

Abstract
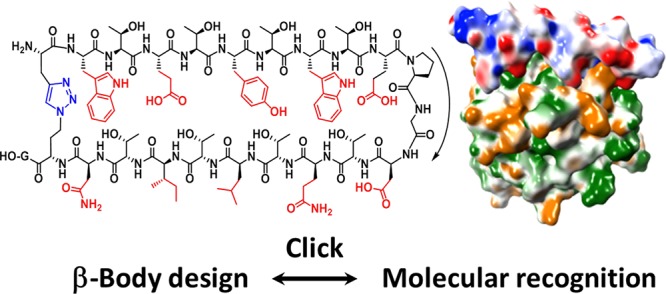
The development of recognition molecules with antibody-like properties is of great value to the biotechnological and bioanalytical communities. The recognition molecules presented here are peptides with a strong tendency to form β-hairpin structures, stabilized by alternate threonines, which are located at one face of the peptide. Amino acids at the other face of the peptide are available for interaction with the target molecule. Using this scaffold, we demonstrate that recognition molecules can efficiently be designed in silico toward four structurally unrelated proteins, GFP, IL-1β, IL-2, and IL-6. On solid support, 10 different antibody-mimetic recognition molecules were synthesized. They displayed high affinity and no cross-reactivity, as observed by fluorescence microscopy. Stabilized variants were readily obtained by incorporation of azido acids and propargylglycine followed by cyclization via the Cu(I)-catalyzed alkyne–azide cycloaddition reaction. As this new class of antibody mimics can be designed toward essentially any protein, the concept is believed to be useful to a wide range of technologies. Here, their use in protein separation and in the detection of proteins in a sandwich-type assay is demonstrated.
Short abstract
Clicked β-hairpins, facially enriched in Thr, with a β-turn, and recognition features at the opposing face, facilitated design of molecules with nanomolar affinity to interleukins and GFP.
Molecular recognition is a fundamental process which is driven by entropy and enthalpy and which precedes all biochemical events.1 This specific recognition between biomolecules has been harvested as a biochemical tool in the form of antibodies and their fragments including single-chain antibodies, nanobodies, and domain-sized nonantibody scaffolds.2−5 Other small scaffolds based on structures isolated from natural sources6,7 such as the antimicrobial tachyplesins,8,9 polyphemusin I10,11 and II,12 and arenicin,13 the β-hairpin protein epitope mimetics (PEMs)14 as well as the small cysteine knot proteins AgRP,15 cyclotides,16 and defensins have been exploited as a biochemical tool as well.17,18 In most of the aforementioned examples, existing protein folds or whole proteins which are already involved in recognition were employed. These entities have been developed to recognize new targets through screening followed by significant efforts in protein engineering and recombinant production in various expression hosts. Alternatively, efficient synthetic receptors have been obtained by extensive screening of phage display libraries or synthetic combinatorial libraries.19 However, although design of hairpins and helical scaffolds using synthetic stapling and constrained turns has been achieved,14,20−22 no general technology platform exists for direct design of endogenous recognition molecules.
The recognition motifs of monoclonal antibodies (mAbs) have been extensively used in synthesis of mimetics. Antibodies are large multifunctional biomolecules, one function of which is the avid and highly specific recognition of molecular surfaces by complementary surface interaction through the CDR region of the Fab (or ScFv) domain.23 This domain in turn is composed of six paratopes, short amino acid sequences looping out from the antibody thereby creating the pit ridges and canyons required for topological and electrostatic complementarity.24 The nanobodies which occur naturally are only half the size of an ScFv molecule and present only three hypervariable paratopes. As described in recent reviews successful attempts to mimic antibody recognition with small molecules have mainly been by connecting such paratopes to molecular scaffolds (templates).25,26
Aptamers are DNA analogs to antibody-mimetic proteins, and they are considered generally nonimmunogenic27 as are a range of antibody-mimetic protein scaffolds presenting paratope-mimetic loops, and derived by combinatorial screening of suitable protein scaffolds from the portfolio of nonimmunogenic proteins, ankyrins, protein A, lipocalin, cyclotides, as well as the very interesting small helical trefoil affibodies.28 It has been demonstrated that a single paratope from a mAb can bind the target antigen with down to sub-mM affinity. Upon the attachment of peptide loops to a calixarene template, the importance of a multivalent paratope display was demonstrated as early as 1997.29 This approach was elegantly refined by using a template technology for both paratope cyclization and attachment to a cyclotriveratrylene scaffold using click chemistry. The affinities of the trivalent products were ∼10 μM, probably demonstrating the need for exact preorganization of the paratopes in the interacting CDR. This problem was addressed in two studies resulting in functionally active antibody-mimetic inhibitors of TNFα30 and of the CD4−gp120 interaction,31 respectively, by computational design of the scaffolding arranging the interacting paratope-loops as in the parent antibody. However, both constructs showed significantly lower affinity for their targets than the antibodies. All of these paratope-based mimetics utilize existing recognition motifs of mAbs and assume that the modular structure of the mAb is essential for recognition. This called for development of a new virtual and evolutionary approach using any structural motif for which structure and recognition would be integrated in a single design process, such as that which was realized in the present work using the β-hairpin motif. An advantage of such an approach would be the independence of preexisting structures and the ability to target any suitable molecular surface.
Thus, we recently invented a new class of peptide scaffolds suitable for de novo design of antibody-mimetic compounds, that we termed β-bodies.32 This invention was based on early work, in which we noted by NMR spectroscopy that threonine-rich rod-shaped mucin glycopeptides were fully extended33 even without glycosylation. By connecting two of such threonine-rich strands by a β-turn,34 it was expected to obtain stable or readily induced35 β-hairpins. Satisfyingly, stable β-hairpin structures were indeed obtained in most cases when threonine (T or Thr) was placed in every second position of each strand. We envisioned that these peptides could be developed into a novel type of protein binder, in which one surface of the molecule is composed of amino acids that induce a β-hairpin or β-sheet structure (such as threonines), while the other surface is composed of amino acid side-chains that specifically and tightly interact in an antibody-like manner with a protein of choice (see Figure 1 and Figure S1).
Figure 1.
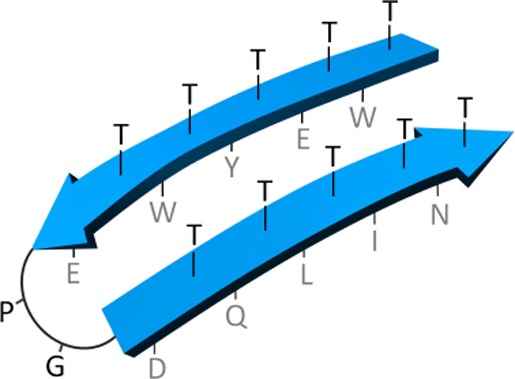
Typical structure of antibody-mimetic β-body: a β-hairpin consisting of two antiparallel β-strands with the polar, β-branched Thr residues pointing upward, and amino acids designed to interact with the target compound pointing downward.
In the present work, we describe the computational design, synthesis and validation of a collection of β-bodies recognizing various proteins of interest. We show that the designed β-bodies are able to recognize their target proteins in a selective manner and with nanomolar affinities, only 1 order of magnitude less than that of DNA-aptamers obtained through selection processes.36 We also demonstrate that β-bodies can be cyclized by click chemistry37 to further stabilize the β-hairpin structure. Finally, we show that β-body pairs can be designed to selectively recognize different sites of the same protein, facilitating their use in a sandwich-type binding assay.
Taking into account the fact that β-bodies can be designed toward a selected surface in essentially any protein structure, we believe that the β-body concept holds great promise in a broad range of applications including molecular diagnostics, protein purification, targeted drug delivery, and even therapeutics. Together with their small size, ease of computational design and synthesis, as well as their versatile recognition domains, the β-bodies constitute a valuable addition to the portfolio of peptide-based aptamers.38 With 10 locations of variable amino acids at the interface between the target protein and the β-body and by exclusion of proline from the 20 natural amino acids, the diversity constitutes ∼6 × 1012 different recognition molecules. With inclusion of the growing number of unnatural amino acids currently available, the number of possible molecules becomes almost infinite.
Results
For proof of concept, two model proteins, green-fluorescent protein (GFP) and interleukin-1β (IL-1β), were selected. GFP was selected because of its intrinsic fluorescence, which would allow for the detection of a binding event using fluorescence techniques without the need to label either of the binding partners with a fluorophore. IL-1β was chosen as a second, biomedically relevant target. This cytokine plays an important role in the inflammatory response and is involved in cell proliferation, differentiation, and apoptosis.39 It is activated by caspase-1 and induces cyclooxygenase-2 (COX2) and fever.40 Because overproduction of IL-1β is associated with a number of severe autoinflammatory syndromes, blocking the activity of IL-1β is an important therapeutic strategy.41,42 Examples of FDA-approved drugs whose mode of action are based on this concept include the IL-1 receptor antagonist anakinra and the antihuman-IL-1β antibody canakinumab.43 We also selected two other interleukins, T-cell-stimulating IL-244 and IL-6,45 associated with chronic inflammation and cancer, to demonstrate the broader perspective of the present technology.
For each interleukin, two β-bodies were designed, which were predicted to bind to different sites of the same protein. The in silico design process was performed in the molecular dynamics (MD) program “Molecular Operating Environment” (MOE from Chemical Computing Group) (see Figure 2).46,47 A structure of the respective protein obtained from the Protein Data Bank (PDB) and determined by either X-ray diffraction or NMR was loaded into MOE, soaked in water, and repaired by the modeling of missing parts while maintaining the PDB structure. Where several high-resolution structures were available in PDB for the same protein, these were compared and combined to increase the fidelity in the generation of the MOE model.
Figure 2.

Schematic of the manual optimization process in the molecular operating environment (MOE) for the generation of high-affinity β-bodies starting from a given protein crystal or NMR structure. After identification of putative positions and hairpin orientations on the protein surface suitable for complex formation, an optimization cycle is entered during which interaction residues are varied in a combinatorial fashion including side-chain orientation and with optimization of topology and surface electrostatic interaction. When the complex is stable for an extended period of MD simulation in water the β-body is accepted for synthesis and determination of protein binding.
The protein structure model was equipped with a molecular surface, color-coded by its electrostatic potential. Then, a generic spatial structure of a two-stranded β-body with the outward sequence Xl(TX)mPG(XT)nXl (or inward sequence Tl(XT)mPG(TX)nTl) was generated (see Figure S1), in which X was initially alanine, with n and m = 4–7 (typically 5) and l = 0 or 1. The spatial structure of the T/A-rich β-body was manually moved and rotated across the entire surface of the protein to identify sites for productive interaction in terms of overall shape fitting and presence of grooves, pits, and patches promising for interaction with amino acid side-chains. Once optimal sites and orientation for β-body interaction had been established, alanine side-chains were replaced with side-chains from other natural amino acids for an initial fit in the selected binding site. Subsequently, a mutational fitting process was performed. This fitting process involved multiple rounds of molecular dynamic simulations with intermediate mutational replacement of the recognition residues taking into account all the additive effects of counter-productive steric interactions, favored amino acid side-chain orientations (the all-important χ-space48), the hydrogen-bond network, hydrophobic interactions, and charge–charge interactions. Even small protrusions such as a hydrophobic methyl group could significantly reduce the interaction of distant amino acids with the target protein and required special attention. Most importantly, shape complementarity and the interaction of electropositive and electronegative patches between the individual surfaces were both optimized at all stages of the calculations. When a mutational step in the MD calculation that locally seemed favorable, eventually presented to be less favorable in the global interaction, it was returned to the previous state, and an alternative mutation was introduced. In this way, the interaction was gradually improved over 1–2 days. About 80% of target surfaces resulted in a single optimal β-body, while about 20% provided two or several equally promising β-bodies. Overall, the modeling procedure aimed at obtaining a maximum uninterrupted overlap of the surfaces of the β-body and target protein, with a spatial complementarity that optimally excluded water molecules from the interface of interaction and which showed optimal overlap of positive with negative and hydrophobic with hydrophobic surface patches. The computationally modeled complexes are presented in Figure S2, while the sequences of the β-bodies are provided in Table 1. Figure 3 illustrates interaction details of the modeled complex between IL-1β and β-body 2.
Table 1. Sequences of Synthesized β-Bodies Designed To Bind GFP and Three Interleukins.
| β-body | target protein | sequence |
|---|---|---|
| 1 | GFP | TETKTVTITRPKMTWTFTHTVTG |
| 2 | IL-1β | ETDTYTETYPGRTITWTITDG |
| 3 | IL-1β | TWTETYTWTEPGDTQTLTITNTG |
| 4 | IL-2 | NTVTNTMTRPGVTETVTQTDG |
| 5 | IL-2 | TRTLTYTEPGITQTKTEAG |
| 6 | IL-6 | HTWTDTLTRPGYTVTHTLTLG |
| 7 | IL-6 | TMTDTDTYPGFTDTLTHAG |
| 8a,b | GFP | c-Pra-ETKTVTITRPKMTWTFTHTV-DabN3-G |
| 9a,b | IL-1β | E-c-Pra-DTYTETYPGRTITWTI-DabN3-DG |
| 10a | IL-1β | TWTDTATEPGYTMTATGTG |
The rationale behind the synthesis of β-bodies 8–10 is described in the second part of the paper.
Pra, l-propargylglycine; DabN3, S-2-amino-4-azidobutyric acid.
Figure 3.
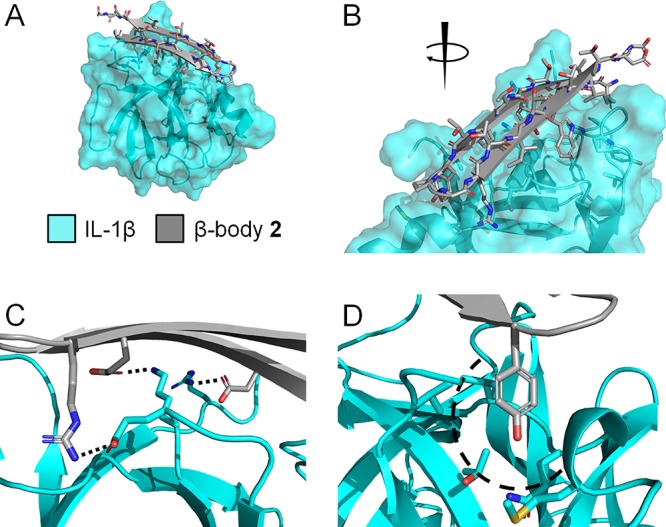
(A, B) Illustration of the interaction details between β-body 2 and IL-1β seen from two different directions. The β-hairpin was placed on the surface of the target protein at a site which contained charged, polar, and hydrophobic residues. Complementary residues were inserted to fit the various chemical characteristics of the target protein in the groove. Only residues of IL-1β predicted to be involved in the binding are shown. (C) Charged groups have been paired with complementary charged residues (dashed lines). (D) An example of an aromatic residue fitted into a hydrophobic pocket (dashed curve), to free the target protein from unfavorable interactions with water. Additional illustrations of the complexes between all β-bodies and their respective target proteins are shown in Figure S2.
All β-bodies were synthesized through standard Fmoc-based solid-phase peptide synthesis using poly[acryloyl-bis(aminopropyl)polyethylene glycol] (PEGA) resin as solid support. The low background fluorescence of PEGA resin, together with the lack of unspecific interactions between polymer resin and biomolecules, and its excellent swelling properties in aqueous solution, allowed for direct, on-bead testing of the β-body–protein interaction. For this purpose, the resin was thoroughly washed with deionized water upon completion of peptide synthesis and removal of the protecting groups. Resin-bound peptides were released from an aliquot of resin for structural characterization. IL-1β was labeled with carboxy-X-rhodamine (ROX) via a conventional N-Hydroxy-succinimide ester coupling and separated from excess of dye. A second aliquot of the beads carrying the capture β-body was transferred to a well of a microtiter plate. Then, the target protein, being either GFP or ROX-IL-1β, was added to the beads, and binding was assessed by fluorescence microscopy. As shown in Figure 4, binding was observed for GFP with β-body 1 and for IL-1β with both β-body 2 and 3, respectively, at protein concentrations as low as 50 nM. N-Acetylated beads acted as a negative control, and no fluorescence was detected on beads in wells where protein had been added to these empty control beads without β-body (see Figure S3). For IL-1β, the on-bead binding assay was also performed in an inverse manner in an independent control experiment. IL-1β was immobilized to Ni-NTA agarose beads via an N-terminal His-tag. The β-bodies designed for IL-1β were labeled with ROX, cleaved from the PEGA resin, and added in nanomolar concentration to the immobilized IL-1β. Also in this case, agarose beads turned fluorescent, confirming that binding was mediated by interactions between the protein and the β-body (see Figure S4).
Figure 4.
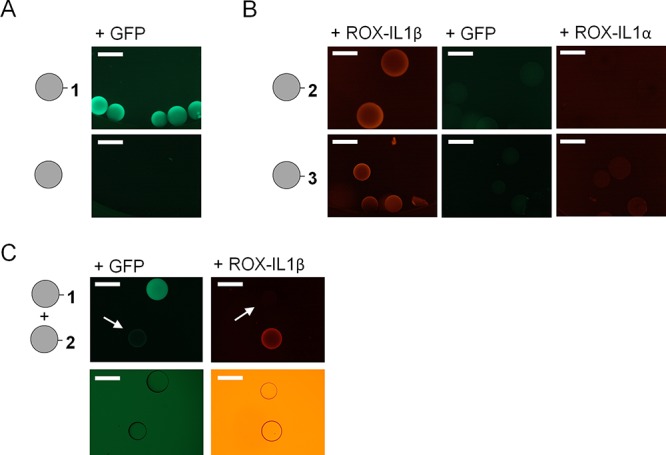
(A) Microscopy images of beads containing β-body 1 and acetylated control beads after incubation with 50 nM of GFP. (B) Microscopy images of beads containing β-bodies 2 and 3 after incubation with 50 nM ROX-labeled IL-1β, GFP, and ROX-labeled IL-1α, respectively. (C) Interaction of a mixture of beads containing 1 or 2 with GFP and ROX-labeled IL-1β, respectively, where the arrows and bright-field images below show the position of the nonlabeled beads. All scale bars represent 500 μm.
For an evaluation of the specificity of binding, GFP was added to beads containing β-bodies designed toward IL-1β. To our delight, no binding was observed in these cases (see Figure 4B). We envisioned that an even better measure of specificity would be obtained by testing whether the β-bodies would bind to a protein with a structure and function similar to the target protein. Therefore, the binding of IL-1α to compounds 2 or 3 was assessed. IL-1α and IL-1β share the same β-trefoil structure, bind to the same receptor (IL-1R1), and have similar biological properties.39 While ROX-labeled IL-1α was found to interact with β-body 3 to a very small degree, no interaction with β-body 2 could be observed (see Figure 4B). This selectivity is similar to that of antibody recognition and indicates that the β-bodies have potential to become prominent members in our toolbox for protein biochemistry, diagnostics, and therapeutics. Thus, β-bodies may be used for specific design of small selective binders, with affinity towards single proteins involved in complex signaling pathways and protein networks.
Next, the affinity of binding was studied in more detail. First, it was assessed whether GFP was able to interact with β-body 1 when present in a bacterial cell lysate. As shown in Figure 5A, even in such a complex mixture of proteins and other biomolecules, binding was observed. The fluorescence was quantitatively transferred from the lysate to the beads modified with β-body 1. Importantly, this result indicated that β-bodies might have the potential to be used in applications such as affinity chromatography and pull-down assays, facilitating the isolation of endogenous proteins free of any affinity tag. For an assessment of the potential of such an application, GFP was purified from a cell lysate using a packed column with PEGA resins containing covalently linked β-body 1. Upon washing the column thoroughly with water, the protein was eluted with PBS. Aliquots before and after purification were analyzed by SDS-PAGE. Despite the fact that this resin is far from optimal for protein purification, a significant up-concentration of GFP was achieved with removal of the majority of other proteins in the cell lysate (see Figure 5B). Overall, this result indicates the good specificity of the β-body.
Figure 5.
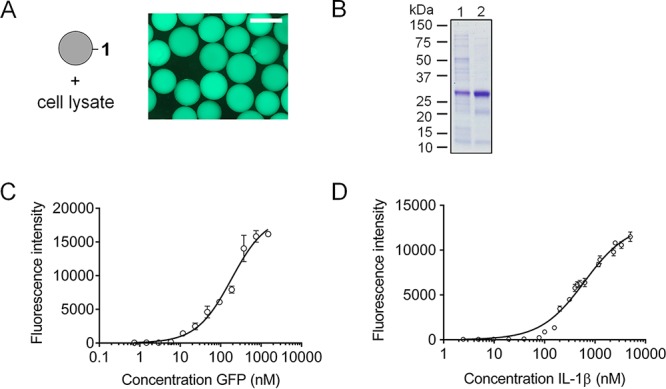
(A) Microscopy image of beads containing β-body 1 after incubation with lysed Escherichia coli cells expressing GFP. The scale bar is 500 μm. (B) SDS-PAGE of the purification of GFP from a whole cell lysate. Lane 1, cell lysate containing expressed GFP; lane 2, GFP washed with water and eluted with PBS from PEGA beads containing GFP-binding β-body 1. (C) Binding curve of the interaction between purified GFP and β-body 1. (D) Binding curve of the interaction between purified IL-1β and β-body 3. The plotted fluorescence intensities in parts C and D are averages of 3–6 measurements per protein concentration.
We investigated the binding of GFP to 1 in an on-bead binding assay, making use of an adapted version of the method we recently reported.49 Equally sized PEGA beads with β-body 1 were distributed in triplicates at three beads per individual well in a 96-well microtiter plate, and incubated with solutions containing increasing amounts of GFP. Binding was allowed to reach the point of equilibration by overnight incubation at room temperature, permitting enough time for the protein molecules to diffuse to the interior of the PEGA beads. Then, the fluorescence intensities of the individual beads were quantified by fluorescence microscopy. The binding curve for the interaction of GFP with β-body 1 is depicted in Figure 5C. Fitting of the data points provided a dissociation constant in the sub-micromolar range (Kd 184.5 ± 28.59 nM). Satisfyingly, IL-1β and β-body 3 showed high affinity to each other not only in water but also in phosphate buffer, providing a dissociation constant Kd of 610 ± 50 nM (Figure 5D).
An important advantage of using designed peptides as antibody mimetics is the fact that they can be chemically synthesized, thus, facilitating the straightforward incorporation of non-canonical and d-amino acids. Not only may such non-canonical moieties facilitate an optimized fit with the target surface, but they can also be used to form a covalent bridge between the two β-strands, hence, stabilizing the β-body structure.8,50,51 For a test of this concept, bridged β-bodies 8 and 9 were prepared using click chemistry (see Figure 6A). Relying on our long-standing experience8,37 with the Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction,52−54 two Thr residues on opposite strands of the same β-body were substituted with propargyl glycine (Pra) and γ-azido-α-aminobutyric acid (Dab-N3), respectively.8,51 Using CuBr as catalyst, triazole bridge formation was quantitatively induced. Intramolecular cyclization was confirmed to have taken place by treating the product with DTT followed by mass spectrometric analysis (see Figures S20 and S21). The lack of the expected loss of 26 Da due to reduction of azide to amine showed that no unreacted azide was left.51 The bridged β-bodies 8 and 9 retained their ability to bind to GFP and IL-1β, as shown in Figure 6B.
Figure 6.
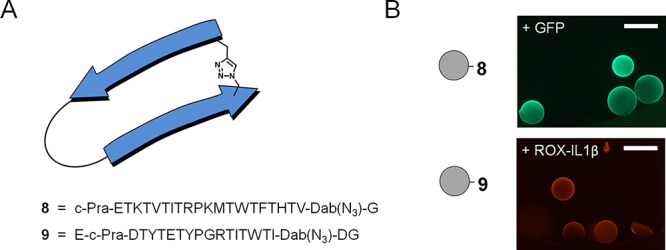
(A) Schematic representation of a triazole-bridged β-body and sequences of the β-bodies 8 and 9. (B) Microscopy images of beads containing bridged β-body 8 or 9 after addition of GFP (50 nM) and ROX-labeled IL-1β (50 nM), respectively. The scale bars are 500 μm.
The structural features of the β-bodies compound 2 and 9 recognizing IL-1β with high specificity were accessed by CD and NMR spectroscopy. The peptides were purified to yield single peaks in LC-MS (Figures S5–S8). For the acquisition of structural information from 1H NMR, solutions of 3.7 mM in H2O/D2O were prepared. It should be noted that this concentration was 37,000-fold higher than that used in the biomolecular interaction studies (100 nM). It was immediately apparent that at the NMR time scale and this elevated concentration both compounds 2 and 9 formed at least three distinct, but equilibrating complexes according to 1D- and 2D-1H NMR spectroscopy. Compound 2 formed a major complex or conformer and two minor conformers. According to an 1H NMR spectrum of 2 recorded at 10-fold dilution the major conformer transformed into another different single conformer. This change was supported by recording a dilution-series of CD spectra of compound 2 (Figure S9), which clearly showed transition from an unconventional CD spectrum at high concentration toward the β-structure upon dilution. However, the recording of NOESY and DQF-COSY required the more concentrated sample, and the resulting spectra were used to assign the major conformer present at 3.7 mM (Tables S1). The transition toward β-structure upon 10-fold dilution was supported by a downfield shift of the amide protons corresponding to a minor conformer also present at the higher concentration (see expansions in Figure S11). For assistance in the assignment, the NOESY spectrum was recorded with a mixing time of 600 ms resulting in some additional spin diffusion. Therefore the inter-residue NOESY cross-peaks that are listed in Table S2 can only be used in qualitative assessment of intramolecular proximities. Of most interest were several interstrand NOESY cross-peaks Y5-I18, Y5-W16, Tyr9-Ile14, and D3-I18. Together these NOE’s provided a strong indication that β-hairpin was present in the mixture even at the high concentration used during recording of the NOESY spectrum. The complexity of the NH-Hα region due to the dynamic mixture of conformers did not allow unambiguous assignment of NH-Hα NOESY cross-peaks in this region. The cyclic compound 9 formed three structurally closely related complexes at high concentration and while significant change was observed upon dilution in both CD and NMR it did not converge to a single conformer (Figure S10), indicating stronger self-association of the cyclic peptide. The two major conformers of 9 were very similar in structure, and protons could be assigned by overlay of COSY and DQF-NOESY spectra (Figure S12 and Tables S3). However, according to the CD study (Figure S10) the trend in the transition of 9 upon dilution progressed toward a β-hairpin. By overlay of the two NOESY spectra of compounds 2 and 9 (not shown) these appeared to be very similar except for signals resulting from the two residues connected to the triazole in compound 9. A curious observation was that while the solution of 2 became slightly turbid at the 3.7 mM concentration, the solution of 9 was clear at this concentration and did not immediately indicate any aggregation. In any event the study shows that the β-bodies are probably predominantly β-hairpins at the bioassay conditions (100 nM), and what remains is that both compounds 2 and 9 present highly specific interaction with exactly the protein for which they were designed.
Another powerful feature of the presented approach is the fact that pairs of β-bodies can directly be designed to bind different sites of the same protein (see Figure 7A). Such pairs of recognition molecules can be used in sandwich-type assays in which one of the β-bodies, while bound to a solid support, acts as a specific capture reagent, and the second β-body, after labeling with a fluorophore, acts as a specific detection reagent. For a demonstration of the feasibility of this powerful concept, ROX was conjugated to the N-terminus of β-body 5, designed to recognize IL-2. ROX-5 was added to a suspension of beads containing covalently attached β-body 4 with no observation of fluorescence on the beads. Satisfyingly, fluorescent beads were then observed exclusively after addition of 50 nM of IL-2 (see Figure 7B). The same experiment was performed using β-bodies designed to recognize IL-1β and IL-6. For IL-6, some unspecific interaction between capture peptide 6 and ROX-labeled detection peptide 7 was observed at the surface of the beads. This could be circumvented, though, by the addition of 0.05–0.1 M urea in the binding and washing buffer. Following this procedure, the fluorescence from beads in a solution without IL-6 was completely removed, while the beads to which IL-6 was added remained strongly fluorescent. With respect to IL-1β, the unspecific binding of ROX-labeled detection peptide 2 to capture peptide 3 could not be prevented without affecting at the same time the binding of IL-1β. Therefore, a third capture β-body was designed (10) which in combination with ROX-2 enabled us to also detect IL-1β in nanomolar concentrations in a sandwich-type assay (see Figure 7B).
Figure 7.
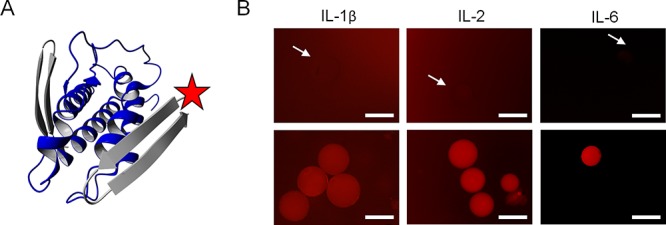
(A) Three-dimensional-ribbon representation of the modeled complex of IL-2 (blue) with β-body 4 in the back and ROX-labeled 5 in the front (both gray). (B) Microscopy images of PEGA1900 beads in sandwich assays for IL-1β, IL-2, and IL-6, in which one IL-specific β-body is attached to the bead and the other, labeled with fluorophore (ROX), added in solution. Top: Mixture of capture and detection β-body prior to the addition of protein. Bottom: After addition of protein. IL-1β: TWTDTATEPGYTMTATGTG-bead (10) and ROX-ETDTYTETYPGYTSTWTITDG (2), 50 nM protein. IL-2: NTVTNTMTRPGVTETVTQTDG-bead (4) and ROX-TRTLTYTEPGITQTKTEAG (5), 50 nM protein. IL-6: HTWTDTLTRPGYTVTHTLTLG-bead (6) and ROX-TMTDTDTYPGFTDTLTHAG (7), 20 nM protein. The arrows point at nonfluorescent beads. The scale bars are 500 μm.
Discussion
Recognition by structured peptides is a research topic currently undergoing rapid development, and a range of proteins and polynucleotides in the size range 10–50 kDa have been described. The templates used are frequently derived from natural counterparts involved in specific recognition, such as antibodies or natural receptor ligands. Because of the complexity of the macromolecules and the fact that residues providing recognition and structure are not separate, the affinity is generally achieved through evolutionary processes involving screening and panning technologies. To be able to use computational design and chemical synthesis for the preparation of recognition molecules, we developed a β-hairpin scaffold in which we spatially separated the structural features from the residues involved in molecular interaction with the target macromolecule. This was successfully achieved by alternate incorporation of β-branched threonines in strands connected by the known turn-inducing structures −Pro–Gly– or −Pro–Lys–. The use of threonines in promoting hairpin formation also served the purpose of providing a favorable hydration shell at the structural face of the hairpin to increase solubility, reduce nonspecific interaction, and promote formation of hairpin monomers.
Interestingly, the β-bodies were shown to have quite high selectivity for their target proteins, and even interleukin-1α and -1β, which share the same protein fold and display high sequence similarity, could be distinguished using two β-bodies targeted toward IL-1β. This selectivity was then manyfold potentiated in a sandwich assay by the application of two β-bodies designed for different sites on the same protein. Since the β-bodies are readily designed to be highly specific to a particular protein surface, they are ideally suited for affinity purification of valuable biologics. We demonstrate this in the tag free isolation of GFP from a cell lysate (Figure 5), but because of the general nature of the β-body technology this should be feasible for any protein with a known structure.
The properties of the designed β-bodies in terms of solubility, structural stability, self-aggregation, and compatibility with buffer ions, pH and salt depended to a large extent on the nature of the recognition site on the protein, which dictated the composition and combination of the recognition residues in the β-body. For future β-body design, a recognition site may be considered optimal when providing a balanced opportunity for charge–charge interactions, hydrogen bonds, aromatic stacking, and hydrophobic interactions, such that the different regions of the interacting surfaces contribute in a balanced manner to the adhesion, depending on the nature of the surrounding solution. High symmetry of β-bodies may occasionally facilitate self-interaction and aggregation. The β-body concept may be extended from the presently described β-hairpins to larger β-sheets for complex interactions with larger surfaces or even multiple proteins.
Peptide chemists have long searched for a platform, which allows for direct computational design of high-affinity molecules in a process which, with high probability, leads to specific molecular recognition. The properly structured peptide β-hairpin constitutes the smallest class of molecules with a stable protein-like fold. Importantly, the designed β-bodies, with a molecular weight of typically 2000–2600 Da, are synthetically tractable allowing incorporation of non-natural amino acids and initial designs to be further improved by combinatorial chemistry. To this end, we recently reported the high-yield flow synthesis of uniform microparticle-encoded macrobeads, which may allow expedient optimization of designed active β-bodies using a one-bead–one-compound (OBOC) technique with microparticle matrix decoding.55
Depending on the register of alternate threonines with respect to the two turn residues, two types of β-bodies exist. In one, side-chains of recognition residues orient outward acting as landing pads for interaction with a somewhat planar surface on the target. In the other arrangement, they are oriented in toward the center of the β-body allowing tight interaction with canyon and clefts at the target site of interaction (see Figure S1). We have designed over 70 β-bodies, many of which have already presented good affinity to their target proteins. These β-bodies are currently under investigation for their ability to control protein–protein interaction, protein fibrillation, enzyme inhibition, and receptor activation. Since β-bodies can be designed and synthesized in the metabolically more stable D-amino acid forms, their possible future use in blocking viral entry or for induction of apoptosis in cancer cells is of particular interest.
Initial experiments in mice (see Figure S27) indicated that tested β-bodies were nonimmunogenic as measured by lack of interleukin response in a broad cytokine assay detecting loss of body mass, INF-γ, TNF-α, IL-1β, IL-2, CXCL-1, IL-5, IL-6, IL-10, IL-12, and IL-4.
The lack of interleukin response holds great promise for β-bodies as therapeutic molecules such as inhibitors of protein–protein interactions or as enzyme inhibitors. Sandwich assays using β-bodies are quite powerful in providing dual specificity of the already specific β-body toward the target protein. This can have important applications in diagnostics and significantly reduce the cost of disease detection. β-Bodies may also be useful for recognition of other types of biopolymers. We are confident that this technology will be of great value to the scientific community in the clinic and as a source of new approaches in medicinal chemistry.
Methods
Materials
PEGA800 and PEGA1900 resins were produced by free-radical polymerization as previously described.55,56 The 9-fluorenylmethyloxycarbonyl (Fmoc) protected amino acids, 1-(mesitylene-2-sulfonyl)-3-nitro-1,2,4-triazole (MSNT), 4-hydroxymethyl benzamide (HMBA), O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TBTU), and 1-methylimidazole (MeIm) were purchased from Bachem AG, Chem-Impex International, Inc., or Honeywell Fluka. N-Ethylmorpholine (NEM), piperidine, trifluoroacetic acid (TFA), N,N-diisopropylethylamine (DIPEA), triisopropylsilane (TIPS), Tris base, phosphate-buffered saline (PBS), imidazole, LB, ampicillin, isopropyl β-d-1-thiogalactopyranoside (IPTG), lysozyme (from chicken egg white), Coomassie Brilliant Blue R-250, and HIS-Select nickel affinity gel were purchased from Sigma-Aldrich. The 5(6)-ROX N-succinimidyl ester was purchased from Sigma-Aldrich and Chemodex. The gene fragment encoding human IL-1β was ordered from Eurofins. Recombinant human IL-2 (Gibco) was purchased from Thermo Scientific. The water used for all experiments was purified using an Ultra Clear water system (Siemens) set at 0.055 μS/cm. IL-2 and IL-6 were purchased from ProSpec, and reconstituted as instructed.
β-Body Evolution by MD Simulation
The in silico design process was performed in the molecular dynamics program “Molecular Operating Environment” (MOE from Chemical Computing Group)46,47 using the Amber14ETH force field. The target protein crystal structures with PDB IDs 4EUL, 2ERJ, and 4CNI were used as models of GFP, IL-2, and IL-6, respectively, while IL-1b was represented by the PDB-NMR structure with PDB ID 6I1B. The structure of the respective protein was loaded into MOE, soaked in water, and repaired by modeling of missing parts while maintaining the PDB structure. Where several high-resolution structures were available in PDB for the same protein, these were compared and, for missing regions, combined, to increase the fidelity in the generation of the MOE model. The protein structure model was equipped with a color-coded surface electrostatic field. Surface electrostatic fields were estimated using the short-range Gaussian-screened portion of the Ewald summation and filtered to remove minor unimportant patches as described in the MOE software. The map obtained described the excess positive or negative charge close to the molecular surface and by estimation the electrical field gradient between proximate surfaces. Then, a generic spatial structure of a two-stranded β-body with the outward sequence Xl(TX)mPG(XT)nXl (or inward sequence Tl(XT)mPG(TX)nTl) was generated (see Figure S1), in which X was initially alanine, with n and m = 4–7 (typically 5) and l = 0 or 1. The spatial structure of this generic T/A-rich β-body was equipped with weak H-bond restraints (weight 0.5) between the strands, solvated in a water droplet, and manually moved and rotated across the entire protein surface with intermediate short MD fittings to the local surface to identify sites for productive interaction in terms of overall shape fitting and presence of grooves, pits, and patches promising for interaction with amino acid side-chains. Once optimal sites and orientation for β-body interaction had been established, β-bodies were added to these locations and (again) solvated in a droplet of 8 layers of water (it was feasible to optimize several β-bodies simultaneously). Under consideration of spatial arrangements, putative electrostatic interaction, H-bonding, and hydrophobicity, alanine side-chains were then replaced with side-chains from other natural amino acids for an initial fit in the selected binding site. The protein residues in direct contact with the β-body were released while all other protein residues remained fixed as defined by the crystal structure.
Subsequently, a mutational fitting process was performed. This fitting process involved multiple rounds of molecular dynamic simulations with intermediate mutational replacement of the recognition residues taking into account all the additive effects of counterproductive steric interactions, favored amino acid side-chain orientations (the all-important χ-space48), the hydrogen-bond network, hydrophobic interactions, and charge–charge interactions. Most importantly, shape complementarity and the interaction of electropositive with electronegative and hydrophobic with hydrophobic patches between the individual surfaces were both optimized at all stages of the calculations. When a mutational step seemed locally favorable during the MD calculation, but eventually presented a less favorable overall interaction, it was returned to the previous state, and an alternative mutation was introduced.
The optimization process was considered to be complete when the β-body would form a long-term stable complex allowing annealing from 450 K without any significant change in structure. Moreover, the optimized complex presented a maximum of uninterrupted overlap of the surfaces of the β-body and target protein, with a spatial complementarity that optimally excluded water molecules from the interface of interaction and optimal overlap of positive with negative and hydrophobic with hydrophobic surface patches.
Solid-Phase Peptide Synthesis
Peptides were synthesized manually at room temperature on the basis of Fmoc solid-phase peptide synthesis. Washings were performed adiabatically with 3 × 3 resin volumes of DMF, and Fmoc groups were removed by repeated treatment with 20% (v/v) piperidine in DMF first for 5 min and then 15 min except for the second amino acid where the resin was treated for only 5 min and immediately coupled with the subsequent amino acid. The base-labile linker HMBA (3 equiv) was coupled to the PEGA resin using standard coupling conditions (TBTU, 2.88 equiv; and NEM, 4 equiv in DMF). Subsequently, Fmoc-Gly-OH (2 × 3 equiv) was esterified on to HMBA in a double MSNT coupling (MSNT, 3 equiv; and MeIm, 2.25 equiv in DCM). The subsequent Fmoc-protected amino acids were coupled using standard TBTU couplings. After the last coupling and Fmoc-cleavage the resin was washed with DMF and DCM. The side-chain protecting groups were removed using a mixture of TFA/DTT/water/TIPS (88/5/5/2). The peptides were cleaved from the resin with 5% TEA in water, and characterized by LC-MS and HPLC.
Peptide Analysis
LC-MS analysis of β-bodies was performed on a Dionex UltiMate 3000 (Thermo Scientific) instrument equipped with an Acclaim RSLC 120 C18 column (2.2 μm, 120 Å, 2.1 × 100 mm) coupled to a Bruker microTOF-QIII mass spectrometer. A linear gradient of CH3CN in H2O with 0.1% formic acid was used, running from 5% to 100% CH3CN, 0.5 mL/min over 10 min.
HPLC analysis was performed on an analytical Agilent 1100 HPLC using a 100 mm XBridge C18 column. A linear gradient of CH3CN in H2O with 0.1% TFA was used, running from 0% to 90% CH3CN, 1 mL/min over 10 min. Signal detection was performed by measuring absorbance at 254 nm.
Conformational Studies on Peptides 2 and 9
The peptides were dissolved in D2O/H2O 1:9 at a concentration of 3.7 mM and 1D-, DQF-COSY, and NOESY were recorded on an 800 MHz Bruker instrument (DTU) NOESY with Watergate and excitation sculpting. Two-dimensional spectra were recorded with 1024 points in the F1 direction and with 4096 points and 4 scans/spectrum in the F2 dimension. The NOESY spectra were recorded with a mixing time of 600 ms. The spectra were processed using TOPSPIN. Spectra were carefully phase-corrected and aligned using the proton ortho to OH in tyrosine as an internal reference at 6.68 ppm. Assignment was problematic because of the presence of several closely similar conformational states but was feasible with appropriate overlay of NOESY and COSY spectra. Assignments are presented in Tables S1 and S3 and intraresidue NOE’s are presented in Table S2.
Labeling of Peptides with ROX-NHS
Peptides 2, 3, 5, 7, and 9 were labeled with ROX-NHS as follows. Following removal of the Fmoc group of the final amino acid, a mixture of ROX-NHS (1.1 equiv), TBTU (1.1 equiv), and NEM (1 equiv) in DMF was added to the resin, which was left to react overnight. The resin was washed with DMF (3 × 3 volumes) and DCM (3 × 3 volumes), and side-chain deprotection and cleavage of the peptide from the resin were performed as described above.
Click Chemistry
A suspension of resin containing deprotected peptides 8 and 9 in 4.5 mL of DMF was degassed overnight by bubbling with N2. The 2,6-lutidine (10 equiv, 10 μL) and DIPEA (10 equiv, 105 μL) were added to the resin followed by the addition of 5 mL of a degassed solution of sodium ascorbate (20 equiv) in DMF. Subsequently, 1% (w/v) solution of CuBr in 1.5 mL of degassed acetonitrile was added. The reaction mixture was agitated under argon at room temperature for 8 h. Reagents were removed by washing with DMF and DCM.
Purification of Peptides
Peptides 1–3 were purified by RP-HPLC on a Gilson 215 liquid handler using a 10 μm 19 × 150 mm XTerra Prep RP18 column. A 25 min gradient from 13.5% to 76.5% CH3CN in H2O with 0.1% TFA and a flow of 15 mL/min was used. Detection of signal was performed by a Gilson 170 diode array measuring the absorbance of UV-light at 210 and 254 nm.
CD Spectroscopy
Measurements in the far-UV region (190–260) were conducted using a 0.1 cm path length cuvette on a J-810 Jasco spectropolarimeter. All measurements were performed at 25 °C. A stock solution of peptide 2 was diluted to 80–100 μg/mL in 10 mM phosphate buffer, pH 7.4. The spectrum shown is an average of 5 scans, and the optical activity is reported as ellipticity per peptide bond.
Protein Expression and Purification
The gene fragment encoding human IL-1β was ordered codon-optimized for E. coli from Eurofins. The plasmid pET15-IL1B was created by digestion of the gene fragment with NcoI and BamHI and ligation into the corresponding sites of plasmid pET15b. The use of plasmid pET15-GFP has been reported previously.57 The plasmid pET20b-IL1A (lacking the pelB leader sequence) was kindly provided by Y. Chin (Department of Chemistry, National Tsing Hua University, Hsinchu, Taiwan).58
GFP, IL-1β, and IL-1α were produced in BL21(DE3) E. coli cells transformed with pET expressions plasmids containing the respective genes fused to an N-terminal (GFP and IL-1β) or C-terminal (IL-1α) His-tag. Cells were cultured in LB medium (0.2–1 L) containing 100 μg/mL ampicillin at 37 °C until the midexponential phase, at which time expression was induced with 1 mM IPTG for 18 h at 20 °C. After harvesting, cells were resuspended in lysis buffer (50 mM Tris, 0.5 M NaCl, pH 8.0). Lysozyme (1 mg/mL) was added, and the lysate was left at room temperature for 20 min. Following sonication, any debris and unbroken cells were removed by centrifugation at 12000 g for 30 min at 4 °C. The supernatant was collected, and the protein was purified by Ni2+-NTA affinity chromatography on an ÄKTA pure system using 1 mL HisTrap HP columns (GE Healthcare). The 50 mM Tris, 0.5 M NaCl, pH 8.0, supplemented with 20 mM imidazole was used during washing of the column. Proteins were eluted with 250 mM imidazole in 50 mM Tris, 0.5 M NaCl, pH 8.0. Elution fractions containing GFP, IL-1β, and IL-1α were additionally purified by size-exclusion chromatography on an Äkta pure system using a superdex 75 Increase 10/300 GL column (GE Healthcare) and phosphate-buffered saline (PBS, 10 mM phosphate buffer, 2.7 mM KCl, 137 mM NaCl, pH 7.4) as eluent.
Protein Characterization and Concentration Determination
Protein purity was assessed by SDS-PAGE gel electrophoresis using any kD Mini-PROTEAN TGX precast protein gels (Biorad). Gels were stained in 0.1% (w/v) Coomassie Brilliant Blue solution [10% (v/v) AcOH, 50% (v/v) MeOH in deionized water] and destained in destaining solution [10% (v/v) AcOH, 50% (v/v) MeOH in deionized water].
The proteins were characterized by LC-MS. Deconvoluted mass spectra of proteins were generated using Compass DataAnalysis software with the Charge Deconvolution option (version 4.1, Bruker). Processing before deconvolution included smoothing using one cycle of the Savitzky Golay smoothing algorithm (0.05 Da) and spectrum baseline subtraction with the flatness parameter set at 0.8.
The concentration of GFP was determined by measuring absorbance intensity at 490 nm using an extinction coefficient of 55 000 M–1 cm–1. The concentrations of IL-1β and IL-1α were determined using the Pierce 660 nm protein assay reagent (Thermo Scientific). A NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific) was used for protein concentration determination.
Labeling of IL-1β and IL-1α with ROX-NHS
A 100 μL portion of protein (1–2 mg/mL in PBS) was reacted with 3 (IL-1β) or 6 (IL-1α) equiv of 5(6)-carboxy-X-rhodamine N-succinimidyl ester (15 mM in DMSO). Samples were incubated for 1 h at room temperature while shaken at 1500 rpm on an Eppendorf thermomixer. Excess of dye was removed using fluorescent dye removal columns (Thermo Scientific) according to the manufacturer’s instructions using Amicon Ultra 0.5 mL centrifugal filters (Millipore) with a 100 kDa cutoff instead of the supplied spin columns. The protein was eluted with PBS. The final concentration of the protein was determined using the Pierce 660 nm protein assay (Thermo Fisher Scientific). The degree of labeling was assessed by MALDI-TOF MS on a Bruker Autoflex speed MALDI-TOF instrument. As for IL-1β, approximately 50% monolabeled protein was obtained. IL-1α was labeled to a larger extent, such that in addition to monolabeled protein, also difunctionalized product was obtained (see Figure S18).
Protein Purification to Remove Excess ROX
The gel filtration Sephadex G-25 (purchased from GE Health) was used for complete removal of nonreacted fluorescent dyes from IL-1β labeling reactions. The G-25 was swollen in phosphate-buffered saline (PBS, pH 7.4) for 20 min before loading samples on the column. Labeling reaction mixture (3 equiv of ROX-NHS ester) was gently applied (400 μL) to the column without disturbing the gel bed. Once loading of the sample was complete, the column was eluted at 0.5 mL/min and 22 °C. The eluted fractions containing protein were collected, and the final concentration of the protein was determined using Pierce 660 nm protein assay (Thermo Fisher).
Fluorescence Microscopy (General)
Fluorescence microscopy images were acquired on an Olympus wide-field fluorescence IX73 inverted microscope equipped with a 4× objective (0.16 numerical aperture) and 10× objective (0.4 numerical aperture), and an X-Cite 120Q excitation light source. GFP fluorescence was detected using a filter cube with a 482/18 nm excitation filter and 520/28 nm emission filter. ROX fluorescence was detected using a filter cube with a 560/14 nm excitation filter and a 605/52 nm emission filter. Olympus CellSens Dimension software was used for quantification of the fluorescence signal.
On-Bead Binding Assay
Binding constants were determined in a microplate bead-binding assay. PEGA resins carrying β-bodies against GFP or IL-1β were evenly distributed into 96-well plates using a bead sorter, by which three beads were disposed in each well. A concentration series of ROX-labeled IL-1β (in 5 mM phosphate buffer, starting from 10 μM and diluted two times continuously in each well) or GFP (in Milli-Q, starting from 20 μM and diluted two times continuously in each well) was added to the relevant beads in 200 μL total volume. Fluorescence intensity was measured for each well after overnight incubation. The exposure time for GFP was 1 ms, and for ROX-labeled IL-1β 2 ms. The experiments were performed in triplicate. Values were plotted and fitted to a one-site specific binding curve with nonlinear regression using GraphPad Prism.
Sandwich Assays
Human recombinant IL-2 (ProSpec), human recombinant IL-6 (ProSpec), and human recombinant IL-1β (Thermo Fisher) were reconstituted in deionized water as suggested by the manufacturer. Sandwich assays for IL-1β and IL-2 were conducted in the same way: Purified labeled detection β-body and beads containing the covalently linked capture β-body were incubated in deionized water in glass vials for 3 h. Then, IL-1β or IL-2 was added to their respective mixture of capture and detection peptides, and the capture and detection of the protein was followed by microscopy. The final concentration of the protein was 50 nM. For IL-6, the labeled detection β-body and beads containing the covalently linked capture β-body were incubated in a solution of 0.1 M urea.This work was supported by aLighthouse Grant in Evolutionary Chemical Biology by University of Copenhagen.
Acknowledgments
University of Copenhagen has supported (Lighthouse grant) the present research. We thank Y. Chin (Department of Chemistry, National Tsing Hua University, Hsinchu, Taiwan) for providing the pET20-IL1A plasmid and Daniel Madsen for providing technical support. We are grateful to Charlotte Held Gotfredsen from the Technical University of Denmark for recording 800 MHz proton spectra of compounds 2 and 9.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.8b00614.
Two- and three-dimensional models of β-body–protein interactions; positive and negative control experiments; structural features of compound 2 and 9 accessed by CD; 1D- and 2D-NMR spectroscopy; sequences and mass spectrometry data of β-bodies 1–10; sequences SDS-PAGE and mass spectrometry data of GFP, IL-1β, and IL-1α; and immunogenicity measured as interleukin response of β-body 2 in Balb-C mice (PDF)
Author Contributions
The present work was designed and supervised by S.S. and M.M. M.M. designed the β-bodies. H.H. and M.L. synthesized and H.H. purified the β-bodies. H.H. performed binding studies with assistance from M.L. H.H. and S.S. expressed and purified proteins. C.K. provided GFP and cell lysates. M.W. recorded and analyzed the CD spectra. M.M., S.S., and C.K. prepared figures for the manuscript. H.H., M.M., and M.L. performed the sandwich assays. C.H.F.H. and A.K.H. tested for immunogenecity in mice. J.G., J.H., and F.D provided technical support and advice. S.S. and M.M. wrote the draft manuscript and performed the conformational studies. All authors edited the manuscript.
This work was supported by aLighthouse Grant in Evolutionary Chemical Biology by University of Copenhagen.
The authors declare the following competing financial interest(s): M.M., H.H., M.L., S.S., and F.D. are listed as coinventors in a patent application with the title Binding Peptides.
Notes
Safety statement: no unexpected or unusual high safety hazards were encountered during this work. Housing and health monitoring for mice used in immunology studies was conducted according to FELASA guidelines.
Supplementary Material
References
- Frederick K. K.; Marlow M. S.; Valentine K. G.; Wand A. J. Conformational entropy in molecular recognition by proteins. Nature 2007, 448 (7151), 325–329. 10.1038/nature05959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X.; Yang Y. P.; Dikici E.; Deo S. K.; Daunert S. Beyond Antibodies as Binding Partners: The role of antibody mimetics in bioanalysis. Annu. Rev. Anal. Chem. 2017, 10 (1), 293–320. 10.1146/annurev-anchem-061516-045205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosse R. J.; Rothe A.; Power B. E. A new generation of protein display scaffolds for molecular recognition. Protein Sci. 2006, 15 (1), 14–27. 10.1110/ps.051817606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa P.; Davey N. E.; Gibson T. J.; Babu M. M. A million peptide motifs for the molecular biologist. Mol. Cell 2014, 55 (2), 161–169. 10.1016/j.molcel.2014.05.032. [DOI] [PubMed] [Google Scholar]
- Vazquez-Lombardi R.; Phan T. G.; Zimmermann C.; Lowe D.; Jermutus L.; Christ D. Challenges and opportunities for non-antibody scaffold drugs. Drug Discovery Today 2015, 20 (10), 1271–1283. 10.1016/j.drudis.2015.09.004. [DOI] [PubMed] [Google Scholar]
- Edwards I. A.; Elliott A. G.; Kavanagh A. M.; Zuegg J.; Blaskovich M. A.; Cooper M. A. Contribution of amphipathicity and hydrophobicity to the antimicrobial activity and cytotoxicity of beta-hairpin peptides. ACS Infect. Dis. 2016, 2 (6), 442–450. 10.1021/acsinfecdis.6b00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozic M.; Fox S. J.; Thomas J. M.; Verma C. S.; Rigden D. J. Large scale ab initio modeling of structurally uncharacterized antimicrobial peptides reveals known and novel folds. Proteins: Struct., Funct., Genet. 2018, 86, 1–18. 10.1002/prot.25473. [DOI] [PubMed] [Google Scholar]
- Holland-Nell K.; Meldal M. Maintaining biological activity by using triazoles as disulfide bond mimetics. Angew. Chem., Int. Ed. 2011, 50 (22), 5204–5206. 10.1002/anie.201005846. [DOI] [PubMed] [Google Scholar]
- Doherty T.; Waring A. J.; Hong M. Peptide-lipid interactions of the beta-hairpin antimicrobial peptide tachyplesin and its linear derivatives from solid-state NMR. Biochim. Biophys. Acta, Biomembr. 2006, 1758 (9), 1285–1291. 10.1016/j.bbamem.2006.03.016. [DOI] [PubMed] [Google Scholar]
- Powers J. P.; Rozek A.; Hancock R. E. Structure-activity relationships for the beta-hairpin cationic antimicrobial peptide polyphemusin I. Biochim. Biophys. Acta, Proteins Proteomics 2004, 1698 (2), 239–250. 10.1016/j.bbapap.2003.12.009. [DOI] [PubMed] [Google Scholar]
- Robinson J. A.; Shankaramma S. C.; Jetter P.; Kienzl U.; Schwendener R. A.; Vrijbloed J. W.; Obrecht D. Properties and structure-activity studies of cyclic beta-hairpin peptidomimetics based on the cationic antimicrobial peptide protegrin I. Bioorg. Med. Chem. 2005, 13 (6), 2055–2064. 10.1016/j.bmc.2005.01.009. [DOI] [PubMed] [Google Scholar]
- DeMarco S. J.; Henze H.; Lederer A.; Moehle K.; Mukherjee R.; Romagnoli B.; Robinson J. A.; Brianza F.; Gombert F. O.; Lociuro S.; Ludin C.; Vrijbloed J. W.; Zumbrunn J.; Obrecht J. P.; Obrecht D.; Brondani V.; Hamy F.; Klimkait T. Discovery of novel, highly potent and selective beta-hairpin mimetic CXCR4 inhibitors with excellent anti-HIV activity and pharmacokinetic profiles. Bioorg. Med. Chem. 2006, 14 (24), 8396–8404. 10.1016/j.bmc.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Shenkarev Z. O.; Balandin S. V.; Trunov K. I.; Paramonov A. S.; Sukhanov S. V.; Barsukov L. I.; Arseniev A. S.; Ovchinnikova T. V. Molecular mechanism of action of beta-hairpin antimicrobial peptide arenicin: oligomeric structure in dodecylphosphocholine micelles and pore formation in planar lipid bilayers. Biochemistry 2011, 50 (28), 6255–6265. 10.1021/bi200746t. [DOI] [PubMed] [Google Scholar]
- Luther A.; Moehle K.; Chevalier E.; Dale G.; Obrecht D. Protein epitope mimetic macrocycles as biopharmaceuticals. Curr. Opin. Chem. Biol. 2017, 38, 45–51. 10.1016/j.cbpa.2017.02.004. [DOI] [PubMed] [Google Scholar]
- Silverman A. P.; Levin A. M.; Lahti J. L.; Cochran J. R. Engineered cystine-knot peptides that bind alpha(v)beta(3) integrin with antibody-like affinities. J. Mol. Biol. 2009, 385 (4), 1064–1075. 10.1016/j.jmb.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poth A. G.; Chan L. Y.; Craik D. J. Cyclotides as grafting frameworks for protein engineering and drug design applications. Biopolymers 2013, 100 (5), 480–491. 10.1002/bip.22284. [DOI] [PubMed] [Google Scholar]
- Ganz T.; Lehrer R. I. Antibiotic peptides from higher eukaryotes: biology and applications. Mol. Med. Today 1999, 5 (7), 292–297. 10.1016/S1357-4310(99)01490-2. [DOI] [PubMed] [Google Scholar]
- Ganz T. Defensins: antimicrobial peptides of vertebrates. C. R. Biol. 2004, 327 (6), 539–549. 10.1016/j.crvi.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Besenius P.; Cormack P. A.; Liu J.; Otto S.; Sanders J. K.; Sherrington D. C. Tailored polymer-supported templates in dynamic combinatorial libraries: simultaneous selection, amplification and isolation of synthetic receptors. Chem. - Eur. J. 2008, 14 (29), 9006–9019. 10.1002/chem.200800326. [DOI] [PubMed] [Google Scholar]
- Garner J.; Harding M. M. Design and synthesis of alpha-helical peptides and mimetics. Org. Biomol. Chem. 2007, 5 (22), 3577–3585. 10.1039/b710425a. [DOI] [PubMed] [Google Scholar]
- Miltschitzky S.; Koenig B. Small peptides with a beta-hairpin structure. Org. Prep. Proced. Int. 2005, 37 (4), 307–336. 10.1080/00304940509354968. [DOI] [Google Scholar]
- Singh Y.; Dolphin G. T.; Razkin J.; Dumy P. Synthetic peptide templates for molecular recognition: recent advances and applications. ChemBioChem 2006, 7 (9), 1298–1314. 10.1002/cbic.200600078. [DOI] [PubMed] [Google Scholar]
- Sela-Culang I.; Kunik V.; Ofran Y. The structural basis of antibody-antigen recognition. Front. Immunol. 2013, 4 (302), 1–13. 10.3389/fimmu.2013.00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilton E. E.; Opyr M. P.; Kailasam S.; Kothe R. F.; Wieden H. J. sdAb-DB: The Single Domain antibody database. ACS Synth. Biol. 2018, 7 (11), 2480–2484. 10.1021/acssynbio.8b00407. [DOI] [PubMed] [Google Scholar]
- Haussner C.; Lach J.; Eichler J. Synthetic antibody mimics for the inhibition of protein-ligand interactions. Curr. Opin. Chem. Biol. 2017, 40, 72–77. 10.1016/j.cbpa.2017.07.001. [DOI] [PubMed] [Google Scholar]
- Simeon R.; Chen Z. In vitro-engineered non-antibody protein therapeutics. Protein Cell 2018, 9 (1), 3–14. 10.1007/s13238-017-0386-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keefe A. D.; Pai S.; Ellington A. Aptamers as therapeutics. Nat. Rev. Drug Discovery 2010, 9 (7), 537–550. 10.1038/nrd3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygren P. A. Alternative binding proteins: affibody binding proteins developed from a small three-helix bundle scaffold. FEBS J. 2008, 275 (11), 2668–2676. 10.1111/j.1742-4658.2008.06438.x. [DOI] [PubMed] [Google Scholar]
- Hamuro Y.; Calama M. C.; Park H. S.; Hamilton A. D. A calixarene with four peptide loops: An antibody mimic for recognition of protein surfaces. Angew. Chem., Int. Ed. Engl. 1997, 36 (23), 2680–2683. 10.1002/anie.199726801. [DOI] [Google Scholar]
- Feng J.; Li Y.; Zhang W.; Shen B. Rational design of potent mimic peptide derived from monoclonal antibody: antibody mimic design. Immunol. Lett. 2005, 98 (2), 311–316. 10.1016/j.imlet.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Haussner C.; Damm D.; Nirschl S.; Rohrhofer A.; Schmidt B.; Eichler J. Peptide paratope mimics of the broadly neutralizing HIV-1 antibody b12. ChemBioChem 2017, 18 (7), 647–653. 10.1002/cbic.201600621. [DOI] [PubMed] [Google Scholar]
- Meldal M.; Hu H.; Li M.; Fischer N. H.; Schoffelen S.; Diness F.. Binding peptides. Patent PCT/EP2017/078766, 2018.
- Kinarsky L.; Suryanarayanan G.; Prakash O.; Paulsen H.; Clausen H.; Hanisch F. G.; Hollingsworth M. A.; Sherman S. Conformational studies on the MUC1 tandem repeat glycopeptides: implication for the enzymatic O-glycosylation of the mucin protein core. Glycobiology 2003, 13 (12), 929–39. 10.1093/glycob/cwg109. [DOI] [PubMed] [Google Scholar]
- Stanger H. E.; Gellman S. H. Rules for antiparallel beta-sheet design: D-Pro-Gly is superior to L-Asn-Gly for beta-hairpin nucleation. J. Am. Chem. Soc. 1998, 120 (17), 4236–4237. 10.1021/ja973704q. [DOI] [Google Scholar]
- Oldfield C. J.; Cheng Y.; Cortese M. S.; Romero P.; Uversky V. N.; Dunker A. K. Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry 2005, 44 (37), 12454–12470. 10.1021/bi050736e. [DOI] [PubMed] [Google Scholar]
- Ren X.; Gelinas A. D.; von Carlowitz I.; Janjic N.; Pyle A. M. Structural basis for IL-1alpha recognition by a modified DNA aptamer that specifically inhibits IL-1alpha signaling. Nat. Commun. 2017, 8 (1), 810–823. 10.1038/s41467-017-00864-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meldal M.; Tornoe C. W. Cu-catalyzed azide-alkyne cycloaddition. Chem. Rev. 2008, 108 (8), 2952–3015. 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- Reverdatto S.; Burz D. S.; Shekhtman A. Peptide aptamers: development and applications. Curr. Top. Med. Chem. 2015, 15 (12), 1082–1101. 10.2174/1568026615666150413153143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocker C.; Thompson D.; Matsumoto A.; Nebert D. W.; Vasiliou V. Evolutionary divergence and functions of the human interleukin (IL) gene family. Hum. Genomics 2010, 5 (1), 30–55. 10.1186/1479-7364-5-1-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters S. L.; Simon A.; Aksentijevich I.; Kastner D. L. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*). Annu. Rev. Immunol. 2009, 27, 621–668. 10.1146/annurev.immunol.25.022106.141627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello C. A.; Simon A.; van der Meer J. W. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discovery 2012, 11 (8), 633–52. 10.1038/nrd3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlanda C.; Dinarello C. A.; Mantovani A. The interleukin-1 family: back to the future. Immunity 2013, 39 (6), 1003–1018. 10.1016/j.immuni.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterba Y.; Ilowite N. Biologics in pediatric rheumatology: Quo vadis?. Curr. Rheumatol. Rep. 2016, 18 (7), 45–51. 10.1007/s11926-016-0593-9. [DOI] [PubMed] [Google Scholar]
- Arenas-Ramirez N.; Zou C.; Popp S.; Zingg D.; Brannetti B.; Wirth E.; Calzascia T.; Kovarik J.; Sommer L.; Zenke G.; Woytschak J.; Regnier C. H.; Katopodis A.; Boyman O. Improved cancer immunotherapy by a CD25-mimobody conferring selectivity to human interleukin-2. Sci. Transl. Med. 2016, 8 (367), 1–12. 10.1126/scitranslmed.aag3187. [DOI] [PubMed] [Google Scholar]
- Hodge D. R.; Hurt E. M.; Farrar W. L. The role of IL-6 and STAT3 in inflammation and cancer. Eur. J. Cancer 2005, 41 (16), 2502–2512. 10.1016/j.ejca.2005.08.016. [DOI] [PubMed] [Google Scholar]
- Vilar S.; Cozza G.; Moro S. Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8 (18), 1555–1572. 10.2174/156802608786786624. [DOI] [PubMed] [Google Scholar]
- Scholz C.; Knorr S.; Hamacher K.; Schmidt B. DOCKTITE-a highly versatile step-by-step workflow for covalent docking and virtual screening in the molecular operating environment. J. Chem. Inf. Model. 2015, 55 (2), 398–406. 10.1021/ci500681r. [DOI] [PubMed] [Google Scholar]
- Hruby V. J.; Cai M. Design of peptide and peptidomimetic ligands with novel pharmacological activity profiles. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 557–580. 10.1146/annurev-pharmtox-010510-100456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.; Hoeck C.; Schoffelen S.; Gotfredsen C. H.; Meldal M. Specific electrostatic molecular recognition in water. Chem. - Eur. J. 2016, 22 (21), 7206–7214. 10.1002/chem.201600231. [DOI] [PubMed] [Google Scholar]
- Ganesh Kumar M.; Mali S. M.; Raja K. M.; Gopi H. N. Design of stable beta-hairpin mimetics through backbone disulfide bonds. Org. Lett. 2015, 17 (2), 230–233. 10.1021/ol503310r. [DOI] [PubMed] [Google Scholar]
- Celentano V.; Diana D.; Di Salvo C.; De Rosa L.; Romanelli A.; Fattorusso R.; D’Andrea L. D. 1,2,3-Triazole bridge as conformational constrain in beta-hairpin peptides: Analysis of hydrogen-bonded positions. Chem. - Eur. J. 2016, 22 (16), 5534–5537. 10.1002/chem.201600154. [DOI] [PubMed] [Google Scholar]
- Tornøe C. W.; Christensen C.; Meldal M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67 (9), 3057–3064. 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- Meldal M.; Tornøe C. W.. Copper(I)-catalyzed 1,3-dipolar cycloadditions on solid-phase. In Peptides: The Wave of the Future; Springer: Dordrecht, 2001; Vol. 7, pp 263–264. [Google Scholar]
- Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective ″ligation″ of azides and terminal alkynes. Angew. Chem., Int. Ed. 2002, 41 (14), 2596–2599. . [DOI] [PubMed] [Google Scholar]
- Hu H.; Nikitin S. V.; Berthelsen A. B.; Diness F.; Schoffelen S.; Meldal M. Sustainable flow synthesis of encoded beads for combinatorial chemistry and chemical biology. ACS Comb. Sci. 2018, 20, 492–498. 10.1021/acscombsci.8b00052. [DOI] [PubMed] [Google Scholar]
- Auzanneau F. I.; Meldal M.; Bock K. Synthesis, characterization and biocompatibility of PEGA resins. J. Pept. Sci. 1995, 1 (1), 31–44. 10.1002/psc.310010106. [DOI] [PubMed] [Google Scholar]
- Vutti S.; Schoffelen S.; Bolinsson J.; Buch-Manson N.; Bovet N.; Nygard J.; Martinez K. L.; Meldal M. Click chemistry mediated functionalization of vertical nanowires for biological applications. Chem. - Eur. J. 2016, 22 (2), 496–500. 10.1002/chem.201504540. [DOI] [PubMed] [Google Scholar]
- Chang H. K.; Mohan S. K.; Chin Y. 1H, 13C and 15N backbone and side chain resonance assignments of human interleukin 1 alpha. Biomol. NMR Assignments 2010, 4 (1), 59–60. 10.1007/s12104-010-9207-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.