

Abstract
Pancreatic cancer is one of the most aggressive human cancers and is associated with a poor prognosis. To develop a novel strategy for pancreatic cancer treatment, it is essential to elucidate the molecular mechanisms underlying the invasion and proliferation of cancer cells. ATPase family AAA domain containing protein 2 (ATAD2) is a highly conserved protein with an AAA+ domain and a bromodomain. Accumulating studies have demonstrated that ATAD2 is associated with the progression of multiple cancers. The present study demonstrated that ATAD2 depletion suppressed cell invasion and migration. In addition, ATAD2 knockdown suppressed anchorage-independent growth of pancreatic cancer cells. Finally, ATAD2 depletion was demonstrated to sensitize pancreatic cancer cells to gemcitabine. The results of the present study indicate that ATAD2 is involved in the malignant characteristics of pancreatic cancer.
Keywords: ATAD2, AAA+, pancreatic cancer, gemcitabine
Introduction
Pancreatic cancer is one of the most aggressive human cancers and is a leading cause of cancer-associated mortality. Surgical resection is the most effective treatment; however, tumors have already advanced locally or metastasized to distant organs at the time of diagnosis in most cases (1,2). Gemcitabine is the current standard treatment for advanced or resected pancreatic cancer (3). However, as a large number of patients are resistant to gemcitabine treatment, the drug provides only modest survival benefits. To develop novel therapeutic strategies for pancreatic cancer, it is necessary to gain further insight into the molecular mechanisms supporting the malignant characteristics of pancreatic cancer.
ATPase family AAA domain containing protein 2 (ATAD2), which is also known as AAA+ nuclear co-regulator cancer associated (ANCCA) is highly conserved in a wide range of species and is associated with multiple biological processes, including cell proliferation, migration and invasion (4). ATAD2 has an AAA+ domain in the central region and a bromodomain in the C-terminal region (5). AAA+ domains mediate protein multimerization and promote ATP hydrolysis to produce chemical energy, which subsequently induces conformational changes of substrate proteins to regulate their functions (6,7). Bromodomains are small helical interacting modules that bind to acetylated lysines in histones or other proteins (8). ATAD2 has been demonstrated to regulate gene expression or chromatin modification in cooperation with multiple transcription factors (9–13). Accumulating studies have demonstrated that ATAD2 is associated with the progression of numerous cancers. For example, high expression of ATAD2 is a strong predictor of poor prognosis in breast cancer (14,15). ATAD2 promotes proliferation and survival of breast cancer cells by regulating the expression of B-Myb, enhancer of zeste 2 polycomb repressive complex 2 subunit or nuclear receptor coactivator 3 (16). High expression of ATAD2 has also been reported in endometrial carcinoma, hepatocellular carcinoma, cervical cancer and lung cancer (17–28). These studies clearly demonstrate that ATAD2 is critical for cancer progression and is a promising target for cancer therapy. The present study examined whether ATAD2 was involved in the progression of pancreatic cancer, and demonstrated that ATAD2 is important for survival, invasion and drug resistance of pancreatic cancer cells.
Materials and methods
Cells and antibodies
The pancreatic cancer cell lines, KP4, PK9, MIAPaCa-2, PK8, RI151, PANC1 and KML1 were cultured in RPMI medium (Wako, Osaka, Japan) with 10% fetal bovine serum (FBS) (Equitec, Hendra, Australia) and antibiotics (penicillin, 50 IU/ml, streptomycine, 50 µg/ml) at 37°C with 5% carbon dioxide (CO2). The cell lines were obtained from RIKEN BioResource Center (Tsukuba, Japan). ATAD2 (AMAB90541) and β-actin (A5316) antibodies were obtained from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany), and cleaved poly (ADP ribose) polymerase antibody (9541) was obtained from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Western blotting
Cells were lysed with Laemmli sample buffer (20% glycerol, 135 mM Tris-HCl, pH 6.8, 4% SDS, 10% 2-Mercaptoethanol, and 0.003% bromophenol blue) and boiled for 5 min. The protein concentrations of the lysates were measured using the RC-DC Protein Assay (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Equal protein quantities (30 µg) were separated by SDS-PAGE (10%) and transferred to PVDF membranes (Merck KGaA). The membranes were blocked with 0.5% non-fat skim milk at 37°C for 30 min, incubated with each primary antibody (1,000 dilution) at room temperature for 1 h, washed with TBS-T buffer (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, and 0.05% Tween-20) and then incubated with HRP-conjugated anti-mouse IgG (31430) or anti-rabbit IgG (31460) secondary antibodies (Thermo Fisher Scientific, Inc., Waltham, MA, USA) in 3,000 dilution at room temperature for 1 h. The proteins were visualized using enhanced chemiluminescence (GE Healthcare Life Sciences, Uppsala, Sweden).
Small interfering RNA (siRNA) transfection
siRNAs were obtained from Sigma-Aldrich; Merck KGaA. The sequences of siRNAs for ATAD2 were as follows: ATAD2-1, 5′-CCAGCUGUCAUUCAUGCUUTT-3′ and ATAD2-2, 5′-GCCUACACCCUCACUUGUUTT-3′. The sequence of the control siRNA targeting luciferase was 5′-CUUACGCUGAGUACUUCGATT-3′. Cells were transfected with 20 nM siRNA using Lipofectamine RNAiMAX (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol.
Proliferation assay
Cells (1,000 cells/well) were cultured in 96-well plates 24 h following transfection with siRNAs, and the number of viable cells at 24, 48 and 72 h were evaluated using a Cell Counting Kit-8 assay, according to manufacturer's protocol (Dojindo Molecular Technologies, Inc., Kumamoto, Japan). To evaluate the proliferation of siRNA-transfected cells in the presence of gemcitabine, siRNA-transfected cells (1,000 cell/well) were cultured in triplicate in 96-well plates with various concentrations of gemcitabine (0.05, 0.1, 0.2, 0.4, 0.6, 0.8 and 1.0 µM) at 37°C for 48 h. Gemcitabine was obtained from Wako (Osaka, Japan). The number of viable cells was evaluated using Cell Counting Kit-8 assays 3 days following this.
TUNEL assay
Cells were fixed with 4% paraformaldehyde and subjected to TUNEL assays using the In Situ Cell Death Detection kit and fluorescein (Roche Diagnostics, Basel, Switzerland) 72 h following transfection with siRNAs, according to the manufacturer's protocol. Cells in five randomly selected fields were evaluated using fluorescent microscope (BX60; Olympus, Tokyo, Japan) at ×100 magnification, and three independent experiments were performed.
Migration and invasion assay
Cell migration and invasion were measured using Boyden chambers (8 µm pore size and 6.5 mm membrane diameter; Corning Incorporated, Corning, NY, USA). To evaluate cell migration, 72 h following transfection with siRNAs, cells (4×104) were placed on the upper surface of a filter coated with fibronectin and then allowed to migrate to the bottom surface. Upper chamber and lower chamber were filled with RPMI medium without serum. Cells were fixed with 70% ethanol and stained with 0.5% crystal violet 6 h subsequent to this. Cells that migrated to the lower surface of the filters were quantified in five randomly selected fields using a microscope (BX60; Olympus) at ×40 magnification, three independent experiments were performed. To evaluate cell invasion, cells were placed on the upper surface of a filter coated with Matrigel (BD Bioscience, San Jose, CA, USA) 72 h following transfection with siRNAs. The upper chamber was filled with RPMI and the lower chamber was filled with RPMI supplemented with 10% FBS. Cells were fixed with 70 % ethanol and stained 24 h subsequent to this to count cells that had invaded to the lower surface of the filter.
Colony formation assay
Cells were transfected with siRNAs, and 24 h subsequent to this, cells (1×104) were mixed with 0.36% agar in RPMI medium supplemented with 10% FBS, and overlaid onto a 0.72% agarose layer in 6-well plates. Following incubation at 37°C for 2 weeks, colonies in five randomly selected fields were counted using a microscope (BX60; Olympus). Three independent experiments were performed.
Statistical analysis
The data were expressed as the mean ± standard deviation. Comparisons between the groups were performed using unpaired Student's t-tests using Excel software (Microsoft Corporation, Redmond, WA, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
ATAD2 knockdown promotes apoptosis
To determine the involvement of ATAD2 in pancreatic cancer progression, the expression of ATAD2 was examined in KP4, PK9, MIAPaCa-2, PK8, RI151, PANC1 and KML1 pancreatic cancer cell lines. ATAD2 was expressed at similar levels in multiple pancreatic cancer cell lines (Fig. 1A). The KP4 and PK9 cell lines were selected for further analysis. Transfection of two different siRNAs sufficiently reduced the level of expression of ATAD2 in KP4 and PK9 cell lines (Fig. 1B). Depletion of ATAD2 significantly reduced the proliferation of either KP4 or PK9 cells (Fig. 1C). To determine whether reduced proliferation resulted from an increase in apoptotic cells, cells transfected with siRNAs were stained for DNA breaks using the TUNEL assay. ATAD2 knockdown promoted apoptosis of both cell lines (Fig. 1D).
Figure 1.

Depletion of ATAD2 induces apoptosis in pancreatic cancer cells. (A) Expression of ATAD2 in pancreatic cancer cell lines was examined by western blotting. (B) KP4 and PK9 cells were transfected with siRNAs and, 72 h later, cells were lysed to undergo western blotting. (C) Cells were transfected with siRNAs, and the number of viable cells at the indicated time points was evaluated using Cell Counting Kit-8 assays. (D) Cells were transfected with siRNAs and, 72 h later, cells were subjected to TUNEL assays. The graph depicts the percentage of TUNEL-positive cells. Three independent experiments were performed, and the data are expressed as the mean ± standard deviation. *P<0.05 vs. Ctrl. ATAD2, ATPase family AAA domain containing protein 2; siRNA/si, small interfering RNA; Ctrl, control.
ATAD2 knockdown suppresses cell migration and invasion
Cell migration and invasion of ATAD2-depleted cells was examined using a modified Boyden chamber. KP4 and PK9 cells were transfected with siRNAs and, 72 h following this, cells were suspended and placed in the upper chambers of the filters. The cells were allowed to migrate to the bottom surface of the filter, which was coated with fibronectin. The migrated cells were counted 6 h subsequent to this to evaluate cell migration. The migration of KP4 and PK9 cells was decreased following ATAD2 knockdown compared with cells transfected with control siRNA (Fig. 2A). To determine cell invasive ability, Matrigel-coated Boyden chambers were used. ATAD2 depletion significantly suppressed the invasion of KP4 and PK9 cells compared with cells transfected with control siRNA (Fig. 2B). These results demonstrated that ATAD2 was associated with the migration and invasion of pancreatic cancer cells.
Figure 2.

ATAD2 knockdown suppresses cell migration and invasion. (A) Representative images and the average number of migrated cells per field, as determined by migration assay. (B) Representative images and the average number of invaded cells per field, as determined by invasion assay. Three independent experiments were performed, and the data are expressed as the mean ± standard deviation. *P<0.05 vs. Ctrl. ATAD2, ATPase family AAA domain containing protein 2; siRNA, small interfering RNA; Ctrl, control.
ATAD2 depletion suppresses anchorage-independent growth
One of the malignant characteristics of cancer cells is the ability to grow in the absence of cell adhesion to the extracellular matrix, known as anchorage-independent growth. To determine the ability of cells to grow in the absence of cell adhesion, siRNA-transfected cells were cultured in soft agar for two weeks, and then the formation of colonies was examined. ATAD2 depletion significantly suppressed anchorage-independent growth of KP4 and PK9 cells compared with cells transfected with control siRNA (Fig. 3).
Figure 3.
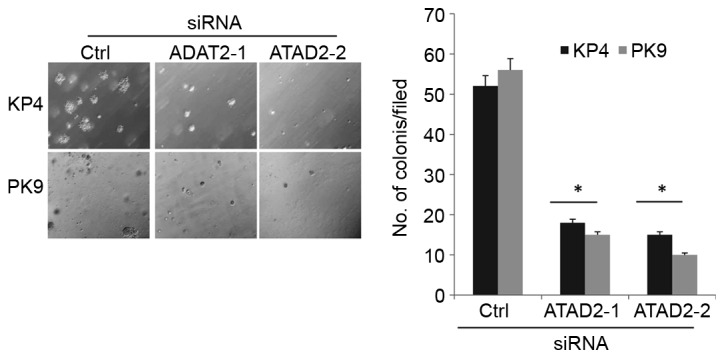
ATAD2 knockdown suppresses anchorage-independent growth. siRNA-transfected cells were cultured in soft agar for 2 weeks. Representative images and the average number of cells per field are depicted. Three independent experiments were performed, and the data are expressed as the mean ± standard deviation. *P<0.05 vs. Ctrl. ATAD2, ATPase family AAA domain containing protein 2; siRNA, small interfering RNA; Ctrl, control.
ATAD2 depletion sensitizes cells to gemcitabine
To determine whether ATAD2 knockdown sensitized pancreatic cancer cells to gemcitabine, which is a nucleoside analog frequently used for pancreatic cancer treatment, siRNA-transfected cells were cultured in the presence of 0–1.0 µM gemcitabine and cell growth was assessed. ATAD2 siRNA-transfected cells were more sensitive to gemcitabine than control siRNA-transfected cells (Fig. 4A). To further confirm this result, TUNEL assays were performed. siRNA-transfected cells were cultured in the presence of 1.0 µM gemcitabine, and the apoptotic cells were examined. More apoptotic cells were observed among the ATAD2-depleted cells than the control cells (Fig. 4B). These results indicated that ATAD2 knockdown sensitized pancreatic cancer cells to gemcitabine.
Figure 4.
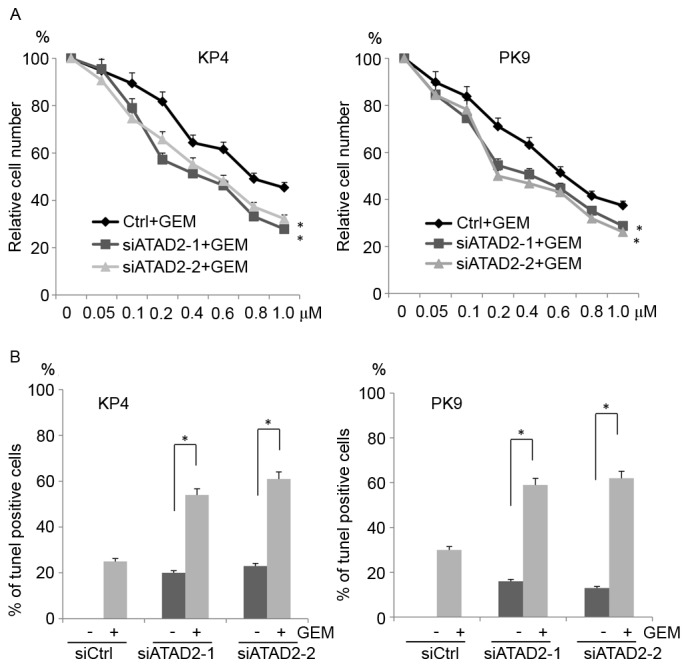
Depletion of ATAD2 sensitizes cells to GEM. (A) siRNA-transfected cells were cultured in the presence of different concentrations of GEM, and the proliferation ratio was examined. The data are presented as the percent growth of cells treated without gemcitabine (B) siRNA-transfected cells were cultured in the presence of 1 µM GEM for 72 h and then subjected to TUNEL assays. The graph depicts the percentage of apoptotic cells from three independent experiments. The data are expressed as the mean ± standard deviation. *P<0.05 vs. Ctrl. ATAD2, ATPase family AAA domain containing protein 2; GEM, gemcitabine; siRNA/si, small interfering RNA; Ctrl, control.
Discussion
The present study provided evidence that ATAD2 is associated with the progression of pancreatic cancer. Western blotting analysis demonstrated that ATAD2 was expressed in all the pancreatic cancer cell lines that were examined. To determine the function of ATAD2 in the malignant characteristics of pancreatic cancer cells, two siRNAs that targeted different regions of the ATAD2 mRNA were used, to exclude the possibility of off-target effects. These siRNAs sufficiently decreased ATAD2 expression. Depletion of ATAD2 by these siRNAs significantly reduced invasion and migration in KP4 and PK9 cells. In addition, ATAD2 knockdown suppressed anchorage-independent growth of pancreatic cancer cells. These results indicated that ATAD2 is associated with the malignancy of pancreatic cancer.
ATAD2 suppression has previously been indicated to facilitate P53 and P38-dependent apoptosis of hepatocellular carcinoma cells (27). ATAD2 is also essential for the survival and proliferation of breast cancer cells. ATAD2 was required for the expression of members of the kinesin families, including kinesin family member (KIF) 4A, KIF20A, and KIF23, that are essential for the progression of mitosis (15). As a result, depletion of ATAD2 induced apoptosis of drug-sensitive and drug-resistant cancer cells (15). ATAD2 depletion was demonstrated to significantly induce apoptosis of pancreatic cancer cells in the present study. These results indicate that ATAD2 is essential for the survival of multiple cancer cells.
ATAD2 depletion increased the sensitivity of pancreatic cancer cells to gemcitabine. Although gemcitabine is often used for pancreatic cancer treatment, its effect is far from satisfactory. Accumulating studies have demonstrated that chemical inhibitors for AAA+ proteins are useful for cancer treatment (29–31). p97, a member of the AAA+ family of proteins, is essential for the regulation of protein homeostasis. Inhibition of p97 by small chemicals induced unfolded protein responses and promoted apoptosis in multiple cancers (32). Thus, chemical inhibitors that disrupt the enzymatic activity of ATAD2 may be useful for the treatment of pancreatic cancer in combination with gemcitabine. In addition to the AAA+ domain, ATAD2 has a conserved domain called the bromodomain, which is essential for the binding of ATAD2 to acetylated histone. Previous studies have reported that certain chemicals specifically inhibit the bromodomain of ATAD2, further demonstrating that ATAD2 may be a promising target for the development of chemical inhibitors (33,34). The results of the present study and other studies indicate that combination therapy of gemcitabine and ATAD2 inhibitors may be a potential therapeutic strategy for the treatment of pancreatic cancers.
Acknowledgements
The authors would like to thank the members of the Division of Cancer Biology, Nagoya University Graduate School of Medicine (Nagoya, Japan) for their helpful discussions and technical assistance. The present study was funded by a grant from the Naito Foundation.
References
- 1.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 3.Berlin J, Benson AB., III Chemotherapy: Gemcitabine remains the standard of care for pancreatic cancer. Nat Rev Clin Oncol. 2010;7:135–137. doi: 10.1038/nrclinonc.2010.16. [DOI] [PubMed] [Google Scholar]
- 4.Boussouar F, Jamshidikia M, Morozumi Y, Rousseaux S, Khochbin S. Malignant genome reprogramming by ATAD2. Biochim Biophys Acta. 2103;1829:1010–1014. doi: 10.1016/j.bbagrm.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 5.Cattaneo M, Morozumi Y, Perazza D, Boussouar F, Jamshidikia M, Rousseaux S, Verdel A, Khochbin S. Lessons from yeast on emerging roles of the ATAD2 protein family in gene regulation and genome organization. Mol Cells. 2014;37:851–856. doi: 10.14348/molcells.2014.0258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ogura T, Wilkinson AJ. AAA+ superfamily ATPases: Common structure-diverse function. Genes Cells. 2001;6:575–597. doi: 10.1046/j.1365-2443.2001.00447.x. [DOI] [PubMed] [Google Scholar]
- 7.Hanson PI, Whiteheart SW. AAA+ proteins: Have engine, will work. Nat Rev Mol Cell Biol. 2005;6:519–529. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 8.Ntranos A, Casaccia P. Bromodomains: Translating the words of lysine acetylation into myelin injury and repair. Neurosci Lett. 2016;625:4–10. doi: 10.1016/j.neulet.2015.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zou JX, Guo L, Revenko AS, Tepper CG, Gemo AT, Kung HJ, Chen HW. Androgen-induced coactivator ANCCA mediates specific androgen receptor signaling in prostate cancer. Cancer Res. 2009;69:3339–3346. doi: 10.1158/0008-5472.CAN-08-3440. [DOI] [PubMed] [Google Scholar]
- 10.Ciró M, Prosperini E, Quarto M, Grazini U, Walfridsson J, McBlane F, Nucifero P, Pacchiana G, Capra M, Christensen J, Helin K. ATAD2 is a novel cofactor for MYC, overexpressed and amplified in aggressive tumors. Cancer Res. 2009;69:8491–8498. doi: 10.1158/0008-5472.CAN-09-2131. [DOI] [PubMed] [Google Scholar]
- 11.Hsia EY, Kalashnikova EV, Revenko AS, Zou JX, Borowsky AD, Chen HW. Deregulated E2F and the AAA+ coregulator ANCCA drive proto-oncogene ACTR/AIB1 overexpression in breast cancer. Mol Cancer Res. 2010;8:183–193. doi: 10.1158/1541-7786.MCR-09-0095. [DOI] [PubMed] [Google Scholar]
- 12.Revenko AS, Kalashnikova EV, Gemo AT, Zou JX, Chen HW. Chromatin loading of E2F-MLL complex by cancer-associated coregulator ANCCA via reading a specific histone mark. Mol Cell Biol. 2010;30:5260–5272. doi: 10.1128/MCB.00484-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duan Z, Zou JX, Yang P, Wang Y, Borowsky AD, Gao AC, Chen HW. Developmental and androgenic regulation of chromatin regulators EZH2 and ANCCA/ATAD2 in the prostate Via MLL histone methylase complex. Prostate. 2013;73:455–466. doi: 10.1002/pros.22587. [DOI] [PubMed] [Google Scholar]
- 14.Caron C, Lestrat C, Marsal S, Escoffier E, Curtet S, Virolle V, Barbry P, Debernardi A, Brambilla C, Brambilla E, et al. Functional characterization of ATAD2 as a new cancer/testis factor and a predictor of poor prognosis in breast and lung cancers. Oncogene. 2010;29:5171–5181. doi: 10.1038/onc.2010.259. [DOI] [PubMed] [Google Scholar]
- 15.Zou JX, Duan Z, Wang J, Sokolov A, Xu J, Chen CZ, Li JJ, Chen HW. Kinesin family deregulation coordinated by bromodomain protein ANCCA and histone methyltransferase MLL for breast cancer cell growth, survival, and tamoxifen resistance. Mol Cancer Res. 2014;12:539–549. doi: 10.1158/1541-7786.MCR-13-0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kalashnikova EV, Revenko AS, Gemo AT, Andrews NP, Tepper CG, Zou JX, Cardiff RD, Borowsky AD, Chen HW. ANCCA/ATAD2 overexpression identifies breast cancer patients with poor prognosis, acting to drive proliferation and survival of triple-negative cells through control of B-Myb and EZH2. Cancer Res. 2010;70:9402–9412. doi: 10.1158/0008-5472.CAN-10-1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fouret R, Laffaire J, Hofman P, Beau-Faller M, Mazieres J, Validire P, Girard P, Camilleri-Bröet S, Vaylet F, Leroy-Ladurie F, et al. A comparative and integrative approach identifies ATPase family, AAA domain containing 2 as a likely driver of cell proliferation in lung adenocarcinoma. Clin Cancer Res. 2012;18:5606–5616. doi: 10.1158/1078-0432.CCR-12-0505. [DOI] [PubMed] [Google Scholar]
- 18.Raeder MB, Birkeland E, Trovik J, Krakstad C, Shehata S, Schumacher S, Zack TI, Krohn A, Werner HM, Moody SE, et al. Integrated genomic analysis of the 8q24 amplification in endometrial cancers identifies ATAD2 as essential to MYC-dependent cancers. PLoS One. 2013;8:e54873. doi: 10.1371/journal.pone.0054873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, Sun Y, Li Y, Fang Z, Wang R, Pan Y, Hu H, Luo X, Ye T, Li H, et al. ANCCA protein expression is a novel independent poor prognostic marker in surgically resected lung adenocarcinoma. Ann Surg Oncol. 2013;20(Suppl 3):S577–S582. doi: 10.1245/s10434-013-3027-1. [DOI] [PubMed] [Google Scholar]
- 20.Wu G, Liu H, He H, Wang Y, Lu X, Yu Y, Xia S, Meng X, Liu Y. miR-372 down-regulates the oncogene ATAD2 to influence hepatocellular carcinoma proliferation and metastasis. BMC Cancer. 2014;14:107. doi: 10.1186/1471-2407-14-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wan WN, Zhang YX, Wang XM, Liu YJ, Zhang YQ, Que YH, Zhao WJ. ATAD2 is highly expressed in ovarian carcinomas and indicates poor prognosis. Asian Pac J Cancer Prev. 2014;15:2777–2783. doi: 10.7314/APJCP.2014.15.6.2777. [DOI] [PubMed] [Google Scholar]
- 22.Wu G, Lu X, Wang Y, He H, Meng X, Xia S, Zhen K, Liu Y. Epigenetic high regulation of ATAD2 regulates the Hh pathway in human hepatocellular carcinoma. Int J Oncol. 2014;45:351–361. doi: 10.3892/ijo.2014.2416. [DOI] [PubMed] [Google Scholar]
- 23.Hwang HW, Ha SY, Bang H, Park CK. ATAD2 as a Poor prognostic marker for hepatocellular carcinoma after curative resection. Cancer Res Treat. 2015;47:853–861. doi: 10.4143/crt.2014.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng L, Li T, Zhang Y, Guo Y, Yao J, Dou L, Guo K. Oncogene ATAD2 promotes cell proliferation, invasion and migration in cervical cancer. Oncol Rep. 2015;33:2337–2344. doi: 10.3892/or.2015.3867. [DOI] [PubMed] [Google Scholar]
- 25.Shang P, Meng F, Liu Y, Chen X. Overexpression of ANCCA/ATAD2 in endometrial carcinoma and its correlation with tumor progression and poor prognosis. Tumour Biol. 2015;36:4479–4485. doi: 10.1007/s13277-015-3089-8. [DOI] [PubMed] [Google Scholar]
- 26.Krakstad C, Tangen IL, Hoivik EA, Halle MK, Berg A, Werner HM, Ræder MB, Kusonmano K, Zou JX, Øyan AM, et al. ATAD2 overexpression links to enrichment of B-MYB-translational signatures and development of aggressive endometrial carcinoma. Oncotarget. 2015;6:28440–28452. doi: 10.18632/oncotarget.4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu WJ, Chua MS, So SK. Suppression of ATAD2 inhibits hepatocellular carcinoma progression through activation of p53- and p38-mediated apoptotic signaling. Oncotarget. 2015;6:41722. doi: 10.18632/oncotarget.6152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang M, Zhang C, Du W, Yang X, Chen Z. ATAD2 is overexpressed in gastric cancer and serves as an independent poor prognostic biomarker. Clin Transl Oncol. 2016;18:776–781. doi: 10.1007/s12094-015-1430-8. [DOI] [PubMed] [Google Scholar]
- 29.Chou TF, Brown SJ, Minond D, Nordin BE, Li K, Jones AC, Chase P, Porubsky PR, Stoltz BM, Schoenen FJ, et al. Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc Natl Acad Sci USA. 2011;108:4834–4849. doi: 10.1073/pnas.1015312108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chou TF, Li K, Frankowski KJ, Schoenen FJ, Deshaies RJ. Structure-activity relationship study reveals ML240 and ML241 as potent and selective inhibitors of p97 ATPase. Chem Med Chem. 2013;8:297–312. doi: 10.1002/cmdc.201200520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Magnaghi P, D'Alessio R, Valsasina B, Avanzi N, Rizzi S, Asa D, Gasparri F, Cozzi L, Cucchi U, Orrenius C, et al. Covalent and allosteric inhibitors of the ATPase VCP/p97induce cancer cell death. Nat Chem Biol. 2013;9:548–556. doi: 10.1038/nchembio.1313. [DOI] [PubMed] [Google Scholar]
- 32.Anderson DJ, Le Moigne R, Djakovic S, Kumar B, Rice J, Wong S, Wang J, Yao B, Valle E, Kiss von Soly S, et al. Targeting the AAA ATPase p97 as an approach to treat cancer through disruption of protein homeostasis. Cancer Cell. 2015;28:653–665. doi: 10.1016/j.ccell.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Demont EH, Chung CW, Furze RC, Grandi P, Michon AM, Wellaway C, Barrett N, Bridges AM, Craggs PD, Diallo H, et al. Fragment-Based Discovery of Low-Micromolar ATAD2Bromodomain Inhibitors. J Med Chem. 2015;58:5649–5673. doi: 10.1021/acs.jmedchem.5b00772. [DOI] [PubMed] [Google Scholar]
- 34.Bamborough P, Chung CW, Furze RC, Grandi P, Michon AM, Sheppard RJ, Barnett H, Diallo H, Dixon DP, Douault C, et al. Structure-based optimization of naphthyridones into PotentATAD2 bromodomain inhibitors. J Med Chem. 2015;58:6151–6178. doi: 10.1021/acs.jmedchem.5b00773. [DOI] [PubMed] [Google Scholar]