

Abstract
Patients with acute myocardial infarction receive a P2Y12 receptor antagonist prior to reperfusion, a treatment that has reduced, but not eliminated, mortality, or heart failure. We tested whether the caspase −1 inhibitor VX-765 given at reperfusion (a requirement for clinical use) can provide sustained reduction of infarction and long- term preservation of ventricular function in a pre-clinical model of ischemia/reperfusion that had been treated with a P2Y12 receptor antagonist. To address, the hypothesis open-chest rats were subjected to 60-min left coronary artery branch occlusion/120-min reperfusion. Vehicle or inhibitors were administered intravenously immediately before reperfusion. With vehicle only, 60.3 ± 3.8% of the risk zone suffered infarction. Ticagrelor, a P2Y12 antagonist, and VX-765 decreased infarct size to 42.8 ± 3.3 and 29.2 ± 4.9%, respectively. Combining ticagrelor with VX-765 further decreased infarction to 17.5 ± 2.3%. Similar to recent clinical trials, combining ticagrelor and ischemic postconditioning did not result in additional cardioprotection. VX-765 plus another P2Y12 antagonist, cangrelor, also decreased infarction and preserved ventricular function when reperfusion was increased to 3 days. In addition, VX-765 reduced infarction in blood-free, isolated rat hearts indicating at least a portion of injurious caspase-1 activation originates in cardiac tissue. While the prodrug VX-765 only protected isolated hearts when started prior to ischemia, its active derivative VRT-043198 provided the same amount of protection when started at reperfusion, indicating that even in blood-free hearts, caspase- 1 appears to exert its injury only at reperfusion. Moreover, VX- 765 decreased circulating IL-1, prevented loss of cardiac glycolytic enzymes, preserved mitochondrial complex I activity, and decreased release of lactate dehydrogenase, a marker of pyroptosis. Our results are the first demonstration of a clinical-grade drug given at reperfusion providing additional, sustained infarct size reduction when added to a P2Y12 receptor antagonist.
Keywords: Cardioprotection, Caspase-1, Ischemia/reperfusion injury, Myocardial infarction, P2Y12 receptor antagonist, VX-765
Introduction
Patients with acute myocardial infarction (AMI) are currently treated with percutaneous coronary intervention (PCI) to limit myocardial cell death by restoration of coronary blood flow and P2Y12 receptor antagonists to maintain patency of the deployed stents. Despite the cardioprotective benefits of PCI combined with P2Y12 receptor antagonists, approximately one out of four patients currently treated for a left anterior descending coronary artery (LAD) thrombus will either die or develop chronic heart failure in the following year [8, 16, 19]. Attempts to translate interventions that reduce infarct size in pre-clinical models of ischemia/reperfusion (I/R) have failed to improve outcomes in patients with AMI treated with PCI [7, 9, 10, 14 ], suggesting that animal hearts might be an inappropriate model of the human heart. However, an alternate explanation could be that those animal protocols did not include any of the comedications AMI patients concurrently receive, and this might have distorted the results. In the present study, we have made our rat model more clinically relevant by including P2Y12 antagonists which in animal models confer direct protection against infarction [4, 15, 37, 39, 41, 42] and, therefore, might mask protection from an additional intervention.
Growing evidence suggests the inflammasome- caspase-1 axis contributes to inflammation-induced cell death during ischemia/reperfusion (I/R) [11, 18, 22, 26, 31, 32, 35, 36]. Inflammasomes are intracellular danger-sensing complexes consisting of nucleotide oligomerization domain-like receptors (NLRs) that trigger processing of pro-caspase-1 into its active form, caspase-1 (Fig. 1) [24, 25]. One such NLR, the NLR Family Pyrin Domain Containing 3 (NLRP3), assembles its inflamma-some in response to damage -associated molecular patterns (DAMPs) [24], some of which are generated during I/R. Beyond the canonical role of caspase-1 in activating inflammation by converting pro -interleukins IL- 1 and IL-18 into their active forms, this cysteine protease cleaves other proteins including glyceraldehyde-3-phosphate dehydrogenase (GAPDH), mitochondrial enzymes affecting ROS generation, and gasdermin D (the executioner of pyroptotic cell death) [23, 33, 34].
Fig. 1.
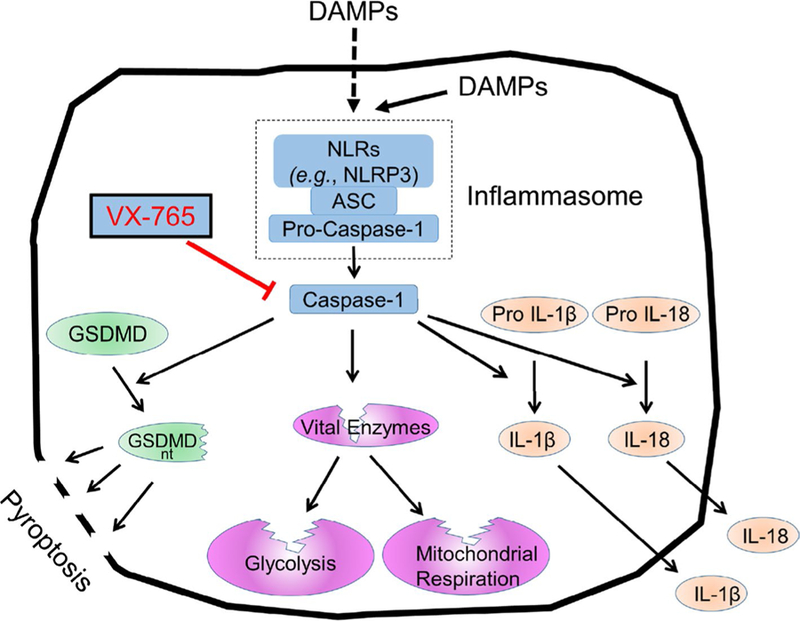
Schematic representing inflammasome activation during I/R. VX-765 blocks caspase-1 activation. ASC apoptosis-associated speck-like protein containing a caspase recruit-ment domain, DAMP damage-associated molecular pattern, GSDMD gasdermin D, GSDMD nt N-terminal fragment of GSDMD, IL-1β, 18 interleu-kin 1, 18, NLR nucleotide oligomerization domain-like receptor
Involvement of caspases in myocardial I/R injury was first implicated in a study in which the pan-caspase inhibitor zVAD reduced infarct size in rats [28]. Evidence for involve-ment of caspase-1 in I/R injury came from studies using the selective inhibitor YVAD, which protected human atrial explants from simulated I/R [31], and reduced myocardial infarct size in rabbits [18]. Furthermore, caspase-1−/− mice had reduced infarct size and attenuated left ventricular remodeling after I/R [11]. Conversely, caspase-1 overexpression in mice increased infarct size [ 35]. However, studies targeting the NLRP3 inflammasome have yielded conflicting results. Acute pharmacologic inhibition of NLRP3 has been cardioprotective, while genetic knockouts have not [20, 26, 32, 36]. A possible explanation for these disparate findings is genetic deletion of NLRP3 and leads to compensatory upregulation of other NLRs that can activate caspase-1 during I/R. Similarly, inhibiting downstream targets of cas-pase-1 such as IL-1β, IL-18, or their receptors ignores any parallel contributions from other potentially injurious targets of caspase- 1 which results in pyroptosis or loss of vital enzymes [1, 23, 33, 34]. Thus, caspase-1 may be at a critical choke point for eliciting injury during I/R.
We recently found that a highly selective caspase-1 inhibitor, VX-765 [38], could further reduce infarct size in rats when administered prior to onset of ischemia along with the P2Y12 inhibitor cangrelor infused immediately before reperfusion [ 40]. While interventions such as hypothermia or cariporide can further protect rat hearts receiving a P2Y12 receptor inhibitor from I/R injury, they only do so when administered prior to ischemia [39]. However, pretreating AMI patients presenting with ischemia in progress is not possible. To be clinically useful, VX-765 must offer protection when given at reperfusion, and to do that it must target a reperfusion injury. Two additional shortcomings of our VX-765 pretreatment study were that we did not determine whether VX-765- mediated protection against infarction is long-lasting, or if the salvaged tissue would be contractile. Thus, in the present study, we assessed the effect of VX-765 treatment at reperfusion on infarct size after 120 min of rep-erfusion and on infarct size and ventricular function after 3 days of reperfusion. In addition to these critical questions, we explored potential mechanisms underlying VX-765-mediated myocardial protection.
Materials and methods
The University of South Alabama Institutional Animal Care and Use Committee approved the animal protocols, and all conformed to published guidelines [29].
The open-chest, LAD coronary artery occlusion, 60-min regional ischemia/120-min reperfusion protocol
Aged, male Sprague–Dawley rats (retired breeders) weighing ~ 500 g were anesthetized with 100 mg/kg intraperitoneal sodium pentobarbital and supplemented as needed to maintain a surgical plane [39]. Rectal temperature was maintained between 37–38 °C with a heating pad. Animals received positive pressure ventilation with 100% oxygen. Catheters were inserted into a carotid artery for measurement of blood pressure, and into a jugular vein for blood sampling and drug administration. The chest was opened in the fourth left intercostal space, and the heart exposed. A suture was placed around the LAD coronary artery to form a snare that was tightened to occlude the artery for 60 min, and then loosened to reperfuse the artery. Rats were euthanized by exsanguination as the heart was removed at the end of the experiment.
Infarct size measurement
The excised heart was mounted on a Langendorff apparatus to perfuse the aortic root with normal saline. The LAD was re-occluded with the snare, and fluorescent microspheres (2–9µm) infused into the aortic perfusate to identify the previously ischemic (risk) zone as the non- fluorescent region. The heart was fast-frozen, sectioned into 1-mm-thick slices, and incubated in triphenyltetrazolium chloride (TTC) at 37 °C to stain live tissue deep red. The areas of infarct (unstained by TTC) and ischemic risk zone (non-fluorescent) were determined in a high-resolution digital photograph of the slices with Image-J software. Infarct was differentiated from red-stained living heart muscle by adjusting Image-J’s color threshold. The total volumes of infarct and risk were calculated as the sum of their areas times the slice thick-ness, and infarct size was expressed as a percent of the total ischemic volume for each heart.
Determining intravenous doses for VX-765, cangrelor, and ticagrelor
Sixteen mg/kg VX-765 gives a dose -independent (maximal) protection when administered 30 min prior to the onset of ischemia [40]. However, in pilot studies, this dose gave less protection when given 5 min prior to reperfusion. Although the pharmacokinetic profile of VX-765 has not been published, we rationalized that the failure may have been because there was incomplete activation of VX-765 which is a prodrug that must be activated by cellular esterases [38 ]. Increasing the dose at reperfusion to 32 mg/kg restored the salvage to a level similar to the dose- independent response with pretreatment. We reasoned that the higher dose increased the amount of active drug available during the critical time when caspase-1 caused cell death. Thus, we tested an intravenous bolus of 32 mg/kg of VX-765. Can-grelor was administered as previously determined [39] and ticagrelor as reported by Ye et al. [42].
Protocols for the 60-min regional ischemia/120-min reperfusion open-chest protocol
We evaluated eight groups of rats ( ≥ 5 animals per group): (1) DMSO, vehicle for VX-765 (1.0 mL × kg body weight intravenous bolus with final volume diluted to 0.9 mL with normal saline); (2) cangrelor intravenous bolus (60 µg/kg) 10 min before reperfusion followed by a continuous infusion (6 µg/kg/min) for 2 h; (3) ticagrelor intraperitoneal bolus (30 mg/kg) 10 min before reperfusion; (4) VX-765 intravenous bolus (32 mg/kg) 5 min before reperfusion; (5) combination of cangrelor and VX-765 as described above; (6) combination of ticagrelor and VX-765 as described above; (7) ischemic postconditioning (IPOC) with three cycles of 30 s of reperfusion followed by 30 s of reocclusion; and (8) IPOC plus cangrelor as described above.
The open-chest LAD coronary artery occlusion, 60-min regional ischemia/3-day reperfusion protocol
Rats were anesthetized with sodium pentobarbital as described above for open-chest rats. Ultrasonography was performed on spontaneously breathing rats with a 60-MHz probe (Vevo 770, FUJIFILM VisualSonics Inc., Toronto, ON, Canada). Six images at the level of the posterior papillary muscle were recorded (short and long axes, three images each). Long- and short-axis images of the left ventricle were displayed, and an M-mode cursor was placed across the images to record motion of the anterior and posterior walls and measure end -systolic and end-diastolic chamber dimensions. Fractional shortening was calculated as the dif-ference between end-systolic and end-diastolic dimensions normalized for end-diastolic dimension, and averaged over six consecutive cardiac cycles.
Upon completion of ultrasonography, animals were intu-bated for ventilation with 100% oxygen, and surgery under semi -sterile conditions was performed as described above. Two groups were evaluated: (1) DMSO (as described above, six surviving rats) and (2) cangrelor combined with VX-765 (as described above, five surviving rats). After 60 min of coronary occlusion, the ligature was released to initiate reperfusion. Following chest closure, the animals were allowed to recover, and received buprenorphine (0.01 mg/kg subcuta-neously) to provide analgesia. After 3 days of recovery, they were re-anesthetized and ultrasonography performed again. Animals were then euthanized, and infarct size measured as described above.
The isolated heart, 40-min global ischemia/90-or 120-min reperfusion protocol
To evaluate the effects of VX-765 on blood-free, isolated hearts, rats were anesthetized as described above. The heart was excised and mounted on a Langendorff apparatus within 1 min of removing it from the chest [41]. The coronary arteries were retroperfused with Krebs buffer through the aortic root, and a latex balloon was placed in the lumen of the left ventricle and inflated with saline to achieve 100-mmHg peak systolic pressure. After 20 min of equilibration, perfusion was stopped for 40 min to induce global ischemia. During ischemia, the heart was immersed in buffer in a water-jacketed chamber at 37 °C. After the ischemic period, perfusion was re-initiated for 120 min, and the left ventricular balloon was deflated to avoid impairment of subendocardial perfusion from increased luminal diastolic pressure should contracture occur. After 120 min of reperfusion, the balloon was re-inflated to a luminal diastolic pressure of 0 mmHg (unstressed ventricular volume), and final developed pressure was measured.
The hearts were perfused with buffer containing either DMSO (0.5 mL/L buffer, six hearts) or VX-765 (30 M final concentration, five hearts) for 10 min before ischemia and throughout reperfusion. After reperfusion (120 min), infarct size was measured with TTC staining as described above.
A second series of isolated hearts received either DMSO (four hearts) or 9 µM VRT-043765 (the active derivative of the prodrug VX-765) present during only the 90 min reperfusion period (four hearts). We collected all of the effluent during the following intervals: 0–1, 1–2, 2–4, 4–6, 6–8, 8–12, 12–16, 16–20, 20–40, 40–60, 60–80, and 80–90 min after the onset of reperfusion. It was necessary to shorten the reperfusion period for these groups to reduce the amount of VRT-043198 used.
The venous effluent was analyzed for lactate dehydrogenase (LDH) as previously described [3]. The amount of LDH released during each collection interval was calculated as the units of LDH washout/min/g of heart tissue. An independent aliquot was concentrated 20-fold using a 3000 MW cut- off ultrafilter (Millipore, Billerica, MA), and assayed for IL-1 by ELISA (ThermoFisher Scientific, Waltham, MA) [2].
The open-chest LAD occlusion, 60-min regional ischemia/30-min reperfusion protocol
The open-chest LAD occlusion protocol was modified to make measurements of heart tissue enzymes and mito-chondrial function after only 30 min of reperfusion. Because infarct size was not different between hearts experiencing 3 days or 120 min of reperfusion, we reasoned that the ultimate infarct size had already been achieved by 120 min. We chose a shorter period of reperfusion, so that we could hopefully harvest heart tissue in the process of dying. Two groups were tested with either an intrave-nous bolus of VX-765 or vehicle as described above. After 30 min of reperfusion, blood was collected for assay of IL-1 by ELISA [2]. The heart was excised and hung on a Langendorff apparatus as described above, the snared coronary artery was re-occluded, and fluorescent micro-spheres were injected into the aortic perfusate to demarcate ischemic and non-ischemic regions. Tissue from the ischemic and non-ischemic zones was harvested, fast-frozen, and stored at − 80 °C for biochemical analysis.
Mitochondrial respirometry
Some of the tissue was immediately prepared for high-res-olution respirometry (Oroboros oxygraph O2 K, Innsbruck, Austria) as previously described [30]. Different states of mitochondrial respiration and electron-transfer complexes were evaluated using the experimental protocol, as out-lined in Fig. 2.
Fig. 2.
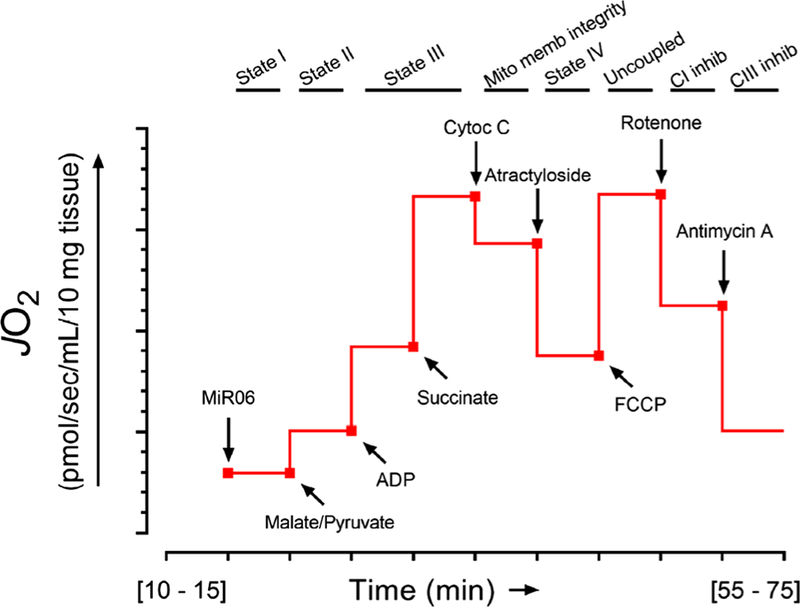
Schematic of high-resolution respirometry protocol to assess mitochondrial respiration from heart biopsies. Thin sections (~ 10 mg weight) were obtained from heart biopsies and dissected to separate the muscle fibers from each other. The tissue was suspended in sterile MiR06 buffer (MiR05 + 280 IU/mL catalase); MiR05 contained (mM): sucrose (110), potassium lactobionate (60), ethylene glycolbis( -aminoethyl ether)-N,N,N’,N’-tetraacetic acid (EGTA, 0.5, magnesium chloride (MgCl2, 3), taurine (20), monopotassium phosphate (KH2PO4, 10), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, 20) adjusted to pH 7.1 with potassium hydroxide at 37 °C and 1 g/L bovine serum albumin (fatty acid free). Tissue was per-meabilized with 1% saponin for 15 min at room temperature and transferred to individual chambers of a respirometer (Oxygraph-2 k, Oroboros Instruments, Innsbruck, Austria) containing 2-mL MiR06 for high-resolution respirometry. The linear rate of oxygen consumption (mitochondrial respiration) was measured in each chamber with continuous stirring. After 10 min of equilibration at 37 °C, oxygen flux began to be reported and the basal oxygen flux (JO2) was assessed (state I), and then, malate (1.5 mM) and pyruvate (5.0 mM) were added as electron donors for complex I followed by the addition of adenosine diphosphate (ADP, 1.0 mM) to assess states II and III. Succinate (9.5 mM) was added as electron donor for complex II to measure state III in the presence of the three electron donors coupled to complexes I and II. Specimens were then treated with cytochrome C (10 µM) to confirm integrity of the mitochondrial membranes, followed by atractyloside (50 µM) to measure state IV. Trifluoro-carbonylcyanide phenylhydrazone (FCCP, 10 µM) was added as an uncoupler to measure maximal respiration (state 3U). Respiratory ratio was calculated by dividing JO2 from state III by state IV. Rote-none (0.1 µM) was added to assess complex I activity followed by the addition of antimycin A (2.5 µM) to determine complex III activity. Overall protocol length ranged between 55 and 75 min
Western blotting
Tissue homogenates/lysates were normalized (90 µg of total protein per lane) and resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (NuPAGE; 4–12% Bis Tris-gradient gels, Invitrogen, Carlsbad, CA), transferred to nitrocellulose membranes, and analyzed by immunoblotting [2] (four rats/group). A total of four gels (labeled Gel 1 –4) were used to resolve all of the samples collected. After transfer, membranes were cut into strips representing desired molecular weight ranges based on a protein standard loaded in the far left lane of each gel [denoted as molecular weight marker (MWM) in kDa], and probed for protein levels by Western blotting. Primary antibodies against cas-pase-1, GAPDH, aldolase-A, and hexokinase II (Santa Cruz Biotechnology, Dallas. TX) were used. Protein transfer was assessed by staining membranes with BLOT Fast Stain (G Biosciences, St. Louis, MO), and total protein levels determined by fluorescence (Odyssey, LI-COR, Lincoln, NE). Total signal from each lane was used to normalize all immunoblot data.
Statistics
Prism v6.1 (GraphPad, La Jolla, CA) was used for all analyses. Data are reported as mean ± standard error. Data were assessed for normality before analysis. Parametric data were evaluated using two-tailed unpaired t test (two groups) or one- or two-way analysis of variance (> 2 groups). Post-hoc analysis was performed with Tukey’s test. Differences with P ≤ 0.05 were considered significant.
Results
Administration of VX- 765 and a P2Y12 receptor antagonist just before reperfusion is highly cardioprotective.
Discovery of new clinically applicable treatments to reduce infarct size following I/R hinges on testing their efficacy in combination with guideline-mandated P2Y12 receptor antagonists. Open-chest rats were subjected to in situ 60-min regional ischemia/120-min reperfusion, and inter-ventions were administered shortly before the onset of reperfusion. In the DMSO vehicle-treated group, 60.3 ± 3.8% of the risk (ischemic) zone suffered infarction (Fig. 3). When administered individually 10 min before reperfusion, P2Y12 receptor antagonist ticagrelor reduced infarction to 42.8 ± 3.3% of the risk zone (Fig. 3). Cangrelor as we previously reported [40] resulted in 43.8 ± 2.4% infarction in the same model which was virtually identical to that obtained herein with ticagrelor. This protection by P2Y12 inhibitors is consistent with the previous reports [37, 40–42]. The ticagrelor group is analogous to patients undergoing PCI that receive a P2Y12 receptor inhibitor. Although the cangrelor data were previously reported [40 ], this group was studied within the same 4 -month interval and by the same investigators as the new groups revealed here and thus belong to the same cohort. The previously presented data are clearly identified in the figure. When VX-765 was administered 5 min before reperfusion or ischemic postconditioning initiated immediately after reperfusion (three cycles of 30-s reperfusion/30-s occlusion), infarction was reduced to 29.2 ± 4.9 and 39.8 ± 4.3%, respectively (Fig. 3).
Fig. 3.
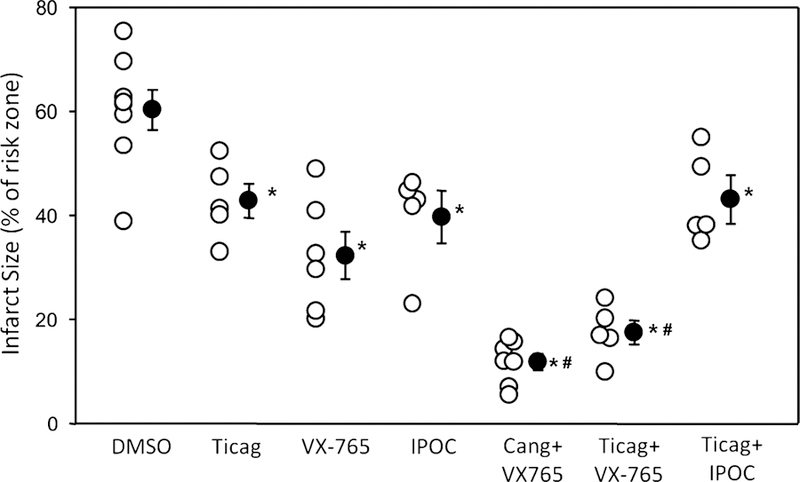
Administration of VX-765 and a P2Y12 receptor antagonist at the onset of reperfusion decreases infarct size. Infarct size as a percentage of risk zone in rats subjected to 60-min regional ischemia/120-min reperfusion. Rats were treated 10 min prior to reperfusion with vehicle (DMSO), ticagrelor (Ticag), and 5 min before reperfusion with VX-765. Ischemic postconditioning (IPOC) was initiated immediately after reperfusion. In other groups, Cang and VX-765, Ticag and VX-765, Ticag and IPOC interventions were combined. Open circles represent individual data points; closed circles designate mean ± SEM. P < 0.001 for one-way ANOVA of all groups. *P 0.02 vs vehicle (DMSO), #P < 0.001 vs ticagrelor alone
When administered in combination, VX-765 and either cangrelor or ticagrelor provided strikingly superior protection when compared to either platelet inhibitor alone. Infarction was further reduced to 11.9 ± 1.9 and 17.5 ± 2.3% of the risk zone, respectively (Fig. 3). Similar to observations in recent clinical trials [7, 9, 10], ischemic postconditioning failed to provide additional protection when combined with the P2Y 12 antagonist ticagrelor in our experiments. There were no significant differences in either baseline heart rate or blood pressure between groups.
VX-765 and cangrelor administration just before reperfusion provides sustained reduction of infarct size and long-term preservation of ventricular function
We next determined whether the cardioprotective effect of combining VX-765 and the P2Y12 receptor antagonist cangrelor was long-lasting. To this end, we modified the in situ I/R protocol, whereby after 60 min of ischemia, drug treatment, and 2 h of reperfusion, the chest was closed and the rat was allowed to recover. Ventricular function was measured before infarction and again after 3 days of reperfusion which is long enough for mechanical stunning to subside, but not long enough for significant post-infarction remodeling. Infarct size was also assessed at 3 days. In the DMSO vehicle-treated group, 63.8 ± 4.8% of the risk (ischemic) zone suffered infarction (Fig. 4a). VX-765 with cangrelor reduced infarction of the risk zone to 17.7 ± 1.4% (Fig. 4a). The infarct sizes observed in acute (120-min reperfusion, Fig. 3) and chronic (3-day reperfusion) protocols were similar suggesting the majority of cell death occurs during the first 120 min of reperfusion, and the combination of a single bolus of VX-765 and 120 -min cangrelor infusion is sufcient to produce sustained reduction of infarction.
Fig. 4.
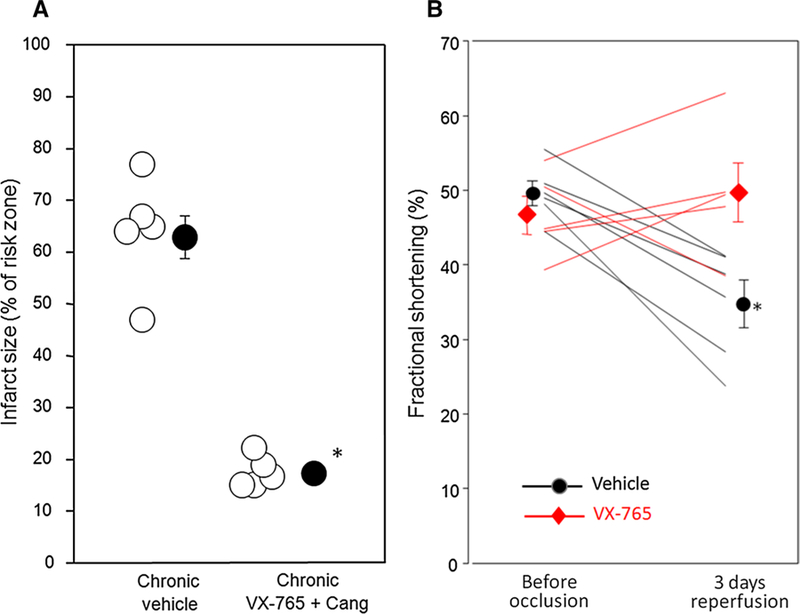
Administration of VX-765 and a P2Y12 receptor antagonist at the onset of reperfusion results in sustained cardioprotection and preservation of left ventricular function 3 days post-reperfusion. a Infarct size as a percentage of risk zone in rats subjected to 60-min regional ischemia/3-day reperfusion, and treated with vehicle (DMSO) or the combination of cangrelor infusion initiated 10 min before reperfusion and VX-765 administered 5 min before reperfusion. Open circles represent individual data points; closed circles designate mean ± SEM. *P < 0.0001. b Echocardiographic left ventricular fractional shortening before 60 min of coronary occlusion and after 3 days of reperfusion. *P = 0.006 for interaction between treatment and time
Ventricular wall motion was measured by ultrasonography in each anesthetized animal prior to coronary occlusion and after 3 days of reperfusion. Figure 4b shows that while vehicle-treated animals displayed a 30% decline in fractional shortening, animals treated with VX-765 and cangrelor retained normal ventricular function.
VX-765 administration is cardioprotective in an ex vivo blood-free, isolated rat heart subjected to global I/R
Cangrelor shows no cardioprotection in ex vivo buffer-perfused, isolated hearts in which platelets and circulating immune cells are absent [5]. We tested whether VX-765-mediated cardioprotection requires platelets and/or circulating immune cells by testing it in blood -free, buffer-perfused isolated rat hearts. Adding VX-765 to the perfusate only during reperfusion had no protective effect against infarction (data not shown), possibly because VX-765 is a prodrug that must be activated by blood esterases [38] that would be missing in this model. We, therefore, tried including VX-765 in the buffer for 10 min before stopping perfusion with the hope that exposing pro-drug to esterases in the cardiac tissue during ischemia might activate it. In isolated hearts subjected to 40-min global ischemia/120 -min reperfusion, vehicle (DMSO) added to the perfusion buffer (0.5 ml/L) starting 10 min before the onset of ischemia and throughout reperfusion resulted in 82.8 ± 3.3% infarction of risk zone (Fig. 5a). Conversely, VX-765 at 30 M, administered with the same schedule, reduced infarct size to 45.8 ± 1.3% of the risk zone (Fig. 5a). These data suggest that VX-765 protects by inhibiting caspase-1 originating in heart tissue, supporting the previous reports that the inflammasome-caspase-1 axis is present in rat cardiac fibroblasts [22, 32].
Fig. 5.
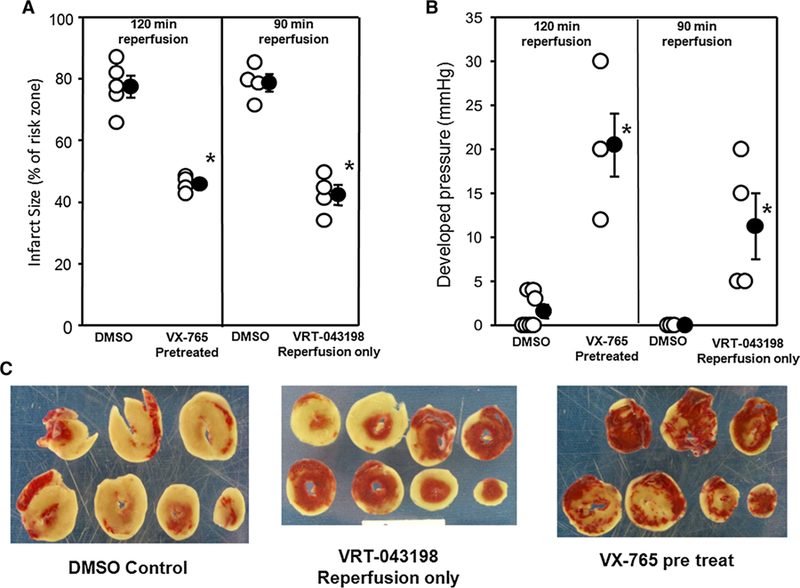
VX-765 starting before ischemia or VRT-043198 at reperfusion is cardioprotective in an ex vivo blood-free, isolated rat heart experiencing global I/R. a Left panel shows infarct size as a percentage of risk zone in isolated rat hearts receiving DMSO or VX-765 for 10 min prior to 40-min global ischemia and again throughout the 120 min of reperfusion. The right panel shows infarcts for hearts treated with DMSO or VRT-043198 started at the initiation of 90 min of reperfusion. Open circles represent individual data points; closed circles designate mean ± SEM. *P < 0.001 vs respective DMSO control. b Final left ventricular developed pressure at the end of reperfusion. *P < 0.05 VX-765- or VRT-043198 vs their respective DMSO control. c Ventricular slices stained with triphenyltetrazolium chloride from three representative hearts treated with either vehicle (DMSO), VX-765 started before ischemia, or VRT-043198 started at reperfusion. The tan areas are infarcted
Since VX-765 given 5 min before reperfusion protected in situ hearts, we suspected that VX-765 also protected isolated hearts by preventing an injury early in reperfusion as suggested by our previous study [ 40]. However, in the absence of blood, VX-765 could not be activated quickly enough to block the injury when it was started at reperfusion. In the blood-perfused model, VX-765 had 5 min of exposure to blood until it was allowed into the ischemic zone at reperfusion, thus providing enough time for the pro- drug VX-765 to be converted to its active component which then protected when introduced into the freshly reperfused myo-cardium. To test our delayed activation hypothesis, we used VRT-043198 (the active derivative of VX-765) only during reperfusion in isolated hearts. Figure 5a shows that VRT-043198 at reperfusion was as protective as VX-765 pretreatment confirming our hypothesis that essentially, all of the caspase-1-dependent injuries occur during reperfusion. A caspase- 1-dependent reperfusion injury is also consistent with our observation that in blood-perfused hearts, VX-765 pretreatment [40] was no more protective than treatment at reperfusion, as shown in Fig. 3. Figure 5b shows that both VX-765 and VRT-043198 caused a significant increase in residual developed pressure at the end of reperfusion compared to control hearts. Figure 5c shows the amount of infarction (tan tissue) in representative DMSO control hearts, hearts treated before ischemia with VX-765, and hearts treated at reperfusion with VRT-043198.
An advantage of the ex vivo isolated heart protocol is that it permits examination of venous effluent. Effluents from these isolated hearts were assayed for two downstream bio-markers of caspase-1 activity, IL-1 (indicating conversion of pro-interleukins), and LDH (a commonly used marker of membrane failure with necrosis or pyroptosis) [23, 25, 34]. IL-1β was undetectable by ELISA in both vehicle- and VX-765-treated heart effluents. In vehicle-treated hearts, LDH release into the effluent peaked within the first 5 min of reperfusion, and steadily diminished thereafter (Fig. 6a). Infusion of VRT- 043198 significantly reduced LDH release during the entire reperfusion period (Fig. 6a). LDH release from hearts treated with VRT-043198 peaked in the first 2 min of reperfusion indicating the caspase-1-dependent component of injury occurs very early in reperfusion. The areas under the curves for control and VRT-treated hearts are significantly different (P = 0.017). Finally, VRT-043198 hearts had much better post-ischemic perfusion than their DMSO counterparts (Fig. 6b).
Fig. 6.
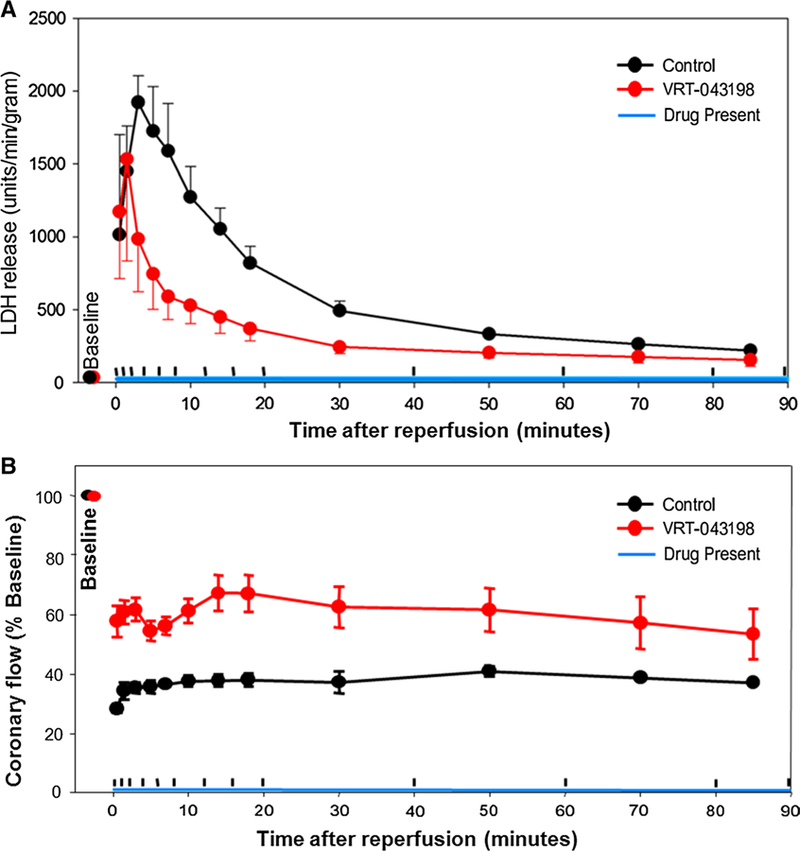
LDH and coronary blood flow time course during reperfusion following ischemia. a Rate of LDH release into effluent before ischemia, and during reperfusion. P = 0.0039 for DMSO (control, n = 3) vs VRT-043198-treated hearts (n = 4). The areas under the two curves were significantly different (P = 0.017). b Coronary flow at various time points during reperfusion for the two groups of hearts. The upward hash marks on the time axis for both panels separate the periods when venous effluent was collected for analysis
VX-765 administration just before reperfusion infuences IL-1β, glycolytic enzymes, and mitochondrial function during I/R
To determine potential mechanisms through which VX-765-mediated caspase-1 inhibition elicits cardioprotection, we revisited the in situ I/R protocol in which hearts were subjected to 60-min regional ischemia/30 -min reperfusion. We shortened reperfusion to 30 min to measure functional downstream effectors, while the myocardium was still viable rather than after cell death had occurred. Blood levels of IL-1 after 30 min of reperfusion were lower in VX-765- treated rats compared to vehicle controls (Fig. 7a). Biopsies from non-ischemic and ischemic left ventricular regions were then assayed for caspase-1 and glycolytic proteins (Fig. 7 b–d). Protein levels of active caspase-1 were increased in ischemic tissue in both vehicle and VX- 765 groups (Fig. 7b). The increased caspase-1 protein in the VX- 765 samples was expected, since VX-765 inhibits only caspase-1 enzymatic activity, but not its autoproteo-lytic activation. This observation is consistent with simi-lar measurements using another less-selective caspase- 1 inhibitor, YVAD [27]. VX-765 preserved GAPDH and to a lesser extent aldolase-A in ischemic zones (Fig. 7c, d). Both are known caspase-1 targets [33]. VX- 765 had no effect on hexokinase II which is not a caspase-1 target (Fig. 7e) [33]. The selective preservation of caspase-1 targets by VX-765 suggests that loss of GAPDH and aldolase-A during reperfusion was not only a result of membrane failure, but may have also been affected by direct proteolysis.
Fig. 7.
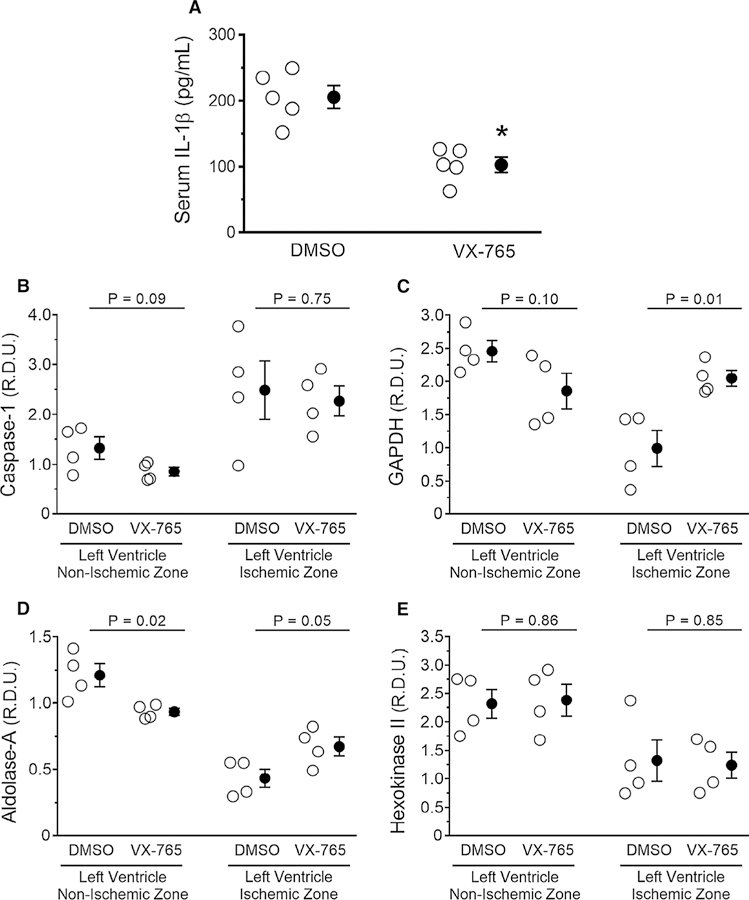
Administration of VX-765 at the onset of reperfusion influences IL −1 and glycolytic enzymes. Rats were subjected to 60-min regional ischemia/30-min reperfusion after treatment with vehicle (DMSO) or VX-765 administered 5 min prior to reperfusion. Open circles represent individual data points; closed circles designate mean ± SEM. Measurements of a serum IL-1 levels and tissue levels of b caspase-1, c glyceraldehyde 3-phosphate dehydrogenase (GAPDH), d aldolase-A, and e hexokinase II by western blotting of biopsies from risk (ischemic) zone and normal myocardium. P values are reported in the figure
We performed high-resolution respirometry on heart biopsies from non-ischemic and ischemic left ventricular regions from vehicle- and VX-765-treated rats, and measured mitochondrial function (i.e., JO2), as depicted in Fig. 2 [30]. Mitochondrial fitness was validated by showing no differences in the respiratory ratio between ischemic and non-ischemic regions in all four groups (Fig. 8a). Interestingly, however, assaying mitochondrial complex activities using target-specific inhibitors revealed that complex I activity in non-ischemic and ischemic tissues was significantly lower in both suggesting an action unrelated to caspase −1 inhibi-tion (Fig. 8b). In addition, VX-765 administration increased complex III activity in ischemic but not in non-ischemic zones (Fig. 8c). No heart tissues from any of the four groups displayed JO2 differences when the various states of mito-chondrial respiration were measured (Fig. 9).
Fig. 8.
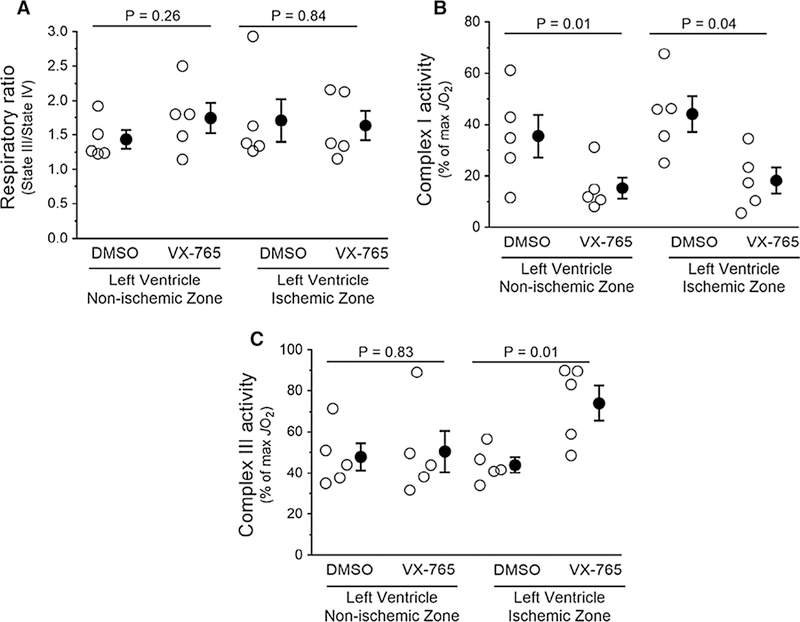
Administration of VX-765 at the onset of reperfusion influences mitochondrial respiration. Rats were subjected to 60-min regional ischemia/30-min reperfusion after treatment with vehicle (DMSO) or VX-765 administered 5 min prior to reperfusion. Respirometry was assessed in biopsies of risk (ischemic) zone and normal myocardium. Open circles represent individual data points; closed circles designate mean ± SEM. a Mitochondrial respiratory ratio (fitness). b Mitochondrial complex I activity. c Mitochondrial complex III activity. P values are reported in the figure
Fig. 9.
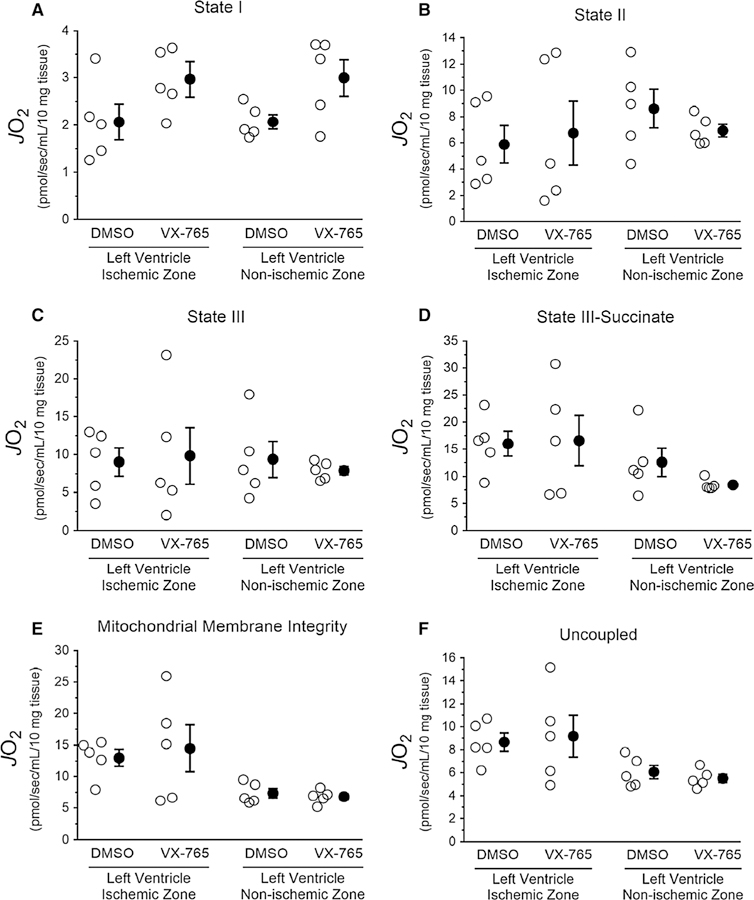
Effect of administration of the selective caspase-1 inhibitor VX-765 on mitochondrial respiratory states assessed in heart biopsies of rats subjected to ischemia/reperfusion. Heart tissues from rats treated with the selective caspase-1 inhibitor VX-765 or its vehicle dimethyl sulfoxide (DMSO) control were prepared and analyzed by respirometry, as described in Fig. 2. Data depicted with open circles represent individual experiments and closed circles represent the mean ± standard error from each intervention. a State I, b state II— alate and pyruvate, c state III—malate, pyruvate and ADP, d state III—malate, pyruvate, ADP, and succinate, e mitochondrial membrane integrity—cytochrome C, and f uncoupled—FCCP. No significant differences were observed between VX-765 groups vs DMSO (unpaired t test)
Discussion
The present study shows that co- administration of VX-765 and a P2Y12 receptor antagonist immediately prior to reperfusion produced additive cardioprotection and preserved long-term ventricular function in a pre-clinical rat model of AMI. Co-administration of VX-765 and ticagrelor or cangrelor reduced myocardial infarction from ~ 60% of the risk zone in untreated rats to ~ 15%, an unprecedented response that was superior to any single intervention. In contrast, ischemic postconditioning did not add to the protection afforded by P2Y12 receptor antagonist, an observation reminiscent of reports from clinical trials of ischemic postconditioning in AMI [7, 9, 10].
Although we previously found that treatment with VX-765 prior to ischemia could add its protection to that from cangrelor [38, 40], the present data reveal that VX-765 does so by preventing a reperfusion injury that occurs during reperfusion rather than during ischemia. That positions VX-765 as a suitable agent for use in AMI patients being treated with PCI who cannot be given the drug prior to the onset of ischemia. In addition, we showed that the reduction of infarct size by a single bolus of VX-765 combined with 120 min of treatment with can-grelor was still evident after 3 days of reperfusion and that ventricular function was markedly preserved. Both findings are indicative of a permanent salvage of contractile myocardium.
These studies provided that some insight into mechanisms involved in VX-765 ʼs protection as well. VX- 765 protected blood-free isolated hearts in the absence of platelets and circulating immune cells, indicating that VX-765-mediated cardioprotection works, at least in part, by inhibiting activated caspase-1 originating in heart tissue, most likely in fibroblasts which do express the inflammasome- caspase-1 axis [22]. Although we found some evidence that caspase-1 inhibition influences inflammatory, glycolytic, mitochondrial, and pyroptotic cell death pathways in a salutary man-ner, we could not determine whether of any of these path-ways actually contributed to myocardial death.
In addition to prevention of thrombosis, P2Y12 receptor antagonists such as clopidogrel, ticagrelor, and cangrelor elicit cardioprotective effects in animal models of AMI [5, 37, 39, 41]. While the beneficial effects of P2Y12 receptor antagonists in PCI are undisputedly attributed to their anticoagulant properties, we hypothesize that their activation of unrecognized postconditioning- like cardioprotection offers a possible explanation for why clinical trials using postconditioning or cyclosporine offered little-to-no additional protection [8–10, 17, 19].
We have previously tested cangrelor in combination with ischemic preconditioning [39] and ischemic post-conditioning [41] in the open-chest rat model and found no more protection against infarction than with cangrelor alone. In the present study, we observed that the same behavior with the much more commonly used P2Y12 receptor inhibitor ticagrelor. Previously, we tested seven signaling inhibitors against cangrelor which are known to block protection from preconditioning and found that while none blocked its anti-aggregatory effect, all blocked its antiinfarct effect [ 41] indicating that cangrelor has a protective mechanism very similar to that seen in pre- and postconditioning. Several of these inhibitors were also tested against clopidogrel [41] and ticagrelor [39] with similar outcomes suggesting a class effect for all P2Y12 receptor inhibitors. Thus, we suspect that P2Y12 receptor inhibitors condition the heart, so that any additional conditioning stimulus becomes redundant.
What sets VX-765 apart from other ant- infarct interventions is its ability to add to the protection from the P2Y12 receptor antagonists. Because ischemic postconditioning or ticagrelor reduce infarct size, but do not eliminate it, some other process must also be killing the reperfused heart muscle. The present study suggest that caspase-1 is killing a considerable amount of heart muscle that platelet inhibitors cannot protect against and does so during early reperfusion.
It is tantalizing to postulate that activation of cas-pase-1 in myocardial tissue during I/R induces pyrop-tosis as a mechanism underlying loss of myocardial viability (Fig. 1). Gasdermin D has been identified as the executioner of pyroptosis, whose cleavage by caspase-1 releases the active subunit that creates pores in the cell membrane leading to lysis and release of cellular contents including LDH [23, 34]. In support of this hypothesis, gasdermin D activation has been recently demonstrated in rat hearts during I/R [26]. LDH washout has traditionally been attributed to membrane failure caused by loss of volume regulation, hypercontracture, and proteolytic activation related to ATP depletion and calcium rise [12, 16]. If pyroptosis was also contributing to cell killing, then gasdermin D knockout mouse [13] should be protected from I/R and VX-765 should be less effective in it.
Our studies have identified additional potential mechanisms, whereby caspase-1 could contribute to ATP depletion. One is proteolysis of vital glycolytic enzymes (e.g., GAPDH), whose dysfunction has been previously implicated during I/R [33]. Another could be increased mitochondrial complex I activity associated with succinate-driven reverse mitochondrial electron transport during I/R [6] caused by proteolysis of critical mitochondrial enzymes. Further studies will be required to fully elucidate the relative contributions of caspase-1 inflammatory, glycolytic, and respiration targets to cell death during I/R.
Conclusion
VX-765 becomes the first clinical-grade drug [21 ] demonstrated to add its cardioprotection to that from a P2Y12 receptor antagonist when both were administered at reperfusion in an animal model of AMI. A single bolus of VX-765 in combination with cangrelor resulted in marked preservation of left ventricular function and an infarct size that continued to be small when measured 3 -day post-reperfusion suggesting a clinically significant long-term salvage of heart muscle.
Acknowledgments
Funding
This work was supported by the National Institutes of Health (Grant #R01HL118334 to DFA and JPA).
Footnotes
Compliance with ethical standards
Conflict of interest There are no relationships to disclose.
References
- 1.Abbate A, Salloum FN, Vecile E, Das A, Hoke NN, Straino S, Biondi-Zoccai GG, Houser JE, Qureshi IZ, Ownby ED, Gustini E, Biasucci LM, Severino A, Capogrossi MC, Vetrovec GW, Crea F, Baldi A, Kukreja RC, Dobrina A (2008) Anakinra, a recombi-nant human interleukin-1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation 117:2670–2683. 10.1161/CIRCULATIONAHA.107.740233 [DOI] [PubMed] [Google Scholar]
- 2.Alvarez DF, Housley N, Koloteva A, Zhou C, O’Donnell K, Audia JP (2016) Caspase-1 activation protects lung endothelial barrier function during infection-induced stress. Am J Respir Cell Mol Biol 55:500–510. 10.1165/rcmb.2015-0386OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Audia JP, Lindsey AS, Housley NA, Ochoa CR, Zhou C, Toba M, Oka M, Annamdevula NS, Fitzgerald MS, Frank DW, Alvarez DF (2013) In the absence of effector proteins, the Pseudomonas aeruginosa type three secretion system needle tip complex con-tributes to lung injury and systemic inflammatory responses. PLoS One 8:e81792 10.1371/journal.pone.0081792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barrabes JA, Inserte J, Mirabet M, Quiroga A, Hernando V, Figueras J, Garcia-Dorado D (2010) Antagonism of P2Y12 or GPIIb/IIIa receptors reduces platelet-mediated myocardial injury after ischaemia and reperfusion in isolated rat hearts. Thromb Haemost 104:128–135. 10.1160/TH09-07-0440 [DOI] [PubMed] [Google Scholar]
- 5.Bell RM, Sivaraman V, Kunuthur SP, Cohen MV, Downey JM, Yellon DM (2015) Cardioprotective properties of the plate-let P2Y12 receptor inhibitor, cangrelor: protective in diabetics and reliant upon the presence of blood. Cardiovasc Drugs Ther 29:415–418. 10.1007/s10557-015-6609-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord EN, Smith AC, Eyassu F, Shirley R, Hu CH, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa AS, Brookes PS, Davidson SM, Duchen MR, Saeb-Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T, Murphy MP (2014) Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515:431–435. 10.1038/nature13909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen MV, Downey JM (2017) The impact of irreproducibility and competing protection from P2Y12 antagonists on the discovery of cardioprotective interventions. Basic Res Cardiol 112:64 10.1007/s00395-017-0653-y [DOI] [PubMed] [Google Scholar]
- 8.Cung TT, Morel O, Cayla G, Rioufol G, Garcia-Dorado D, Angoulvant D, Bonnefoy-Cudraz E, Guerin P, Elbaz M, Delarche N, Coste P, Vanzetto G, Metge M, Aupetit JF, Jouve B, Motreff P, Tron C, Labeque JN, Steg PG, Cottin Y, Range G, Clerc J, Claeys MJ, Coussement P, Prunier F, Moulin F, Roth O, Belle L, Dubois P, Barragan P, Gilard M, Piot C, Colin P, De Poli F, Morice MC, Ider O, Dubois-Rande JL, Unterseeh T, Le Breton H, Beard T, Blanchard D, Grollier G, Malquarti V, Staat P, Sudre A, Elmer E, Hansson MJ, Bergerot C, Boussaha I, Jossan C, Derumeaux G, Mewton N, Ovize M (2015) Cyclosporine before PCI in patients with acute myocardial infarction. N Engl J Med 373:1021–1031. 10.1056/NEJMoa1505489 [DOI] [PubMed] [Google Scholar]
- 9.Engstrom T, Kelbaek H, Helqvist S, Hofsten DE, Klovgaard L, Clemmensen P, Holmvang L, Jorgensen E, Pedersen F, Saunamaki K, Ravkilde J, Tilsted HH, Villadsen A, Aaroe J, Jensen SE, Raun-gaard B, Botker HE, Terkelsen CJ, Maeng M, Kaltoft A, Krusell LR, Jensen LO, Veien KT, Kofoed KF, Torp-Pedersen C, Kyhl K, Nepper-Christensen L, Treiman M, Vejlstrup N, Ahtarovski K, Lonborg J, Kober L, Third Danish study of optimal acute treat-ment of patients With STEMI-IPI (2017) Effect of Ischemic post-conditioning during primary percutaneous coronary intervention for patients with ST-segment elevation myocardial infarction: a randomized clinical trial. JAMA Cardiol 2:490–497. 10.1001/jamacardio.2017.0022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Favaretto E, Rof M, Frigo AC, Lee MS, Marra MP, Napodano M, Tarantini G (2014) Meta-analysis of randomized trials of post-conditioning in ST-elevation myocardial infarction. Am J Cardiol 114:946–952. 10.1016/j.amjcard.2014.06.026 [DOI] [PubMed] [Google Scholar]
- 11.Frantz S, Ducharme A, Sawyer D, Rohde LE, Kobzik L, Fukazawa R, Tracey D, Allen H, Lee RT, Kelly RA (2003) Targeted deletion of caspase-1 reduces early mortality and left ventricular dilatation following myocardial infarction. J Mol Cell Cardiol 35:685–694. 10.1016/S0022-2828(03)00113-5 [DOI] [PubMed] [Google Scholar]
- 12.Fröhlich GM, Meier P, White SK, Yellon DM, Hausenloy DJ (2013) Myocardial reperfusion injury: looking beyond primary PCI. Eur Heart J 34:1714–1722. 10.1093/eurheartj/eht090 [DOI] [PubMed] [Google Scholar]
- 13.Fujii T, Tamura M, Tanaka S, Kato Y, Yamamoto H, Mizushina Y, Shiroishi T (2008) Gasdermin D (Gsdmd) is dispensable for mouse intestinal epithelium development. Genesis 46:418–423. 10.1002/dvg.20412 [DOI] [PubMed] [Google Scholar]
- 14.Hausenloy DJ, Botker HE, Engstrom T, Erlinge D, Heusch G, Ibanez B, Kloner RA, Ovize M, Yellon DM, Garcia-Dorado D (2017) Targeting reperfusion injury in patients with ST-segment elevation myocardial infarction: trials and tribulations. Eur Heart J 38:935–941. 10.1093/eurheartj/ehw145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hausenloy DJ, Garcia-Dorado D, Botker HE, Davidson SM, Downey J, Engel FB, Jennings R, Lecour S, Leor J, Madonna R, Ovize M, Perrino C, Prunier F, Schulz R, Sluijter JPG, Van Laake LW, Vinten-Johansen J, Yellon DM, Ytrehus K, Heusch G, Ferdinandy P (2017) Novel targets and future strategies for acute cardioprotection: position paper of the European society of cardi-ology working group on cellular biology of the heart. Cardiovasc Res 113:564–585. 10.1093/cvr/cvx049 [DOI] [PubMed] [Google Scholar]
- 16.Heusch G, Gersh BJ (2017) The pathophysiology of acute myo-cardial infarction and strategies of protection beyond reperfu-sion: a continual challenge. Eur Heart J 38:774–784. 10.1093/eurheartj/ehw224 [DOI] [PubMed] [Google Scholar]
- 17.Heusch G, Rassaf T (2016) Time to give up on cardioprotection? critical appraisal of clinical studies on ischemic pre-, post-, and remote conditioning. Circ Res 119:676–695. 10.1161/CIRCRESAHA.116.308736 [DOI] [PubMed] [Google Scholar]
- 18.Holly TA, Drincic A, Byun Y, Nakamura S, Harris K, Klocke FJ, Cryns VL (1999) Caspase inhibition reduces myocyte cell death induced by myocardial ischemia and reperfusion in vivo. J Mol Cell Cardiol 31:1709–1715. 10.1006/jmcc.1999.1006 [DOI] [PubMed] [Google Scholar]
- 19.Ibáñez B, Heusch G, Ovize M, Van de Werf F (2015) Evolving therapies for myocardial ischemia/reperfusion injury. J Am Coll Cardiol 65:1454–1471. 10.1016/j.jacc.2015.02.03220 [DOI] [PubMed] [Google Scholar]
- 20.Jong WM, Leemans JC, Weber NC, Juffermans NP, Schultz MJ, Hollmann MW, Zuurbier CJ (2014) Nlrp3 plays no role in acute cardiac infarction due to low cardiac expression. Int J Cardiol 177:41–43. 10.1016/j.ijcard.2014.09.148 [DOI] [PubMed] [Google Scholar]
- 21.Kaufman R. (Last updated December 3, 2007) Phase 2 clinical study in psoriasis with oral investigational drug VX-765. ClinicalTrials.gov Web site: https://clinicaltrials.gov/ct2/show/NCT00205465. Accessed 5 Oct 2017.
- 22.Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J, Hongo M, Noda T, Nakayama J, Sagara J, Taniguchi S, Ikeda U (2011) Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 123:594–604. 10.1161/CIRCULATIONAHA.110.982777 [DOI] [PubMed] [Google Scholar]
- 23.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC, Dixit VM (2015) Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526:666–671. 10.1038/nature15541 [DOI] [PubMed] [Google Scholar]
- 24.Man SM, Kanneganti TD (2015) Regulation of inflammasome activation. Immunol Rev 265:6–21. 10.1111/imr.12296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinon F, Burns K, Tschopp J (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-1b. Mol Cell 10:417–426. 10.1016/S1097-2765(02)00599-3 [DOI] [PubMed] [Google Scholar]
- 26.Mastrocola R, Penna C, Tullio F, Femminó S, Nigro D, Chi-azza F, Serpe L, Collotta D, Alloatti G, Cocco M, Bertinaria M, Pagliaro P, Aragno M, Collino M (2016) Pharmacological inhibition of NLRP3 inflammasome attenuates myocardial ischemia/reperfusion injury by activation of RISK and mito-chondrial pathways. Oxid Med Cell Longev 2016:11 10.1155/2016/5271251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitra S, Wewers MD, Sarkar A (2015) Mononuclear phagocyte-derived microparticulate caspase-1 induces pulmonary vascular endothelial cell injury. PLoS One 10:e0145607 10.1371/journal.pone.0145607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mocanu MM, Baxter GF, Yellon DM (2000) Caspase inhibition and limitation of myocardial infarct size: protection against lethal reperfusion injury. Br J Pharmacol 130:197–200. 10.1038/sj.bjp.0703336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.National Research Council (2011) Guide for the care and use of laboratory animals, vol 8 The National Academies Press, Washington: 10.17226/12910 [DOI] [Google Scholar]
- 30.Pesta D, Gnaiger E (2012) High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol Biol 810:25–58. 10.1007/978-1-61779-382-0_3 [DOI] [PubMed] [Google Scholar]
- 31.Pomerantz BJ, Reznikov LL, Harken AH, Dinarello CA (2001) Inhibition of caspase 1 reduces human myocardial ischemic dys-function via inhibition of IL-18 and IL-1b. Proc Natl Acad Sci USA 98:2871–2876. 10.1073/pnas.041611398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sandanger O, Gao E, Ranheim T, Bliksoen M, Kaasboll OJ, Alf-snes K, Nymo SH, Rashidi A, Ohm IK, Attramadal H, Aukrust P, Vinge LE, Yndestad A (2016) NLRP3 inflammasome activation during myocardial ischemia reperfusion is cardioprotective. Biochem Biophys Res Commun 469:1012–1020. 10.1016/j.bbrc.2015.12.051 [DOI] [PubMed] [Google Scholar]
- 33.Shao W, Yeretssian G, Doiron K, Hussain SN, Saleh M (2007) The caspase-1 digestome identifies the glycolysis pathway as a target during infection and septic shock. J Biol Chem 282:36321–36329. 10.1074/jbc.M708182200 [DOI] [PubMed] [Google Scholar]
- 34.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526:660–665. 10.1038/nature15514 [DOI] [PubMed] [Google Scholar]
- 35.Syed FM, Hahn HS, Odley A, Guo Y, Vallejo JG, Lynch RA, Mann DL, Bolli R, Dorn GW (2005) Proapoptotic effects of cas-pase-1/interleukin-converting enzyme dominate in myocardial ischemia. Circ Res 96:1103–1109. 10.1161/01.RES.0000166925.45995.ed [DOI] [PubMed] [Google Scholar]
- 36.van Hout GPJ, Bosch L, Ellenbroek GH, de Haan JJ, van Solinge WW, Cooper MA, Arslan F, de Jager SC, Robertson AA, Pas-terkamp G, Hoefer IE (2016) The selective NLRP3-inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac func-tion in a pig model of myocardial infarction. Eur Heart J 10.1093/eurheartj/ehw247 [DOI] [PubMed]
- 37.Vilahur G, Gutiérrez M, Casani L, Varela L, Capdevila A, Pons-Lladó G, Carreras F, Carlsson L, Hidalgo A, Badimon L (2016) Protective effects of ticagrelor on myocardial injury after infarc-tion. Circulation 134:1708–1719. 10.1161/CIRCULATIONAHA.116.024014 [DOI] [PubMed] [Google Scholar]
- 38.Wannamaker W, Davies R, Namchuk M, Pollard J, Ford P, Ku G, Decker C, Charifson P, Weber P, Germann UA, Kuida K, Randle JC (2007) (S)-1-((S)-2-{[1-(4-amino −3-chloro-phenyl)-methanoyl]-amino}−3,3-dimethyl-butanoy l)-pyrroli-dine-2-carboxylic acid ((2R,3S)-2-ethoxy-5-oxo-tetrahydro-furan-3-yl)-amide (VX-765), an orally available selective interleukin (IL)-converting enzyme/caspase-1 inhibitor, exhibits potent anti-inflammatory activities by inhibiting the release of IL-1b and IL-18. J Pharmacol Exp Ther 321:509–516. 10.1124/jpet.106.111344 [DOI] [PubMed] [Google Scholar]
- 39.Yang XM, Cui L, Alhammouri A, Downey JM, Cohen MV (2013) Triple therapy greatly increases myocardial salvage dur-ing ischemia/reperfusion in the in situ rat heart. Cardiovasc Drugs Ther 27:403–412. 10.1007/s10557-013-6474-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang XM, Downey JM, Cohen MV, Housley NA, Alvarez DF, Audia JP (2017) The highly selective caspase-1 inhibitor VX-765 provides additive protection against myocardial infarction in rat hearts when combined with a platelet inhibitor. J Cardiovasc Pharmacol Ther 22:574–578. 10.1177/1074248417702890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang XM, Liu Y, Cui L, Yang X, Liu Y, Tandon N, Kambayashi J, Downey JM, Cohen MV (2013) Platelet P2Y12 blockers confer direct postconditioning-like protection in reperfused rabbit hearts. J Cardiovasc Pharmacol Ther 18:251–262. 10.1177/1074248412467692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ye Y, Birnbaum GD, Perez-Polo JR, Nanhwan MK, Nylander S, Birnbaum Y (2015) Ticagrelor protects the heart against reperfusion injury and improves remodeling after myocardial infarc-tion. Arterioscler Thromb Vasc Biol 35:1805–1814. 10.1161/atvbaha.115.305655 [DOI] [PubMed] [Google Scholar]