

Abstract
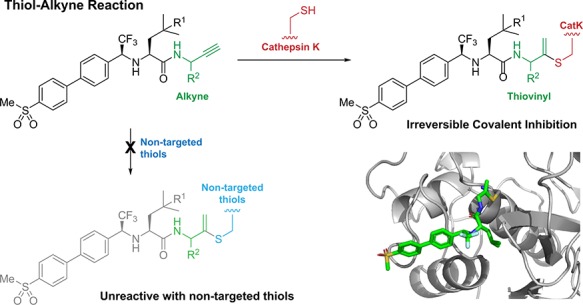
Irreversible covalent inhibitors can have a beneficial pharmacokinetic/pharmacodynamics profile but are still often avoided due to the risk of indiscriminate covalent reactivity and the resulting adverse effects. To overcome this potential liability, we introduced an alkyne moiety as a latent electrophile into small molecule inhibitors of cathepsin K (CatK). Alkyne-based inhibitors do not show indiscriminate thiol reactivity but potently inhibit CatK protease activity by formation of an irreversible covalent bond with the catalytic cysteine residue, confirmed by crystal structure analysis. The rate of covalent bond formation (kinact) does not correlate with electrophilicity of the alkyne moiety, indicative of a proximity-driven reactivity. Inhibition of CatK-mediated bone resorption is validated in human osteoclasts. Together, this work illustrates the potential of alkynes as latent electrophiles in small molecule inhibitors, enabling the development of irreversible covalent inhibitors with an improved safety profile.
Introduction
Irreversible covalent inhibition of a target protein minimizes the required systemic drug exposure as protein activity can only be restored by de novo protein synthesis, resulting in a prolonged therapeutic effect long after the compound is cleared from the blood.1,2 Strategically placing an electrophilic moiety on the inhibitor will allow it to undergo attack by a nucleophilic amino acid residue upon binding to the target protein, forming an (ir)reversible bond that is much stronger than typical noncovalent interactions. However, the ability to form a covalent bond with the target enzyme has raised concerns about indiscriminate reactivity with off-target proteins,3−5 even though some of the most prescribed drugs are covalent irreversible binders.6,7 This led to the disfavor of covalent modifiers as drug candidates until the recent successful development of irreversible covalent kinase inhibitors ibrutinib and afatinib, which form an irreversible covalent bond between an acrylamide warhead and a nonconserved cysteine residue on the ATP-binding site2,8−10 but also with nontargeted cellular thiols.11 The ability to form covalent adducts with off-target proteins has been linked to an increased risk of unpredictable idiosyncratic toxicity along with the daily drug dose administered to patients.11−14 This risk can be reduced by incorporating less reactive electrophilic moieties into irreversible covalent inhibitors.
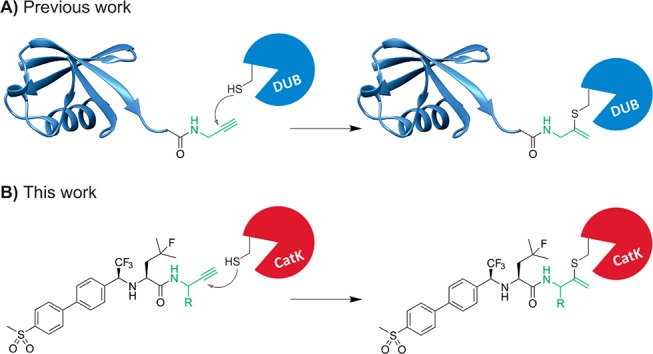
Terminal alkynes are generally considered “inert” toward cellular components in the absence of radical initiators and are therefore often used in bioorthogonal approaches as chemoselective “Click” handles.15,16 However, our group has shown a C-terminal propargyl moiety on ubiquitin to react in an activity-based manner with the catalytic cysteine residue in deubiquitinating enzymes (DUBs), forming an irreversible thioether bond via an in situ thiol–alkyne addition (Scheme 1).17 Markovnikov hydrothiolation of (terminal) alkynes with aliphatic thiols has been described for metal-catalyzed reactions18−21 but has not been reported to occur outside the active-site of a cysteine protease under physiological conditions. The alkyne moiety on ubiquitin did not react with cysteine residues present in nontargeted proteins nor with excess thiol. Work by Sommer et al. revealed that the catalytic triad does not have to be intact for covalent bond formation, indicating a proximity-driven reactivity.22 Although it was believed that the reactivity of the alkyne resulted from a template effect: recognition of (large) protein fragments driving the formation of the thermodynamically unfavored Markovnikov-type thiovinyl product,23 here we show that strong enough binding can be achieved with a small molecule recognition part. This study highlights the potential of alkynes as latent electrophiles in irreversible covalent small molecule inhibitors, as demonstrated for cathepsin K (CatK).
Scheme 1. Terminal Alkyne Moiety as Latent Electrophile for Thiol–Alkyne Addition in (A) Ubiquitin-Based Activity Probes Targeting DUB Proteases and (B) Irreversible Covalent Small Molecule Inhibitors of Cysteine Protease CatK.
CatK is a cysteine protease that is highly expressed in osteoclasts and is the most important protease in bone degradation.24 Implicated in diseases such as osteoporosis, its inhibition has been of therapeutic interest for the past decade.25 The most promising small molecule CatK inhibitor to date was odanacatib (ODN),26 a nonlysosomotropic inhibitor with a nitrile moiety as reversible covalent warhead that binds to catalytic Cys25 (Figure S1). ODN has a high selectivity for CatK versus other cathepsins and only has to be taken once weekly because of its very long half-life of 66–93 h.27 The development was terminated after phase III clinical trials showed side effects including increased stroke risks and cardiovascular events.28−30 It is currently unclear whether this is due to inhibition of nonskeletal degradation properties of CatK or because of off-target inhibition.31 Nonetheless, the close proximity of the nitrile moiety relative to Cys25 made it a suitable model to incorporate an alkyne moiety as electrophile.
Results and Discussion
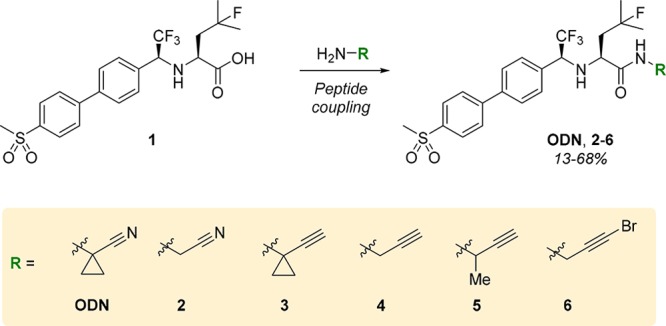
Derivatives of ODN were obtained by functionalization of precursor 1, as reported previously (Scheme 2 and Scheme S1).32,33 Replacing the nitrile with an alkyne led to compromised solubility in aqueous media for alkyne 3, which could be overcome by removal of the hydrophobic cyclopropane in nitrile 2, propargyl 4, and monomethylated propargyl 5. The cyclopropane moiety is not essential for CatK inhibition but was introduced in the development of ODN to reduce metabolic liabilities.26 Alkyne electrophilicity increases if an electron-withdrawing substituent is introduced on the terminal position,19,34 while remaining less electrophilic than acrylamides, and therefore, electron-deficient alkyne 6 was taken along to investigate the effect of electrophilicity on the inhibitor selectivity. Conjugate addition of cysteine to electron-deficient internal alkynes in aqueous medium has been reported and has recently been utilized in the irreversible covalent kinase inhibitor acalabrutinib.19,35,36
Scheme 2. Synthesis of ODN, Nitrile 2, Alkynes 3–5, and Electron-Deficient Alkyne 6.
Indiscriminate Thiol Reactivity
Indiscriminate thiol reactivity was assessed following an established protocol in which nitrile-based inhibitors form an irreversible covalent adduct with cysteine37 (Table 1, Figure S2). Briefly, compounds were incubated with cysteine for 23 h after which they were analyzed by LC–MS. The adduct formation was quantified from the UV trace. ODN and nitrile 2 show adduct formation that increases upon increasing the pH of the buffer, as do acrylamide-based inhibitors ibrutinib and afatinib and irreversible pan-cathepsin inhibitor E-64.38 Adduct formation was not detected for alkyne-based inhibitors 3–5, which supports our hypothesis that the unactivated alkyne is not reactive toward cysteine residues in nontargeted proteins. As expected,36 adduct formation with electron-deficient alkynes 6 and acalabrutinib was observed, underlining the importance of alkyne electrophilicity in indiscriminate thiol reactivity.
Table 1. Indiscriminate Thiol Reactivitya.

Adduct formation quantified from LC–MS UV trace after 23 h incubation with 10 mM cysteine or 5 mM GSH at 37 °C in buffer at different pH values.
Reversible adduct formation.
Glutathione (GSH), a tripeptide with a cellular concentration of 0.5–10 mM,39 is a commonly used biological thiol to assess the risk of iodiosyncratic toxicity. GSH adduct was observed for acrylamides and electron-deficient alkynes, as reported,11,36 but not for inhibitor 4 upon incubation with 5 mM GSH.
In Vitro Inhibition
A recurring issue in CatK drug development is the difference in amino acids at the active-site for rodentCatK compared to humanCatK, thus reducing the apparent potency of ODN up to 182-fold in mice and rats.40 We therefore assessed the potency of our inhibitors in an in vitro inhibition assay on recombinant human cathepsins (Table 2). As reported, ODN is selective for hCatK with an IC50 below 1 nM. Noncovalent interactions have been optimized for ODN, and we anticipated that replacing the polarized nitrile moiety by a nonpolarized alkyne moiety would decrease the interaction with active-site residues, reducing the noncovalent complex formation (kon), This is indeed reflected in increased IC50 values for all alkyne-based inhibitors. Selectivity for CatK over related human cathepsins was conserved for alkynes 4 and 5, while all selectivity is lost for electron-deficient alkyne 6. Inhibition of hCatK activity was validated in a gel-based probe labeling experiment with quenched activity-based probe BMV109 (Figure S3).
Table 2. In Vitro IC50 Values (M) against Proteolytic Activity of Cysteine Proteasesa.

Incubation of cysteine protease and inhibitor for 30 min prior to addition of fluorogenic substrate. Protease concentrations: hCatK (150 pM), hCatL (5 pM), hCatS (1 nM), hCatV (25 pM), hCatB (1 nM), and papain (3 nM). Mean ± SD for a single representative experiment (triplicate measurement). NA = not available. More details are available in the SI.
Binding Mode of Alkynes Is Irreversible and Covalent
Reversibility of hCatK inhibition was assessed in a jump dilution assay.41 Recombinant hCatK was incubated with inhibitors at high concentration to allow full active-site occupation and subsequently diluted 300-fold into fluorogenic substrate (Z-FR-AMC) solution resulting in an increase of substrate hydrolysis in case of reversible inhibition (Figure 1). The progress curve shows that ODN is a (fast) reversible inhibitor, while inhibition by alkynes 4–6 is irreversible.
Figure 1.
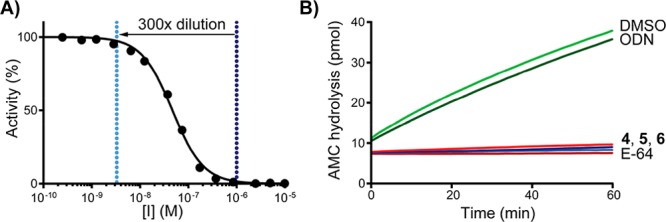
Jump dilution assay (A) 300-fold dilution of inhibitor concentration from full inhibition to full activity. (B) Progress curves for hCatK proteolytic activity after dilution in Z-FR-AMC. Control: E-64 is an irreversible pan-cathepsin inhibitor.
The nature of the inhibitor–cathepsin interaction was elucidated by LC–MS analysis of intact CatK and intact CatK–inhibitor complexes (Figure 2). Recombinant hCatK was incubated with inhibitor for 6 h to allow full covalent bond formation and submitted for measurement. An increase in the deconvoluted mass corresponding to addition of the inhibitor to hCatK was observed for alkynes 4–6, confirming the formation of a covalent hCatK–inhibitor complex.
Figure 2.
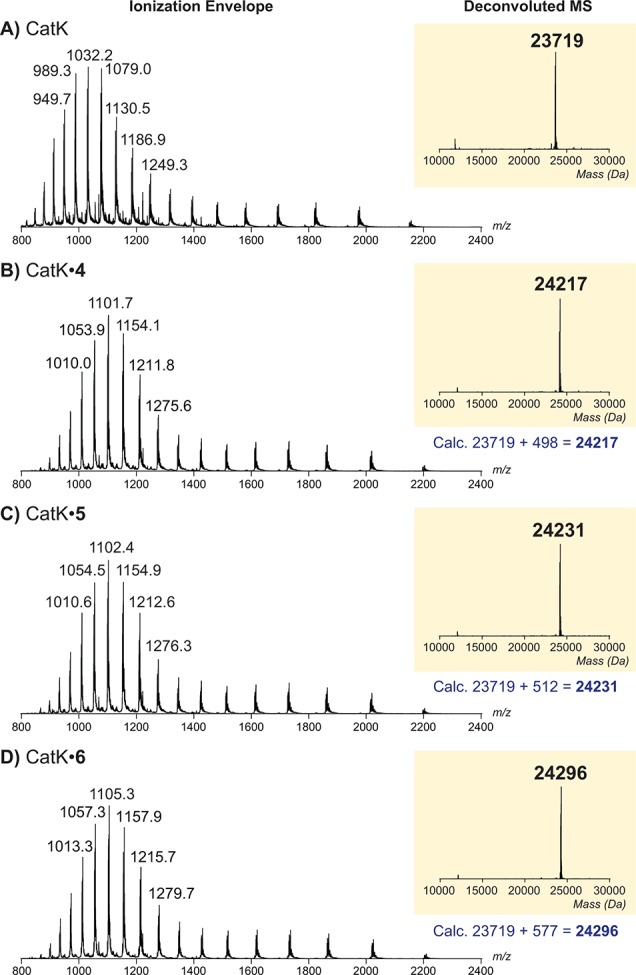
Representative ionization envelope (left) and deconvoluted electrospray ionization mass spectrum (right) of (A) intact hCatK or intact covalent complex with (B) inhibitor 4, (C) inhibitor 5, or (D) inhibitor 6 upon incubation with excess inhibitor.
The apparent potency of irreversible covalent inhibitors increases upon longer incubation with the enzyme, since the interaction of inhibitor with enzyme is not at equilibrium.1,42−45 As a result, the potency of these compounds can better be assessed by comparison of the kinact/KI ratio, which can be derived from the progress curve of substrate hydrolysis when the reaction is initiated by addition of the enzyme (Table 3; more details in the SI).42 Interestingly, the maximum rate of covalent bond formation (kinact) did not correlate with reactivity of the alkyne, as kinact for alkynes 4 and 5 is faster than for electron-deficient alkyne 6. We hypothesize that halogen bonding by the terminal bromine with the thiol moiety on hCatK positions the alkyne less optimal relative to the cysteine residue thus reducing the rate of proximity-driven C–S bond formation.46 The rate of covalent bond formation for ODN (k5) is faster than for the alkynes, also when correcting for the reverse reaction (k6).
Table 3. In Vitro Kinetic Evaluation of CatK Inhibitiona.
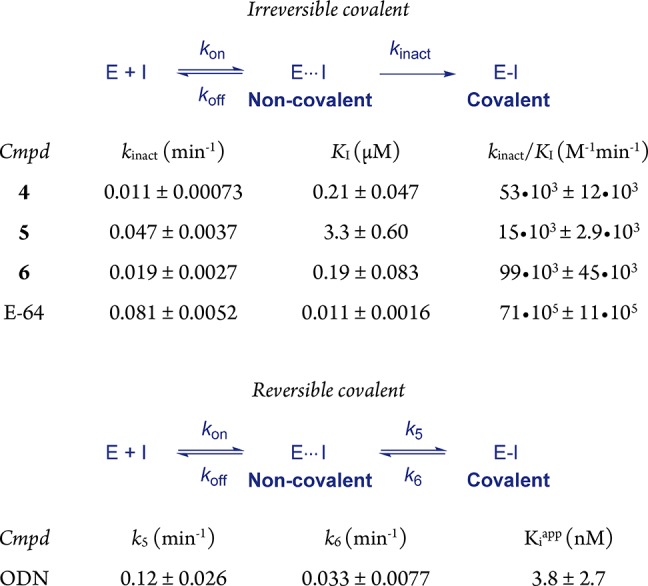
100 pM recombinant hCatK, 4 μM fluorogenic substrate Z-FR-AMC, 26 °C. Reaction initialization by addition of CatK. Mean ± SD for a single representative experiment (triplicate measurement). Information on data fitting to obtain kinetic constants is provided in the SI.
Alkynes Form a Covalent Thiovinyl Bond with Catalytic Cysteine Residue
Covalent CatK–alkyne 4 complex was submitted to bottom-up proteomic analysis to identify which amino acid residue is modified. In the tryptic digestion of unreacted CatK, the various length variant peptides containing the NQGQCGSCW-stretch have both Cys22 and Cys25 labeled with a carbamidomethyl group due to the alkylation reaction with iodoacetamide during the sample processing (Figure S4A,B). After reaction with alkyne 4, these peptides disappear, but various peptides containing the NQGQCGSCW-stretch appear labeled with one carbamidomethyl group and one inhibitor. Tandem mass spectrometric analysis by HCD and EThcD analysis of peptide NQGQCGSCWAFSSVGALEGQLKKK indicates inhibitor 4 is on the second cysteine residue (Cys25, Figure S4C). Together, this clearly shows that one of these cysteine residues is labeled, most likely catalytic Cys25.
The formation of a vinyl thioether linkage between catalytic Cys25 on hCatK and the internal carbon of the alkyne moiety was confirmed by solving the crystal structure of CatK–inhibitor 7 complex (Figure 3). Mature CatK was inactivated with MMTS for purification and storage and reactivated with DTT in the presence of alkyne inhibitors at high concentration (200 μM) to prevent self-degradation of CatK. Solubility of alkynes 4 and 5 was not sufficient, which can be contributed to the fluoroleucine moiety. We therefore synthesized alkyne 7, a closely related derivative in which the fluorine on the l-leucine building block was replaced by a proton to improve solubility (Scheme S2). The resulting CatK–inhibitor 7 complex was crystallized using a sitting drop method, and the structure could be solved at 1.7 Å resolution using maximum-likelihood free-kick (ML FK) electron density map47 (Figure S5). The refined structure unambiguously revealed the presence of a bond between the thiol atom of Cys25 and the internal carbon in alkyne 7, with a C–S distance of 1.8 Å.
Figure 3.
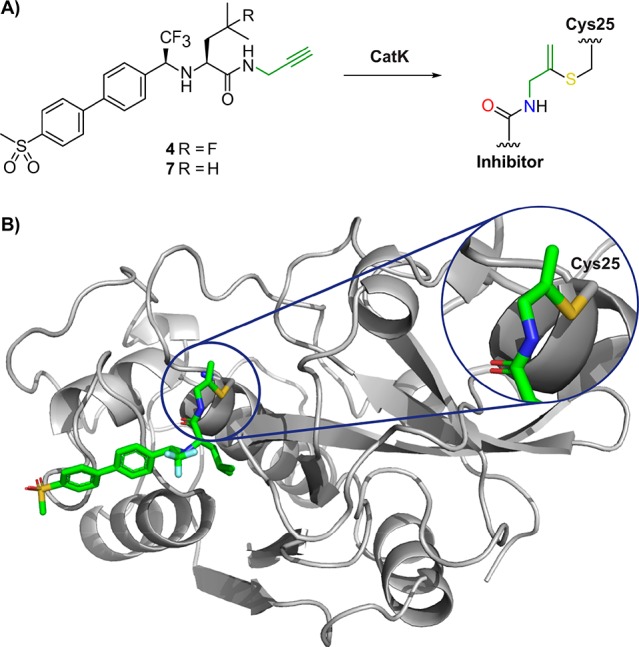
Crystal structure of alkyne 7 bound covalent to catalytic Cys25 in CatK. (A) Structure of inhibitor 7 before and after covalent bond formation with CatK. (B) X-ray structure of inhibitor 7 bound to Cys25 in CatK. PDB: 6QBS.
Inhibition of Bone Resorption Activity in Osteoclasts
Having established the covalent, irreversible inhibition of CatK on purified recombinant enzyme, we decided to test the inhibitory properties in a biologically relevant setting; inhibition of bone resorption by osteoclasts (OCs). OCs are the cells that degrade the bone matrix by secretion of acid and CatK into the resorption lacunae, resulting in the cleavage of collagen type I. OCs are essential in bone repair, and aberrant activity is observed in numerous diseases including osteoporosis, rheumatoid arthritis, giant cell tumor of the bone, and bone metastases.48−50
Inhibition of osteoclastic CatK was studied by culturing OCs on cortical bone slices in the presence of inhibitor. Mature OCs were obtained by treatment of CD14+ monocytes with M-CSF (macrophage colony stimulating factor) and RANKL (receptor activator of nuclear factor κB ligand) to stimulate differentiation to mature OCs (Figure 4).51 Mature OCs are formed by merging of mononuclear osteoclast precursors to form large multinucleated cells, a process that continued until the end of the culturing period. When the culturing medium was refreshed (every 3 days), inhibitor was freshly added to make sure there always is inhibitor present to inhibit CatK in the newly formed mature OCs. The OCs were cultured on bone slices for sufficient time to clearly observe bone resorption. After culturing for 21 days, the OCs were washed off the bone slices and the resorption pits were stained to visualize bone resorption activity. OCs with normal CatK activity form trenches, resorbing the bone while they move over the surface of the bone. Previously published observations in OCs from CatK–/– mice show that OCs lacking CatK are still able to form shallow pits, but unable to form trenches, with accumulation of collagen I fragments in the lysosomes.52
Figure 4.

Inhibition of CatK activity in human osteoclasts (OCs). (A) Maturation of OCs from monocytes. (B) CD14+ monocytes on bone slices were treated with M-CSF (day 0) and RANKL (day 3) to stimulate differentiation to mature OCs. Medium containing either inhibitor or DMSO was refreshed on day 7, 10, 13, and 16. At day 21, OCs were washed away and lysed, and bone slices were stained to visualize bone resorption. (C) Bone resorption visualized by staining of resorption pits with Coomassie Brilliant Blue. More staining means more resorption pits and, thus, more bone resorption activity. Normal OCs predominantly form deep trenches (paths), while OCs lacking CatK form small pits (circular dots). (D) Schematic overview of CatK activation. (E) CatK activity and expression in OC lysates. Top: fluorescence scan of CatK bound to irreversible activity-based probe BMV109 shows mature, active CatK. Middle/bottom: Western blotting against CatK shows total amount of CatK present in OC lysates. Darker bands indicate more CatK activity/expression.
Staining of bone slices for bone resorption showed formation of deep trenches for samples treated with 3 nM ODN, while 15 nM ODN resulted in the formation of shallow pits (Figure 4C), corresponding to an effective dose of around 15 nM.53 Treatment with 4 successfully inhibited bone resorption at concentrations from 80 nM, while inhibition with 5 was nonconclusive; we observed trenches as well as pits at all tested concentrations. Quantification of the total resorption area confirmed these observations, even though it is not possible to distinguish between shallow pits and deep trenches (Figure S6). From this experiment, we concluded that alkynes 4 and 5 are inhibitors of bone resorption with a higher potency than expected based on their potency to inhibit recombinant CatK.
Next, we treated the OC lysates with activity-based cathepsin probe BMV109 to assess whether the observed inhibition of bone resorption could be correlated with CatK activity (Figure 4D,E). CatK activity for OCs treated with DMSO is low, which is expected because mature CatK in its uninhibited form is self-degrading,54 and the observed bone resorption is the result of secreted mature CatK activity. Additionally, we expect that intracellular CatK is predominantly catalytic inactive pro-CatK, which is activated by cleavage of the activation peptide, an autoproteolytic event that requires an environment with a low pH for example lysosomes and the resorption lacunae.55 Interestingly, we observe a strong increase of mature CatK activity in all samples treated with ODN, while samples with inhibitor 4 or 5 do not show any CatK activity. The observed increase in mature CatK activity for ODN-treated samples does not reflect the actual intracellular proteolytic activity, but is the result of displacement of reversibly bound ODN by excess of irreversible probe BMV109. Alkynes 4 and 5 form an irreversible covalent bond with CatK, and can thus not be displaced by BMV109. Western blotting for CatK revealed an increase in the intracellular levels of mature CatK for OCs that were treated with high concentration of any inhibitor, which could be the result of inhibition of proteolytic CatK activity, which would normally degrade mature CatK.
Counting OCs that were cultured on plastic revealed an increase in the number of OCs for the highest concentrations of ODN, 4 and 5 (Figure S7). This is in agreement with previous reports that observed an increase of OC maturation as a response to CatK activity loss; the same number of bone marrow cells from CatK–/– mice led to a greater number of active OCs compared to bone marrow cells from the control mice.52 A significant increase in CatK expression upon 100 nM ODN treatment has been reported, without an increase in the number of OCs.56 We hypothesize that complete inhibition of CatK activity stimulates the maturation of OCs, and further investigations to identify the feedback mechanism are ongoing.
Conclusion
To conclude, alkynes are not only suitable as latent electrophiles in (large) peptides but also in small molecule inhibitors, as shown here for inhibition of cysteine protease cathepsin K (CatK). Alkyne-based covalent inhibitors do not show indiscriminate thiol reactivity but do form an irreversible covalent bond formation with CatK, as confirmed by MS analysis of the (intact) CatK–inhibitor complexes. X-ray crystallography confirmed the formation of the Markovnikov-type product between the active-site cysteine thiol and the internal carbon of the alkyne moiety. Kinetic evaluation shows that the rate of covalent bond formation (kinact) does not correlate with electrophilicity of the alkyne, supporting our hypothesis of proximity-driven reactivity. Optimization of the alkyne position relative to the cysteine residue could result in more potent compounds with faster covalent bond formation while not compromising on indiscriminate thiol reactivity. Treatment of human osteoclasts (OCs) with alkynes 4 and 5 showed a potent inhibition of CatK-mediated bone resorption activity, with only a 5-fold difference in effective dose between ODN and inhibitor 4. Further investigations into the biological effect of irreversible inhibition of CatK are ongoing.
Finally, we urge everyone using the alkyne moiety as a Click handle to be careful with the assumption that the alkyne is truly bioorthogonal; the binding of a small molecule inhibitor can be strong enough to initiate a thiol–alkyne reaction when the alkyne moiety is positioned in close proximity to a cysteine residue. More importantly, based on the proof-of-concept studies described herein, we foresee latent electrophiles such as the alkyne to be of great value in future development of cysteine-targeting covalent inhibitory drugs with a reduced risk of idiosyncratic toxicity.
Acknowledgments
We thank Patrick Celie at The Netherlands Cancer Institute (NKI) protein facility for the expression and purification of pro-CatK and Stephanie Hoppe from the NKI for the gift of ibrutinib and afatinib. This work was supported by a VICI grant from The Netherlands Organization for Scientific Research N.W.O. (H.O.) and by P1-0140 (B.T.) and P1-0048 (D.T.) grants from Slovene Research Agency.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b11027.
Detailed experimental procedures, figures, and crystallographic data (PDF)
The authors declare the following competing financial interest(s): E.M., H.O., and C.A.A.v.B. are inventors on a pending patent application on the use of the herein described cathepsin K inhibitors.
Supplementary Material
References
- Singh J.; Petter R. C.; Baillie T. A.; Whitty A. The resurgence of covalent drugs. Nat. Rev. Drug Discovery 2011, 10 (4), 307–317. 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Bauer R. A. Covalent inhibitors in drug discovery: from accidental discoveries to avoided liabilities and designed therapies. Drug Discovery Today 2015, 20 (9), 1061–1073. 10.1016/j.drudis.2015.05.005. [DOI] [PubMed] [Google Scholar]
- Silva D. G.; Ribeiro J. F. R.; De Vita D.; Cianni L.; Franco C. H.; Freitas-Junior L. H.; Moraes C. B.; Rocha J. R.; Burtoloso A. C. B.; Kenny P. W.; Leitão A.; Montanari C. A. A comparative study of warheads for design of cysteine protease inhibitors. Bioorg. Med. Chem. Lett. 2017, 27 (22), 5031–5035. 10.1016/j.bmcl.2017.10.002. [DOI] [PubMed] [Google Scholar]
- Lonsdale R.; Burgess J.; Colclough N.; Davies N. L.; Lenz E. M.; Orton A. L.; Ward R. A. Expanding the Armory: Predicting and Tuning Covalent Warhead Reactivity. J. Chem. Inf. Model. 2017, 57 (12), 3124–3137. 10.1021/acs.jcim.7b00553. [DOI] [PubMed] [Google Scholar]
- Barf T.; Kaptein A. Irreversible Protein Kinase Inhibitors: Balancing the Benefits and Risks. J. Med. Chem. 2012, 55 (14), 6243–6262. 10.1021/jm3003203. [DOI] [PubMed] [Google Scholar]
- Baillie T. A. Targeted Covalent Inhibitors for Drug Design. Angew. Chem., Int. Ed. 2016, 55 (43), 13408–13421. 10.1002/anie.201601091. [DOI] [PubMed] [Google Scholar]
- González-Bello C. Designing Irreversible Inhibitors—Worth the Effort?. ChemMedChem 2016, 11 (1), 22–30. 10.1002/cmdc.201500469. [DOI] [PubMed] [Google Scholar]
- Solca F.; Dahl G.; Zoephel A.; Bader G.; Sanderson M.; Klein C.; Kraemer O.; Himmelsbach F.; Haaksma E.; Adolf G. R. Target Binding Properties and Cellular Activity of Afatinib (BIBW 2992), an Irreversible ErbB Family Blocker. J. Pharmacol. Exp. Ther. 2012, 343 (2), 342. 10.1124/jpet.112.197756. [DOI] [PubMed] [Google Scholar]
- Pan Z.; Scheerens H.; Li S.-J.; Schultz B. E.; Sprengeler P. A.; Burrill L. C.; Mendonca R. V.; Sweeney M. D.; Scott K. C. K.; Grothaus P. G.; Jeffery D. A.; Spoerke J. M.; Honigberg L. A.; Young P. R.; Dalrymple S. A.; Palmer J. T. Discovery of Selective Irreversible Inhibitors for Bruton’s Tyrosine Kinase. ChemMedChem 2007, 2 (1), 58–61. 10.1002/cmdc.200600221. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Sabnis Y.; Zhao Z.; Zhang T.; Buhrlage S. J.; Jones L. H.; Gray N. S. Developing irreversible inhibitors of the protein kinase cysteinome. Chem. Biol. 2013, 20 (2), 146–159. 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata Y.; Chiba M. The Role of Extrahepatic Metabolism in the Pharmacokinetics of the Targeted Covalent Inhibitors Afatinib, Ibrutinib, and Neratinib. Drug Metab. Dispos. 2015, 43 (3), 375. 10.1124/dmd.114.061424. [DOI] [PubMed] [Google Scholar]
- Nakayama S.; Atsumi R.; Takakusa H.; Kobayashi Y.; Kurihara A.; Nagai Y.; Nakai D.; Okazaki O. A Zone Classification System for Risk Assessment of Idiosyncratic Drug Toxicity Using Daily Dose and Covalent Binding. Drug Metab. Dispos. 2009, 37 (9), 1970. 10.1124/dmd.109.027797. [DOI] [PubMed] [Google Scholar]
- Schwöbel J. A. H.; Koleva Y. K.; Enoch S. J.; Bajot F.; Hewitt M.; Madden J. C.; Roberts D. W.; Schultz T. W.; Cronin M. T. D. Measurement and Estimation of Electrophilic Reactivity for Predictive Toxicology. Chem. Rev. 2011, 111 (4), 2562–2596. 10.1021/cr100098n. [DOI] [PubMed] [Google Scholar]
- Zhao Z.; Bourne P. E. Progress with covalent small-molecule kinase inhibitors. Drug Discovery Today 2018, 23 (3), 727–735. 10.1016/j.drudis.2018.01.035. [DOI] [PubMed] [Google Scholar]
- Wright M. H.; Sieber S. A. Chemical proteomics approaches for identifying the cellular targets of natural products. Nat. Prod. Rep. 2016, 33 (5), 681–708. 10.1039/C6NP00001K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exceptions are published for MAO inhibitors in which the terminal alkyne acts as a nucleophile and for terminal alkynes that are transformed into more reactive ketene species by CYP450 enzymes.; a Sadler N. C.; Nandhikonda P.; Webb-Robertson B.-J.; Ansong C.; Anderson L. N.; Smith J. N.; Corley R. A.; Wright A. T. Hepatic Cytochrome P450 Activity, Abundance, and Expression Throughout Human Development. Drug Metab. Dispos. 2016, 44 (7), 984. 10.1124/dmd.115.068593. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Fan P. W.; Gu C.; Marsh S. A.; Stevens J. C. Mechanism-Based Inactivation of Cytochrome P450 2B6 by a Novel Terminal Acetylene Inhibitor. Drug Metab. Dispos. 2003, 31 (1), 28. 10.1124/dmd.31.1.28. [DOI] [PubMed] [Google Scholar]; c Borštnar R.; Repič M.; Kržan M.; Mavri J.; Vianello R. Irreversible Inhibition of Monoamine Oxidase B by the Antiparkinsonian Medicines Rasagiline and Selegiline: A Computational Study. Eur. J. Org. Chem. 2011, 2011 (32), 6419–6433. 10.1002/ejoc.201100873. [DOI] [Google Scholar]
- Ekkebus R.; van Kasteren S. I.; Kulathu Y.; Scholten A.; Berlin I.; Geurink P. P.; de Jong A.; Goerdayal S.; Neefjes J.; Heck A. J. R.; Komander D.; Ovaa H. On Terminal Alkynes That Can React with Active-Site Cysteine Nucleophiles in Proteases. J. Am. Chem. Soc. 2013, 135 (8), 2867–2870. 10.1021/ja309802n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlov N. V. Metal Catalysis in Thiolation and Selenation Reactions of Alkynes Leading to Chalcogen-Substituted Alkenes and Dienes. ChemistryOpen 2015, 4 (6), 682–697. 10.1002/open.201500137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castarlenas R.; Di Giuseppe A.; Pérez-Torrente J. J.; Oro L. A. The Emergence of Transition-Metal-Mediated Hydrothiolation of Unsaturated Carbon–Carbon Bonds: A Mechanistic Outlook. Angew. Chem., Int. Ed. 2013, 52 (1), 211–222. 10.1002/anie.201205468. [DOI] [PubMed] [Google Scholar]
- Lowe A. B. Thiol-yne ‘click’/coupling chemistry and recent applications in polymer and materials synthesis and modification. Polymer 2014, 55 (22), 5517–5549. 10.1016/j.polymer.2014.08.015. [DOI] [Google Scholar]
- Jayasree E. G.; Reshma S. A computational study on the reaction mechanism and energetics of Markovnikov and anti-Markovnikov addition in alkyne hydrothiolation reactions. Comput. Theor. Chem. 2016, 1098, 13–21. 10.1016/j.comptc.2016.10.012. [DOI] [Google Scholar]
- Sommer S.; Weikart N. D.; Linne U.; Mootz H. D. Covalent inhibition of SUMO and ubiquitin-specific cysteine proteases by an in situ thiol–alkyne addition. Bioorg. Med. Chem. 2013, 21 (9), 2511–2517. 10.1016/j.bmc.2013.02.039. [DOI] [PubMed] [Google Scholar]
- Arkona C.; Rademann J. Propargyl Amides as Irreversible Inhibitors of Cysteine Proteases—A Lesson on the Biological Reactivity of Alkynes. Angew. Chem., Int. Ed. 2013, 52 (32), 8210–8212. 10.1002/anie.201303544. [DOI] [PubMed] [Google Scholar]
- Turk V.; Stoka V.; Vasiljeva O.; Renko M.; Sun T.; Turk B.; Turk D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta, Proteins Proteomics 2012, 1824 (1), 68–88. 10.1016/j.bbapap.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizovišek M.; Fonović M.; Turk B. Cysteine cathepsins in extracellular matrix remodeling: Extracellular matrix degradation and beyond. Matrix Biol. 2018, 10.1016/j.matbio.2018.01.024. [DOI] [PubMed] [Google Scholar]
- Gauthier J. Y.; Chauret N.; Cromlish W.; Desmarais S.; Duong L. T.; Falgueyret J.-P.; Kimmel D. B.; Lamontagne S.; Léger S.; LeRiche T.; Li C. S.; Massé F.; McKay D. J.; Nicoll-Griffith D. A.; Oballa R. M.; Palmer J. T.; Percival M. D.; Riendeau D.; Robichaud J.; Rodan G. A.; Rodan S. B.; Seto C.; Thérien M.; Truong V.-L.; Venuti M. C.; Wesolowski G.; Young R. N.; Zamboni R.; Black W. C. The discovery of odanacatib (MK-0822), a selective inhibitor of cathepsin K. Bioorg. Med. Chem. Lett. 2008, 18 (3), 923–928. 10.1016/j.bmcl.2007.12.047. [DOI] [PubMed] [Google Scholar]
- Stoch S. A.; Zajic S.; Stone J.; Miller D. L.; Van Dyck K.; Gutierrez M. J.; De Decker M.; Liu L.; Liu Q.; Scott B. B.; Panebianco D.; Jin B.; Duong L. T.; Gottesdiener K.; Wagner J. A. Effect of the Cathepsin K Inhibitor Odanacatib on Bone Resorption Biomarkers in Healthy Postmenopausal Women: Two Double-Blind, Randomized, Placebo-Controlled Phase I Studies. Clin. Pharmacol. Ther. 2009, 86 (2), 175–182. 10.1038/clpt.2009.60. [DOI] [PubMed] [Google Scholar]
- Brömme D.; Lecaille F. Cathepsin K inhibitors for osteoporosis and potential off-target effects. Expert Opin. Invest. Drugs 2009, 18 (5), 585–600. 10.1517/13543780902832661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brömme D.; Panwar P.; Turan S. Cathepsin K osteoporosis trials, pycnodysostosis and mouse deficiency models: Commonalities and differences. Expert Opin. Drug Discovery 2016, 11 (5), 457–472. 10.1517/17460441.2016.1160884. [DOI] [PubMed] [Google Scholar]
- Mullard A. Merck & Co. drops osteoporosis drug odanacatib. Nat. Rev. Drug Discovery 2016, 15 (10), 669. 10.1038/nrd.2016.208. [DOI] [PubMed] [Google Scholar]
- Selent J.; Kaleta J.; Li Z.; Lalmanach G.; Brömme D. Selective Inhibition of the Collagenase Activity of Cathepsin K. J. Biol. Chem. 2007, 282 (22), 16492–16501. 10.1074/jbc.M700242200. [DOI] [PubMed] [Google Scholar]
- Dolman S. J.; Gosselin F.; O’Shea P. D.; Davies I. W. Selective metal-halogen exchange of 4,4′-dibromobiphenyl mediated by lithium tributylmagnesiate. Tetrahedron 2006, 62 (21), 5092–5098. 10.1016/j.tet.2006.03.039. [DOI] [Google Scholar]
- O’Shea P. D.; Chen C.-y.; Gauvreau D.; Gosselin F.; Hughes G.; Nadeau C.; Volante R. P. A Practical Enantioselective Synthesis of Odanacatib, a Potent Cathepsin K Inhibitor, via Triflate Displacement of an α-Trifluoromethylbenzyl Triflate. J. Org. Chem. 2009, 74 (4), 1605–1610. 10.1021/jo8020314. [DOI] [PubMed] [Google Scholar]
- Liu G.; Kong L.; Shen J.; Zhu G. A regio- and stereoselective entry to (Z)-β-halo alkenyl sulfides and their applications to access stereodefined trisubstituted alkenes. Org. Biomol. Chem. 2014, 12 (14), 2310–2321. 10.1039/C4OB00103F. [DOI] [PubMed] [Google Scholar]
- Shiu H.-Y.; Chan T.-C.; Ho C.-M.; Liu Y.; Wong M. K.; Che C.-M. Electron-Deficient Alkynes as Cleavable Reagents for the Modification of Cysteine-Containing Peptides in Aqueous Medium. Chem. - Eur. J. 2009, 15 (15), 3839–3850. 10.1002/chem.200800669. [DOI] [PubMed] [Google Scholar]
- Barf T.; Covey T.; Izumi R.; van de Kar B.; Gulrajani M.; van Lith B.; van Hoek M.; de Zwart E.; Mittag D.; Demont D.; Verkaik S.; Krantz F.; Pearson P. G.; Ulrich R.; Kaptein A. Acalabrutinib (ACP-196): A Covalent Bruton Tyrosine Kinase Inhibitor with a Differentiated Selectivity and In Vivo Potency Profile. J. Pharmacol. Exp. Ther. 2017, 363 (2), 240. 10.1124/jpet.117.242909. [DOI] [PubMed] [Google Scholar]
- Oballa R. M.; Truchon J.-F.; Bayly C. I.; Chauret N.; Day S.; Crane S.; Berthelette C. A generally applicable method for assessing the electrophilicity and reactivity of diverse nitrile-containing compounds. Bioorg. Med. Chem. Lett. 2007, 17 (4), 998–1002. 10.1016/j.bmcl.2006.11.044. [DOI] [PubMed] [Google Scholar]
- Siklos M.; BenAissa M.; Thatcher G. R. J. Cysteine proteases as therapeutic targets: does selectivity matter? A systematic review of calpain and cathepsin inhibitors. Acta Pharm. Sin. B 2015, 5 (6), 506–519. 10.1016/j.apsb.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lushchak V. I. Glutathione Homeostasis and Functions: Potential Targets for Medical Interventions. J. Amino Acids 2012, 2012, 1–26. 10.1155/2012/736837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law S.; Andrault P.-M.; Aguda A. H.; Nguyen N. T.; Kruglyak N.; Brayer G. D.; Brömme D. Identification of mouse cathepsin K structural elements that regulate the potency of odanacatib. Biochem. J. 2017, 474, 851–864. 10.1042/BCJ20160985. [DOI] [PubMed] [Google Scholar]
- Copeland R. A.; Basavapathruni A.; Moyer M.; Scott M. P. Impact of enzyme concentration and residence time on apparent activity recovery in jump dilution analysis. Anal. Biochem. 2011, 416 (2), 206–210. 10.1016/j.ab.2011.05.029. [DOI] [PubMed] [Google Scholar]
- Strelow J. M. A Perspective on the Kinetics of Covalent and Irreversible Inhibition. SLAS Discovery 2017, 22 (1), 3–20. 10.1177/1087057116671509. [DOI] [PubMed] [Google Scholar]
- Holdgate G. A.; Meek T. D.; Grimley R. L. Mechanistic enzymology in drug discovery: a fresh perspective. Nat. Rev. Drug Discovery 2017, 17, 115. 10.1038/nrd.2017.219. [DOI] [PubMed] [Google Scholar]
- Strelow J. M.; Dewe W.; Iversen P. W.; Brooks H. B.; Radding J. A.; McGee J.; Weidner J.. Mechanism of Action Assays for Enzymes. https://www.ncbi.nlm.nih.gov/books/NBK92001/.
- Copeland R. A.Irreversible Enzyme Inactivators. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, 2nd ed.; John Wiley & Sons, Inc., 2013; Chapter 9. [PubMed] [Google Scholar]
- Cavallo G.; Metrangolo P.; Milani R.; Pilati T.; Priimagi A.; Resnati G.; Terraneo G. The Halogen Bond. Chem. Rev. 2016, 116 (4), 2478–2601. 10.1021/acs.chemrev.5b00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pražnikar J.; Turk D. Free kick instead of cross-validation in maximum-likelihood refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2014, 70, 3124–3134. 10.1107/S1399004714021336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake M. T.; Clarke B. L.; Oursler M. J.; Khosla S. Cathepsin K Inhibitors for Osteoporosis: Biology, Potential Clinical Utility, and Lessons Learned. Endocr. Rev. 2017, 38 (4), 325–350. 10.1210/er.2015-1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindeman J. H. N.; Hanemaaijer R.; Mulder A.; Dijkstra P. D. S.; Szuhai K.; Bromme D.; Verheijen J. H.; Hogendoorn P. C. W. Cathepsin K Is the Principal Protease in Giant Cell Tumor of Bone. Am. J. Pathol. 2004, 165 (2), 593–600. 10.1016/S0002-9440(10)63323-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgington-Mitchell L. E.; Rautela J.; Duivenvoorden H. M.; Jayatilleke K. M.; van der Linden W. A.; Verdoes M.; Bogyo M.; Parker B. S. Cysteine cathepsin activity suppresses osteoclastogenesis of myeloid-derived suppressor cells in breast cancer. Oncotarget 2015, 6 (29), 27008–27022. 10.18632/oncotarget.4714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson G. C.; Malakellis M.; Collier F. M.; Cameron P. U.; Holloway W. R.; Gough T. J.; Gregorio-King C.; Kirkland M. A.; Myers D. E. Induction of osteoclasts from CD14-positive human peripheral blood mononuclear cells by receptor activator of nuclear factor κB ligand (RANKL). Clin. Sci. 2000, 99 (2), 133. 10.1042/CS19990355. [DOI] [PubMed] [Google Scholar]
- Kiviranta R.; Morko J.; Alatalo S. L.; NicAmhlaoibh R.; Risteli J.; Laitala-Leinonen T.; Vuorio E. Impaired bone resorption in cathepsin K-deficient mice is partially compensated for by enhanced osteoclastogenesis and increased expression of other proteases via an increased RANKL/OPG ratio. Bone 2005, 36 (1), 159–172. 10.1016/j.bone.2004.09.020. [DOI] [PubMed] [Google Scholar]
- Pirapaharan D. C.; Søe K.; Panwar P.; Madsen J. S.; Bergmann M. L.; Overgaard M.; Brömme D.; Delaisse J.-M. A Mild Inhibition of Cathepsin K Paradoxically Stimulates the Resorptive Activity of Osteoclasts in Culture. Calcif. Tissue Int. 2019, 104 (1), 92–101. 10.1007/s00223-018-0472-7. [DOI] [PubMed] [Google Scholar]
- Thompson S. K.; Halbert S. M.; Bossard M. J.; Tomaszek T. A.; Levy M. A.; Zhao B.; Smith W. W.; Abdel-Meguid S. S.; Janson C. A.; D’Alessio K. J.; McQueney M. S.; Amegadzie B. Y.; Hanning C. R.; DesJarlais R. L.; Briand J.; Sarkar S. K.; Huddleston M. J.; Ijames C. F.; Carr S. A.; Garnes K. T.; Shu A.; Heys J. R.; Bradbeer J.; Zembryki D.; Lee-Rykaczewski L.; James I. E.; Lark M. W.; Drake F. H.; Gowen M.; Gleason J. G.; Veber D. F. Design of potent and selective human cathepsin K inhibitors that span the active site. Proc. Natl. Acad. Sci. U. S. A. 1997, 94 (26), 14249. 10.1073/pnas.94.26.14249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQueney M. S.; Amegadzie B. Y.; D’Alessio K.; Hanning C. R.; McLaughlin M. M.; McNulty D.; Carr S. A.; Ijames C.; Kurdyla J.; Jones C. S. Autocatalytic Activation of Human Cathepsin K. J. Biol. Chem. 1997, 272 (21), 13955–13960. 10.1074/jbc.272.21.13955. [DOI] [PubMed] [Google Scholar]
- Leung P.; Pickarski M.; Zhuo Y.; Masarachia P. J.; Duong L. T. The effects of the cathepsin K inhibitor odanacatib on osteoclastic bone resorption and vesicular trafficking. Bone 2011, 49 (4), 623–635. 10.1016/j.bone.2011.06.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.