‐1‐(1‐(1 ‐(2,2,2‐trifluoroethyl)piperidin‐4‐yl)ethyl)‐1H‐indole‐3‐carboxamide (CPI‐1205), a potent and selective inhibitor of histone methyltransferase EZH2, suitable for phase I clinical trials for B‐cell lymphomas. J Med Chem 59: 9928–9941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, Thomas GV, Li G, Roy‐Burman P, Nelson PS, Liu X, Wu H (2003) Prostate‐specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 4: 209–221 [DOI] [PubMed] [Google Scholar]
- Wang L, Wang S, Li W (2012) RSeQC: quality control of RNA‐seq experiments. Bioinformatics 28: 2184–2185 [DOI] [PubMed] [Google Scholar]
- Wang L, Zeng X, Chen S, Ding L, Zhong J, Zhao JC, Wang L, Sarver A, Koller A, Zhi J, Ma Y, Yu J, Chen J, Huang H (2013) BRCA1 is a negative modulator of the PRC2 complex. EMBO J 32: 1584–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Ding L, Wang L, Zhao Y, Sun Z, Karnes RJ, Zhang J, Huang H (2015) LncRNA MALAT1 enhances oncogenic activities of EZH2 in castration‐resistant prostate cancer. Oncotarget 6: 41045–41055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Paucek RD, Gooding AR, Brown ZZ, Ge EJ, Muir TW, Cech TR (2017) Molecular analysis of PRC2 recruitment to DNA in chromatin and its inhibition by RNA. Nat Struct Mol Biol 24: 1028–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson PA, Ellwood‐Yen K, King JC, Wongvipat J, Lebeau MM, Sawyers CL (2005) Context‐dependent hormone‐refractory progression revealed through characterization of a novel murine prostate cancer cell line. Cancer Res 65: 11565–11571 [DOI] [PubMed] [Google Scholar]
- Weingarten‐Gabbay S, Khan D, Liberman N, Yoffe Y, Bialik S, Das S, Oren M, Kimchi A (2014) The translation initiation factor DAP5 promotes IRES‐driven translation of p53 mRNA. Oncogene 33: 611–618 [DOI] [PubMed] [Google Scholar]
- Weissmueller S, Manchado E, Saborowski M, Morris JPT, Wagenblast E, Davis CA, Moon SH, Pfister NT, Tschaharganeh DF, Kitzing T, Aust D, Markert EK, Wu J, Grimmond SM, Pilarsky C, Prives C, Biankin AV, Lowe SW (2014) Mutant p53 drives pancreatic cancer metastasis through cell‐autonomous PDGF receptor beta signaling. Cell 157: 382–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Jin X, Yang J, Yang Y, He Y, Ding L, Pan Y, Chen S, Jiang J, Huang H (2016) Inhibition of EZH2 by chemo‐ and radiotherapy agents and small molecule inhibitors induces cell death in castration‐resistant prostate cancer. Oncotarget 7: 3440–3452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, Wu X, Stack EC, Loda M, Liu T, Xu H, Cato L, Thornton JE, Gregory RI, Morrissey C, Vessella RL, Montironi R, Magi‐Galluzzi C, Kantoff PW, Balk SP et al (2012) EZH2 oncogenic activity in castration‐resistant prostate cancer cells is Polycomb‐independent. Science 338: 1465–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Shao Z, Li D, Xie H, Kim W, Huang J, Taylor JE, Pinello L, Glass K, Jaffe JD, Yuan GC, Orkin SH (2015) Developmental control of polycomb subunit composition by GATA factors mediates a switch to non‐canonical functions. Mol Cell 57: 304–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada A, Fujii S, Daiko H, Nishimura M, Chiba T, Ochiai A (2011) Aberrant expression of EZH2 is associated with a poor outcome and P53 alteration in squamous cell carcinoma of the esophagus. Int J Oncol 38: 345–353 [DOI] [PubMed] [Google Scholar]
- Yang DQ, Halaby MJ, Zhang Y (2006) The identification of an internal ribosomal entry site in the 5′‐untranslated region of p53 mRNA provides a novel mechanism for the regulation of its translation following DNA damage. Oncogene 25: 4613–4619 [DOI] [PubMed] [Google Scholar]
- Yap DB, Chu J, Berg T, Schapira M, Cheng SW, Moradian A, Morin RD, Mungall AJ, Meissner B, Boyle M, Marquez VE, Marra MA, Gascoyne RD, Humphries RK, Arrowsmith CH, Morin GB, Aparicio SA (2011) Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 117: 2451–2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue X, Zhao Y, Liu J, Zhang C, Yu H, Wang J, Zheng T, Liu L, Li J, Feng Z, Hu W (2015) BAG2 promotes tumorigenesis through enhancing mutant p53 protein levels and function. Elife 4: e08401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Zhang J, Chen X, Cho SJ, Chen X (2013) Glycogen synthase kinase 3 promotes p53 mRNA translation via phosphorylation of RNPC1. Genes Dev 27: 2246–2258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chen X, Liu Q, Zhang S, Hu W (2017) Translation repression via modulation of the cytoplasmic poly(A)‐binding protein in the inflammatory response. Elife 6: e27786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT (2008) Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322: 750–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J, Ding L, Bohrer LR, Pan Y, Liu P, Zhang J, Sebo TJ, Karnes RJ, Tindall DJ, van Deursen J, Huang H (2014) p300 acetyltransferase regulates androgen receptor degradation and PTEN‐deficient prostate tumorigenesis. Cancer Res 74: 1870–1880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Sammons MA, Donahue G, Dou Z, Vedadi M, Getlik M, Barsyte‐Lovejoy D, Al‐awar R, Katona BW, Shilatifard A, Huang J, Hua X, Arrowsmith CH, Berger SL (2015) Gain‐of‐function p53 mutants co‐opt chromatin pathways to drive cancer growth. Nature 525: 206–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 7
Data Availability Statement
High‐throughput EZH2 RIP‐seq data have been deposited to the GEO database with the GEO number GSE63230.