

Abstract
N-myristoyltransferase (NMT) attaches the fatty acid myristate to the N-terminal glycine of proteins to sort them into soluble and membrane-bound fractions. Function of the energy-sensing AMP-activated protein kinase (AMPK) is myristoylation-dependent. In rheumatoid arthritis (RA), pathogenic T cells shift glucose away from ATP production towards synthetic and proliferative programs, promoting proliferation, cytokine production, and tissue invasion. We found that RA T cells have a defect in NMT1 function, which prevented AMPK activation and enabled unopposed mTORC1 signaling. Lack of the myristate lipid tail disrupted the lysosomal translocation and activation of AMPK. Instead, myristoylation-incompetent RA T cells hyperactivated the mTORC1 pathway and differentiated into pro-inflammatory TH1 and TH17 T helper cells. In vivo, NMT1 loss caused robust synovial tissue inflammation, whereas forced NMT1 overexpression rescued AMPK activation and suppressed synovitis. Thus, NMT1 has tissue-protective functions by facilitating lysosomal recruitment of AMPK and dampening of mTORC1 signaling.
Introduction
T lymphocytes are key pathogenic effector cells in rheumatoid arthritis (RA)1, 2. In the inflamed joint, CD4+ T cells promote lymphoid neogenesis, autoantibody production, macrophage and osteoclast activation, and pannus formation. The organotropism of RA may reflect autoantigen availability, but recent data indicate cell-intrinsic abnormalities in RA T cells that foster tissue-invasive and pro-inflammatory behavior3.
A key feature of RA T cells is the reprogramming of cellular metabolism, which redirects energy sources towards a cell building program4, 5. RA T cells transcriptionally repress 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3)6 and upregulate glucose-6-phosphate dehydrogenase (G6PD)7, shifting glucose away from glycolysis and ATP production to the pentose phosphate pathway (PPP) and biosynthesis7, 8. RA T cells are low in ATP, pyruvate, and reactive oxygen species (ROS) but accumulate NADPH, acetyl-CoA and fatty acids5, 7. Low availability of ROS prevents activation of ataxia telangiectasia mutated (ATM)9, impairs DNA repair and, together with deficient nuclease MRE11A10, accelerates T cell aging.
Favoring anabolic over catabolic conditions leads to a tissue-invasive, hypermobile, pro-inflammatory phenotype driving chronic-destructive tissue inflammation11. Excess acetyl-CoA and NADPH promote lipogenesis5 and lipid droplet accumulation; supplying building blocks for membranes. Comparable to tumor cell invadosomes, RA T cells form membrane ruffles, become hypermobile, and rapidly intrude into tissue sites to organize inflammatory infiltrates. Lipid droplets accumulate due to insufficient mitochondrial β-oxidation, ordinarily triggered in energy-oversupplied cells12. Lipid accumulation in ATPlow conditions breaks the bioenergetics’ rule that the energy-sensing 5’-AMP-activated protein kinase (AMPK) registers low ADP/AMP concentrations, switches on ATP production and halts ATP consumption13, 14. To restore energy homeostasis, decreasing ATP should result in reduced lipid synthesis, mTORC1 deactivation and proliferative arrest. Instead, increased mitochondrial biogenesis should provide ATP, ROS, and metabolic intermediates fueling cataplerotic reactions15. The coexistence of lipogenesis and ATP deficiency in RA T cells implies a fundamental abnormality in energy sensing and utilization.
Upon sensing AMP, AMPK complexes with AXIN-LKB1, translocates to lysosomal surfaces and assembles into the v-ATPase-Ragulator-AXIN/LKB1-AMPK super-complex, where AMPKα-Thr172 is phosphorylated by LKB116. Also, AMPK monitors glucose availability independently of changes in adenine nucleotides17, 18. Once activated AMPK directs glucose-sensitive metabolic checkpoints and mitochondrial metabolism to support effector T cell bioenergetics and viability13, 19. The mammalian target of rapamycin (mTOR) integrates growth factor and nutrient signals for biosynthetic pathways and suppresses catabolic processes, programing T cell differentiation into functional lineages20, 21.22. AMPK and mTORC1 share the lysosomal v-ATPase-Ragulator complex as an activator16, 23, interconnecting the AMPK and mTORC1 systems. Under conditions of energy shortage, AMPK phosphorylates Raptor and TSC2, thus inactivating mTORC113, 24.
Localization of the v-ATPase-Ragulator-AXIN/LKB1-AMPK super-complex to lysosomal membranes requires membranous anchoring, for which the C14-fatty acid myristic acid is covalently attached to AMPKβ1 and β225. N-myristoylation is necessary for AMPK activation at lysosomal surfaces where the hydrophobic myristoyl group is buried in the phospholipid biolayer25. N-myristoylation is a co/post-translational protein-lipid modification catalyzed by N-myristoyltransferase (NMT)26. The two isozymes NMT1 and NMT2 are functionally non-redundant26, 27. NMT1 is critical for tumor cell proliferation, early mouse development and proper monocytic differentiation of mouse bone marrow cells27, 28, 29.
Given the resistance of RA T cells to activate catabolic processes despite low AMP/ATP ratios, the current study examined activation, partitioning and subcellular localization of the energy sensor AMPK. To understand trafficking and subcellular distribution of AMPK, we focused on its myristic acid tail that confers membrane localization. We found that RA T cells have a defect in NMT1 function, disrupting AMPK’s lipidic modification. Loss-of-function and gain-of-function experiments connected NMT1 to T cell differentiation, but more importantly, NMT1 controlled disease-inducing T cell functions in a humanized mouse model of synovitis. Mechanistic studies linked NMT1 deficiency to a defect in lysosomal recruitment of AMPK. Impaired AMPK activation endowed RA T cells with unopposed mTORC1 activation and biased them to differentiate into pro-inflammatory effector cells. Two interventions corrected the AMPK-dependent functional abnormalities in RA T cells; NMT1 overexpression and drug-induced AMPK activation; adding interference with protein post-translational modification and trafficking as potential anti-inflammatory strategies.
Results
NMT1 deficiency in RA CD4 T cells
CD4+ T cells from RA patients are prematurely aged, metabolically reprogrammed, tissue-invasive, and pro-inflammatory5, 7, 30. To examine whether the trafficking, membrane association and subcellular localization of proteins is intact, we compared protein myristoylation in patient-derived and healthy control CD4+ T cells. We purified CD4+CD45RA+ T cells from peripheral blood mononuclear cells (PBMCs) and differentiated them into effector cells by anti-CD3/CD28 bead stimulation for 72 h. Isolated CD4+CD45RA+ populations had no significant contamination with stem-like memory T cells; CD95+ interferon-γ (IFN-γ)-producing CD4+ T cells were distinctly infrequent. Amounts of myristoylated proteins were 45% lower in stimulated CD4+ RA T cells than in controls (Fig. 1a,b). Concentrations of intracellular free fatty acids, including the myristoylation substrate myristic acid, were even higher in RA T cells, excluding substrate availability as the limiting factor (Fig. 1c). To better understand the myristoylation defect, we quantified protein expression of NMT1 in CD4+ T cells from 4 patient cohorts [RA, psoriatic arthritis (PsA), systemic lupus erythematosus (SLE), granulomatosis with polyangiitis (GPA)] and age-matched controls (Fig. 1d,e). Healthy T cells and PsA, SLE and GPA T cells had indistinguishable NMT1 protein expression, in RA T cells the enzyme was reduced up to 50% of controls. The NMT1lo status in RA T cells was confirmed by immunoblotting (Fig. 1f,g). In contrast, NMT2 expression was indistinguishable in RA and control T cells (Supplementary Fig. 1a). NMT1-specific transcripts were not different in RA and control T cells (Supplementary Fig. 1b), assigning reduced NMT1 expression to post-translational events. In a cohort of 24 RA patients, NMT1 protein expression in activated CD4+ T cell correlated inversely with clinical disease activity (Fig. 1h).
Fig. 1. N-myristoyltransferase 1 (NMT1) deficiency in T cells from patients with RA.

CD4+CD45RA+ T cells from patients and age-matched healthy individuals were stimulated for 72 h. (a, b) Quantification of global protein myristoylation in CD4+ T cells by flow cytometry. Myristoylated proteins were labeled with Click-iT myristic acid azide and stained with Alexa Fluor® 594 alkyne. Representative histograms and collective MFIs (mean ± SEM) from 12 controls and 17 RA patients. Unpaired Mann-Whitney-Wilcoxon rank test. (c) Free fatty acids were quantified in cellular extracts from CD4+ T cells stimulated for 72 h. Data (mean ± SEM) from 12 healthy-RA pairs. Unpaired Mann-Whitney-Wilcoxon rank test. (d, e) Expression of NMT1 protein in CD4+ T cells quantified by flow cytometry. Representative histograms and collective MFIs (mean ± SEM). Healthy individuals (n = 9); rheumatoid arthritis (RA, n = 9); psoriatic arthritis (PsA, n = 9); systemic lupus erythematosus (SLE, n = 6); granulomatosis with polyangiitis (GPA, n = 6). One-way ANOVA and post-ANOVA pair-wise two-group comparisons conducted with Tukey’s method. (f, g) Immunoblotting for NMT1 protein in activated CD4+ T cells. Representative immunoblots and quantification from 6 individuals in each group. One-way ANOVA and post-ANOVA pair-wise two-group comparisons conducted with Tukey’s method. (h) Correlation of clinical disease activity (measured by CDAI) and NMT1 protein expression in CD4+ T cells determined by flow cytometry in n = 24 RA patients. Spearman correlation coefficient is shown. ***p < 0.001.
To examine NMT1 expression in different immune cell populations, we applied multi-color flow cytometry in PBMCs from RA and PsA patients (Supplementary Fig. 1c–h). Low NMT1 expression was a feature of naïve CD4+CD45RA+ T cells, memory CD4+CD45RA– T cells and CD19+ B cells. CD8+CD45RA+ and CD8+CD45RA– T cells as well as CD14+ monocytes were unaffected.
To test the hypothesis that NMT1 regulates T cell effector functions, we chose a gain-of-function and loss-of function approach. NMT1 was restored in NMT1lo RA T cells by transfection with an NMT1 overexpression construct (Supplementary Fig. 2a, Fig. 2a–f). NMT1 in healthy T cells was partially silenced with siRNA (Supplementary Fig. 2b, Fig. 2g–l). To determine the frequencies of pro-inflammatory TH1 and TH17 cells, we measured the expression of the lineage-determining transcription factors T-bet and RORγt and the cytoplasmic stores for IFN-γ and interleukin 17 (IL-17). The TH2-associated transcription factor GATA-3 served as a control. RA CD4+ T cells typically deposit neutral lipid droplets5. We therefore quantified cellular neutral lipid content by Bodipy 493/503 staining (Supplementary Fig. 3a). NMT1 overexpression in RA T cells reduced T-bet and RORγt expression and diminished IFN-γ+ and IL-17+ T cell frequencies (Fig. 2a–d) but left GATA-3 unaffected (Fig. 2e). Neutral fatty acids declined significantly (Fig. 2f, Supplementary Fig. 3b). NMT1 loss promptly induced T-bet and RORγt expression and forced T cells towards TH1 and TH17 differentiation (Fig. 2g–j). GATA-3 expression was unaffected (Fig. 2k) but cytoplasmic lipid droplets increased (Fig. 2l, Supplementary Fig. 3c).
Fig. 2. NMT1 controls proinflammatory T cell differentiation.
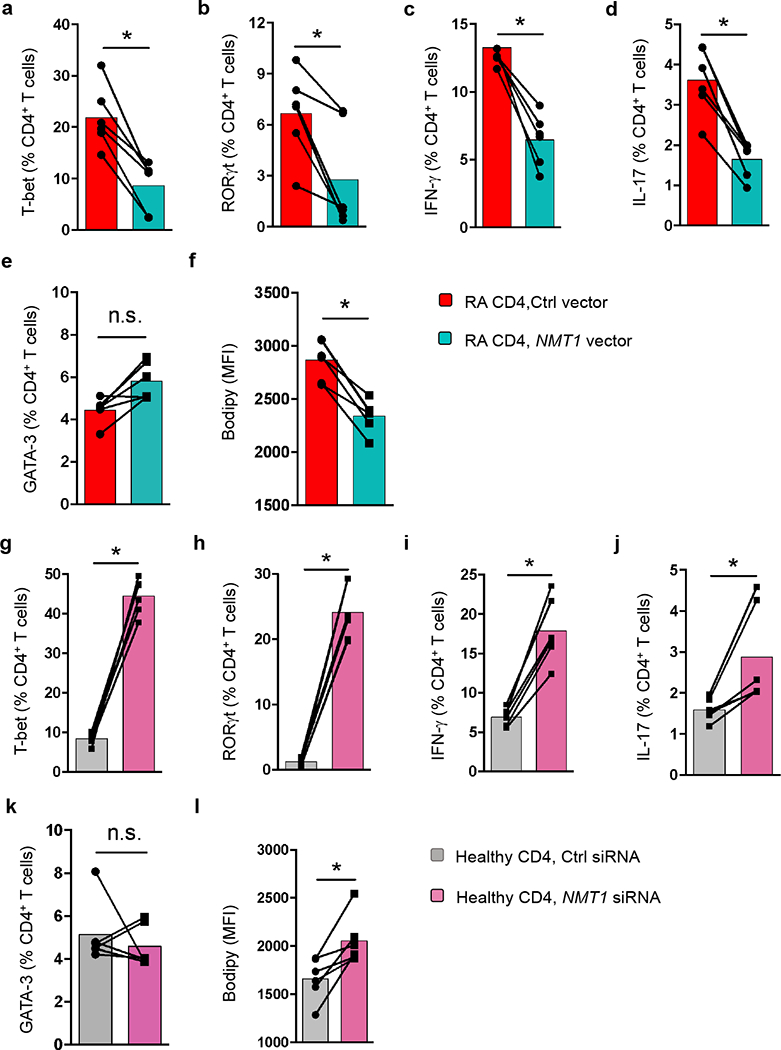
CD4+CD45RA+ T cells from patients with RA and age-matched healthy individuals were stimulated for 72 h. Lineage-determining transcription factors (T-bet, RORγt, GATA-3) were analyzed 4 days after stimulation. IFN-γ and IL-17 were measured by intracellular cytokine staining on day 6. Neutral lipid droplets were detected with BODIPY 493/503. (a-f) CD4+ T cells from RA patients were transfected with an NMT1 expression vector or a control vector. Data (mean ± SEM) from 6 RA patients compared by paired Mann-Whitney-Wilcoxon rank test. (g-l) CD4+ T cells from healthy individuals were transfected with NMT1 siRNA or control siRNA. Data (mean ± SEM) from 6 healthy donors. Paired Mann-Whitney-Wilcoxon rank test. *p < 0.05.
Given the distinguishing metabolic signature of RA T cells3, we explored whether metabolic signals can alter NMT1 protein expression (Supplementary Fig. 4). We screened the impact of a series of metabolic interventions and measured NMT1 and NMT2 protein expression cytometrically. The myristoylating enzymes proved insensitive to changing metabolic conditions in and around the T cells. In summary, protein myristoylation in RA T cells is impaired; attributable to defective function of N-myristoyltransferase 1.
N-myristoylation protects against synovial inflammation
To investigate whether and how protein myristoylation affects inflammatory disease, we made use of a humanized mouse model in which synovitis is induced in engrafted human synovial tissue after adoptive transfer of human PBMCs5, 10 (Supplementary Fig. 5a). After synovitis induction, explanted tissues were analyzed for gene expression profiles and tissue-residing cells. Synovial grafts from chimeras injected with control RA CD4+CD45RO– T cells developed aggressive tissue inflammation (Fig. 3a) with dense T cell infiltrates (Fig. 3a,b) and about 60% of tissue-resident T cells producing IFN-γ (Fig. 3c). Enforced overexpression of NMT1 was strongly anti-inflammatory; density of T cell infiltrates fell by 50% and frequencies of IFN-γ-producing T cells declined by 35% (Fig. 3b,c). Tissue transcriptome analysis revealed marked suppression of inflammatory markers (TBX21, RORC, IFNG, IL17A, TNF), whereas GATA3, IL4 and FOXP3 remained unaffected (Fig. 3d).
Fig. 3. N-myristoylation protects against synovial inflammation.
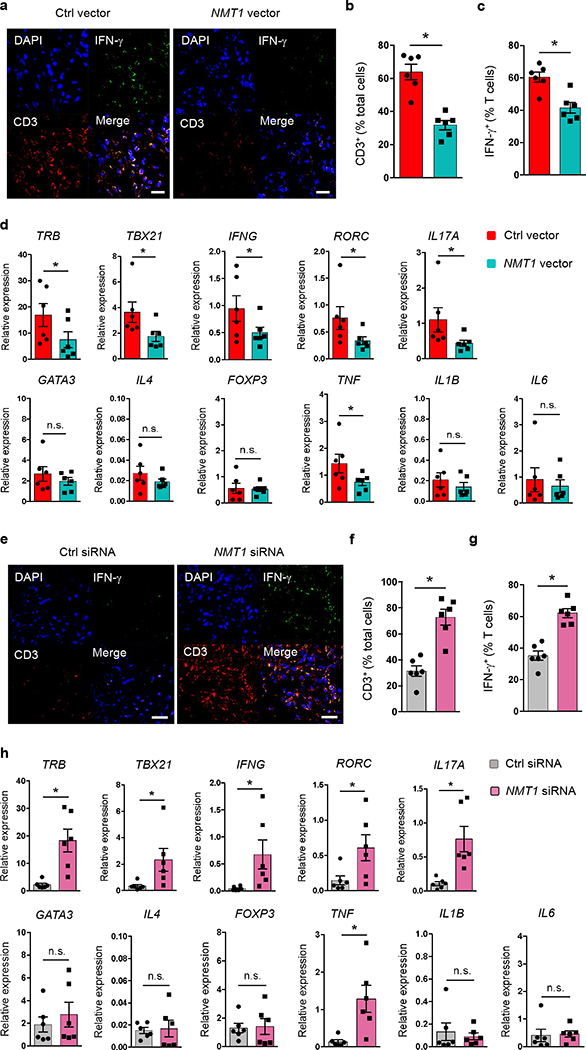
(a-d) RA CD4+CD45RO– T cells were transfected with an NMT1 or control expression vector and injected into NSG mice engrafted with human synovial tissue. Synovial tissues were harvested for transcriptome analysis and immunostaining. (a) Tissue sections stained with anti-IFN-γ (green) and anti-CD3 (red). Nuclei marked with DAPI. Representative images from 6 grafts. Scale bars 20 μm. (b, c) Frequencies of CD3+ T cells and of CD3+IFN-γ+ T cells in tissue sections. (d) Gene transcripts measured in tissue extracts by qPCR. (e-h) Healthy CD4+CD45RO– T cells were transfected with NMT1 siRNA or control siRNA and adoptively transferred into NSG mice engrafted with human synovial tissue. Synovial explants were analyzed for gene expression and by immunohistochemical staining. (e) Tissue-infiltrating human T cells evaluated by dual-color immunostaining of CD3 (red) and IFN-γ (green). Nuclei marked with DAPI. Representative images from 6 grafts. Scale bars 20 μm. (f, g) Frequencies of tissue-residing CD3+ T cells and CD3+IFN-γ+ T cells. (h) Tissue transcriptome of inflammation-associated genes determined by qPCR.Data are mean ± SEM from 6 synovial grafts. Paired Mann-Whitney-Wilcoxon rank test. *p < 0.05.
Conversely, healthy CD4+CD45RO– T cells rendered NMT1lo before adoptive transfer gained inflammatory capacity (Fig. 3e–h). Histologic evaluation demonstrated denser inflammatory infiltrates after NMT1 knockdown (Fig. 3e). Frequencies of tissue-invading T cells and tissue-residing IFN-γ+ T cells increased by about 2-fold (Fig. 3f,g). Tissue transcriptome analysis yielded unaffected expression of GATA3, IL4 and FOXP3 but marked induction of inflammatory molecules (Fig. 3h). Specifically, mRNA expression of TRB, TH1 and TH17 lineage markers and the inflammatory cytokine TNF were upregulated by NMT1 knockdown.
The anti-inflammatory effect of NMT1 overexpression and the pro-inflammatory effects of NMT1 loss were confirmed in human synovium-NSG mice reconstituted with transfected PBMCs (Supplementary Fig. 5b–i). Together, these data identified NMT1 as a tissue-protective enzyme, controlling inflammatory properties of effector T cells.
NMT1lo T cells insufficiently phosphorylate AMPKα
Myristoylation is a lipid modification of proteins, changing their physical properties and their sub-cellular distribution and adding diversity to the proteome26. Due to the membrane targeting effects of the myristic acid tail, signaling pathways occurring at membrane surfaces are particularly dependent on lipidic modification. Considering the metabolic reprogramming of NMT1low RA T cells, we explored whether AMPK, known to be myristoylated at its β-subunit N-terminus25, is functionally affected by NMT1 deficiency.
We first examined whether AMPK signaling is intact in RA T cells. RA T cells had lower concentrations of ATP, accumulated AMP and had increased ratios of ADP/ATP and AMP/ATP (Fig. 4a–d). Rising ratios should trigger AMPKα phosphorylation and enzyme activation14, 17. Phosflow examination of RA, PsA and control T cells for p-AMPKα (Thr172) demonstrated robust phosphorylation in healthy and PsA T cells, but low abundance of p-AMPKα in RA T cells (Fig. 4e–f). Overall, fluorescence intensity signals for p-AMPKα were 2-fold higher in healthy versus RA T cells. Immunoblotting experiments confirmed that protein concentrations of AMPKα, β and γ were similar in healthy, PsA and RA T cells, but RA T cells failed to phosphorylate AMPKα to the same degree as controls (Fig. 4g–h). AMPK initiates inhibitory phosphorylation of acetyl-CoA carboxylase (ACC), an upstream enzyme in the lipogenic program. By immunoblotting, p-ACC was detectable in control T cells, but essentially absent in RA T cells (Supplementary Fig. 6a). Quantification of mRNA transcripts for the AMPK complex components revealed intact transcription of all elements in RA T cells (Supplementary Fig. 6b), suggesting that the defect in AMPK activation occurs post-transcriptionally.
Fig. 4. Impaired activation of the energy sensor AMPK in RA T cells.
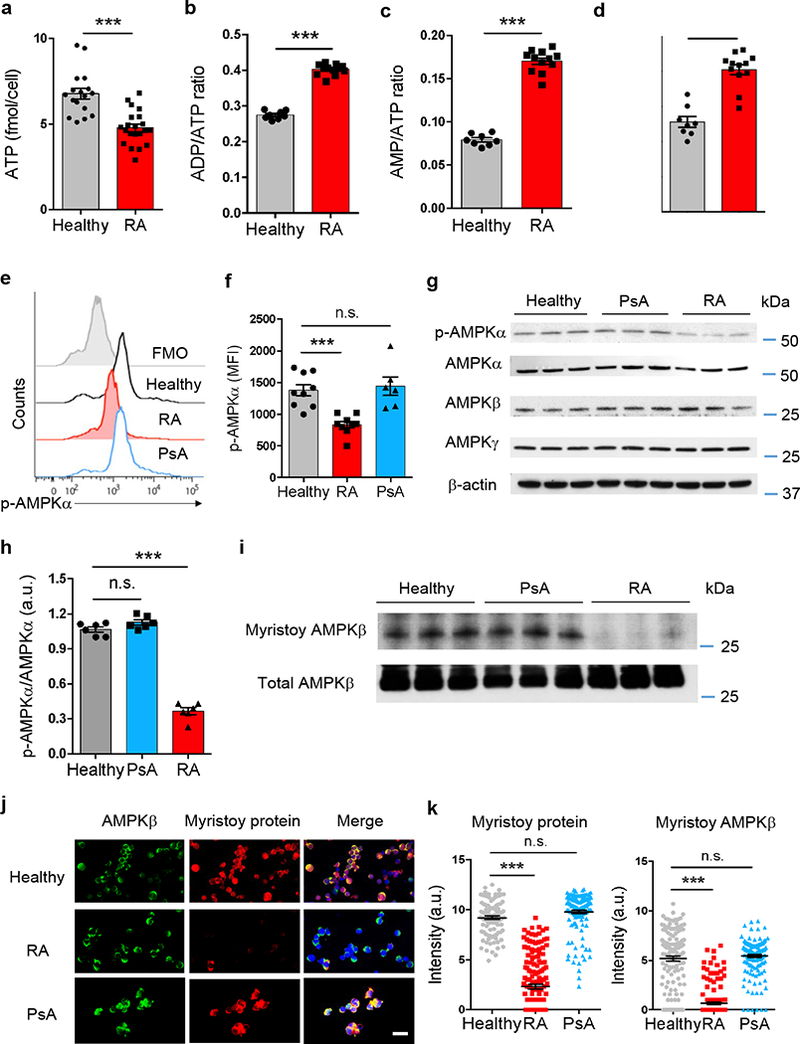
CD4+CD45RA+ T cells from RA patients, PsA patients and age-matched healthy controls were stimulated for 72 h. (a-d) Quantification of intracellular ATP concentrations, ADP/ATP ratios, AMP/ATP ratios and AMP concentrations in T cells from RA patients and healthy controls. Each dot represents one individual. Unpaired Mann-Whitney-Wilcoxon rank test. (e, f) Phosphorylated AMPKα determined with phosflow cytometry. Representative histograms and mean ± SEM from 9 RA patients, 6 PsA patients and 9 controls. Each dot represents the result from one individual. One-way ANOVA and pair-wise comparison using Tukey’s method. (g, h) Protein expression of AMPK subunits and phosphorylated AMPKα analyzed by immunoblotting. Representative and collective data (mean ± SEM) from 6 individuals in each group. One-way ANOVA and pair-wise comparison using Tukey’s method. (i) Immunoblotting of myristoylated AMPKβ. Click-iT myristic acid azide was used to label myristoylated proteins. AMPKβ protein was immunoprecipitated and detected with Biotin-alkyne plus HRP-streptavidin. Representative immunoblots from 6 individuals in each group. (j) Dual-color immuno-staining of myristoylated AMPK. The AMPK β-subunit was marked with Fluor 488 (green) and myristoylated protein was detected by click chemistry with myristic acid azide (Alexa Fluor® 594 alkyne, red). Scale bars 20 μm. (k) Staining intensities of myristoylated AMPKβ quantified by confocal microscopy. Mean ± SEM of the myristoylated AMPKβ intensities from 10 RA patients, 6 PsA patients and 10 healthy controls. One-way ANOVA and pair-wise comparison using Tukey’s method. ***p < 0.001.
Secondly, we quantified myristoylated AMPKβ protein using click chemistry and immunoblotting (Fig. 4i). Compared to healthy controls and PsA patients, RA T cells were essentially deficient in myristoylated AMPKβ. We confirmed the myristoylation defect by single-cell analysis. Applying click chemistry, we quantified the co-localization signal for myristoylated protein and AMPKβ. AMPKβ localized to the cytoplasm of healthy, PsA and RA T cells, with similar expression intensities (Fig. 4j). The overall load of myristoylated protein was markedly lower in RA T cells (Fig. 4j–k). Signal intensities for AMPK-myristoylated protein co-localization were 7.7-fold higher in control than in RA T cells (Fig. 4j–k).
To understand how NMT1 affects AMPKβ myristoylation, we applied a loss-of-function and a gain-of-function approach (Fig. 5a–d). NMT1 knockdown in healthy T cells left AMPKβ expression unaffected, but markedly suppressed lipid-modified proteins (Fig. 5a,b). AMPKβ/myristoylated protein co-localization declined significantly, mimicking conditions in RA T cells. Conversely, NMT1 overexpression in NMT1lo RA T cells restored protein myristoylation with AMPKβ amounts unchanged (Fig. 5c,d). Immunoblotting experiments confirmed that the concentrations of myristoylated AMPKβ protein were NMT1-dependent (Fig. 5e,f). NMT1 silencing in healthy T cells markedly reduced the amount of myristoylated AMPKβ and vice versa, forced overexpression of NMT1 in RA T cells rescued formation of myristoylated AMPKβ
Fig. 5. AMPK activation is myristoylation-dependent.
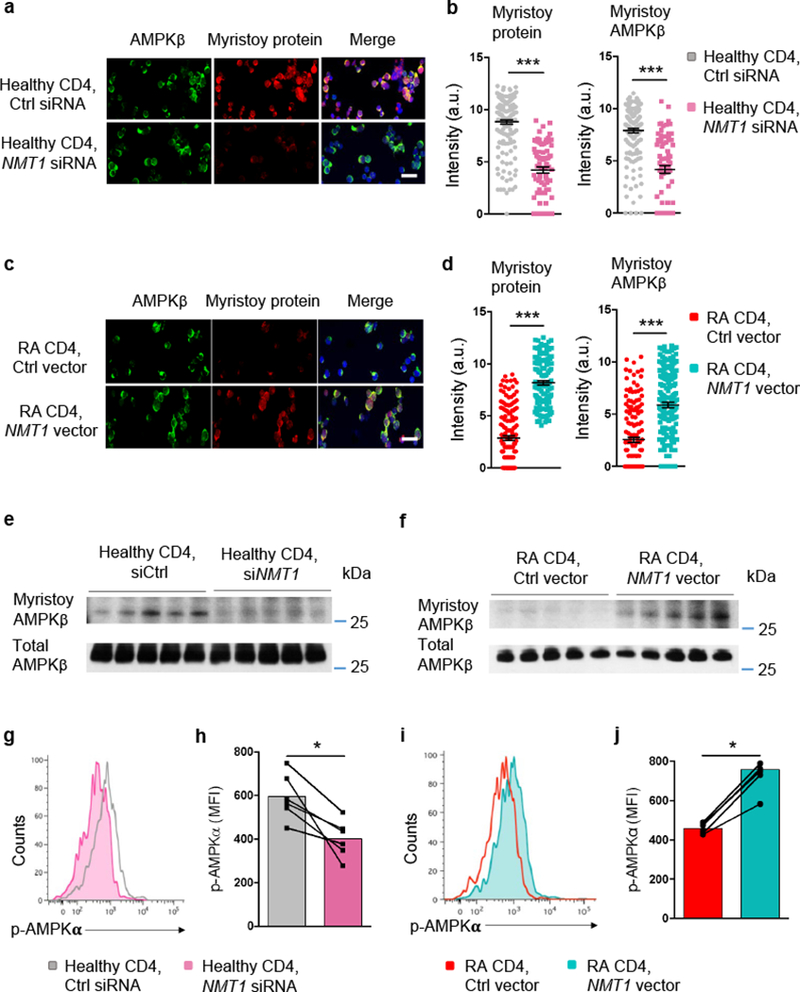
CD4+CD45RA+ T cells from RA patients and age-matched healthy controls were stimulated for 72 h. (a-d) Healthy T cells were transfected with NMT1 siRNA or control siRNA (a, b). RA T cells were transfected with a NMT1 expression vector or a control vector (c, d). Shown are representative stains for AMPKβ (green) and myristoylated proteins (red). Confocal microcopy analysis of myristoylated AMPKβ in individual cells. Mean ± SEM from 6 different individuals in each group. Scale bars 20 μm. Unpaired Mann-Whitney-Wilcoxon rank test. (e, f) Quantification of myristoylated AMPKβ after silencing or overexpression of NMT1. Healthy T cells were transfected with NMT1 siRNA or control siRNA (e). RA T cells were transfected with a NMT1 expression vector or a control vector (f). Myristoylated proteins were labeled with click chemistry and myristoylated AMPKβ was immunoprecipitated and immunoblotted. Immunoblots from 5 individuals in each group. (g, h) T cells from healthy individuals were transfected with NMT1 siRNA or control siRNA and stimulated for 72 h. Phosphorylated AMPKα was quantified by phosflow cytometry. Representative histogram and MFIs from 6 experiments are shown. Paired Mann-Whitney-Wilcoxon rank test. (i, j) T cells from RA patients were transfected with a NMT1 expression vector or a control vector and stimulated for 72 h. Expression of phosphorylated AMPKα was determined by phosflow cytometry. Representative histogram and MFIs from 6 experiments. Paired Mann-Whitney-Wilcoxon rank test. *p < 0.05. ***p < 0.001.
We verified these results by genetic manipulation of NMT1 in T cells, using phosphorylation of AMPKα as a readout of kinase activation. Flow cytometry of p-AMPKα confirmed the reduction of phosphorylated AMPKα in healthy T cells after NMT1 knockdown (Fig. 5g,h). Conversely, phosphorylated AMPKα increased significantly in RA T cells transfected with an NMT1 expression vector (Fig. 5i,j). Together, these studies identified NMT1 as a critical regulator of AMPK function and implicated the process of co/post-translational lipid modification in the failed activation of AMPK in energy-starved RA T cells.
NMT1 controls lysosomal recruitment of AMPK
AMPKα phosphorylation depends on relocation of the kinase to the cytoplasmic surface of lysosomes where AMPK is embedded into the v-ATPase-Ragulator-AXIN-LKB1 super-complex16, 17. Lysosomal retention relies on the N-terminal myristic acid tail being buried in the membrane. To elucidate whether failed AMPK activation in RA T cells is a consequence of altered subcellular distribution, we mapped AMPK localization in myristoylation-deficient RA T cells and in myristoylation-competent healthy T cells and examined whether genetic manipulation of NMT1 affected the kinase’s subcellular localization. Lysosomes were identified by immunostaining for lysosomal-associated membrane protein 1 (LAMP1); AMPK localization was visualized with AMPKα-specific antibodies. Staining intensities for LAMP1 and AMPKα were similar in RA and healthy T cells (Fig. 6a). In healthy T cells, AMPKα colocalized with lysosomal LAMP1. In contrast, RA T cells had significantly reduced LAMP1/AMPKα co-localization (Fig. 6a,b).
Fig. 6. NMT1 controls lysosomal recruitment of AMPK.
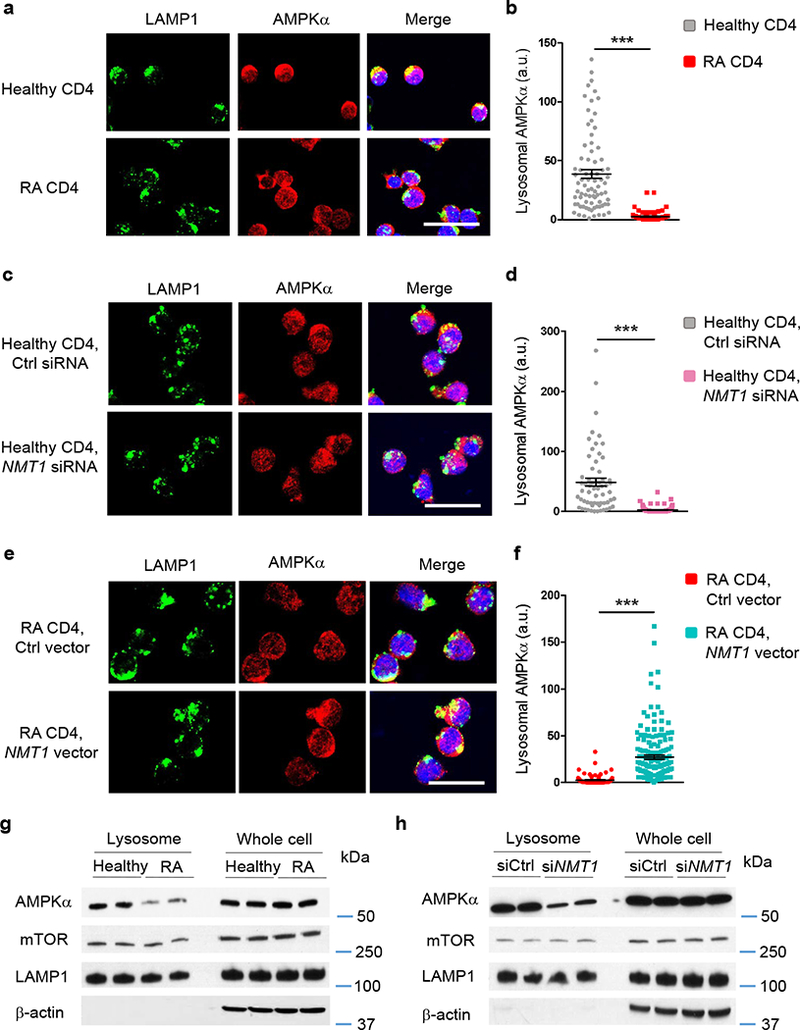
CD4+CD45RA+ T cells from RA patients and healthy individuals were stimulated for 72 h. (a-f) Lysosome was identified by staining with rabbit anti-human LAMP1 (green). AMPKα was localized with mouse anti-human AMPKα (red). Individual cells were analyzed for LAMP1-AMPKa co-localization by confocal microscopy and each dot represents one T cell. Scale bars 20 μm. (a, b) Lysosomal localization of AMPK in RA and control T cells. Representative immuno-stains and quantification of the co-localization signal in individual cells. Data are from 6 RA patients and 6 healthy controls. (c, d) T cells from 6 healthy individuals were transfected with NMT1 siRNA or control siRNA and stimulated for 72 h. (e, f) T cells from 6 RA patients were transfected with a NMT1 expression vector or a control vector and stimulated for 72 h. Data (mean ± SEM) collected from 6 samples in each group. ***p < 0.001 by unpaired Mann-Whitney-Wilcoxon rank test. (g) Lysosomes were isolated from stimulated CD4+ T cells and immunoblotted for AMPKα and mTOR. Whole cell lysates were used as controls. Representative immunoblots from a total of 4 RA patients and 4 healthy controls. (h) CD4+CD45RA+ T cells from healthy individuals were transfected with NMT1 siRNA or control siRNA and stimulated for 72 h. AMPKα and mTOR expression was quantified in lysosomal fractions and in whole cell lysates. Shown are representative immunoblots from a total of 4 individuals.
To relate loss of lysosomal AMPK recruitment in RA T cells to NMT1 deficiency, we rendered healthy T cells NMT1lo by silencing (Fig. 6c,d). Alternatively, we attempted to rescue lysosomal relocation of AMPK in RA T cells by overexpressing NMT1 (Fig. 6e,f). Repression of NMT1 in healthy T cells had no effect on LAMP1 or AMPKα expression intensity (Fig. 6c). However, AMPK’s subcellular distribution changed markedly, no longer overlapping with perinuclear lysosomes (Fig. 6c,d). Colocalization of the LAMP1 and AMPKα signal in NMT1 siRNA-transfected T cells resembled results in RA T cells. NMT1 overexpression did not change the signal intensity for LAMP1 or AMPKα but moved the kinase into LAMP1+ sites (Fig. 6e,f). RA T cells with forced NMT1 overexpression efficiently colocalized LAMP1 and AMPKα, strongly suggestive for successful lysosomal recruitment.
Abnormalities in AMPKα’s lysosomal recruitment was confirmed by immunoblotting experiments (Fig. 6g,h). Lysosomes were isolated and AMPKα, mTOR and LAMP1 were quantified in the lysosomal fraction and whole cell lysate by immunoblotting. Lysosomal AMPKα loading was low in RA compared to healthy T cells (Fig. 6g). Control and RA T cells had similar amounts of lysosomal mTOR (Fig. 6g, Supplementary Fig. 7a). NMT1 knockdown reduced AMPKα in the lysosomal fraction of healthy T cells (Fig. 6h) but did not affect the amount of lysosomal mTOR. To examine whether AMPK determines mTOR loading/retention on the lysosomal surface, we silenced AMPKα in healthy T cells and quantified mTOR in the whole cell and lysosomal fractions. Knockdown of AMPKα left lysosomal mTOR unaffected (Supplementary Fig. 7b,c). Overall, these data confirmed the crucial role of NMT1 in regulating AMPK trafficking between subcellular compartments and mechanistically linked the failure of AMPK activation in RA T cells to its displacement from lysosomal membranes.
NMT1 regulates mTORC1 activation in RA T cells
Regulation of both AMPK and mTORC1 by the lysosomal v-ATPase-Ragulator complex provides a powerful switch between catabolism and anabolism16.
To investigate the status of the mTORC1 signaling pathway, we quantified the phosphorylation of S6 kinase (S6K1) and S6 Ribosomal Protein (S6RP). Flow cytometry revealed markedly higher phosphorylated S6RP (p-S6RP) in RA T cells (Fig. 7a,b); verified by immunoblotting, which demonstrated higher concentrations of p-S6K1 and p-S6RP in RA versus controls (Fig. 7c,d). By pretreating with the mTORC1 inhibitor rapamycin, we estimated that about 50% of p-S6RP in healthy T cells resulted directly from mTOR activation; in RA T cells two-thirds of p-S6RP were rapamycin-sensitive (Fig. 7a,b).
Fig. 7. NMT1 deficiency promotes mTORC1 hyperactivity in RA T cells.

CD4+CD45RA+ T cells from RA patients, PsA patients and healthy controls were stimulated for 72 h. (a, b) Expression of phospho-S6RP protein was analyzed by phosflow cytometry. Cells were pre-treated with rapamycin (10 μM, 1 h) as indicated. Representative histograms and collective MFIs (mean ± SEM) from 6 individuals in each group. One-way ANOVA and pair-wise comparison using Tukey’s method. (c, d) Quantification of the mTORC1 targets phospho-S6K1 and phospho-S6RP by immunoblotting. Representative and collective data from 6 donors in each group. One-way ANOVA and pair-wise comparison using Tukey’s method. (e, f) CD4+CD45RA+ T cells from healthy donors were transfected with NMT1 siRNA or control siRNA and stimulated for 72 h. mTORC1 activity was assessed by phosflow cytometry of phospho-S6RP protein. Representative histograms and collective MFIs (mean ± SEM) from 6 individuals compared by paired Mann-Whitney-Wilcoxon rank test. (g, h) CD4+CD45RA+ T cells from RA patients were transfected with an NMT1 expression vector or a control vector and stimulated for 72 h, followed by quantification of phospho-S6RP with phosflow cytometry. Representative histograms and collective MFIs (mean ± SEM) from 6 patients. Paired Mann-Whitney-Wilcoxon rank test. (i, j) CD4+CD45RA+ T cells from RA patients were stimulated in the absence or presence of the AMPK activators A769662 (10 μM) or metformin (50 μM). Expression of phospho-S6RP protein was determined 72 h after stimulation. Median MFI and interquartile range are indicated. Each dot represents the data from one donor. One-way ANOVA and pair-wise comparison using Tukey’s method. *p < 0.05. ***p < 0.001.
To investigate whether NMT1 not only regulates AMPK activity but also controls mTORC1 signaling, we repressed NMT1 expression in healthy CD4+ T cells (Fig. 7e,f). NMT1 repression resulted in mTORC1 activation, measured by p-S6RP abundance. In parallel, we attempted to revert the mTORC1 hyperactivity in RA T cells by rescuing NMT1 function (Fig. 7g,h). Forced NMT1 overexpression corrected p-S6RP concentrations in RA T cells.
AMPK opposes mTORC1 via multiple mechanisms31, 32, 33. We explored whether restoring AMPK function could suppress mTORC1 hyperactivity. To activate AMPK, we applied A769662, an allosteric AMPK activator that acts independently of the lysosome16, and metformin, an indirect AMPK activator that depends on lysosomal pathways34. A769662 sharply reduced p-S6RP amounts in RA T cells (Fig. 7i,j). p-S6RP concentrations were unchanged after metformin treatment, indicating persistent mTORC1 hyperactivity (Fig. 7i,j). Pharmacologic inhibition of AMPK with Compound C increased p-S6RP, demonstrating mTORC1 activation (Supplementary Fig. 7d). Together, these data associated loss-of-function in AMPK signaling with a gain-of-function in mTORC1 signaling. NMT1 emerged as an upstream regulator for both nutrient sensors, mechanistically connecting lipid modification with energy homeostasis and T cell differentiation.
AMPK activation is tissue protective
To determine whether suppressed AMPK and exuberant mTORC1 signaling influenced T cell differentiation, we compared frequencies of CD4 T cells expressing T-bet, RORγt, IFN-γ and IL-17 after stimulation under non-polarizing conditions. RA T cells produced higher frequencies of TH1-committed effector cells, whereas IL-4-producing TH2 cells were similar in all three study cohorts (Fig. 8a–f). Mal-differentiation of naive CD4+ T cells into pro-inflammatory TH1 and TH17 cells was a feature exclusively for RA patients. Pharmacologic interference targeting AMPK with A769662 normalized frequencies of T-bet+ and IFN-γ+ T cells (Fig. 8g,h); metformin failed to oppose the differentiation bias of RA T cells.
Fig. 8. AMPK activation corrects arthrogenic T cell effector functions in vitro and in vivo.

CD4+CD45RA+ T cells from RA patients, PsA patients and healthy controls were stimulated for 4 days and expression of the lineage-determining transcription factors T-bet and RORγt was assessed by flow cytometry. On day 6 after stimulation, intracellular cytokines (IFN-γ, IL-17, IL-4) were quantified. All data are mean ± SEM. (a-f) Representative contour plots and cumulative data from 7 patient-control pairs. One-way ANOVA and post-ANOVA pair-wise two-group comparisons with Tukey’s method. (g, h) RA T cells were stimulated in the absence or presence of the AMPK activators A769662 (10 μM) or metformin (50 μM). T-bet and RORγt were analyzed on day 4, IFN-γ and IL-17 on day 6. Median frequencies and interquartile ranges are indicated. One-way ANOVA and post-ANOVA pair-wise two-group comparisons were conducted with Tukey’s method. (i-p) NSG mice were engrafted with human synovial tissue and reconstituted with PBMCs from RA patients. Such chimeras were treated with vehicle (i-p), A769662 (i-l) or rapamycin (m-p). Tissue transcriptomes and tissue T cells were analyzed in synovial explants after 7 days of treatment. (i, m) T cell infiltrate in the synovial tissue shown by dual-color immunostaining of CD3 (red) and IFN-γ (green). Nuclei marked with DAPI. Representative images from 6 grafts. Scale bar 20 μm. (j, k and n, o) Frequencies of tissue CD3+ T cells and of CD3+IFN-γ+ T cells. (l, p) Tissue transcriptome of inflammation-associated genes from 6 synovial grafts in each group. Paired Mann-Whitney-Wilcoxon rank test. *p < 0.05. **p < 0.01. ***p < 0.001.
To test whether forced AMPK activation translated into tissue protection in vivo, we explored A769662 treatment in the human-synovium chimera model (Fig. 8i–l). The chimeras tolerated A769662 well, without obvious toxic effects. A769662 treatment efficiently suppressed synovitis (Fig. 8i–l). The density of synovium-infiltrating T cells and of IFN-γ+ tissue-resident T cells were reduced by > 50% (Fig. 8i–k). A769662 therapy downregulated expression of TH1 and TH17 cell-associated genes (TBX21, RORC, IFNG, IL17A) and of those encoding innate inflammatory cytokines (TNF, IL1B, IL6) (Fig. 8l). GATA3, IL4 and FOXP3 gene expression remained unaffected (Supplementary Fig. 8a).
A769662’s anti-inflammatory effect was comparable to that of rapamycin. Rapamycin treatment in human synovium-NSG chimeras efficiently reduced IFN-γ production of tissue-resident T cells (Fig. 8m–o). Inflammation-related genes were highly sensitive to rapamycin-induced immunosuppression (Fig. 8p). Conversely, tissue expression of TH2 and regulatory T cell lineage markers were unaffected (Supplementary Fig. 8b). In vitro studies confirmed that activating AMPK with A769662 and inhibiting mTORC1 with rapamycin had equal potency to suppress pro-inflammatory T cells (Fig. 8g,h, Supplementary Fig. 8c). Treatment with either A769662 or rapamycin normalized frequencies of TH1 and TH17 cells. Frequencies of CD4+FoxP3+ T cells tripled in the presence of rapamycin (Supplementary Fig. 8c) and doubled in A769662-treated cultures (Supplementary Fig. 8d). Overall, these data connected the AMPK/mTORC1 balance to pro-inflammatory conditioning of adaptive immune responses, revealed the sensitivity of the pathway to pharmacotherapy and identified the endomembrane placement of kinases as critical in pharmacologic strategies.
Discussion
Cellular bioenergetics shape T cell effector functions and thus direct adaptive immune responses as exemplified in the autoimmune disease RA. Pathogenic T cells in RA patients have a distinctive metabolic signature, separating them from healthy activated T cells and from T cells in other inflammatory conditions, such as SLE3, 8, 11. RA T cells restrict glycolytic breakdown, freeing glucose for PPP entry to supply NADPH and biosynthetic precursors. Excessive lipogenesis supports formation of invasive membrane structures, promoting tissue inflammation5. In search for upstream defects, we explored how such T cells monitor AMP/ATP ratios; the most sensitive variable used to adjust energy production to demand. We localized the primary defect to N-myristoyltransferase, a highly specific enzyme responsible for the co/post-translational modification of proteins by attaching the C-14 fatty acid myristate to the N-terminal glycine. Deficient N-fatty acylation profoundly affected the subcellular distribution of AMPK, disrupting its translocation to membrane-bound compartments on lysosomal surfaces. Insufficient AMPK activation left mTORC1 activation unopposed, promoting cellular proliferation and progressive differentiation. Myristoylation-incompetent T cells favored IFN-γ and IL-17 production and drove tissue inflammation. T cell-induced synovitis was preventable by NMT1 overexpression, identifying myristoylation as a tissue-protective mechanism.
Several lines of evidence provided clues for AMPK deficiency in RA T cells. AMPK inhibits fat synthesis and promotes fat oxidation35, 36. Yet, RA T cells accumulate lipid droplets, most likely due to insufficient mitochondrial β-oxidation5. AMPK signaling enhances mitochondrial biogenesis via regulating PGC1α and coordinating mitophagy via ULK1/215. Reduced ATP in RA T cells should prompt AMPK activation, switching off ATP-consuming processes and promoting ATP generation. However, this feedback loop depends on the cell’s ability to assign AMPK to relevant subcellular compartments. RA T cells retained the kinase in a diffuse cytoplasmic distribution, failing to anchor AMPK at lysosomal surfaces. Lysosomal recruitment embeds AMPK into the v-ATPase-Ragulator-LKB1-Axin super-complex, co-localizing it with mTORC1. Experiments using pharmacologic AMPK activators suggested additional extra-lysosomal activation sites for the kinase. Metformin failed to rescue AMPKα phosphorylation, in line with its proposed role in lysosome-dependent AMPK activation by inhibiting mitochondrial complex 134. A769662-enforced AMPK activation did not require lysosomal docking. These data suggest multiple subcellular compartments enabling AMPK activation, opening opportunities for select therapeutic manipulations.
NMT1 influences a series of cellular signal transduction pathway by redirecting proteins from the soluble to the membrane-bound fraction. Interaction with calnexin retains NMT1 at the ER membrane37, close to proteins trafficking out of the ER. NMT1 regulates replication of human immunodeficiency virus38 and determines cell-cycle progression and malignant growth of cancer cells39. The global N-myristoylated proteome in human cells includes more than 100 proteins40. Most of the myristoylated proteins are undescribed, suggesting that the NMT1lo RA T cells may have abnormalities beyond AMPK misdistribution. Being shorter than palmitoyl groups, myristoyl groups promote weaker and reversible membrane-protein interactions, but AMPK’s spatiotemporal distribution seems particularly sensitive to this co/post-translational modification. AMPK’s myristate tail was first described more than two decades ago, predicting that the kinase would position at endomembranes. Besides guiding the enzyme to lysosomes, the lipidic modification has also been implicated in non-canonical AMPK functions. Mitochondrial translocation of AMPK regulates mitophagy and cellular survival41. The NMTs are overexpressed in malignant cells and are possible therapeutic targets in cancer treatment. Conversely, autoimmune tissue inflammation appears associated with deficient NMT1 function, assigning a tissue-protective role to this transferase.
Deficient N-myristoylation profoundly affected T cell differentiation. Loss-of function and gain-of-function experiments mechanistically connected lineage commitment to the pro-inflammatory TH1 and TH17 lineage with NMT1 abundance. In vivo, siRNA-mediated NMT1 knockdown induced accumulation of tissue TH1 and TH17 cells. NMT1 overexpression in spontaneously NMT1lo RA T cells was strongly anti-inflammatory. T cell mal-differentiation was amendable to pharmacologic AMPK activation and resulted from excessive activation of the mTORC1 kinase. The activation status of mTORC1 was tightly connected to AMPK activation. Given that mTORC1 is responsible for promoting cellular growth42, mTORC1 activation would only seem appropriate under conditions of energy surplus, when AMPK is deactivated. RA T cells break this rule and hyper-proliferate despite low intracellular ATP pools.
The current study has immediate clinical consequences, including a more refined understanding of how metabolic reprogramming, cellular protein trafficking and tissue inflammation are interlinked. Data presented will allow the identification of therapeutic targets to hinder the uncontrolled expansion of energy-deprived and arthritogenic T cells. By monitoring AMPK’s activation state, predictions can be made about inflammatory propensity, providing a useful screening system for potential therapeutic agents. The current study tested three small molecule reagents. Metformin, a lysosomal AMPK activator widely applied to manage type 2 diabetes mellitus and believed to have anti-inflammatory activity43, 44, failed to revert AMPK mislocalization; in line with the minimal anti-inflammatory effects in autoimmune disease45. The direct AMPK activator A76966216, 46 proved effective in preventing T cell mal-differentiation and tissue inflammation in vivo. The most successful intervention directly targeted mTORC1; rapamycin was potently anti-inflammatory, confirming recent reports that rapamycin is an effective disease-modulatory therapy in SLE47, 48. However, mTORC1 inhibition cannot restore AMPK’s N-myristoylation, achievable only by restoring the expression of functionally intact NMT1.
Methods
Patients
Patients with a diagnosis of RA (n = 155) who were positive for rheumatoid factor and/or anti-cyclic citrullinated peptide autoantibodies were recruited into the study. Patients with a diagnosis of psoriatic arthritis (PsA, n = 33), systemic lupus erythematosus (SLE, n = 6, 49.7±13.9 years old and 100% female) and granulomatosis with polyangiitis (GPA, n = 6, 42.8±19.8 years old, 83.3% female, 50% positive for anti-myeloperoxidase and 50% positive for anti-proteinase 3 antibodies) served as controls. Individuals with cancer, uncontrolled medical disease or any other inflammatory syndrome were excluded. Age-matched healthy donors without a personal or family history of cancer or autoimmune disease were enrolled as healthy controls. Characteristics of RA and PsA patients are summarized in Supplementary Table 1. Appropriate informed consent was obtained and all studies were approved by the Stanford Institutional Review Board.
Cell preparation and cell culture
Peripheral blood mononuclear cells (PBMCs) were isolated by gradient centrifugation with Lymphocyte Separation Medium (Lonza) and cultured in RPMI 1640 medium supplemented with 10% FCS (Hyclone) plus Pen/Strep/Glutamine. Naïve CD4+CD45RA+ T cells were purified from fresh PBMCs using an EasySep™ Human naïve CD4 T Cell Enrichment Kit (STEMCELL Technologies). Purity of cell populations was consistently >95%.
To induce effector CD4+ T cells, CD4+CD45RA+ T cells were stimulated with anti-CD3/anti-CD28-coated beads (Life Technologies AS) at a ratio of 2:1 for 72 h.
Reagents
The AMPK activator A769662 was purchased from APExBIO. The AMPK activator metformin, the mTORC1 inhibitor rapamycin and the metabolite succinate were obtained from Sigma-Aldrich. The fatty acid synthase (FAS) inhibitor C75 and the AMPK inhibitor Compound C were purchased from Cayman Chemical. The PFKFB3 inhibitor 3PO was from EMD Millipore. The human NMT1 cDNA ORF Clone and the appropriate control were purchased from Origene. Human NMT1 siRNA and control siRNA, human AMPKα siRNA and control siRNA, the pyruvate kinase M2 (PKM2) activator ML265, the PKM2 inhibitor Shikonin, the metabolites pyruvate and malic acid, were bought from Santa Cruz Biotechnology. Nucleofection kits from Lonza were used for cell transfections. Lysosomes were isolated with the Lysosome enrichment kit for cultured cells (Thermo Fisher Scientific). Concentrations of cellular free fatty acids were determined with the Free Fatty Acid Quantification Assay Kit (Abcam). All reagents were used according to the manufacturers’ instructions.
Flow cytometry
For cell surface staining, cells were stained with fluorochrome-labeled antibodies to the following antigens: CD4 (BioLegend, Clone RPA-T4), CD8 (BioLegend, Clone SK1), CD19 (BioLegend, Clone HIB19), CD14 (BD, Clone M5E2), CD45RA (BioLegend, Clone HI100), CD28 (BioLegend, Clone CD28.2) and CD95 (BioLegend, Clone DX2). For intracellular staining, cells were fixed with Fix Buffer I (BD Biosciences) and permeabilized with Perm Buffer III (BD Biosciences). Multiparametric flow cytometry panels were assembled with antibodies to T-bet, RORγt, FoxP3, p-AMPKα, p-S6RP, NMT1, NMT2 and neutral lipids, combined with anti-CD4 as follows: FITC or PE-Cy7 anti-human CD4 (BioLegend, Clone RPA-T4), PE or PE-Cy7 anti-human T-bet (eBioscience, Clone eBio4B10), APC anti-human RORγt (eBioscience, Clone AFKJS-9), PE anti-human FoxP3 (BioLegend, Clone 259D), Phospho-AMPK alpha-1,2 (Thr172) (Thermo Fisher Scientific, 44–1150G) plus Alexa Fluor® 488 anti-rabbit IgG (Thermo Fisher Scientific, A-11034), PE anti-human phospho-S6 protein (Cell Signaling, Clone D57.2.2E), rabbit anti-human NMT1 (Abcam, ab84666) plus Alexa Fluor® 488 anti-rabbit IgG (Thermo Fisher Scientific, A-11034), mouse anti-human NMT2 (Novus Biologicals, NBP2–01676) plus Alexa Fluor® 594 anti-mouse IgG (Thermo Fisher Scientific, A-11032), and BODIPY™ 493/503 (Thermo Fisher Scientific, D3922), respectively. For quantification of IFN-γ and IL-17-expressing CD4+ T cells, cells were treated with PMA (50 ng/ml, Tocris) plus Ionomycin (500 ng/ml, Tocris) in the presence of Brefeldin A (5 μg/ml, BioLegend) for 5 h, fixed, permeabilized and stained with FITC anti-human CD4 (BioLegend, Clone RPA-T4), PerCP-Cy5.5 anti-human IFN-γ (BioLegend, Clone 4S.B3) plus APC anti-human IL-17 (eBioscience, Clone eBio64DEC17). Cells were stained for 45 min at 4 °C. Flow cytometry was performed on an LSR II flow cytometer (BD Biosciences). Data were analyzed with FlowJo software (Tree Star Inc.).
Real-time PCR
Total RNA was extracted using Trizol (Thermo Fisher Scientific) and Direct-zol™ RNA MiniPrep Kit (ZYMO Research). cDNA was synthesized with Maxima First Strand cDNA Synthesis Kits for RT-qPCR (Thermo Fisher Scientific). Quantitative PCR analyses were carried out using SYBR Green qPCR Master Mix (Bimake) following previously reported protocols. Primers were used as previously described5, 10, 49. Gene expression was normalized to 18S ribosomal RNA.
Immunoblotting
Experimental conditions for immunoblotting experiments have been previously published5, 7. Primary antibodies used were as follows: anti-NMT1 (Abcam, ab84666), anti-phospho-AMPKα (Thr172) (Cell Signaling Technology, 40H9), anti-AMPKα−1, 2 (Abcam, 34.2), anti-AMPKβ−1,2 (Thermo Fisher Scientific, E.427.6), anti-AMPKγ−1 (Labome, Y308), anti-mTOR (7C10) (Cell Signaling Technology, 2983S), anti-phospho-p70 S6 Kinase (Thr389) (Cell Signaling Technology, 9205S), anti-p70 S6 Kinase (Santa Cruz Biotechnology, B-5), anti-phospho-S6 ribosomal protein (Ser235/236) (Santa Cruz Biotechnology, 50.Ser 235/236) and anti-S6 ribosomal protein (Santa Cruz Biotechnology, C-8). β-actin expression detected with anti-β-actin (Cell Signaling Technology, 8H10D10) served as the internal control.
Determination of cellular ATP and AMP concentrations
Cellular ADP/ATP ratios were analyzed with ADP/ATP Ratio Assay Kit (Sigma). AMP/ATP ratios were determined by assuming AMP/ATP = Keq*(ADP/ATP)2, where Keq = 1.05, as previously described14. Cellular ATP concentrations were quantified with Luminescent ATP Detection Assay (Abcam) and AMP concentrations were calculated from the values of ATP concentrations and AMP/ATP ratios.
Click chemistry
Protein myristoylation was determined by click chemistry using the Click-iT Metabolic Labelling kit (Thermo Fisher Scientific) as previously published41. Briefly, cells were cultured with Click-IT® Myristic Acid Azide (50 μM, Thermo Fisher Scientific, C10268) for 5 h to metabolically label the myristoylated proteins. After that, cells were washed, fixed and permeabilized for the detection reaction with Alexa Fluor® 594 alkyne (Thermo Fisher Scientific, A10275) using the Click-iT® Cell Reaction Buffer Kit (Thermo Fisher Scientific, C10269). N-myristoylation of AMPKβ was detected by costaining with rabbit anti-human AMPKβ−1 (Abcam, Y367) plus Alexa Fluor® 488 anti-rabbit IgG (Thermo Fisher Scientific, A-11034) after cellular fixation and permeabilization, and visualized by a LSM710 confocal microscope (Carl) with a Plan-Neofluar 40×/1.3-NA oil objective lens. For immunoblotting experiments AMPKβ protein was immunoprecipitated from Myristic Acid Azide-labeled cells with anti-human AMPKβ−1 (1:50, Cell Signaling Technology, 12063S), followed by click reaction with Biotin-alkyne (Thermo Fisher Scientific, B10185) using Click-iT® Protein Reaction Buffer Kit (Thermo Fisher Scientific, C10276) and immunoblot with HRP-Streptavidin (1:2000, Cell Signaling Technology, 3999S).
Immunofluorescence
Subcellular localization of AMPK on lysosomal surfaces was determined with immunofluorescence staining5, 10. Lysosomes were labeled with rabbit anti-human lysosomal-associated membrane protein 1 (LAMP1) (1:100, Cell Signaling Technology, D2D11) plus Alexa Fluor® 488 anti-rabbit IgG (1:200, Thermo Fisher Scientific, A-11034). AMPK was detected with mouse anti-human AMPKα−1,2 (1:100, Abcam, 34.2) plus Alexa Fluor® 594 anti-mouse IgG (1:200, Thermo Fisher Scientific, A-11032). Lysosomal AMPK was determined by co-localizing lysosome green staining and AMPKα red staining and visualized with a LSM710 confocal microscope (Carl Zeiss) with a Plan-Neofluar 40×/1.3-NA oil objective lens.
Immunohistochemistry
Protein expression of CD3 and IFN-γ in tissue sections was performed with immunohistochemistry5, 10, 49. Briefly, frozen sections of synovial tissues were stained with mouse anti-human CD3 (1:50; DAKO, Clone F7.2.38) and rabbit anti-human IFN-γ (1:100, Abcam, ab25101). Antibody binding was visualized with Alexa Fluor 594® anti-mouse IgG (1:200, Thermo Fisher Scientific, A-11032) and Alexa Fluor 488® anti-rabbit IgG (1:200, Thermo Fisher Scientific, A-11034) as secondary antibodies. The LSM710 confocal microscope (Carl Zeiss) with a Plan-Neofluar 40×/1.3-NA oil objective lens was used to acquire images.
Human synovium-SCID mouse chimeras
Human synovium-SCID mouse chimeras were generated as previously described5, 10. NSG mice from the Jackson Laboratory were kept in pathogen-free facilities and used at the age of 8 to 12 weeks. Seventy-eight mice (50 female and 28 male) were subcutaneously implanted with pieces of synovial tissues. Chimeric mice carrying synovial tissues from the same donor were randomly assigned to parallel treatment arms. Seven days post-engraftment, mice were reconstituted with CD45RO– PBMCs (15 × 106 cells/mouse) from patients with active RA. CD4+ T cells were sorted by flow cytometry from CD45RO– PBMCs derived from healthy donors, transfected with NMT1 siRNA or appropriate control siRNA, and adoptively transferred into the chimeric mice together with the CD4+ T cell-depleted CD45RO– population. Similarly, CD4+ T cells were FACS sorted from CD45RO– PBMCs from RA patients, transfected with an NMT1 expression vector or control vector, added back into the CD4+ T cell-depleted CD45RO– PBMCs and injected into the chimeric mice. For some experiments, prior to adoptive transfer, CD45RO– PBMCs from either healthy donors or RA patients were transfected with NMT1 siRNA or the NMT1 expression vector, as appropriate. For treatment experiments, chimeras were reconstituted with RA PBMCs and intraperitoneally injected with A769662 (30 mg/kg/mouse, bid)46 for 7 days or with rapamycin (5 mg/kg/mouse, every other day)50 for 9 days. Synovial grafts were harvested, shock frozen for RNA isolation and OCT-embedded for immunostaining. All protocols were approved by the Stanford University Institutional Animal Care and Use Committee.
Statistics
Data are presented as mean ± standard error of the mean (SEM). Paired and unpaired Mann-Whitney-Wilcoxon rank tests were used for two-group comparisons as appropriate. The Hochberg’s step-up procedure was applied for multiple testing to control the family-wise error rate (at a level of 0.05). ANOVA was used for comparison of more than two groups and post-ANOVA pair-wise two-group comparisons were conducted with Tukey’s method. Spearman correlation was used for testing correlations. Heatmaps were generated by z-scores. For each gene, the z-score was obtained by standardizing the gene expression level relative to the 18S ribosomal RNA to have zero mean and unit variance. All statistical analyses were conducted by using PRISM 6.0 (GraphPad Software Inc.). P < 0.05 was considered significant. All data analyses were overseen by the Department of Biomedical Data Science at Stanford University.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (R01 AR042527, R01 HL 117913, R01 AI108906 and P01 HL129941 to C.M.W. and R01 AI108891, R01 AG045779, U19 AI057266, and I01 BX001669 to J.J.G.).
Footnotes
Competing interests
The authors declare no conflicts.
Data availability
The data that support the findings of this study are available from the corresponding author upon request.
References
- 1.Rao DA et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature 542, 110–114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weyand CM & Goronzy JJ T-cell-targeted therapies in rheumatoid arthritis. Nat Clin Pract Rheumatol 2, 201–210 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Weyand CM & Goronzy JJ Immunometabolism in early and late stages of rheumatoid arthritis. Nat Rev Rheumatol 13, 291–301 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weyand CM, Shen Y & Goronzy J Redox-Sensitive Signaling in Inflammatory T cells and in Autoimmune Disease. Free Radic Biol Med (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shen Y et al. Metabolic control of the scaffold protein TKS5 in tissue-invasive, proinflammatory T cells. Nat Immunol 18, 1025–1034 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang Z, Fujii H, Mohan SV, Goronzy JJ & Weyand CM Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J Exp Med 210, 2119–2134 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang Z et al. Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Sci Transl Med 8, 331ra338(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsokos GC Metabolic control of arthritis: Switch pathways to treat. Sci Transl Med 8, 331fs338(2016). [DOI] [PubMed] [Google Scholar]
- 9.Shao L et al. Deficiency of the DNA repair enzyme ATM in rheumatoid arthritis. J Exp Med 206, 1435–1449 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y et al. Deficient Activity of the Nuclease MRE11A Induces T Cell Aging and Promotes Arthritogenic Effector Functions in Patients with Rheumatoid Arthritis. Immunity 45, 903–916 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsokos GC Fat T cells go to the joint. Nat Immunol 18, 955–956 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Wang CW Lipid droplets, lipophagy, and beyond. Biochim Biophys Acta 1861, 793–805 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Ma EH, Poffenberger MC, Wong AH & Jones RG The role of AMPK in T cell metabolism and function. Curr Opin Immunol 46, 45–52 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Gowans GJ, Hawley SA, Ross FA & Hardie DG AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab 18, 556–566 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herzig S & Shaw RJ AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol 19, 121–135 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang CS et al. The lysosomal v-ATPase-Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab 20, 526–540 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Lin SC & Hardie DG AMPK: Sensing Glucose as well as Cellular Energy Status. Cell Metab 27, 299–313 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Zhang CS et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 548, 112–116 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blagih J et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity 42, 41–54 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Kim J, Kundu M, Viollet B & Guan KL AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13, 132–141 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chi H Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol 12, 325–338 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delgoffe GM et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol 12, 295–303 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sancak Y et al. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hardie DG AMPK and Raptor: matching cell growth to energy supply. Mol Cell 30, 263–265 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Oakhill JS et al. beta-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK). Proc Natl Acad Sci U S A 107, 19237–19241 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Udenwobele DI et al. Myristoylation: An Important Protein Modification in the Immune Response. Front Immunol 8, 751(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ducker CE, Upson JJ, French KJ & Smith CD Two N-myristoyltransferase isozymes play unique roles in protein myristoylation, proliferation, and apoptosis. Mol Cancer Res 3, 463–476 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang SH et al. N-myristoyltransferase 1 is essential in early mouse development. J Biol Chem 280, 18990–18995 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Shrivastav A et al. Requirement of N-myristoyltransferase 1 in the development of monocytic lineage. J Immunol 180, 1019–1028 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Weyand CM, Yang Z & Goronzy JJ T-cell aging in rheumatoid arthritis. Curr Opin Rheumatol 26, 93–100 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Finlay D & Cantrell DA Metabolism, migration and memory in cytotoxic T cells. Nat Rev Immunol 11, 109–117 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Navarro MN & Cantrell DA Serine-threonine kinases in TCR signaling. Nat Immunol 15, 808–814 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gwinn DM et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30, 214–226 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang CS et al. Metformin Activates AMPK through the Lysosomal Pathway. Cell Metab 24, 521–522 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Fullerton MD et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat Med 19, 1649–1654 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lochner M, Berod L & Sparwasser T Fatty acid metabolism in the regulation of T cell function. Trends Immunol 36, 81–91 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Dudek E et al. N-Myristoyltransferase 1 interacts with calnexin at the endoplasmic reticulum. Biochem Biophys Res Commun 468, 889–893 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Ohta H, Takamune N, Kishimoto N, Shoji S & Misumi S N-Myristoyltransferase 1 enhances human immunodeficiency virus replication through regulation of viral RNA expression level. Biochem Biophys Res Commun 463, 988–993 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Kim S et al. Blocking Myristoylation of Src Inhibits Its Kinase Activity and Suppresses Prostate Cancer Progression. Cancer Res 77, 6950–6962 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thinon E et al. Global profiling of co- and post-translationally N-myristoylated proteomes in human cells. Nat Commun 5, 4919(2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liang J et al. Myristoylation confers noncanonical AMPK functions in autophagy selectivity and mitochondrial surveillance. Nat Commun 6, 7926(2015). [DOI] [PubMed] [Google Scholar]
- 42.Hardie DG AMPK--sensing energy while talking to other signaling pathways. Cell Metab 20, 939–952 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kelly B, Tannahill GM, Murphy MP & O’Neill LA Metformin Inhibits the Production of Reactive Oxygen Species from NADH:Ubiquinone Oxidoreductase to Limit Induction of Interleukin-1beta (IL-1beta) and Boosts Interleukin-10 (IL-10) in Lipopolysaccharide (LPS)-activated Macrophages. J Biol Chem 290, 20348–20359 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O’Neill LA, Kishton RJ & Rathmell J A guide to immunometabolism for immunologists. Nat Rev Immunol 16, 553–565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perl A Review: Metabolic Control of Immune System Activation in Rheumatic Diseases. Arthritis Rheumatol 69, 2259–2270 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cool B et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab 3, 403–416 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Lai ZW et al. Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: a single-arm, open-label, phase 1/2 trial. Lancet 391, 1186–1196 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang N & Perl A Metabolism as a Target for Modulation in Autoimmune Diseases. Trends Immunol 39, 562–576 (2018). [DOI] [PubMed] [Google Scholar]
Methods-only References
- 49.Wen Z et al. The microvascular niche instructs T cells in large vessel vasculitis via the VEGF-Jagged1-Notch pathway. Sci Transl Med 9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fang Y et al. Duration of rapamycin treatment has differential effects on metabolism in mice. Cell Metab 17, 456–462 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.