

Summary
MCM2-7 proteins provide essential helicase functions in eukaryotes at chromosomal DNA replication forks. During the G1 phase of the cell cycle they remain loaded on DNA but are inactive. We have used recombinant methods to show that the Drosophila MCM2-7 helicase is activated in complex with Cdc45 and the four GINS proteins (CMG complex). Biochemical activities of the MCM AAA+ motor are altered and enhanced through such associations: ATP hydrolysis rates are elevated by two orders of magnitude, helicase activity is robust on circular templates, and affinity for DNA substrates is improved. The GINS proteins contribute to DNA substrate affinity and bind specifically to the MCM4 subunit. All pair-wise associations between GINS, MCMs and Cdc45 were detected but tight association takes place only in the CMG. The onset of DNA replication and unwinding may thus occur through allosteric changes in MCM2-7 affected by the association of these ancillary factors.
Introduction
The initiation of genomic DNA replication is an ordered biochemical pathway that follows the same general principles in all organisms. In a first step, DNA associates with initiator proteins that prepare it for the binding of other factors that will eventually create a replication fork (Kornberg and Baker, 1992). A critical early part of this process is the engagement of a DNA helicase that unwinds the two strands of genomic DNA to allow the DNA polymerases to use these strands as templates for the synthesis of new daughter DNA molecules (Enemark and Joshua-Tor, 2008; Singleton et al., 2007). Many of these processes are tightly regulated by cell-cycle signals that dictate the timing of DNA synthesis, but the commitment to DNA replication is determined once the unwinding step is achieved. In this context, it is intriguing that in eukaryotes the helicase proteins are loaded on chromatin early in the G1 phase of the cell cycle in what is likely an inactive form. How such “licensed chromatin” is activated for DNA replication is currently poorly understood. Solving this problem would contribute significantly towards deciphering the logic of the pre-S phase program in eukaryotes.
In eukaryotes, the replicative DNA helicase ‘core’ is formed by six different polypeptides (MCM2 - MCM7) (Forsburg, 2004). Individual MCM subunits are highly homologous and assemble into a hexameric ring with nearest neighbors around the ring following the same organization in different organisms (Crevel et al., 2001; Davey et al., 2003; Schwacha and Bell, 2001). All the MCM proteins belong to a family of AAA+ proteins (ATPases Associated with various cellular Activities) and carry well-conserved sequence elements for ATP binding and hydrolysis, which provide the energy for the helicase motor (Forsburg, 2004).
The loading of the MCMs on initiation sites requires the concerted contribution of several proteins, most notably the ORC complex, Cdc6, and Cdt1 (Bell and Dutta, 2002). The switch from an inactive to an active helicase is dependent on two critical cell-cycle dependent kinases: a cyclin-dependent kinase (CDK) and Cdc7-Dbf4 (DDK) (Sclafani and Holzen, 2007; Stillman, 2005). The action of these kinases may relieve a repressive conformation, allow for the association of other critical factors to activate a latent helicase activity, or allow for an isomerization of the pre-assembled complexes to achieve such a switch. Consistent with many of these scenarios, the helicase activity of the purified eukaryotic MCM2-7 complex has been difficult to demonstrate. It has been shown recently that the Saccharomyces cerevisae MCM2-7 is able to unwind double-stranded DNA with single-strand entry sites if the reaction conditions are appropriately modified. Such modified conditions may induce structural changes in the MCM ring that would, in a physiological setting, occur as a result of some activating factors or modifications (Bochman and Schwacha, 2008).
Studies of MCM associated complexes in various eukaryotes have provided clues to the possible mechanism of the MCM2-7 helicase activation. We have shown previously that MCM2-7 co-purify through stringent fractionation steps from Drosophila melanogaster embryo extracts in complex with Cdc45 and the four GINS proteins (Sld5, Psf1, Psf2, and Psf3) (Moyer et al., 2006). Cdc45 - MCM2-7 - GINS (CMG) complex members also co-purified from S. cerevisiae lysates (Gambus et al., 2006) and associated with the replication fork in Xenopus laevis egg extracts, when the DNA helicase activity was uncoupled from active DNA polymerase (Pacek et al., 2006). Both Cdc45 and GINS are required for the initiation step as well as elongation stages of DNA replication (Aparicio et al., 1997; Aparicio et al., 2006; Kanemaki et al., 2003; Kubota et al., 2003; Labib and Gambus, 2007; Pacek and Walter, 2004; Takayama et al., 2003; Tercero et al., 2000). All this suggests a universal and critical role for the GINS and Cdc45 as co-factors of the eukaryotic MCM2-7 complex.
We have now explored this idea by reconstituting Drosophila MCM2-7 and CMG complexes with recombinant proteins, and testing them side-by-side in various activity assays. We show here that the MCM2-7 helicase complex is activated when associated with Cdc45 and GINS and that this activation involves multiple aspects of the unwinding process, including enhanced ATP hydrolysis and better DNA substrate recognition. We suggest that Cdc45 and GINS allosterically form a scaffold on the MCM ring that assists in the proper coordination of different subunits in the MCM2-7 motor, thus leading to its efficient unwinding of DNA strands. We provide evidence that the MCM4 subunit, a distinct and important target of regulatory cell-cycle dependent kinases, is the main binding partner for the GINS complex. Our data support the idea that the controlled binding of Cdc45 and GINS co-factors in vivo to the otherwise inactive, but DNA associated, MCM2-7 core enzyme provides the necessary activation step for the replicative helicase and the concomitant initiation of DNA synthesis.
Results
Reconstitution of the Drosophila Cdc45-MCM2-7-GINS (CMG) complex
The protocol that we used for purification of the Drosophila CMG is outlined in Figure 1A and described in detail in the Supplemental Experimental Procedures section. The same protocol was used for reconstituting the Drosophila MCM2-7 complex, except that acetate replaced chloride as the anion in the chromatography buffers. This change enhanced the stability of the full MCM2-7 heterohexamer and improved separation from closely eluting incomplete MCM complexes in the Mono Q fractionation step (Figure S1). The purified MCM2-7 and CMG showed the expected patterns of subunit bands on the standard SDS-polyacrylamide gel electrophoresis (SDS-PAGE) (Figure 1B) (Moyer et al., 2006) and the presence of all subunits was verified by mass-spectrometry of both the total trypsin digested samples as well as protein bands cut from the SDS-PAGE gel (Figure S2). Each of the subunits had equal stoichiometry in the reconstituted six-component MCM2-7 and eleven-component CMG, as demonstrated by the results of laser densitometry analysis of SYPRO-red stained SDS-PAGE protein gels (Figure 1C) and by mass spectrometry data (Figure S2). The MCM2-7 and CMG preparations behaved as a single homogenous population through glycerol gradient centrifugation and there were no detectable higher order complexes or large aggregates (Figures 1D and 1E). The anticipated molecular weight for the CMG complex is 711 kD and 545 kD for the MCM2-7. The peak of CMG sedimented with the 670 kD ferritin marker during glycerol gradient centrifugation, while the MCM2-7 peak was approximately two fractions slower. These data and the results of stoichiometry analysis are consistent with both MCM2-7 and CMG forming stable monomeric complexes in solution in accordance with the gel filtration measurements we have previously reported for the CMG complex isolated from Drosophila embryo extracts (Moyer et al., 2006).
Figure 1. Reconstitution of CMG and MCM2-7 complexes.
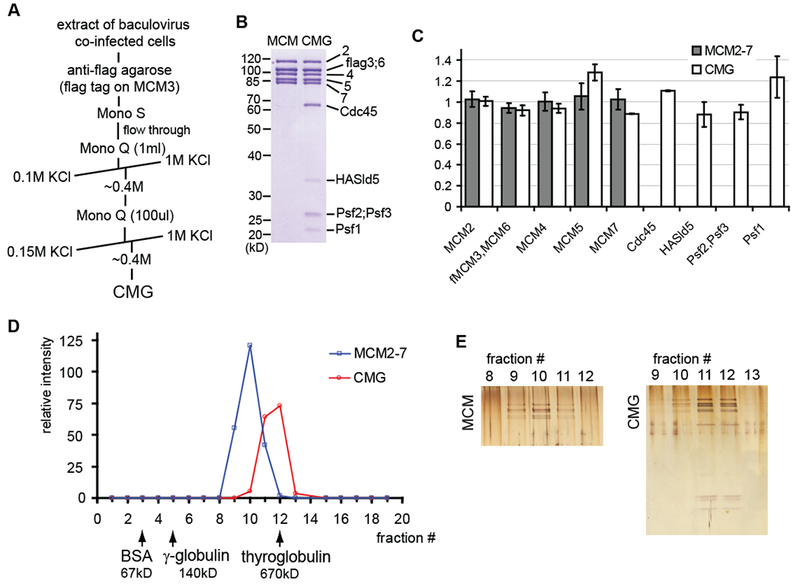
(A) Purification protocol for the Drosophila CMG
(B) Coomassie blue stained 10% SDS-PAGE gels of the purified MCM2-7 and CMG complexes
(C) Stoichiometry of subunits in reconstituted MCM2-7 (dark columns) and CMG (white columns), based on laser densitometry scanning of SYPRO-red stained SDS-PAGE protein gels. Four independent MCM2-7 preparations and 3 CMG preparations were analyzed. The error bars in all the figures of this paper correspond to standard deviation, unless indicated otherwise.
(D and E) Glycerol-gradient centrifugation analysis of the MCM2-7 and CMG. Western blotting with antibodies against MCM2, MCM4, and MCM5 subunits was used to detect protein in the fractions and quantification of these blots through densitometry using ImageJ software is shown in (D). Peak fractions were separated on 12% SDS-PAGE and silver stained (E).
The CMG displays helicase activity on circular and fork substrates
We examined the DNA helicase activity of the CMG and MCM2-7 complexes with two different DNA substrates. One substrate had a radiolabeled oligonucleotide annealed to an M13 single-stranded circle, the other was a forked DNA molecule with 50 base pair double-stranded region and 40 nucleotides long single-stranded poly-T ‘arms’. The incubation of either of these substrates with the CMG complex resulted in the ATP-dependent release of the radiolabeled oligo from the double strands in a protein concentration dependent manner (Figures 2A and 2B, quantified in 2C and 2D; ‘CMG’). The MCM2-7 complex was inactive on the M13 substrate (Figures 2A and 2C; ‘MCM’) and showed barely detectable helicase activity with the forked substrate in side-by-side assays (Figures 2B and 2D; ‘MCM’). Interestingly, the only published report thus far for DNA helicase activity associated with an MCM2-7 complex used this forked DNA substrate. This activity was shown to be dependent on the anionic composition of reaction buffer, with glutamate supporting the highest helicase activity (Bochman and Schwacha, 2008). The replacement of acetate with glutamate in our helicase reactions led to approximately 50% increase of the MCM2-7 helicase activity on the fork (Figures S3A and S3B), but not on the M13 substrate. Glutamate buffer enhanced the CMG helicase by about 30-50% on M13 substrate (Figures S3C and S3D).
Figure 2. DNA helicase assays with purified MCM2-7 and CMG.
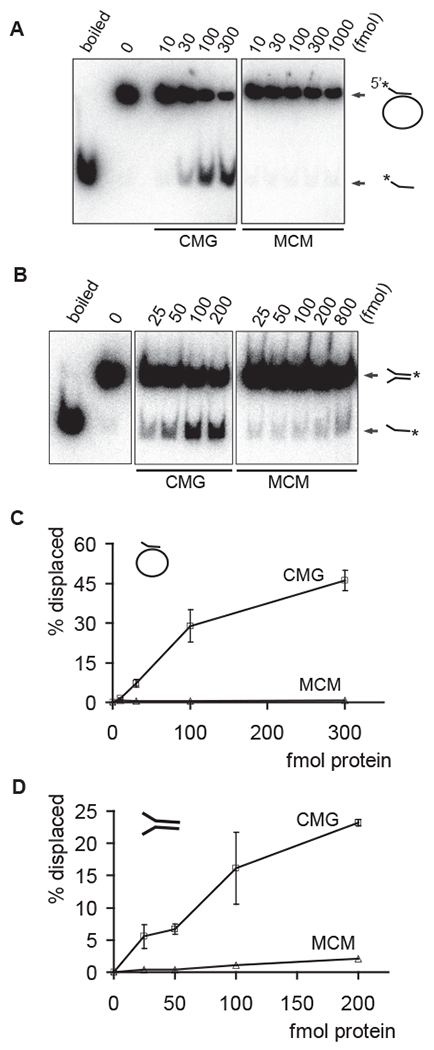
(A and B) Autoradiographs of the reaction products separated on PAGE. Indicated amounts of CMG or MCM2-7 proteins in femtomoles were added to either a circular (A) or a forked substrate (B). Positions of double-stranded substrate and displaced oligo are indicated with arrows, ‘boiled’ and ‘0’ correspond to control lanes with heat denatured substrate or without protein, respectively.
(C and D) Quantified results for two independent helicase assays with circular (C) or forked (D) DNA substrate. The graphs show percentage of displaced oligonucleotide as a function of protein added to reaction.
The Drosophila CMG helicase translocates in a 3’ to 5’ direction on single-stranded DNA (Moyer et al., 2006). This polarity is a general feature of related MCM helicase complexes, as it has been reported in the case of archaeal homohexameric MCM helicases (Chong et al., 2000; Kelman et al., 1999; Shechter et al., 2000) and the eukaryotic MCM4-6-7 sub-complex (Ishimi, 1997). For the M13 based DNA substrate, the oligo that was annealed to circular M13 DNA lacks a single-stranded overhang on its 3’ end. Hence for efficient utilization of such a substrate, the CMG helicase must load on the circular single-stranded part of the substrate and engage at the fork for activity. The MCM2-7 complex, on the other hand, is inactive on this substrate and seems to display weak helicase activity only on the forked substrate, which has a free 3’ single-stranded end. Hence, the interaction of MCM2-7 with Cdc45 and GINS in CMG activates it as a helicase and likely enables its loading on DNA substrates without a free end for entry.
The ATPase activity of MCM2-7 is activated in complex with Cdc45 and GINS.
The MCM2-7 helicase ‘motor’ is fueled by cycles of binding and hydrolysis of ATP, thus we asked if the observed activation of the MCM helicase in the CMG complex is in part a result of enhanced ability to function as an ATPase. We measured the initial ATP hydrolysis rate as a function of ATP concentration using radiolabeled ATP and thin layer chromatography to separate and quantify hydrolysis products (Figure 3). Similar to a previous report for the yeast MCM2-7 complex (Schwacha and Bell, 2001), ATP hydrolysis by Drosophila MCM2-7 and CMG complexes did not follow simple Michaelis-Menten kinetics, as illustrated by respective Eadie-Hofstee plots. Single Michaelis-Menten rates would yield a straight line and in contrast at least three different regions, each with respective kinetic modes, were detected in the Eadie-Hofstee plots for the ATPase rates of MCM2-7 (Figure 3A) and CMG (Figure 3B). These can be described as low, medium, or high ATP concentration modes (indicated by I, II, and III, respectively). Such concentration-dependent changes in the kinetic modes of activity suggest cooperative switching on the MCM ring as a function of bound ATP. While these transitions occurred at approximately the same concentrations of ATP for both CMG and MCM2-7, the shapes of the curves were significantly different.
Figure 3. Analysis of the ATP hydrolysis by MCM2-7 and CMG.
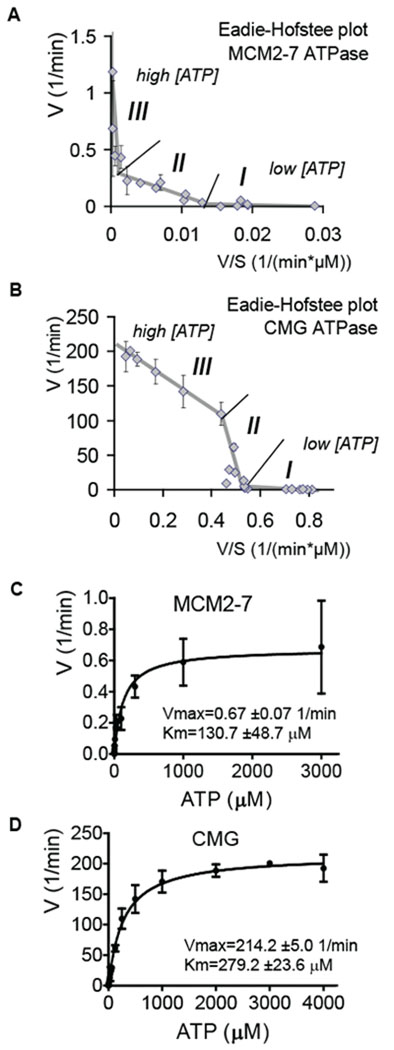
(A and B) Eadie-Hofstee plots of initial in vitro ATP hydrolysis kinetics for MCM2-7 and CMG (B). The regions that correspond to different kinetic modes at low (I), medium (II), or high (III) ATP concentrations are indicated.
(C and D) Initial ATP hydrolysis rate kinetics for MCM2-7 (C) and CMG (D) plotted as a function of ATP concentration. Data were collected from 4 independent experiments, covering the entire tested ATP concentration range (0.15 μM – 4 mM for CMG, 0.03 μM – 3 mM for MCM2-7) with at least 3 overlapping datasets.
With sub-micromolar ATP concentrations, both complexes worked at a relatively low rate but with high affinity for ATP. At about 2 μM ATP a major switch occurred towards a significantly higher rate but lower substrate affinity (from I to II on Figures 3A and 3B). Estimating the kinetic parameters based on the Eadie-Hofstee plots (Km – the negative slope; Vmax – the intercept with Y-axis) shows that an apparent Vmax for MCM2-7 changed from 0.01 to 0.25 mol ATP per min per mol enzyme (1/min) with Km changing from 0.14 to 13μM. The Vmax of CMG changed from 3.8 to 470 1/min and Km from 4.4 to 880μM. The second transition (from II to III) occurred at approximately 150 μM ATP. As with the first switch, the rate for MCM2-7 ATPase increased (Vmax from 0.25 to 0.66) and substrate affinity decreased (Km from 13 to 190 μM). For the CMG, by contrast, this transition resulted in a lower rate (Vmax from 470 to 210) and higher substrate affinity (Km from 880 to 230 μM).
The estimated total ATP concentration in eukaryotic cells is in the low millimolar range (Traut, 1994). Thus, even though CMG was activated relative to MCM2-7 at all the tested ATP concentrations, the high ATP concentration mode is likely to describe the process as it occurs in vivo. Re-plotting the data as an ATPase rate against the entire tested ATP concentration range results in a saturation curve (Figures 3C and 3D), the shape of which is dominated by the high concentration mode (>100μM ATP). Non-linear regression analysis of these data also yield kinetic parameters for the CMG (± standard error): Vmax=214.2 ± 5.0 1/min; Km=279.2 ± 23.6 μM, and for the MCM2-7: Vmax=0.67 ± 0.07 1/min; Km=130.7 ± 47.9 μM. As expected, these numbers are close to the projected values for the high ATP concentration mode provided by the Eadie-Hofstee plot. The apparently similar Km values of the CMG and MCM2-7 at physiological ATP concentrations may reflect similar affinities for ATP in these conditions, but the ATP hydrolysis rates by the MCM2-7 proteins are significantly enhanced in complex with Cdc45 and GINS, as indicated by the roughly 300× higher Vmax value for CMG.
The MCM2-7 ring of CMG complex is functionally asymmetric
The data described above indicate that association with co-factors lacking canonical ATP binding activities alters and activates both the intrinsic ATPase and helicase activities of the MCM proteins. To directly test the roles of individual MCM proteins required for these activities, we changed one conserved residue in each MCM - the invariant lysine in the P-loop of Walker A domain, critical for ATP binding - to an alanine, and purified six different CMG complexes with this single substitution. In each case, the complexes formed with identical purification properties and stoichometry as wild type (wt) CMG (Figure 4A).
Figure 4. Effect of ATPase site mutations in MCM subunits on ATPase and DNA helicase activity of CMG.
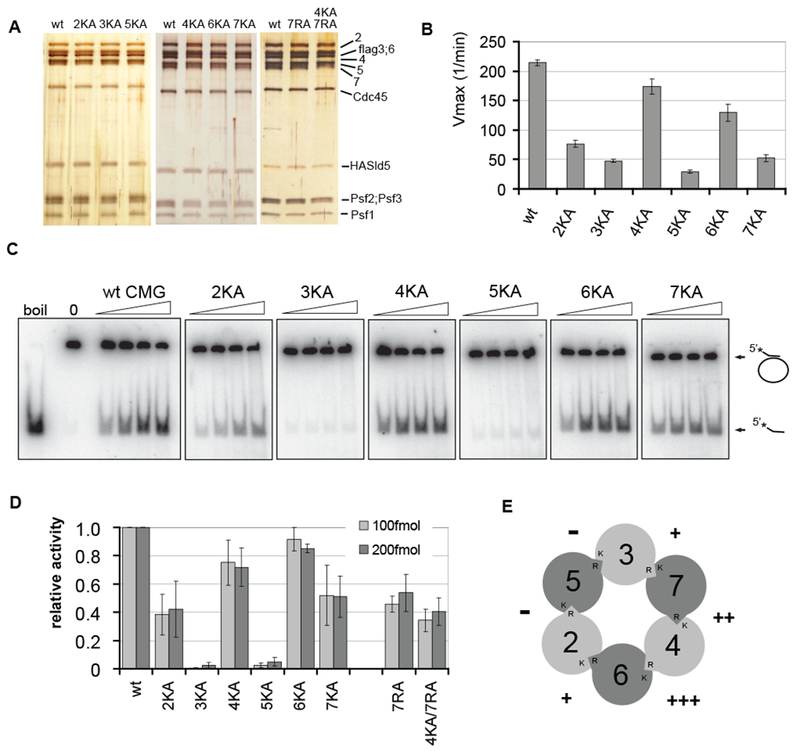
(A) Silver-stained 10% SDS-PAGE gels showing the individual protein complexes. ‘KA’ indicates a K to A substitution in the Walker A box of a given MCM subunit and ‘RA’ an R to A substitution in arginine finger domain, ‘wt’ indicates wild type CMG.
(B) Vmax values (± standard error) for ATP hydrolysis reaction by wt CMG compared to mutant complexes. The values for MCM 2, 3, and 5 KA complexes are based on data from 5 independent series, for MCM 6 and 7 KA – from 2, and for MCM 4KA – from 1 series.
(C and D) DNA helicase assays with wt or mutant CMG complexes. Autoradiographs of products separated on PAGE are shown in (C). Increasing amounts (25, 50, 100, or 200 fmol) of CMG complexes as indicated were added to the circular DNA substrate. In (D), the helicase activity of each mutant CMG complex is expressed relative to the wt as determined from the percent of oligo displaced by 100 or 200 fmol of protein (light grey and dark grey columns, respectively). The data for individual mutant complexes was collected from following number of independent series: 4KA – 9; 5KA - 5; 2KA, 6KA, 7KA, and 7RA – 4; 3KA, and 4KA/7RA – 2.
(E) Graph illustrating the relative effects of individual Walker A box mutations around the MCM ring to the helicase activity of the CMG. The percent of activity retained after mutation is shown next to targeted ATPase sites, with 0-25% of wt indicated by ‘-‘ 25-50% - ‘+’, 50-75% - ‘++’, and 75-100% - ‘+++’.
We found that all six mutant complexes showed defects in the ATPase activity, as indicated by decreased Vmax values for the reaction (Figure 4B). The substitutions in MCM 2, 3, 5, and 7 subunits each resulted in a decrease of more than 1/6th of the activity of wt CMG. Thus as anticipated from the switches in the rates of hydrolysis with titration of ATP, the hexameric MCM motor of the CMG does not function as a simple sum of six independent and equally contributing ATPase domains. Surprisingly, mutations in MCM 4 and 6 had clearly less effect on the ATPase activity of CMG than the substitutions in other subunits.
We tested each of the purified mutant complexes at different protein concentrations for DNA helicase activity. The ATP concentration was kept at 300μM, close to the ‘physiological’ Kd (high ATP concentration mode) of the CMG ATPase. All mutant complexes had reduced activity in the helicase assay, but the extent of defect varied again significantly from complex to complex (Figures 4C and 4D). The CMG complexes harboring the MCM3 and MCM5 KA mutations were the most severely affected, with activity only 1-5% of wt. The mutations in MCM4 and 6, which had the weakest effect on the ATPase activity, also had the weakest effect on the helicase activity of the CMG, retaining 70-80% of wt function. The mutations in MCM2 and MCM7 were intermediate in defect, retaining about half of the wt activity.
The active ATPase sites in the MCM2-7 ring are formed through the interaction surfaces of two neighboring MCM subunits such that a critical “arginine finger” is provided in trans relative to the invariant lysine residue in Walker A box of adjacent subunit (Davey et al., 2003). Our pull-down studies as well as the subunit composition of various incomplete MCM sub-complexes that we detected in our reconstitution experiments confirm the nearest neighbor order for the Drosophila MCM2-7 as reported earlier (Crevel et al., 2001)(data not shown, see Figure 4E for a model). To confirm that the modest effect of the Walker A box mutation in MCM 4 on the CMG helicase activity is due to the specific structure of this region, we introduced an arginine to alanine substitution in the corresponding MCM7 arginine finger. As anticipated, the effect of this mutation on CMG helicase was comparable to that of the corresponding MCM4 KA mutation. The double substitution (4KA/7RA) had an effect similar to the single mutations, retaining about 40% of wt function (Figure 4D).
MCM4, 6, and 7 proteins are capable of forming a hexameric complex with in vitro helicase activity and have thus often been considered as the ‘true catalytic’ subunits of the MCM helicase (Ishimi, 1997). Our results are not consistent with this notion, yet it was surprising to measure relatively small effects with the ATPase mutations in MCM4, 6, and 7 for the CMG helicase. We emphasize that even the defects that are small in vitro may be deleterious in vivo. Furthermore, the highly conserved Walker A motifs within these MCM subunits may play critical roles in vivo either by-passed by our recombinant expression system (e.g. assembly) or not assayed at all (e.g. disassembly at DNA replication termini).
We also note that the quantitative effects of the mutations in the MCM subunits follow an intriguing pattern. The substitutions that had the strongest effect on the CMG helicase were in the ATPase sites surrounding the MCM5 subunit and the substitutions with the weakest effects surround the MCM4 subunit (Figure 4E). Hence, the most crippling mutations are found on the side of MCM ring opposite to the side containing the least deleterious mutations.
Interactions between Cdc45, MCM2-7, and GINS
A component of the observed asymmetry with regard to mutational sensitivity might involve the positions of each of the ancillary factors on the MCM ring. To determine how these proteins interact, we used combinations of baculoviruses expressing the GINS, Cdc45, and MCM subunits to co-infect cells and performed immunoaffinity pull-down (IP) experiments from the extracts of infected cells. Sld5 was expressed with an HA epitope tag as this allowed for a one step co-precipitation of the tetrameric GINS complex. Affinity purified polyclonal Cdc45 antibody covalently linked to Protein A beads was used to pull down Cdc45 and associated complexes.
In the experiments where all six MCM subunits were co-expressed, the ancillary factors GINS and Cdc45 each were able to pull down all the MCMs (Figures 5A and 5B; ‘G+M’ and ‘C+M’). GINS was also able to pull down Cdc45 from co-infected cell extracts (Figure 5C; ‘G+C’). Therefore, each of the three components of the CMG complex associates in all pair-wise combinations. We have also detected and immunoprecipitated co-purifying MCM+GINS and MCM+Cdc45 complexes from the side fractions of Mono Q chromatography step while purifying the full CMG (Figure S1A and data not shown). Interestingly, we were never able to co-purify satisfactory amounts of MCM+GINS and MCM+Cdc45 complexes through the complete ion exchange chromatography steps, suggesting that the stability of these incomplete sub-complexes is not comparable to that of the full CMG.
Figure 5. Interactions between the subunits of CMG.
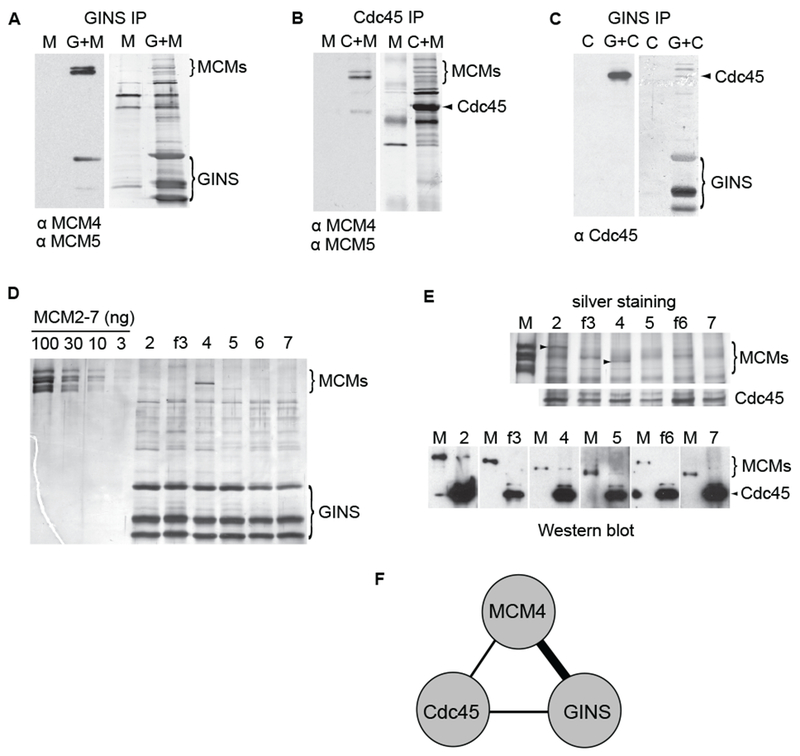
(A - C) Western blots of the immunoprecipitated samples. Left panels show the probed blots with used antibodies indicated below, the right panels show the total protein staining of the same blots by colloidal gold. HA-tagged Sld5 subunit was used to precipitate the intact GINS complex and associated proteins in (A) as well as in (C); Cdc45 pull-down was performed in (B). CMG components in starting extract are shown on top (‘C’ – Cdc45; ‘M’ – MCM2-7; ‘G’ – GINS).
(D) Silver-stained SDS-PAGE gel showing immunoaffinity pull-down of individual MCM subunits (as indicated on top) by GINS (HA-tag on Sld5). First four lines carry indicated amounts of MCM2-7 concentration marker.
(E) Silver-stained SDS-PAGE gel showing the pull-down of individual MCM subunits by Cdc45. Arrowheads show the position of MCM2 and MCM4 bands. Western blotting analysis of the same samples is shown below. Each blot was probed with polyclonal antibodies detecting Cdc45 and individual MCMs; anti flag antibodies were used to probe flag-tagged MCM3 and MCM6. 100 ng of purified MCM2-7 (flag tag on MCM3) was loaded in each pair (‘M’) to compare different blots.
(F) Interactions between the components of CMG complex based on data presented above. The thick line connecting GINS to MCM4 indicates a higher specificity than those indicated for the other pairs.
We then asked if the ancillary factors showed a specific interaction with individual MCMs by performing IP experiments with either GINS or Cdc45 co-expressed with single MCM subunts. All MCM proteins were expressed at comparable levels, as determined by immunoblotting of starting extracts with specific antibodies (Figure S4A). Of the six MCMs, MCM4 reproducibly showed a specific affinity for the GINS (Figure 5D, lane ’4’). This interaction was also the strongest under less stringent washing conditions (250mM KCI in washing buffer instead of 1M), although with the exception of MCM2, all of the MCMs were now able to bind GINS with some affinity (Figure S4B). Thus both shared and unique epitopes on MCM4 may mediate the targeting of the GINS to MCM4. We did not detect any enhanced association of GINS when MCM4 was in sub-complex with either one of its neighboring subunits from the MCM ring - MCM6 or MCM7 (data not shown). The binding of Cdc45 to individual MCM subunits, on the other hand, appeared to be less specific, with both MCM 2 and 4 showing the strongest interaction in Cdc45 pull-down experiments (Figure 5E). All of the individual MCM subunits were able to bind Cdc45 in less stringent conditions (Suppl. Figure 4D). Thus the single copy of Cdc45 that binds to the MCM hexamer in CMG may use a motif present in both MCM4 and MCM2 and related sequences in other subunits. We note that the GINS / MCM4 and the Cdc45 / MCM2 or 4 interactions were also detected in 1M KCI plus ethidium bromide and are not mediated by DNA (Figures S4C and S4E). Hence it seems reasonable to suggest that combinatorial interactions involving MCM4, Cdc45 and members of the GINS complex are each required for the strong associations found in the CMG (Figure 5F).
CMG has higher affinity for DNA than does MCM2-7
We used electrophoretic mobility shift assays (EMSA) to compare the DNA binding properties of the MCM2-7 complex with CMG. The single shifted species identified with each complex over the range of protein concentrations is consistent with a homogeneous population with a unique binding mode (Figure 6A). Both CMG and MCM2-7 bound preferentially to a forked DNA substrate, as the single-stranded DNA, or probes lacking one single-stranded arm, were bound much less efficiently (Figure S5). The DNA-binding activity of CMG was significantly greater than that of MCM2-7 (Figures 6A and 6B). Estimates from half saturation points yield a Kd = 10-20 nM for the CMG and 200-300 nM for the MCM2-7. These binding affinities are within the range previously reported for yeast MCM2-7 and MCM4-6-7 (30-50 nM) (Bochman and Schwacha, 2007). The affinity of CMG to DNA remained unchanged when glutamate buffer was used instead of acetate in the binding reactions (Figures S3E and S3F). Interestingly, the binding of CMG to DNA was ATP dependent, while the binding of MCM2-7 was not. The binding affinity of CMG was decreased approximately tenfold in the absence of non-hydrolysable ATP homologue ATP-γS, to levels similar to the ATP-independent DNA binding of MCM2-7 (Figure 6C).
Figure 6. MCM2-7 binds DNA more efficiently in complex with Cdc45 and GINS.
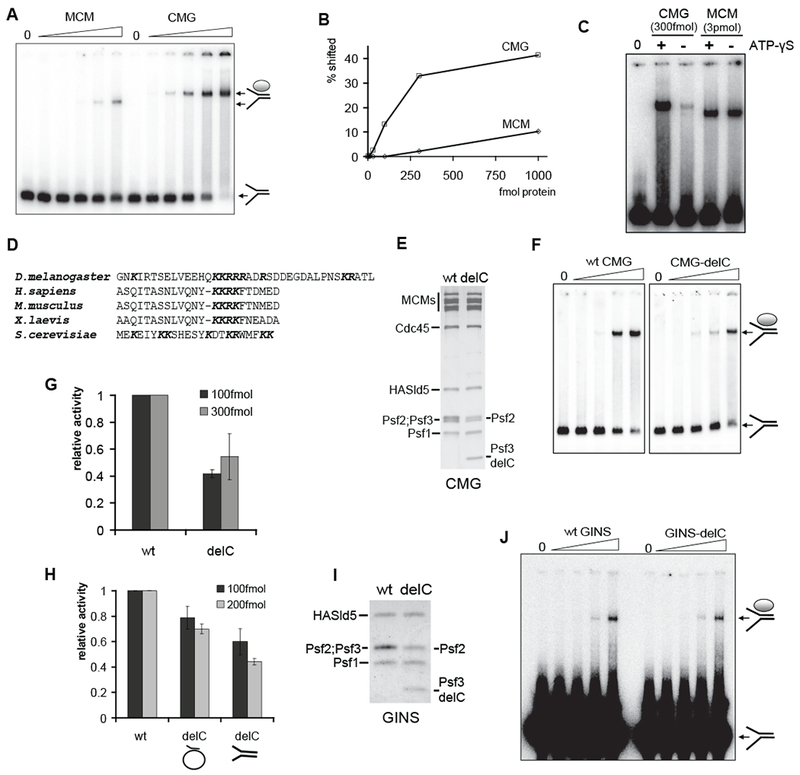
(A) Autoradiograph of the EMSA measuring DNA binding by MCM2-7 and CMG. Arrows show free forked probe and protein : DNA complexes. Increasing amounts (10, 30, 100, 300, and 1000 fmol) of MCM2-7 or CMG was added to the DNA substrate in the presence of ATP-γS. On all panels, ‘0’ corresponds to a control with no protein.
(B) The quantification of data from (A), expressed as a percent of DNA probe that was shifted into a discreet slower moving band. The free probe signal from a control lane was set as 100%
(C) EMSA showing that enhanced DNA biding by CMG is ATP dependent. ATP-γS was added as indicated to the binding reactions containing either 300 fmol CMG or 3 pmol of MCM2-7.
(D) Alignment of C-terminal tails of the Psf3 protein in various organisms.
(E) Silver-stained 12% SDS-PAGE gel of purified wt and CMG-delC complexes. Psf2 runs as a doublet because of an internal start codon (see Supplemental Experimental Procedures for details).
(F and G) DNA binding by wt CMG and CMG-delC. Shown is an autoradiograph of EMSA with 10, 30, 100, and 300 fmol of proteins as indicated (F) and the quantification of data from three independent experiments with two different CMG-delC protein preparations (G). The activity was calculated as in (B) and expressed relative to wt activity in the same series for reactions with 100 fmol (dark grey columns) or 300 fmol (light grey) of protein.
(H) Quantified results of DNA helicase assays with wt and CMG-delC complexes. Two different protein preparations were tested in duplicates on circular or forked substrates as indicated. Columns show the fraction of substrate displaced relative to wt activity by either 100 fmol or 200 fmol of protein (dark grey and light grey columns, respectively).
(I) Coomassie-stained 12% SDS-PAGE gel of purified wt and GINS-delC complexes.
(J) EMSA of DNA binding by increasing amounts (0.3, 1, 3, and 8 pmol per reaction) of wt GINS and GINS-delC.
GINS assists the CMG in DNA-binding
Cdc45 and GINS may improve the DNA binding activity of the CMG relative to MCM2-7 by two non-exclusive and perhaps synergistic mechanisms. Indirectly, these activating factors may cause structural changes in the MCM ring that lead to better accessibility of otherwise masked motifs or promote an organization of elements unstructured in the solo MCM2-7 that could play a role in the DNA binding of CMG. Second, the ancillary factors of the CMG complex may work through binding directly to the template. The latter possibility is suggested by a previous observation that human GINS can bind to DNA substrates in EMSA (Boskovic et al., 2007).
We scanned the amino acids of the four GINS proteins and Cdc45 for conserved clusters of positively charged amino acids, particularly in potentially exposed surface residues that might provide a DNA interaction domain(s). The extreme C-terminus of Psf3 contains several positively charged residues in all examined eukaryotes and maintains in metazoans a short conserved stretch of lysines and arginines (Figure 6D). We reconstituted a mutant CMG that lacks these C-terminal 38 amino acids of Psf3 (CMG-delC). The biochemical yield and the subunit stoichiometry of the mutant complex are identical to that of the wt CMG (Figure 6E), and the deletion does not induce structural changes with potential effect on the ATPase activity of CMG-delC (apparent Vmax=188.0±6.7 1/min; Km=193.7±29.7μM). The mutant complex had 50-60% reduced affinity for the forked DNA substrate compared to the wt CMG in EMSA (Figures 6F and 6G). When analyzed for helicase activity on the same substrate, the same 50% reduction relative to wt activity was observed (Figure 6H, right columns). Reproducible reduction in helicase activity was also apparent when we used the M13 based closed circular substrate (Figure 6H, middle columns).
We purified GINS alone containing either full-length Psf3 or Psf3 with C-terminal truncation (Figure 6I) and compared DNA binding activity of these complexes to the forked DNA substrate by EMSA (Figure 6J). The mutant GINS bound DNA with an apparent affinity equal to the wt complex (Figure 6J). Therefore, while the Psf3 mutation illustrates that the GINS contributes to the enhanced DNA binding and helicase activity of the CMG, the contribution of Psf3 tail to the DNA binding of CMG likely occurs in concert with other subunits of the CMG complex.
Discussion
We have shown here that the MCM2-7 complex is activated as a DNA helicase when associated with auxiliary factors Cdc45 and GINS. These factors do not associate with MCM2-7 in the pre-replication complex in vivo until regulatory kinase signals are executed (Labib and Gambus, 2007) and such tightly regulated binding of critical factors provides an elegant mechanism to explain how the timed activation of the DNA helicase holoenzyme would occur to trigger the G1 to S switch. Several features of the CMG complex that we have characterized provide some insights into the mechanism of activation of the MCM helicase and provide an example of a dramatic, orders of magnitude stimulation of core AAA+ activities by ancillary factors.
Activation of MCM2-7 helicase by Cdc45 and GINS
We found that the rate of ATP hydrolysis by the MCM2-7 ring at physiological ATP concentrations is about 300 fold higher in complex with Cdc45 and GINS. Neither GINS nor Cdc45 are ATPases, nor do we suggest that they contribute residues that participate directly in the catalytic sites at the interfaces between the MCM subunits. The activation by GINS and Cdc45 is thus most likely achieved through allosteric remodeling of the MCM ring to optimize subunit interactions for enhanced hydrolysis rates. Prior studies with the S. cerevisiae MCM2-7 have established the cooperative nature of the ATPase activity of these motors by demonstrating that single point mutations in one subunit can dramatically diminish the rates for the entire complex (Schwacha and Bell, 2001). Similarly we have found that single mutation in critical residues of MCM 2, 3, 5, or 7 decreases ATPase rates of the entire CMG to well below the 1/6th lower level expected for a stochastic model. These data demonstrate that the MCM subunits do not work independently from each other. One might posit that such mutational data results because an order of hydrolysis or occupancy of bound and unbound nucleotide around the ring dictates reaction velocities and if a particular subunit is mutant that might “hang up” or even kill the entire activity, an extreme case being the sequential model for hydrolysis as proposed for the T7 helicase (Crampton et al., 2006). The details of the Eadie-Hofstee plot for both the Drosophila MCM2-7 complex and CMG indicate cooperative physical changes around the MCM ring as ATP binding occupies more subunits. Significantly, the different shapes of these plots, comparing MCM2-7 and CMG, are consistent with the model that Cdc45 and GINS alter the relationships and interactions of the MCM subunits for hydrolysis. The ATPase activity of the CMG complex is not stimulated by DNA (data not shown), a result not anticipated for AAA+ helicases, and this might be explained if the ATPase active sites already have an optimal conformation in the CMG.
We report that the association of GINS and Cdc45 to the MCM2-7 significantly improves the DNA affinity of the resulting complex to both forked substrates and single-stranded DNA relative to the MCM complex alone. Atomic models derived from crystallographic and electron microscopy studies of the archeal MCM complexes provide important clues as to how the eukaryote MCM2-7 may bind DNA (for review, see Costa and Onesti, 2009; Sakakibara et al., 2009). The six archeal MCM monomers form a two-tiered structure where the C-terminal AAA+ domains form a ring upon which the N-terminal domains are stacked forming a second ring. While important details of action may be divergent between all of these super-family VI AAA+ protein complexes, it is clear both from the structures and mutational data, that an internal channel provides a possible path for at least one strand of the DNA duplex. Furthermore, translocation of substrate through the channel is provided in part by beta-hairpins in the ATP hydrolyzing AAA+ domain that touch DNA during the power stroke that moves DNA through the channel. That the Cdc45 and GINS-dependent enhancement of DNA affinity by eukaryotic MCM2-7 is manifest upon binding of nucleotide, leads us to posit that this effect is caused in part because of a more effective alignment of the internal beta-hairpin loops in the ATP-binding AAA+ domain and potentially other domains connected internally to the AAA+ domain. This enhanced affinity does not require the ATP hydrolysis dependent translocation per se as the enhancement was measured with a non-hydrolysable nucleotide. The improved binding of DNA substrate is therefore coupled in some way to the same conformational changes induced by the GINS and Cdc45 that enhance the rates of ATP hydrolysis by MCMs.
The GINS complex binds to DNA in EMSA experiments and our data would also be consistent with an indirect effect of the MCM ring on how GINS directly touches DNA in the CMG. For example, ATP binding by the MCM subunits might induce a change in the GINS conformation to enhance the GINS affinity for a fork. We do not favor this idea as other data to be published elsewhere indicate that the GINS binds DNA in the CMG similar to its binding as a tetramer. We have analyzed by mass spectrometry methods the changes in the hydrogen/deuterium exchange patterns of the GINS proteins in solution with or without DNA and compared such protections plus or minus DNA in the CMG and found these patterns to be very much the same and unaffected by bound nucleotide (Pesavento, Ilves, Petojevic, and Botchan; unpublished observations). The deletion of Psf3 C-terminal tail has no measureable effect upon the DNA binding of the free GINS but does have effect on the helicase and DNA binding activity of the CMG and thus may have an indirect role on the DNA binding properties of the CMG. For example, this domain may allow for proper positioning of the GINS along an exit site for one or another tail on the DNA fork as it leaves the central cavity.
Enhanced stability and structural asymmetry of the CMG
The MCM2-7 hexamer dissociates into stable incomplete sub-combinations of the subunits in high salt conditions, particularly in chloride buffers, which was the reason why we isolated the MCM2-7 complex in acetate buffers. In contrast, both the recombinant CMG complex (our present data) and the CMG purified from Drosophila embryo extracts can survive multiple washes with buffers containing 1M KCl (Moyer et al., 2006). We have detected all the pair-wise interactions between the Cdc45, GINS, and MCM2-7 components of the CMG, but none of the corresponding incomplete subcomplexes is as stable as the full CMG. We attribute this stability and consistent 1:1 stoichiometric association of the MCMs within the CMG to the associations with the ancillary factors GINS and Cdc45. We suggest that the presence of both Cdc45 and GINS is required to stabilize the MCM2-7 ring and perhaps these proteins provide some sort of ‘backbone’, which helps to properly align individual MCM subunits for the efficient functioning of the helicase motor.
The results of our helicase assays with circular substrates indicate that the interaction of MCM2-7 with Cdc45 and GINS in CMG enables its loading on circular DNA. This in turn implies that the MCM2-7 ring has the ability to open and pass a DNA strand into its central channel and after binding track along the M13 circle only in the context of the CMG. The presence of such a ‘gate’ between the MCM2 and 5 subunits has been proposed earlier based on DNA binding studies with S. cerevisae MCM2-7 (Bochman and Schwacha, 2007; Bochman and Schwacha, 2008). Such a gate that may both open and close on templates may be a common feature of all eukaryotic CMG complexes.
The specific interaction of GINS with MCM4 that we identified in pull-down assays places GINS binding to a specific region on the MCM2-7 ring. Cdc45 interacts with both GINS and MCM4, and we speculate that all of these interactions are required for the stability of CMG and uniquely place Cdc45 close to the GINS. Interestingly, this situates these ancillary factors opposite to the putative MCM2-5 ‘gate’. This asymmetry also correlates well with the effects of Walker A mutations on the helicase activity of CMG, as the mutations with weakest effect localized around the MCM4 subunit and the ones with strongest effect around the MCM5 subunit on the opposite sides of the MCM ring. This sensitivity to mutation may result because of the precarious organization of the interactions at this part of the ring or because of a hidden sequential movement to helicase activity starting at a unique position in the CMG. Unfortunately, we could never measure a sufficiently robust helicase activity from the MCM2-7 complex to ask if associations in the CMG change the mutational sensitivity that might exist in the MCM2-7 complex.
Helicases in various viral and prokaryote systems provide pivotal interaction functions linking DNA unwinding to leading strand synthesis and both Cdc45 and GINS have been shown to interact – even though perhaps not always directly - with several replication factors at a fork (Labib and Gambus, 2007). GINS and Cdc45 may therefore translate the functionally relevant intrinsic asymmetry of the heterohexameric MCM2-7 complex into the coordinated interactions between the helicase motor, forked template DNA, and other enzymatic and regulatory complexes in the replisome.
Activation of unwinding at a pre-initiation complex
Over-expression of the CMG subunits in the recombinant system likely by-passed the pathway normally required to create the active helicase at a bona fide origin in vivo. In this context it is important to note that we have found by mass-spectrometry analysis that the amino terminal tails of MCM 2 and 4 are not hyper-phosphorylated in the CMG and the modified residues mapped are the same in the free pool of MCMs and in the CMG complex (Pesavento and Botchan, unpublished observations). Under the more stringent conditions of a cycling cell, the phosphorylation of residues in such amino-terminal tails by regulatory CDK and DDK kinases that occurs specifically at a pre-replication complex may be critical to allow GINS and Cdc45 to associate appropriately with the MCMs (Francis et al., 2009). Furthermore, at limiting levels of the ancillary factors found in vivo, chaperones and the precise conformations of the MCMs bound at a pre-RC may allow activation of the unwinding activity. What information do we now have regarding the molecular disposition of the MCMs on duplex in the pre-RC?
Recent work has revealed that ORC, Cdc6 and Cdt1 can load cooperatively onto DNA two copies of the MCM ring where the duplex passes through the double hexamer (Evrin et at., 2009; Remus et al., 2009). Such assisted loading of MCM2-7 rings onto duplex DNA is carried out in a manner that cannot be achieved with the CMG alone, as the CMG has in our hands no detectable affinity for duplex (Figure S5E). Once bound to the fork in an ATP dependent conformation, the CMG has no measureable off rate in our assays (data not shown), while the loaded double hexameric MCM2-7 was reported to slide along the duplex DNA in an ATP independent manner. Thus relative to the “licensed” MCM2-7 double hexamer, the MCM ring of the active CMG helicase may have a narrower channel or DNA binding residues inside the MCM rings may be positioned in functionally different ways.
We suggest that two events might follow the G1 loading of the ‘latent’ MCMs to allow for a switch to unwind the template DNA (Figure 7). These events are dependent upon the action of cell cycle kinases and involve direct phosphorylation of MCM proteins that may expose binding surfaces for GINS and Cdc45 association. We find that the CMG behaves as a discrete monomeric complex in solution and importantly on a forked DNA - its preferred binding partner - no indication of double hexamer formation was detected in EMSAs. Thus in a concerted manner, the binding of ancillary factors may break apart the MCM2-7 double hexamers leading to the formation of substrates for fork assembly. In the hypothetical scheme suggested, the internal channel of MCM ring closes and deforms the duplex DNA by constricting, brought about by the binding of the GINS and Cdc45, which might produce a pre-melted or melted DNA conformation within the channel. This ‘scrunching’ (Kapanidis et al., 2006) of the channel coupled perhaps to subtle changes in the rotational symmetry of the individual MCM subunits may convert to a state where the internal channel binds tightly to a single tracking strand. We further suggest an intriguing possibility that the proposed “gate” in the MCM ring may function in the putative isomerization event allowing for the topological conversions required to achieve the strand arrangements diagrammed in the model figure. A single GINS complex uniquely situated on the ring may use its affinity to DNA to assist in guiding the displaced strand after it is moved out of the internal channel. This geometry with regard to the tracking and displaced strand might also ensure optimal positioning relative to both the active helicase motor and other members of the replisome complex that associate with the CMG. This heuristic model leads to a conventional fork structure, where the CMG would operate naturally on the type of substrates that other well-defined helicases work, and makes predictions that are readily tested.
Figure 7. Activation of the MCM helicase requires binding of GINS and Cdc45.
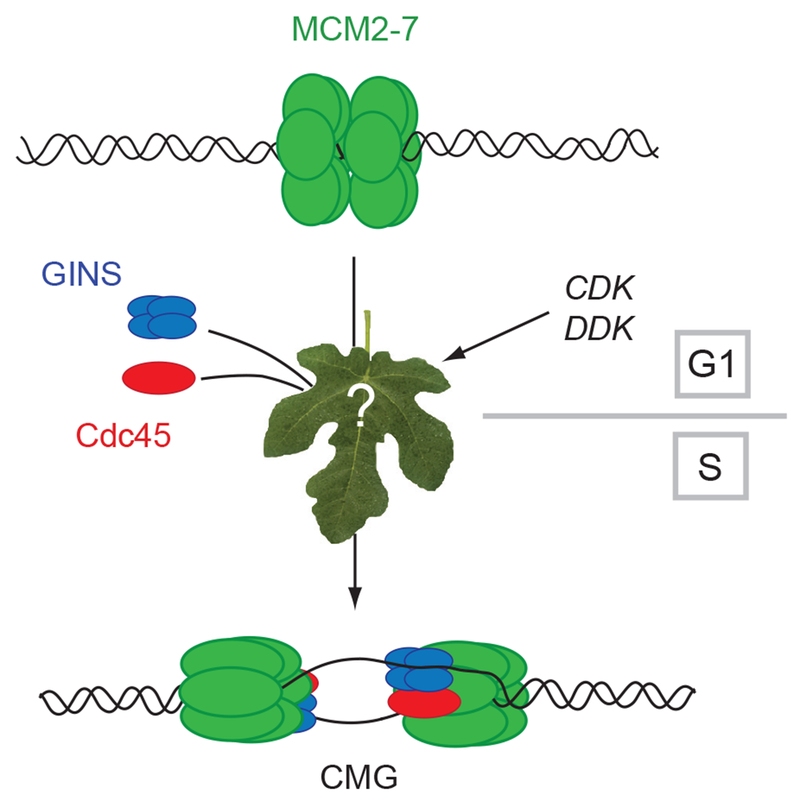
Double hexamers of MCM2-7 are loaded to DNA in G1 and the best available data suggests that these MCM rings are probably on pathway for DNA replication. The subsequent events require CDK regulated formation of potential chaperone complexes such as those associated with Dpb11 (See Botchan, 2007, for review) and the DDK modification of the amino-terminal tails of the MCMs. In the model discussed, these events lead to: A) release of the double hexamers to enable binding of activating GINS and Cdc45 proteins, and B) coupled to the binding of the activators, the MCMs isomerize so that DNA strands are organized to create a bidirectional replication fork at the time of initiation.
Experimental procedures
Protein purification and immunoprecipitation (IP) experiments
All the proteins were purified using the baculovirus expression system. The details for the virus construction, protein purification, glycerol gradient centrifugation and IP experiments are included in Supplemental Experimental Procedures.
ATPase assays
The measurements of initial ATPase rate were performed with 200-400 fmol of wt or mutant CMG in 20 μl of buffer A (25 mM Hepes pH 7.6, 10% glycerol, 50 mM sodium acetate, 10 mM magnesium acetate, 0.2 mM PMSF, 1 mM DTT, 250 μg/ml insulin) in the presence of desired concentration of cold ATP (GE Healthcare) spiked with γ-32P ATP (MP Biomedicals, 1 μCi in the reactions with highest ATP concentration). The reactions were incubated for 20 minutes at 30°C and stopped by adding 2 μl of 0.5 M EDTA. 1.1 μl samples were spotted on cellulose-PEI thin layer chromatography plates and developed in 0.4 M LiCl / 1 M formic acid. For MCM2-7 assays, the reactions were performed in 10 μl volume with 4 pmol of protein. The nonlinear regression analysis of the data was performed with GraphPad Prism software.
DNA helicase and EMSA assays
The preparation of M13 based helicase substrate has been described previously (Moyer et al., 2006). The forked DNA probe corresponds to the 233+235 substrate described in (Bochman and Schwacha, 2007) and prepared following modified protocol (see Supplemental Experimental Procedures).
Both the helicase and EMSA assays were performed in buffer A. The helicase reactions were done with 5 fmol DNA and 0.3 mM ATP in 10 μl of total volume for 30 minutes at 30°C, then stopped by adding SDS to 0.1% and EDTA to 20 mM and products were separated on 8% acrylamide / TBE / 0.1%SDS gel. The EMSA reactions were done with 20 fmol of DNA in 15 μl total volume, 1 mM ATP-γS was added to the MCM2-7 and CMG reactions unless indicated otherwise. The reactions were incubated at 30°C for 20 minutes and loaded on 4% acrylamide (60:1 acrylamide:bis-acrylamide) with 5% glycerol gel run on 4°C in glycine buffer supplemented with 6 mM magnesium acetate.
Supplementary Material
Acknowledgements
We thank Anthony T. lavarone for help with mass spectrometry analysis and members of the Botchan lab for their support, especially Stephen Moyer for advice and discussions and Alyssia Oh for technical assistance in the early stages of this project. This work was supported by National Institutes of Health grants CA R37-30490 to M.R.B. and NRSA GM821972 to J.J.P, also by a PhD fellowship from the Boehringer Ingelheim Fonds to T.P.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aparicio OM, Weinstein DM, and Bell SP (1997). Components and dynamics of DNA replication complexes in S. cerevisiae: redistribution of MCM proteins and Cdc45p during S phase. Cell 91, 59–69. [DOI] [PubMed] [Google Scholar]
- Aparicio T, Ibarra A, and Mendez J (2006). Cdc45-MCM-GINS, a new power player for DNA replication. Cell Div 1, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell SP, and Dutta A (2002). DNA replication in eukaryotic cells. Annual Review of Biochemistry 71, 333–374. [DOI] [PubMed] [Google Scholar]
- Bochman ML, and Schwacha A (2007). Differences in the single-stranded DNA binding activities of MCM2-7 and MCM467: MCM2 and 5 define a slow ATP-dependent step. J. Biol. Chem 282, 33795–33804. [DOI] [PubMed] [Google Scholar]
- Bochman ML, and Schwacha A (2008). The MCM2-7 complex has in vitro helicase activity. Mol. Cell 31, 287–293. [DOI] [PubMed] [Google Scholar]
- Boskovic J, Coloma J, Aparicio T, Zhou M, Robinson CV, Mendez J, and Montoya G (2007). Molecular architecture of the human GINS complex. EMBO Reports 8, 678–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botchan M (2007). Cell biology: a switch for S phase. Nature 445, 272–274. [DOI] [PubMed] [Google Scholar]
- Costa A, and Onesti S (2009). Structural biology of MCM helicases. Crit Rev Biochem Mol Biol 44, 326–42. [DOI] [PubMed] [Google Scholar]
- Crampton DJ, Mukherjee S, and Richardson CC (2006). DNA-induced switch from independent to sequential dTTP hydrolysis in the bacteriophage T7 DNA helicase. Mol. Cell 21, 165–174. [DOI] [PubMed] [Google Scholar]
- Crevel G, Ivetic A, Ohno K, Yamaguchi M, and Cotterill S (2001). Nearest neighbour analysis of MCM protein complexes in Drosophila melanogaster. Nucleic Acids Res 29, 4834–4842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey MJ, Indiani C, and O’Donnell M (2003). Reconstitution of the Mcm2-7p Heterohexamer, subunit arrangement, and ATP site architecture. J. Biol. Chem 278, 4491–4499. [DOI] [PubMed] [Google Scholar]
- Enemark EJ, and Joshua-Tor L (2008). On helicases and other motor proteins. Curr Opin Struct Biol 18, 243–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evrin C, Clarke P, Zech J, Lurz R, Sun J, Uhle S, Li H, Stillman B, and Speck C (2009). A double-hexameric MCM2-7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proc. Natl. Acad. Sci. USA 106, 20240–20245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsburg SL (2004). Eukaryotic MCM proteins: Beyond replication initiation. Microbiol. Mol. Biol. Rev 68, 109–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis LI, Randell JC, Takara TJ, Uchima L, and Bell SP (2009). Incorporation into the prereplicative complex activates the Mcm2-7 helicase for Cdc7-Dbf4 phosphorylation. Genes Dev 23, 643–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambus A, Jones RC, Sanchez-Diaz A, Kanemaki M, van Deursen F, Edmondson RD, and Labib K (2006). GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat. Cell Biol 8, 358–U341. [DOI] [PubMed] [Google Scholar]
- Ishimi Y (1997). A DNA helicase activity is associated with an MCM4, -6, and -7 protein complex. J Biol Chem 272, 24508–24513. [DOI] [PubMed] [Google Scholar]
- Kanemaki M, Sanchez-Diaz A, Gambus A, and Labib K (2003). Functional proteomic identification of DNA replication proteins by induced proteolysis in vivo. Nature 423, 720–724. [DOI] [PubMed] [Google Scholar]
- Kapanidis AN, Margeat E, Ho SO, Kortkhonjia E, Weiss S, and Ebright RH (2006). Initial transcription by RNA polymerase proceeds through a DNA-scrunching mechanism. Science 314, 1144–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelman Z, Lee JK, and Hurwitz J (1999). The single minichromosome maintenance protein of Methanobacterium thermoautotrophicum Delta H contains DNA helicase activity. Proc. Natl. Acad. Sci. USA 96, 14783–14788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg A, and Baker T (1992). DNA Replication (New York, W.H. Freeman and Co.). [Google Scholar]
- Kubota Y, Takase Y, Komori Y, Hashimoto Y, Arata T, Kamimura Y, Araki H, and Takisawa H (2003). A novel ring-like complex of Xenopus proteins essential for the initiation of DNA replication. Genes Dev 17, 1141–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labib K, and Gambus A (2007). A key role for the GINS complex at DNA replication forks. Trends in Cell Biol 17, 271–278. [DOI] [PubMed] [Google Scholar]
- Moyer SE, Lewis PW, and Botchan MR (2006). Isolation of the Cdc45/Mcm2-7/GINS (CMG) complex, a candidate for the eukaryotic DNA replication fork helicase. Proc. Natl. Acad. Sci. USA 103, 10236–10241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacek M, Tutter AV, Kubota Y, Takisawa H, and Walter JC (2006). Localization of MCM2-7, Cdc45, and GINS to the site of DNA unwinding during eukaryotic DNA replication. Mol Cell 21, 581–587. [DOI] [PubMed] [Google Scholar]
- Pacek M, and Walter JC (2004). A requirement for MCM7 and Cdc45 in chromosome unwinding during eukaryotic DNA replication. EMBO J 23, 3667–3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remus D, Beuron F, Tolun G, Griffith JD, Morris EP, and Diffley JF (2009). Concerted loading of Mcm2-7 double hexamers around DNA during DNA replication origin licensing. Cell 139,719–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakibara N, Kelman LM, and Kelman Z (2009). Unwinding the structure and function of the archaeal MCM helicase. Mol Microbiol 72, 286–296. [DOI] [PubMed] [Google Scholar]
- Schwacha A, and Bell SP (2001). Interactions between two catalytically distinct MCM subgroups are essential for coordinated ATP hydrolysis and DNA replication. Mol. Cell 8, 1093–1104. [DOI] [PubMed] [Google Scholar]
- Sclafani RA, and Holzen TM (2007). Cell cycle regulation of DNA replication. Annual Review of Genetics 41, 237–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter DF, Ying CY, and Gautier J (2000). The intrinsic DNA helicase activity of Methanobacterium thermoautotrophicum delta H minichromosome maintenance protein. J Biol Chem 275, 15049–15059. [DOI] [PubMed] [Google Scholar]
- Singleton MR, Dillingham MS, and Wigley DB (2007). Structure and mechanism of helicases and nucleic acid translocases. Annu Rev Biochem 76, 23–50. [DOI] [PubMed] [Google Scholar]
- Stillman B (2005). Origin recognition and the chromosome cycle. FEBS Letters 579, 877–884. [DOI] [PubMed] [Google Scholar]
- Takayama Y, Kamimura Y, Okawa M, Muramatsu S, Sugino A, and Araki H (2003). GINS, a novel multiprotein complex required for chromosomal DNA replication in budding yeast. Genes Dev 17, 1153–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tercero JA, Labib K, and Diffley JFX (2000). DNA synthesis at individual replication forks requires the essential initiation factor Cdc45p. EMBO J 19, 2082–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traut TW (1994). Physiological concentrations of purines and pyrimidines. Mol Cell Biochem 140, 1–22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.