

Abstract
Despite the high frequency of KRAS mutations in non-small cell lung cancer (NSCLC), therapeutic modalities targeting KRAS-mutated NSCLC have not been established. Based on our previous findings that mutant KRAS knockdown sensitized NSCLC cells to a p38 inhibitor, the growth-inhibitory effect of dual MEK and p38 inhibition on tumor growth in NSCLC cells harboring KRAS mutations was investigated. In KRAS-mutated NSCLC cells, the MEK inhibitor, selumetinib, inhibited cell growth in a dose-dependent manner, and its growth-inhibitory effect was enhanced by combined treatment with the p38 inhibitor LY2228820. Similarly, another pair of MEK and p38 inhibitors also exhibited antitumor activity. Small interfering RNAs (siRNAs) against MAPK14, which encodes p38α MAPK, enhanced the growth-inhibitory effect of the MEK inhibitors in NSCLC cells with KRAS mutations. Notably, MEK inhibitors reduced p38 expression levels but increased p38 phosphorylation levels, resulting in sensitization to p38 inhibitors in KRAS-mutated NSCLC cells. These results provide evidence that dual MEK and p38 inhibition could be a potent therapeutic strategy against oncogenic KRAS-driven NSCLC.
Keywords: non-small cell lung cancer, v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog, mitogen activated protein kinase, mitogen-activated protein kinase kinase, p38
Introduction
Lung cancer is a commonly diagnosed cancer, and it is the leading cause of cancer deaths worldwide (1). Lung cancer is divided into two major histological types: Small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). NSCLC represents 80–85% of all lung cancers, and the most common subtype of NSCLC is adenocarcinoma (2). Recent advances in the molecular biology of lung cancer have revealed many genetic and epigenetic alterations, and some of them have been found to be druggable oncogenic drivers, such as EGFR mutations, BRAF V600E mutations and fusions of ALK, ROS1 or RET (3,4). The proto-oncogene KRAS is one of the common driver mutations in lung adenocarcinoma, and it is mutated in approximately 25% of these patients (5). KRAS encodes a small GTP-binding protein that is involved in many cellular processes by regulating multiple signaling cascades (6). Wild-type KRAS has intrinsic GTPase activity because it catalyzes the hydrolysis of GTP bound to GDP; however, KRAS mutations impair GTPase activity, resulting in the dysregulation of its downstream pathways and effectors when it is in the GTP-bound form. Given that meta-analyses have shown that KRAS mutations are associated with an unfavorable prognosis in patients with NSCLC (7,8), targeting oncogenic KRAS-driven NSCLC is highly important. Moreover, there are no clinical trials indicating therapeutic efficacy against KRAS-mutated NSCLC (9). For instance, a randomized phase II study indicated that compared with docetaxel, the mitogen-activated protein kinase kinase (MEK) inhibitor trametinib did not improve progression-free survival for previously treated patients with KRAS-mutated NSCLC (10). Additionally, a phase III study of the MEK inhibitor selumetinib plus docetaxel did not show preferred clinical activity compared with docetaxel alone in NSCLC patients with KRAS mutations (11). Thus, the KRAS mutation remains undruggable, and developing therapeutic strategies against oncogenic KRAS-driven NSCLC is urgently needed.
We previously assessed the growth-inhibitory effect of short hairpin RNA (shRNA)-mediated knockdown of mutant KRAS in combination with various molecular inhibitors; we found that mutant KRAS knockdown sensitized NSCLC cells to a p38 inhibitor (12). In the current study, we adopted MEK inhibitors as alternatives to mutant KRAS knockdown in combination with p38 inhibitors to evaluate the impact of dual MEK and p38 inhibition on the tumor growth of KRAS-mutated NSCLC cells.
Materials and methods
Cell lines and reagents
KRAS mutant NSCLC cell lines NCI-H23, NCI-H157, NCI-H460 and NCI-H1792 were kindly provided by Drs John D. Minna and Adi F. Gazdar of the University of Texas Southwestern Medical Center at Dallas. The cancer cells were cultured in RPMI-1640 medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 5% fetal bovine serum. The reagents selumetinib (Selleck Chemicals, Houston, TX, USA), LY2228820 (Selleck Chemicals), PD0325901 (Sigma-Aldrich), and p38 MAP Kinase Inhibitor V (Calbiochem, San Diego, CA, USA) were purchased from commercial suppliers.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
The mRNA expression levels of MAPK14 and GAPDH were determined by real-time RT-PCR as previously described (13). TaqMan probe and primer sets for these genes were purchased from Applied Biosystems (Carlsbad, CA, USA). Total RNA was extracted using an RNeasy mini kit (QIAGEN, Valencia, CA, USA), and cDNA was synthesized using 2 µg of total RNA with Superscript VILO MasterMix (Invitrogen, Carlsbad, CA, USA) and the oligo (dT) primer system (Invitrogen). qPCR was performed using a LightCycler 480 system (Roche Diagnostics, Tokyo, Japan). For quantitative analysis, the GAPDH gene was used as an internal reference gene to normalize the input cDNA. The comparative Ct method was used to compute the relative expression values.
Use of synthetic small interfering RNA
siRNAs targeting MAPK14 were obtained from the siGENOME library (Dharmacon Inc., Lafayette, CO, USA). An siRNA against Tax was used as a non-targeting control as previously described (13). The cells were transfected with 10 nM siRNA using Lipofectamine RNAiMAX transfection reagent (Invitrogen) according to the manufacturer's protocol. After 48 h, the cells were harvested to verify target gene silencing.
Cell proliferation/viability assays
Eighteen h after plating 1.5×105 trypan-negative cells per well on 6-well plates, the cells were treated with the inhibitors or DMSO alone. After 24, 48 and 72 h, trypan-negative cells were counted by a TC10 Automated Cell Counter (Bio-Rad, Richmond, CA, USA). In addition, 18 h after plating 5,000 trypan-negative cells per well on 96-well plates, these cells were treated with the inhibitors or DMSO alone. After 48 or 72 h, the cell viabilities were evaluated by a CellTiter-Glo luminescent cell viability assay (Promega, Madison, WI, USA).
Colony formation assay
Colony formation assays were performed as described previously (12). Briefly, 24 h after siRNA transfection, the cells were harvested, and 1,000 trypan blue-negative cells were then replated for colony formation in liquid culture. After 24 h, the cells were treated with the inhibitors or DMSO alone. The culture media with the inhibitors was exchanged every 3 days during culture, and the colonies were stained with methylene blue 14 days after the initial treatment.
DNA fragment detection by ELISA
After plating in 96-well plates in replicates of 6, 10,000 trypan blue-negative cells were treated with the inhibitors or DMSO alone. Forty-eight h after the treatment, the cells were assayed by the cytoplasmic histone-associated DNA fragment method using a Cell Death Detection ELISA Plus Kit (Roche Diagnostics, Tokyo, Japan) according to the manufacturer's protocol.
Apoptotic cell detection by Annexin V-fluorescein staining
Four days after siRNA transfection, the cells were double-stained using an Annexin V-FLUOS kit (Roche Diagnostics) and Hoechst 33342 solution (Molecular Probes, Eugene, OR, USA) as previously described (13). The stained cells were viewed immediately using a fluorescence microscope (Keyence, Osaka, Japan; Model BZ-8100), and the cells positive for annexin-V were considered apoptotic. The cells visualized by Hoechst staining were counted in 12 randomly selected microscopic fields, and the percentage of apoptotic cells was calculated by dividing the number of Annexin V-positive cells by the total number of cells. The results were obtained from two independent experiments.
Western blotting
Western blotting was performed as described previously (14). Briefly, after serum starvation for 12 h, cells were treated with selumetinib or U0126 for 12 h, and then whole cell lysates were prepared using RIPA lysis buffer (Santa Cruz Biotechnology, Santa Cruz, CA, USA), separated on SDS/polyacrylamide gels, and electroblotted onto nitrocellulose membranes (Bio-Rad). The membranes were incubated with a phospho-p38 MAPK (Thr180/Tyr182) XP rabbit antibody, p38 MAPK rabbit antibody, phospho-p44/42 MAPK (Thr202/Tyr204) rabbit antibody and p44/42 MAPK rabbit antibody, all of which were purchased from Cell Signaling Technology (Beverly, MA, USA). The membranes were also incubated with anti-Actin mouse monoclonal antibody (Sigma-Aldrich), which was used as a loading control. The membranes were developed with peroxidase-labeled anti-mouse or anti-rabbit IgG antibodies (Amersham Pharmacia, Piscataway, NJ, USA) and Super Signal chemiluminescence substrate (Thermo Scientific, Rockford, IL, USA). The densitometry data were obtained from three independent experiments using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
The data were statistically analyzed with GraphPad Prism 7 for Mac OS X (GraphPad Software, San Diego, CA). Differences between three or more unmatched groups were analyzed by one-way ANOVA with Bonferroni's multiple comparisons or nonparametric Kruskal-Wallis test with Dunn's multiple comparisons. Furthermore, differences between three or more matched groups were analyzed by two-way repeated measures ANOVA with Dunnett's multiple comparisons. P<0.05 was considered to indicate a statistically significant difference.
Results
In our previous study, mutant KRAS knockdown sensitized NSCLC cells to a p38 inhibitor (12). This prompted us to investigate whether MEK inhibitors, as alternatives to mutant KRAS knockdown, could efficiently impair KRAS-mutated NSCLC cell growth in combination with p38 inhibitors. Treatment with the p38 inhibitor p38 V (p38 V) alone had no significant effect on the viability of NCI-H1792 NSCLC cells harboring KRAS mutations; however, the MEK inhibitor PD0325901 significantly reduced cell viability, and combined treatment with PD0325901 plus p38 V further enhanced this inhibitory effect (Fig. 1A). To confirm this finding, we tested another pair: The MEK inhibitor selumetinib and the p38 inhibitor LY2228820. LY2228820 alone modestly impaired cell viability, while selumetinib enhanced its effect in a dose-dependent manner (Fig. 1B). In addition, compared to LY2228820 or selumetinib treatment alone, combined treatment with selumetinib plus LY2228820 significantly reduced NCI-H1792 cell proliferation (Fig. 1C).
Figure 1.
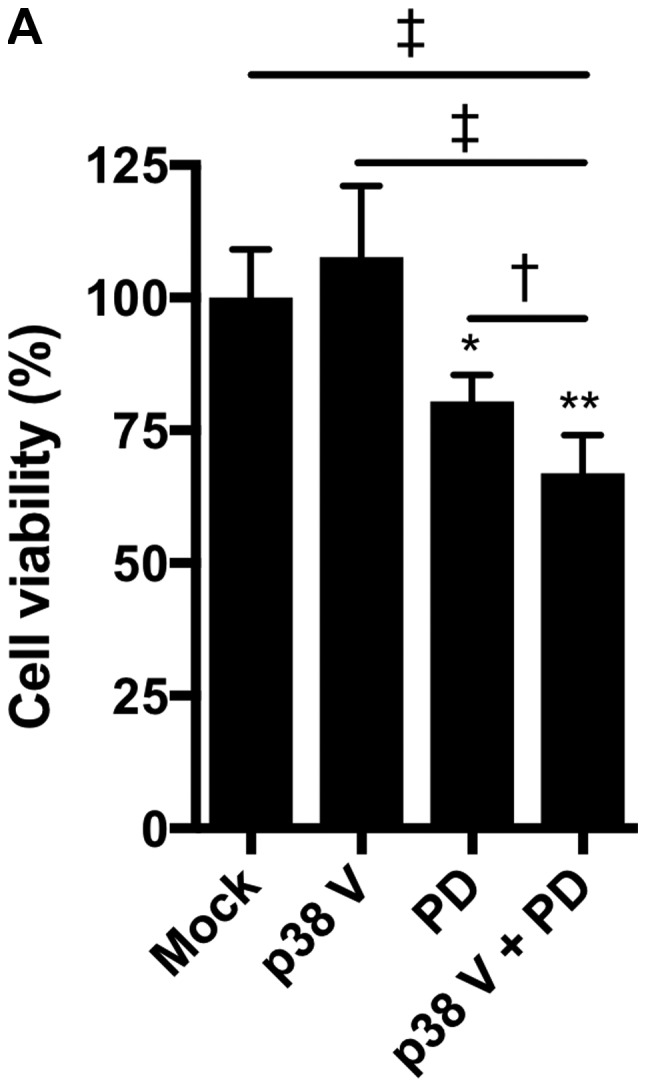
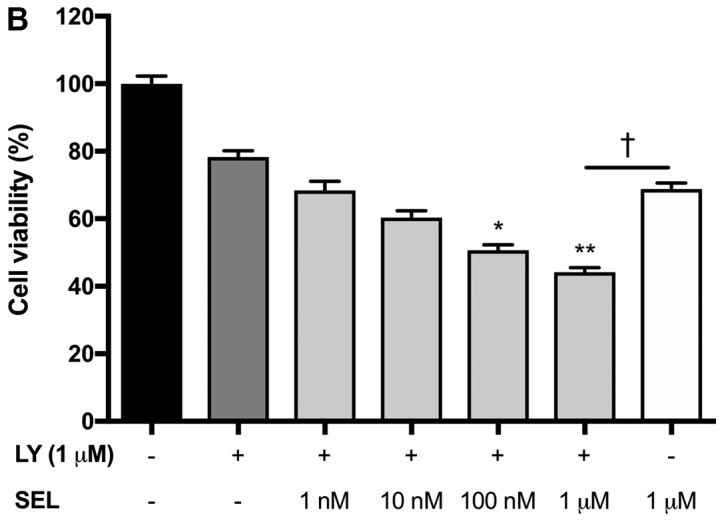
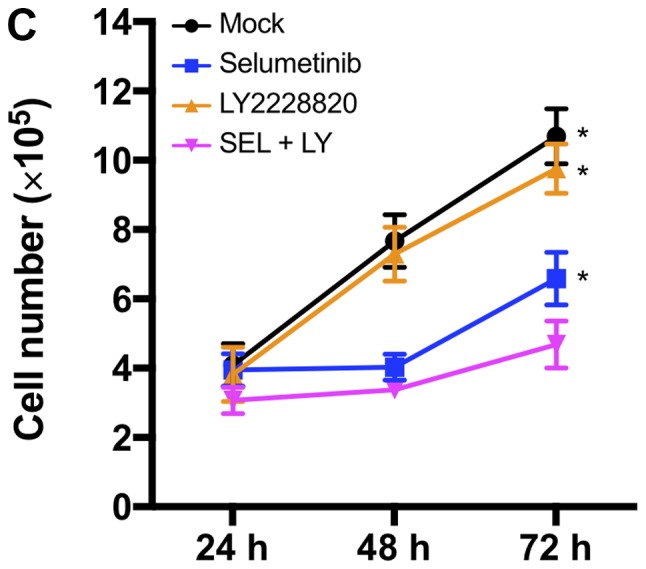
Impaired cell growth by combined treatment of MEK and p38 inhibitors in NCI-H1792 cells. (A) NCI-H1792 cells were treated with p38 V (10 µM), PD0325901 (10 µM) and a combination of both for 48 h. *P<0.01 and **P<0.001 vs. Mock; †P<0.05 and ‡P<0.001 as indicated. One-way analysis of variance with Bonferroni's multiple comparisons was used. (B) NCI-H1792 cells were treated with various concentration (1 nM to 1 µM) of selumetinib plus LY2228820 (1 µM) for 48 h. *P<0.01 and **P<0.001 vs. LY2228820 alone and LY2228820 plus selumetinib; †P<0.05 as indicated. Kruskal-Wallis tests with Dunn's multiple comparisons were used. (C) Combined treatment with selumetinib (1 µM) plus LY2228820 (1 µM) reduced cell proliferation of NCI-H1792 compared with Mock, LY2228820 alone and selumetinib alone. *P<0.001 vs. LY2228820 plus selumetinib. Two-way repeated measures of analysis of variance with Dunnett's multiple comparisons. Mock, treatment with DMSO alone; PD, PD0325901; SEL, selumetinib; LY, LY2228820.
We next assessed whether the combination of MEK and p38 inhibitors induces apoptosis in NCI-H1792 cells. Selumetinib alone, but not LY2228820 alone, increased the number of Annexin V-positive apoptotic cells, and the combination of selumetinib plus LY2228820 further increased the number of apoptotic cells (Fig. 2A). The DNA fragmentation assays confirmed that the induction of apoptosis was enhanced by the combination of both inhibitors (Fig. 2B).
Figure 2.
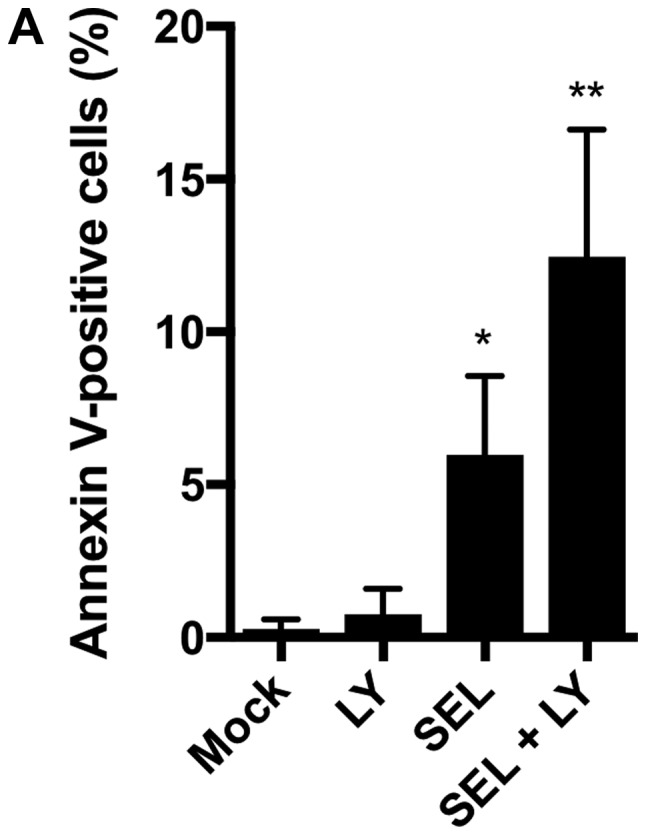
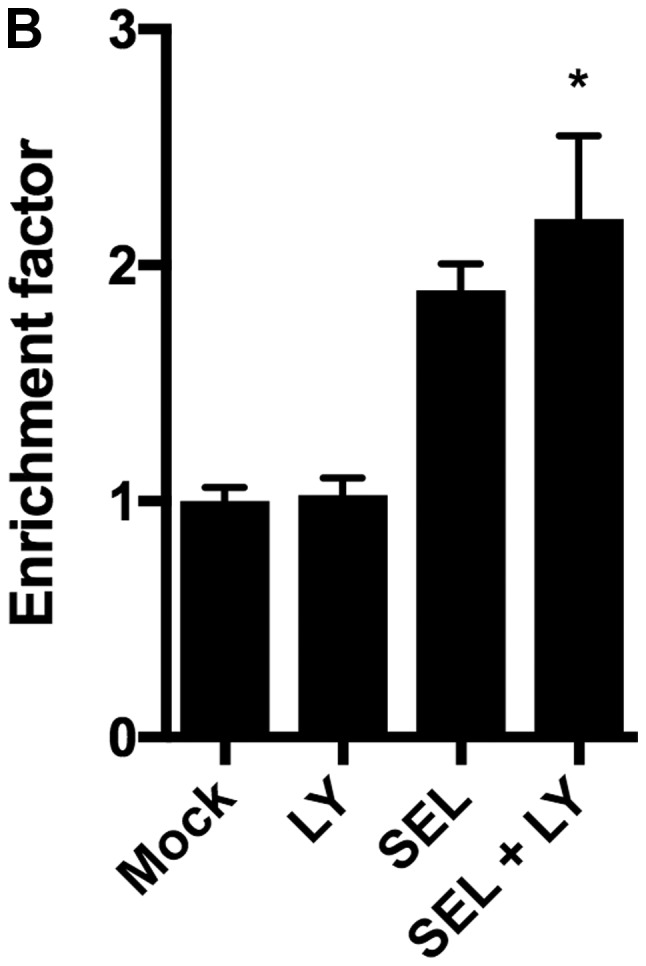
(A) Percentage of Annexin V-positive apoptotic cells and (B) fragmented DNA content, designated as an enrichment factor, increased with combined treatments with selumetinib (1 µM) plus LY2228820 (1 µM) compared with LY2228820 (1 µM) alone or selumetinib (1 µM) alone. *P<0.01 and **P<0.001 vs. Mock. Kruskal-Wallis tests with Dunn's multiple comparisons were applied. Mock, treatment with DMSO alone; LY, LY2228820; SEL, selumetinib.
To evaluate whether combined treatment with MEK and p38 inhibitors impaired cell growth in other KRAS-mutated NSCLC cell lines, we examined the growth-inhibitory effect of selumetinib and LY2228820 in NCI-H23, NCI-H157 and NCI-H460 cells. In all cell lines, compared with single inhibitor treatments, combined treatment with both reagents reduced cell viabilities (Fig. 3). Thus, the combined treatment of MEK and p38 inhibitors appears to be universally effective for inhibiting the growth of KRAS-mutated NSCLC cells.
Figure 3.

Growth-inhibitory effect of combined treatment with LY2228820 plus selumetinib in KRAS-mutated NSCLC cell lines, NCI-H23, NCI-H157 and NCI-H460. Cells were treated with LY2228820 (1 µM) and/or selumetinib (1 µM) for 72 h. *P<0.001 vs. Mock. Kruskal-Wallis tests with Dunn's multiple comparisons were used. Data were obtained from four independent experiments. Mock, treatment with DMSO alone; LY, LY2228820; SEL, selumetinib.
We further investigated whether knocking down MAPK14, which encodes p38α, enhanced the growth-inhibitory effect of MEK inhibitors in KRAS-mutated NSCLC cells. MAPK14 siRNAs markedly reduced MAPK14 mRNA expression in NCI-H1792 cells (Fig. 4A). In addition, the colony formation of NCI-H1792 cells was significantly inhibited by the MAPK14 siRNAs or MEK inhibitors (U0126 or selumetinib), and these inhibitory effects were more prominent upon combined treatment with MAPK14 siRNAs plus MEK inhibitors (Fig. 4B and C). These findings indicate that p38α loss confers hypersensitivity to MEK inhibitors in KRAS-mutated NSCLC cells.
Figure 4.

(A) MAPK14 mRNA expression reduced by siRNAs against MAPK14 (siMAPK14-1 and siMAPK14-2) in NCI-H1792 cells. *P<0.001 vs. siControl (one-way analysis of variance with Bonferroni's multiple comparisons test). Colony formation of NCI-H1792 cells by MAPK14 siRNAs with (B) U0126 (1 µM) or (C) selumetinib (1 µM) †P<0.05 vs. siCont plus U0126 group by one-way ANOVA with Bonferroni's multiple comparisons. *P<0.001 vs. siCont plus DMSO treatment group; ‡P<0.001 vs. siCont plus selumetinib group by one-way ANOVA with Bonferroni's multiple comparisons. siCont, treatment with control small interfering RNA; si, small interfering.
To elucidate the regulatory mechanism by which dual MEK and p38 inhibition efficiently inhibits cell growth, we examined the effects of MEK inhibition on the expression levels of p38 and extracellular regulating kinase (ERK). The MEK inhibitors (U0126 and selumetinib) increased phosphorylated p38 levels but decreased total p38 levels, which were accompanied by decreased phosphorylated ERK levels in NCI-H1792 cells (Fig. 5A-C). These results support the idea that MEK inhibition efficiently sensitizes KRAS-mutated NSCLC cells to p38 inhibitors. We also assessed whether the MEK inhibitor-induced p38 protein repression was due to transcriptional down-regulation. Treatment with selumetinib did not affect MAPK14 mRNA expression levels in NCI-H1792 cells (Fig. 5D), suggesting that p38 protein expression is post-transcriptionally modulated by MEK inhibition. Taken together, the present study indicates that dual inhibition of MEK and p38 has a synergistic effect on cell growth of KRAS-mutated NSCLC.
Figure 5.


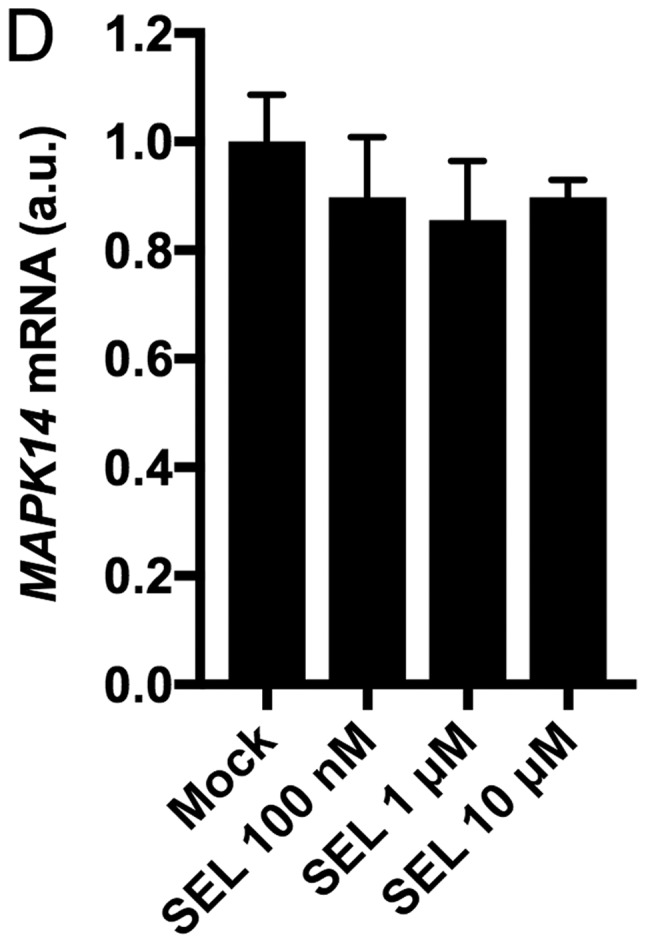
(A) Western blot data of p-ERK, ERK, p-p38 and p38 protein in NCI-H1792 cells treated with selumetinib. (B) Western Blot data in NCI-H1792 cells treated with selumetinib or U0126. (C) The densitometry data obtained from three independent experiments using ImageJ software. For Western Blotting, 15 µg of cell lysates was loaded per lane. Actin protein was used as a loading control. (D) MAPK14 mRNA expression by treatment with selumetinib at different concentrations in NCI-H1792 cells. Mock, treatment with DMSO alone; SEL, treatment with selumetinib; ERK, extracellular-signal-regulated kinase.
Discussion
Mitogen-activated protein kinase (MAPK) signaling pathways are highly conserved among eukaryotes and play essential roles in the transduction of extracellular signals to diverse cellular responses (15). MAPK signaling comprises three major pathways, ERK, c-Jun N-terminal kinase (JNK) and p38 MAPK, all of which mediate a large number of molecules through regulating gene expression and interacting with each other (15). The ERK pathway has a central role in NSCLC tumorigenesis (16), which is supported by the fact that this pathway comprises several oncogenic drivers of NSCLC, such as KRAS and BRAF (5). The p38 MAPK pathway is also involved in tumor development and maintenance by regulating numerous molecules that control tumor growth and survival, but it appears to have opposing roles as either a tumor inhibitor or a tumor promoter, depending on the cellular environment (17–19). Thus, the role of p38 MAPK in tumorigenesis is controversial and has not been fully defined in NSCLC.
In the present study, we showed that dual MEK and p38 inhibition has the potential to suppress KRAS-mutated NSCLC tumor growth. In agreement with our observations, previous studies have shown that blocking both the ERK and p38 pathways efficiently suppresses colorectal cancer growth (20,21). In a study by van Houdt et al, oncogenic KRAS activated p38α to maintain cell proliferation during MEK inhibition in KRAS-mutated colorectal cell lines (21). These authors found that MEK/ERK inhibition induced oncogenic KRAS-dependent p38 phosphorylation in colorectal cancer cells (21), which is consistent with our findings that MEK inhibitors increased phosphorylated p38 levels in KRAS-mutated NSCLC cells. Additionally, it was found that phosphorylated p38 levels were elevated in oncogenic RAS-transformed keratinocytes when EGFR signaling was abrogated, and blocking both EGFR and p38, but not EGFR or p38 alone, impaired cell proliferation (22); these results suggest that oncogenic RAS-driven tumors depend on p38 pathway activation to survive when the signaling pathways downstream of EGFR are blocked. Collectively, these findings suggest that oncogenic KRAS-driven NSCLC tumors could switch survival signaling to the p38 pathway when the ERK pathway is unavailable.
Intriguingly, in addition to increased phosphorylated p38 expression levels, the total p38 expression levels were decreased by MEK inhibitor treatment in KRAS-mutated NSCLC cells. This effect may underlie our observation that the growth-inhibitory effects of the combination of siRNA-mediated p38α knockdown with MEK inhibitors were more prominent than those of combined treatment with MEK and p38 inhibitors. These data support the idea that targeting MEK efficiently sensitizes KRAS-mutated NSCLC tumors to p38 inhibitors. We also found that treatment with a MEK inhibitor did not affect MAPK14 expression, indicating that MEK inhibitor-induced p38 down-regulation may be a post-transcriptional event. Further investigation is needed to clarify the precise mechanism regarding how MEK regulates p38 expression.
Despite the recent development of therapeutic strategies such as molecular targeted therapy and immunotherapy in NSCLC (3,4,23), targeting oncogenic KRAS-driven NSCLC remains a challenge. While many molecular targeted monotherapy drugs have failed to improve outcomes in KRAS-mutated NSCLC, combinatorial approaches with MEK inhibitors plus other inhibitors, such as AKT, CDK4/6, HSP90 or FGFR1 inhibitors, have recently provided hopeful results in preclinical and clinical studies (9,24–28). Our findings may provide a novel combinatorial approach of dual MEK and p38 inhibition, and further studies including in vivo experiments will elucidate its therapeutic significance in oncogenic KRAS-driven NSCLC.
Acknowledgements
The authors would like to thank Dr Junichi Okada, Dr Shuichi Okada and Dr Masanobu Yamada of the Division of Endocrinology and Metabolism, Department of Internal Medicine, Gunma University Graduate School of Medicine, and Dr Yasuhiko Koga and Dr Haruka Aoki-Saito of the Department of Respiratory Medicine, Gunma University Graduate School of Medicine for technical support and advice.
Funding
This study was supported by Grants-in-Aid for Scientific Research (C) (grant no. 26461181) from the Japan Society for the Promotion of Science.
Availability of data and materials
All data generated or analyzed in the current study are included in this article. Data sharing is not applicable to this article as no public datasets were generated or analyzed during the current study.
Authors' contributions
Conceptualization, methodology, funding acquisition and writing of the original draft was performed by NS. Investigation, validation and acquisition of data was performed by NS, YM, YT, NK and TM. Analysis and interpretation of data was performed by NS, YM, YT, NK, TM, RS, KK and TH. Resources were obtained by NS, KK and TH; writing, reviewing and editing the manuscript was performed by NS, YM, YT, NK, TM, RS, KK and TH; supervision and project administration was provided by NS and TH.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Houston KA, Henley SJ, Li J, White MC, Richards TB. Patterns in lung cancer incidence rates and trends by histologic type in the United States, 2004–2009. Lung Cancer. 2014;86:22–28. doi: 10.1016/j.lungcan.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553:446–454. doi: 10.1038/nature25183. [DOI] [PubMed] [Google Scholar]
- 4.Saito M, Shiraishi K, Kunitoh H, Takenoshita S, Yokota J, Kohno T. Gene aberrations for precision medicine against lung adenocarcinoma. Cancer Sci. 2016;107:713–720. doi: 10.1111/cas.12941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kris MG, Johnson BE, Berry LD, Kwiatkowski DJ, Iafrate AJ, Wistuba, Varella-Garcia M, Franklin WA, Aronson SL, Su PF, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA. 2014;311:1998–2006. doi: 10.1001/jama.2014.3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: Weaving a tumorigenic web. Nat Rev Cancer. 2011;11:761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meng D, Yuan M, Li X, Chen L, Yang J, Zhao X, Ma W, Xin J. Prognostic value of K-RAS mutations in patients with non-small cell lung cancer: A systematic review with meta-analysis. Lung Cancer. 2013;81:1–10. doi: 10.1016/j.lungcan.2013.03.019. [DOI] [PubMed] [Google Scholar]
- 8.Ying M, Zhu XX, Zhao Y, Li DH, Chen LH. KRAS mutation as a biomarker for survival in patients with non-small cell lung cancer, a meta-analysis of 12 randomized trials. Asian Pac J Cancer Prev. 2015;16:4439–4445. doi: 10.7314/APJCP.2015.16.10.4439. [DOI] [PubMed] [Google Scholar]
- 9.Matikas A, Mistriotis D, Georgoulias V, Kotsakis A. Targeting KRAS mutated non-small cell lung cancer: A history of failures and a future of hope for a diverse entity. Crit Rev Oncol Hematol. 2017;110:1–12. doi: 10.1016/j.critrevonc.2016.12.005. [DOI] [PubMed] [Google Scholar]
- 10.Blumenschein GR, Jr, Smit EF, Planchard D, Kim DW, Cadranel J, De Pas T, Dunphy F, Udud K, Ahn MJ, Hanna NH, et al. A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC)†. Ann Oncol. 2015;26:894–901. doi: 10.1093/annonc/mdv072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jänne PA, van den Heuvel MM, Barlesi F, Cobo M, Mazieres J, Crinò L, Orlov S, Blackhall F, Wolf J, Garrido P, et al. Selumetinib plus docetaxel compared with docetaxel alone and progression-free survival in patients with KRAS-mutant advanced non-small cell lung cancer: The SELECT-1 randomized clinical trial. JAMA. 2017;317:1844–1853. doi: 10.1001/jama.2017.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sunaga N, Shames DS, Girard L, Peyton M, Larsen JE, Imai H, Soh J, Sato M, Yanagitani N, Kaira K, et al. Knockdown of oncogenic KRAS in non-small cell lung cancers suppresses tumor growth and sensitizes tumor cells to targeted therapy. Mol Cancer Ther. 2011;10:336–346. doi: 10.1158/1535-7163.MCT-10-0750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sunaga N, Kaira K, Imai H, Shimizu K, Nakano T, Shames DS, Girard L, Soh J, Sato M, Iwasaki Y, et al. Oncogenic KRAS-induced epiregulin overexpression contributes to aggressive phenotype and is a promising therapeutic target in non-small-cell lung cancer. Oncogene. 2013;32:4034–4042. doi: 10.1038/onc.2012.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sunaga N, Imai H, Shimizu K, Shames DS, Kakegawa S, Girard L, Sato M, Kaira K, Ishizuka T, Gazdar AF, et al. Oncogenic KRAS-induced interleukin-8 overexpression promotes cell growth and migration and contributes to aggressive phenotypes of non-small cell lung cancer. Int J Cancer. 2012;130:1733–1744. doi: 10.1002/ijc.26164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12:9–18. doi: 10.1038/sj.cr.7290105. [DOI] [PubMed] [Google Scholar]
- 16.Heigener DF, Gandara DR, Reck M. Targeting of MEK in lung cancer therapeutics. Lancet Respir Med. 2015;3:319–327. doi: 10.1016/S2213-2600(15)00026-0. [DOI] [PubMed] [Google Scholar]
- 17.Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochem J. 2010;429:403–417. doi: 10.1042/BJ20100323. [DOI] [PubMed] [Google Scholar]
- 18.Loesch M, Chen G. The p38 MAPK stress pathway as a tumor suppressor or more? Front Biosci. 2008;13:3581–3593. doi: 10.2741/2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sui X, Kong N, Ye L, Han W, Zhou J, Zhang Q, He C, Pan H. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014;344:174–179. doi: 10.1016/j.canlet.2013.11.019. [DOI] [PubMed] [Google Scholar]
- 20.Chiacchiera F, Grossi V, Cappellari M, Peserico A, Simonatto M, Germani A, Russo S, Moyer MP, Resta N, Murzilli S, Simone C. Blocking p38/ERK crosstalk affects colorectal cancer growth by inducing apoptosis in vitro and in preclinical mouse models. Cancer Lett. 2012;324:98–108. doi: 10.1016/j.canlet.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 21.van Houdt WJ, de Bruijn MT, Emmink BL, Raats D, Hoogwater FJ, Borel Rinkes IH, Kranenburg O. Oncogenic K-ras activates p38 to maintain colorectal cancer cell proliferation during MEK inhibition. Cell Oncol. 2010;32:245–257. doi: 10.3233/CLO-2010-0521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wright LN, Ryscavage A, Merlino G, Yuspa SH. Modeling the transcriptional consequences of epidermal growth factor receptor ablation in Ras-initiated squamous cancer. Clin Cancer Res. 2012;18:170–183. doi: 10.1158/1078-0432.CCR-11-1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miura Y, Sunaga N. Role of immunotherapy for oncogene-driven non-small cell lung cancer. Cancers (Basel) 2018;10(pii):E245. doi: 10.3390/cancers10080245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tolcher AW, Khan K, Ong M, Banerji U, Papadimitrakopoulou V, Gandara DR, Patnaik A, Baird RD, Olmos D, Garrett CR, et al. Antitumor activity in RAS-driven tumors by blocking AKT and MEK. Clin Cancer Res. 2015;21:739–748. doi: 10.1158/1078-0432.CCR-14-1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tao Z, Le Blanc JM, Wang C, Zhan T, Zhuang H, Wang P, Yuan Z, Lu B. Coadministration of trametinib and palbociclib radiosensitizes KRAS-mutant non-small cell lung cancers in vitro and in vivo. Clin Cancer Res. 2016;22:122–133. doi: 10.1158/1078-0432.CCR-15-0589. [DOI] [PubMed] [Google Scholar]
- 26.Park KS, Oh B, Lee MH, Nam KY, Jin HR, Yang H, Choi J, Kim SW, Lee DH. The HSP90 inhibitor, NVP-AUY922, sensitizes KRAS-mutant non-small cell lung cancer with intrinsic resistance to MEK inhibitor, trametinib. Cancer Lett. 2016;372:75–81. doi: 10.1016/j.canlet.2015.12.015. [DOI] [PubMed] [Google Scholar]
- 27.Manchado E, Weissmueller S, Morris JP IV, Chen CC, Wullenkord R, Lujambio A, de Stanchina E, Poirier JT, Gainor JF, Corcoran RB, et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nature. 2016;534:647–651. doi: 10.1038/nature18600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kitai H, Ebi H, Tomida S, Floros KV, Kotani H, Adachi Y, Oizumi S, Nishimura M, Faber AC, Yano S. Epithelial-to-mesenchymal transition defines feedback activation of receptor tyrosine kinase signaling induced by MEK inhibition in KRAS-mutant lung cancer. Cancer Discov. 2016;6:754–769. doi: 10.1158/2159-8290.CD-15-1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed in the current study are included in this article. Data sharing is not applicable to this article as no public datasets were generated or analyzed during the current study.