

Abstract
Nanodomains are intracellular foci which transduce signals between major cellular compartments. One of the most ubiquitous signal transducers, the ryanodine receptor (RyR) calcium channel, is tightly clustered within these nanodomains. Super-resolution microscopy has previously been used to visualize RyR clusters near the cell surface. A majority of nanodomains located deeper within cells have remained unresolved due to limited imaging depths and axial resolution of these modalities. A series of enhancements made to expansion microscopy allowed individual RyRs to be resolved within planar nanodomains at the cell periphery and the curved nanodomains located deeper within the interiors of cardiomyocytes. With a resolution of ∼ 15 nm, we localized both the position of RyRs and their individual phosphorylation for the residue Ser2808. With a three-dimensional imaging protocol, we observed disturbances to the RyR arrays in the nanometer scale which accompanied right-heart failure caused by pulmonary hypertension. The disease coincided with a distinct gradient of RyR hyperphosphorylation from the edge of the nanodomain toward the center, not seen in healthy cells. This spatial profile appeared to contrast distinctly from that sustained by the cells during acute, physiological hyperphosphorylation when they were stimulated with a β-adrenergic agonist. Simulations of RyR arrays based on the experimentally determined channel positions and phosphorylation signatures showed how the nanoscale dispersal of the RyRs during pathology diminishes its intrinsic likelihood to ignite a calcium signal. It also revealed that the natural topography of RyR phosphorylation could offset potential heterogeneity in nanodomain excitability which may arise from such RyR reorganization.
Keywords: expansion microscopy, signaling nanodomains, site-specific phosphorylation, ryanodine receptor, computational modeling of intracellular calcium
Intracellular calcium (Ca2+) nanodomains are the structural units of fast intracellular second messenger signaling mechanisms in eukaryotic cell types. Muscle,1−3 neuronal,4,5 and secretory cell types6 all rely on nanodomains, which allow coordinated release of Ca2+ from intracellular compartments to evoke cellular responses such as contraction, modulation of electrical excitability, gene transcription, and secretion. Typical nanodomains can span up to the order of 102 nanometers and can include the principal Ca2+ release channels: the ryanodine receptors (RyR) and/or inositol triphosphate receptors (IP3R). These channels are often strategically co-clustered to allow concerted opening and may be triggered both by their neighbors (via Ca2+-induced Ca2+ release; CICR) or local signal transducers such as voltage-gated L-type calcium channels (LCC) and phospholipase-c (PLC). Junctions between the plasmalemma and the sarcoplasmic reticulum (SR) in cardiac muscle cells (also called cardiomyocytes) are among the most extensively studied nanodomain types. Mounting evidence that the molecular constituents of nanodomains may be reorganized7 or remodelled8,9 in life-threatening pathologies has emphasized the need for imaging modalities which are capable of visualizing their molecular components.
The earliest measurements of the three-dimensional (3D) topology of nanodomains were made using transmission and scanning electron microscopy (EM).10,11 These data laid the foundation for the current paradigm of signal transduction at the nanodomain (see review on muscle).12 The large size (∼2 MDa) and square shape of the RyR tetramer is distinctly identifiable with newer EM techniques,13−15 albeit in larger nanodomains. Fluorescence modalities, including super-resolution microscopy (e.g., techniques known by acronyms STED16 and dSTORM17) have been instrumental in characterizing the clustering properties of RyRs.18,19 DNA-based point accumulation for imaging in nanoscale topography (DNA-PAINT), implemented with total internal reflection fluorescence (TIRF) microscopy, has resolved the positions of individual RyRs within these clusters located near the cell surface.20 It has allowed molecular counts to be made of the regulatory protein Junctophilin-2 which co-clusters with RyR.20 These visual insights now present new opportunities to build geometrically realistic computational models of the local spatiotemporal patterns of Ca2+ signaling.21
A few key features in contemporary super-resolution techniques limit their utility in characterizing nanodomains in primary cell types. The majority of nanodomains (>80%) in cells such as cardiomyocytes are found in deeper regions of the cell interiors, as deep as 5–15 μm from the cell surface (see Supplementary Figure S1 for a schematic illustration). Most single-molecule imaging techniques suffer from poor localization accuracy at these depths. An added complication is the complex curvatures of nanodomains located deeper within the cell interiors (with diameters of 100–200 nm),9,15 as they wrap around tubular invaginations of the plasmalemma (known as “t-tubules”; see Supplementary Figure S1). Techniques such as STED8,19 and dSTORM have previously been used to map these interior nanodomains. 3D dSTORM (with an axial resolution of ∼65 nm) has shown the close relationship of these nanodomains with the t-tubules;22 however, they have only resolved the overall shapes of RyR clusters23,24 and not the intrinsic patterns of channel organization within these clusters. The poorer axial resolutions of these modalities have prevented the visualization of more intricate features and geometries of these nanodomains. Quantification of RyRs and computational modeling of RyR clusters have therefore relied upon assumptions that they form continuous, crystalline arrays8,21−23 as seen in vitro. Recent DNA-PAINT and tomographic EM data from near-surface nanodomains have shown that RyR self-assembly may be amorphous, less continuous, and potentially dynamic depending on the cell’s physiology.13,20 A modality which offers both high in-plane and axial resolution is therefore required. 3D implementations of techniques like dSTORM have been used very effectively to resolve cellular features which are much finer than these (e.g., 3D microtubule networks,25,26 actin rings in neuronal axons,27 and centrioles26,28). However, the anisotropic refractive index (RI; which varies around 1.38 and 1.414) in muscle,29 heavy intrinsic autofluorescence, shallow imaging depth of these techniques, and suboptimal labeling densities have limited their utility for 3D super-resolution mapping of nanodomains.
Recent developments have presented a few potential solutions. Combination of adaptive optics,30 structured illumination (SIM),31 or light-sheet techniques32 with 3D single marker localization approaches have yielded sub-50 nm axial resolution while preserving in-plane super-resolution. An alternative approach is the principle of expanding the fluorescence marker topography of a labeled sample using a self-expanding hydrogel, called expansion microscopy (ExM).33 This allows features which were smaller than the diffraction-limited resolution of the microscopes to be resolved with no additional optical modifications or marker localization protocols. The departure from localization microscopy, compatibility with traditional far-field imaging techniques (e.g., confocal microscopy) and conventional marker probes (with protocols such as protein-retention ExM)34 have allowed biologists to consistently achieve a resolution-improvement by a factor of ∼4.34,35 Combining ExM with STED, SIM, or STORM has allowed this improvement to be further enhanced.36−41 More recent reports have provided refined ExM protocols (called X10 ExM42 and iterative-ExM43) which now allow >10-fold improvement in resolution by improving the volumetric expansion of the sample beyond the standard expansion factor of 4. Over a short period of four years, ExM has been applied in a wide range of studies, such as visualization of the cytoskeletal and membrane structures of isolated and/or cultured mammalian cells,35,44 RNA mapping with the use of expansion-fluorescence in situ hybridization,45 detection of pathological biomarkers in human biopsy tissues,46 neural circuitry in whole brains,47 microbial systems,38,44 and model organisms.41,48−51 Such diversity in applications underscores a number of key improvements that have been made to the hydrogel and probe chemistries and the protocol of expansion,34 which makes the principle of ExM adaptable for a range of cell and tissue types. The more recent applications of ExM for making highly precise measurements on true molecular-scale structures (e.g., 3D tubulin organization in centrioles,52 and subunit organization of the nuclear pore complex53) now present this technique as a validated tool for making nanometer scale measurements.
We aimed to develop an approach, based on ExM, to resolve not only the positions of the individual RyRs but also their individual chemical identities (site-specific phosphorylation) in cardiomyocytes. This paper describes how we combined X10 microscopy with Airyscan (a protocol which we call “enhanced expansion microscopy” or EExM) to achieve a working resolution which we estimate to be ∼15 nm in-plane and ∼35 nm axially. We have exploited this 3D super-resolution to map both the positions and the phosphorylation state of RyRs within the three-dimensionally complex nanodomains, both at the surface and interiors of cardiomyocytes at single-protein precision. This approach has revealed nanometer-scale rearrangements of individual RyRs, deterioration of nanodomain sizes, and the spatial patterns of phosphorylation of RyR (at the residue Ser2808, which enhances RyR excitability and open probability)54 in cardiomyocytes of a rodent model with right ventricular failure. We detail how this level of positional and biochemical information on individual RyRs can be exploited for in silico inquiry of the structural basis of nanodomain Ca2+ signaling at a spatial and temporal resolution which has never been achieved experimentally.
Results
Evaluation of EExM for Super-Resolution Imaging of Cell Interior
For evaluating expansion microscopy as a method for imaging cell interiors, we examined lattices of α-actinin called “z-discs” (red-hot; Figure 1A) and networks of microtubules (green) in the interior of cardiac muscle cells. The highly uniform α-actinin lattices and their span across the entire width of the cell (15–30 μm in thickness) made the z-discs a useful intrinsic standard for comparing the resolution of deconvolved confocal microscopy, two-dimensional (2D) dSTORM (under HiLo oblique illumination),55 DNA-PAINT (in TIRF), 4× EExM, and 10× EExM (Figure 1B, left to right). In longitudinal view of the cells, each modality revealed a highly regular z-disc arrangement. Magnified views showed a double-banded morphology within each z-disc which was resolvable only with DNA-PAINT, 10× EExM, and, to a lesser extent, with 4× EExM (Figure 1C). Line profiles of the α-actinin distribution across the z-discs (as indicated in Figure 1C) in each type of data (Figure 1D) revealed three key observations: The higher resolution techniques (e.g., dSTORM compared to confocal) generally reported the α-actinin lattices to be narrower. Despite offering in-plane resolution comparable to 4× EExM (∼40–50 nm), dSTORM did not detect the double-banded morphology of α-actinin. Intensity profiles of DNA-PAINT images were virtually identical to those acquired in the cell interior with 10× EExM. The longitudinal separation between the peaks (illustrated in red in the right panel of Figure 1D) was very similar between DNA-PAINT and 10× EExM (Figure 1E; Means: 74.0 vs 70.1 nm). This confirmed that the resolution achieved in these two modalities is comparable (we estimate ∼15 nm in-plane). Based on a contemporary model of the cardiac z-disc featuring up to six longitudinally arranged parallel lattices of α-actinin56 (Figure 1F), we simulated a likely antibody (Ab) labeling pattern in these cells (Supplementary Figure S2). A double-banded morphology closely mimicking the experimental data was observed when DNA-PAINT and EExM images of a gradient of Ab penetration at the z-disc lattice were modeled. The simulation confirmed that the principal determinants of the observed labeling pattern were the effective lateral and axial resolution of ∼15 and 35 nm, respectively (Supplementary Figure S3).
Figure 1.

Adaptation of ExM for imaging nanoscale intracellular structures in optically thick cells. (A) Overview of the shape and size of rat ventricular myocytes labeled for α-actinin (red hot) and α-tubulin (green). (B) Comparison of z-disc α-actinin immunolabeling in the cell interior mapped with deconvolved confocal microscopy, dSTORM implemented with HiLo illumination, DNA-PAINT implemented in TIRF, 4× EExM (i.e., ExM images acquired with the Airyscan protocol), and 10× EExM. (C) Magnified view of the respective images revealed only DNA-PAINT and 10× EExM could resolve the double-layer morphology of the z-disc reported by the anti-α-actinin Ab. (D) Line profiles taken across the z-discs in the respective images illustrating a bimodal intensity profile with a separation of ∼70 nm at the peaks in DNA-PAINT, 10× EExM, and, to a lesser extent, in 4× EExM data. (E) Dotplots of the measured separation between the α-actinin double-peaks as measured through 10× EExM and DNA-PAINT (Mean ± SEM: 74.05 ± 3.12 nm and 70.10 ± 2.22 nm respectively; n = 21 and 17; p = 0.31 in two-tailed t test). Overlaid box and whisker plots illustrate the 5th, 25th, 50th, 75th, and 95th percentiles. (F) This double-layer morphology was consistent with a model of the z-disc consisting of six parallel layers of α-actinin (green) anchoring actin filaments (blue); the two outermost layers are optimally labeled with Abs (orange; see Supplementary Figure S2). (G) Maximum intensity projections of a 3D 10× EExM volume of α-actinin (gray) and α-tubulin (colored for depth, indicated in μm) acquired near the center of the cell (at a sample depth of ∼ 50 μm) illustrate the capability of 10× EExM to image cellular regions far from the surface. (H) Magnified region of the same data illustrates the tessellation between microtubule bundles with the z-discs within ∼50 nm, illustrated with (I) overlaid intensity profiles of a region (dotted line in inset). Scale bars: (A) 5 μm; (B and H) 1 μm; (C) 400 nm; and (G) 2 μm (EExM scale bars adjusted to expansion factor).
To demonstrate the enhancement that 10× EExM offers in imaging cell interiors, we illustrate the nanoscale 3D relationship between the α-actinin lattices of the z-discs (gray) and the microtubule network (color-coded 3D geometry traced by α-tubulin in Figure 1G; magnified view in 1H) throughout a 5 μm-thick volume of the cell, at a depth of ∼5 μm from the cell surface. A line profile illustrates how two intertwined microtubule bundles (black line) are tessellated at ∼50 nm of the two outer layers of the α-actinin lattices of the z-disc. We used the regularity of the z-disc spacing (a feature known as “sarcomeric length”; SL) along the length of the cell to confirm isotropy of expansion in these hydrogels (Supplementary Figure S4A–C). The consistency in the double-banded morphology in α-actinin labeling was used as evidence that the spatial measurements were unaffected by gel-to-gel variations in the expansion factor (Supplementary Figure S4D–F).
RyR Arrays within Intracellular Nanodomains Resolved with 10× EExM
The RyR organization within near-surface nanodomains was examined with 10× EExM and compared with DNA-PAINT as the current benchmark.20 A clustered labeling pattern was observed across the surface regions visualized with both techniques (Figure 2A-i). In magnified view, 10× EExM revealed punctate labeling densities (Figure 2A-ii) with strong resemblance to the patterns seen in DNA-PAINT. In dSTORM images at depths of ∼10 μm inside the cells, the RyR labeling appeared more clearly banded (Figure 2B-i). Closer examination of both dSTORM and 10× EExM data revealed clusters which were curved and/or elongated (Figure 2B-ii), however dSTORM data did not reveal any substructure within the cluster regions (left). The 10× EExM images reported a punctate labeling morphology of RyRs (right), similar to that seen near the cell surface. With a simulation, we established that the punctate appearance of clusters was enabled by and consistent with the superior in-plane and axial resolution offered by 10× EExM of ∼ 15 nm and 35–40 nm, respectively (Supplementary Figure S5). A maximum-intensity projection of RyR labeling, color-coded for depth throughout a 1 μm-deep cell volume, illustrates the complexity of the 3D topography of RyRs visualized now with 10× EExM (Figure 2C). Figure 2D-i illustrates the flat topology of cell surface RyR arrays (left) with 3D isosurface visualization (middle), color-coded for the axial (z) depth. This topology was consistent with an RyR (orange in schematic, Figure 2D-i, right) alignment that is parallel to the surface plasmalemma (cyan). The RyR clusters in the cell interior regions (maximum-intensity projections in Figure 2D-ii and -iii, left) revealed nanodomains which curved around local t-tubule membranes. Depending on whether the cluster was imaged either end-on (Figure 2D-ii) or side-on (Figure 2D-iii), relative to the image planes, the depth-encoded 3D visualization showed distinct gradation of the colors of the isosurface (Figure 2D-ii and -iii, middle). The approximated orientations of the RyR array (orange) and the tubular plasmalemma (cyan) are shown schematically on the right.
Figure 2.
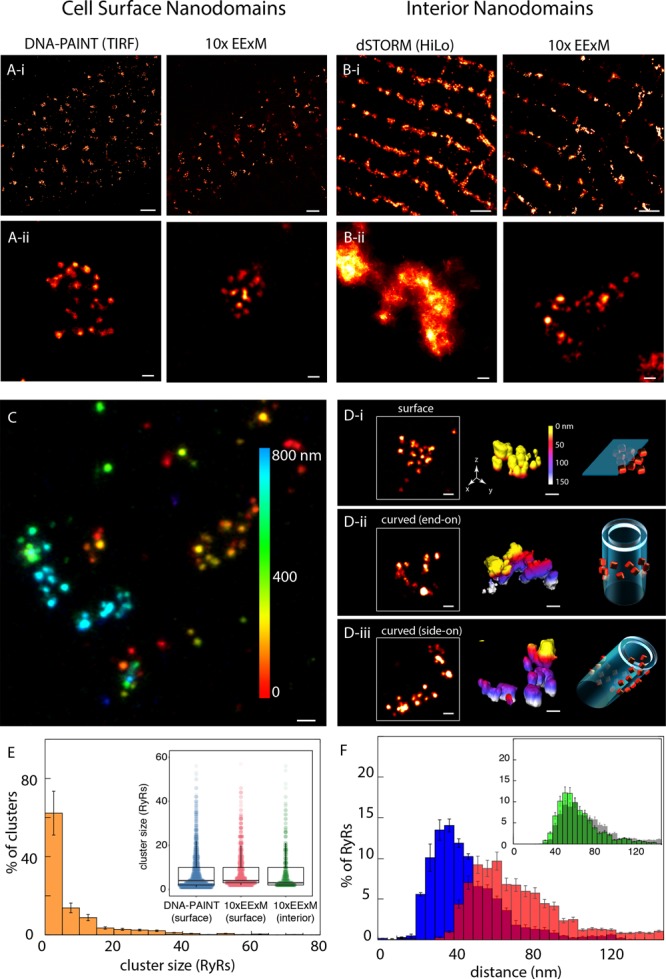
Adaptation of 10× EExM for imaging RyR nanodomain in cardiac muscle cells. (A-i) Comparisons of DNA-PAINT (in TIRF; left) and 10× EExM (right) images of the RyR labeling near the cell surface of rat ventricular cardiac muscle cells, both illustrate domains of RyR labeling whose width is <500 nm. (A-ii) Magnified views illustrate clearly resolved punctate labeling densities, each corresponding to an individual RyR, in both DNA-PAINT and 10× EExM images. (B-i) Comparisons of dSTORM (with HiLo illumination) and 10× EExM images of RyR nanodomains located in the cell interiors. (B-ii) Magnified views of the respective images illustrate unresolved cluster substructure in dSTORM (left) which contrasts with the well-resolved RyR arrays in 10× EExM in cell interior (right). (C) A depth coded (color scale in nm) maximum-intensity projection of a 1 μm-deep cell volume with RyR labeling. Comparison of RyR cluster 3D topographies identified with 10× EExM raw data include (D-i) flat RyR arrays at the cell surface and curved nanodomains in cell interior visualized either (D-ii) end-on, along their axis of curvature, or (D-iii) side-on, orthogonal to the axis of curvature. Shown are the in-plane view of the nanodomains (left), surface rendered 3D 10× EExM data, color coded for depth along the optical axis (z) in nm (middle) and schematic illustration of unique topologies of RyR (orange) arrangement relative to the nearest plasmalemmal membranes (cyan). (E) Percentage histogram of the RyR cluster sizes in the cell interior nanodomains show a Mean ± SEM of 8.23 ± 0.51 RyRs/cluster; n = 11 cells, 1912 clusters. The distributions of the surface cluster sizes, as estimated with both DNA-PAINT (Mean ± SEM of 7.87 ± 0.39 RyRs/cluster; n = 12 cells, 3209 clusters) and the surface and interior clusters imaged 10× EExM, were similar as illustrated by the dot-plots overlaid with the box and whiskers plots (inset; p > 0.05 in Bonferroni-adjusted Mann–Whitney tests). (F) The distribution of NND in interior nanodomains with ≥5 RyRs (blue) showed a mean of 45.41 ± 0.75 nm. The distribution of 3ND (red) for the same RyR nanodomains had a long rightward tail (mean ± SD: 73.74 ± 14.28 nm). Overlaid with the 3ND histogram of the cell interior (gray, inset) is the equivalent distribution for subsurface RyR nanodomains (green), which was more left-shifted. Scale bars: (A-i and B-i) 1 μm; (A-ii, B-ii, C, and D) 50 nm. Error bars in plots: SD.
For further analysis of the positions of the punctate RyR labeling densities, we used an algorithm which determined the in situ 3D centroids of each RyR punctum. Supplementary Figure S5 visually illustrates the RyR puncta of a curved cluster and their corresponding 3D centroids. The spatial organization of these puncta (see 3D data in Supplementary Figure S6) were similar to the selection of 3D-rendered tomographic EM data published previously from rat cardiomyocytes.14 In the interior clusters, an approximately exponential distribution of RyR cluster size was observed (Figure 2E, main panel; mean of ∼8.23 RyRs/cluster). Many of the puncta were observed either as solitary or small (<5 RyRs) clusters both in the cell interior and near-surface regions. The cluster sizes in the cell interior strongly resembled the size distribution in near-surface nanodomains (mean of ∼8.98 RyR/cluster; Figure 2E, inset). A dot-plot analysis illustrated that the counts of RyR in each near-surface cluster were very similar between the 10× EExM and DNA-PAINT data (mean ∼7.84 RyR/cluster; blue in Figure 2E inset). We further examined the RyR assembly pattern in larger (≥4 RyRs) clusters with histograms of the nearest-neighbor distance (NND; blue in Figure 2F) and the average distance between the 3 nearest neighbors for each RyR (3ND; red in Figure 2F). The NNDs, a measure of the co-clustering between individual receptors, followed an approximately Gaussian distribution with a mean of ∼45 nm which was consistent with previous measurements of center-to-center spacings between RyRs made with tomographic EM.13 The 3NDs, reporting the average spacings between RyRs in a given array, were more variable and produced a histogram with a rightward tail and a mean of ∼73 nm. This contrasted with the 3NDs histogram for near-surface nanodomains (gray barplot overlaid with the interior 3ND histogram, shown in green, in the inset of Figure 2F) which showed less variability and a shorter tail (mean ∼64 nm). The NND and the 3ND distributions for near-surface clusters provided an additional point of comparison between the measurements between DNA-PAINT and 10× EExM and showed that the shapes of the two distributions were independent of the method (Supplementary Figure S7).
Adaptation of EExM for Visualizing RyR Rearrangement in Pathology
RyR clusters are susceptible to remodelling, particularly in pathologies of the heart such as atrial fibrillation.8 Exploiting the enhanced resolution of 10× EExM, we examined changes in the structure of RyR nanodomains in right ventricular (RV) myocytes of rats displaying RV failure, following treatment with monocrotaline (MCT). At the cell surface, the clustering pattern of RyR labeling was broadly comparable between control (Figure 3A) and MCT-RV cells (Figure 3B), however, solitary puncta were more abundantly observed in the latter (arrowheads). Compared to control cells (Figure 3C), RyR clusters near the surface of MCT-RV cells (Figure 3D) visually appeared smaller; many regions contained single puncta. Closer examination allowed us to characterize changes within the nanodomains which accompany this pathology. In the interiors of both MCT-RV and control cells, the RyR clusters followed a more banded organization which reflected the organization of the sarcomeres (Figure 3E,F). However, clusters in MCT-RV were smaller and contained visibly fewer RyRs (Figure 3G,H).
Figure 3.

Adaptation of 10× EExM to visualize nanoscale RyR rearrangement in pathology. Comparison of near-surface RyR organization in right ventricular cardiomyocytes isolated from (A) control and (B) MCT-RV, which featured noticeably more frequent solitary RyR puncta (arrowheads). Magnified views of (C) control and (D) MCT-RV nanodomains at the cell surface revealed a visibly sparse dispersed RyR arrangement within the nanodomain areas. RyR nanodomains in the interior of (E) control cells and (F) MCT-RV cells. Magnified view of (G) RyR nanodomains in control cells featured closely clustered RyRs tracing out their curved topography, which contrasted with (H) regions in MCT-RV cell featuring smaller and less discernible nanodomains. (I) NND histogram of interior nanodomains in MCT-RV cells (gray-shaded) featured a larger mean ± SEM (51.65 ± 4.45 nm), a right-shifted primary maximum at ∼45 nm, and a secondary maximum at ∼85 nm (black arrows) compared to control (blue; mode in blue arrow; mean: 45.41 ± 0.76 nm; p = 0.001, df = 19 cells; Bonferroni-adjusted Mann–Whitney test). (J) Histograms showing a rightward shift in the mode for the 3NDs for MCT-RV RyRs (gray-shaded; black arrow; mean 91.02 ± 12.88 nm) compared to control (red; red arrow; mean: 73.74 ± 14.27 nm; p = 0.0002, df = 19, Bonferroni-adjusted Mann–Whitney test). (K) Dot-plots comparing the cluster size distributions in near-surface and interior nanodomains show a diminished mean and the range of RyR counts in individual clusters in both near-surface (p < 0.0001; df = 20) and interior (p < 0.0001; df = 19) of MCT-RV cells compared to control. (L) Schematic illustrating how cluster fragmentation in MCT-RV may coincide with the increased NND and 3ND (green arrows) for some RyRs. Scale bars: (A, B, E, and F) 1 μm and (C, D, G, and H) 50 nm. Error bars in plots: SD.
To assess the nature of the mutual reorganization of RyR channels, we compared the NNDs and the 3NDs of RyRs in the larger clusters (≥4 RyRs) of control and MCT-RV cells. In contrast to the narrow unimodal NND distribution for RyRs in interior nanodomains of control cells (mode ∼ 40 nm; Figure 3I, blue), the MCT-RV NND distribution (gray, overlaid) featured two peaks at ∼45 nm and ∼90 nm, respectively. A similar trend was observed in RyRs in near-surface nanodomains of MCT-RV cells (Supplementary Figure S8). This observation suggested that at least a small subset of RyRs had dissociated from their default clustering pattern. This hypothesis was further supported by the longer rightward tails observed in the 3ND histograms (Figure 3J and Supplementary Figure S7), which related to RyR clusters with looser arrangement of receptors.
Confirming the visual observations of smaller RyR clusters, coupled with higher abundance of solitary RyRs in MCT-RV, we observed an ∼40% reduction in the mean number of RyRs in MCT-RV clusters compared to control in both cell interior and near-surface nanodomains (Figure 3K). The smaller cluster size is also compatible with the hypothesis of nanodomain fragmentation during the disease (Figure 3L), leading to greater variability in the RyR-RyR distances.
Mapping Phosphorylation Status of RyRs
In addition to spatial remodelling, RyR nanodomains are also known to undergo types of biochemical remodelling both in healthy physiology and in disease. These include the phosphorylation of RyR at Ser2808. With two-color 10× EExM, we mapped the Ser2808 phosphorylation state of RyR (with an anti-pSer2808 Ab; purple in Figure 4A) relative to the positions of individual RyRs (gray) in near-surface and interior nanodomains of rat cardiac muscle cells. Magnified views of both types of nanodomains showed that punctate pSer2808 labeling densities either overlapped or closely tessellated with RyRs consistently (Figure 4B). These data (inset, Figure 4C) were used to localize the centroids of the RyR (gray circles) and pSer2808 puncta (purple). Individual RyRs were recorded as “phosphorylated” if their centroid consisted of any detectable pSer2808 labeling within a 30 nm 3D radius (Figure 4C). Approximately 1–3% of the pSer2808 puncta were not detected in association with RyRs (arrowhead).
Figure 4.

10× EExM visualization of variable phosphorylation of RyRs during pathology and acute stimulation. (A) Overlay of 10× EExM images of RyR (gray) and pS2808 (purple) at the surface of a rat ventricular cardiac myocyte. Inset shows the equivalent view in a region ∼5 μm below the cell surface. (B) Magnified view of nanodomains near (i) the cell surface and (ii) cell interior visually illustrates that punctate pS2808 (purple) labeling densities, observed in fewer numbers, follow either close overlap or proximity with RyR puncta (gray). (C) The centroids of the RyR (gray) and pS2808 puncta (purple) were localized and paired with the nearest RyR within a 30 nm 3D distance (dashed line) to identify RyRs which were phosphorylated at the Ser2808 site. Raw data of region illustrated in inset. An example of unpaired pS2808 marker is shown with an arrowhead. Magnified views of the overlays of subsurface nanodomains in cells from the right ventricles of (D) control and (E) MCT rats show a minor fraction of RyRs which associate pS2808 labeling. The same comparison in the cell interior regions illustrates a similar fraction of RyRs with Ser2808 phosphorylation in (F) control nanodomains, while a greater proportion of RyRs, despite smaller cluster size, coincide with pS2808 labeling in (G) MCT-RV nanodomains. (H) Percentage histograms of the mean fraction of RyRs determined to be phosphorylated at Ser2808 (Pphos) in the subsurface nanodomains containing ≥4 RyRs illustrate near-identical distributions (mean ± SD of Pphos of 0.31 ± 0.10 and 0.30 ± 0.12; p > 0.05; df = 20 cells; Bonferroni-corrected Mann–Whitney t test) for both control (purple) and MCT-RV (blue). (I) Cell interior nanodomains in MCT-RV (blue; 0.45 ± 0.07) showed a rightward shifted Pphos distribution compared to control (purple; 0.27 ± 0.09 ; p = 0.022, df = 19). Pphos distributions for both subsurface and interior nanodomains appeared to show a right-shift in control cells stimulated with isoproterenol and simultaneous electrical pacing at 1 Hz (insets). (J) Histograms of the 2D pS2808 localization density as a function of the distance within the subsurface nanodomain relative to its boundary in control (purple line), stimulated (black), and MCT-RV (blue) cells. (K-i) In control cells, RyRs phosphorylated at Ser2808 are located, on average, uniformly throughout the nanodomain. (K-ii) Stimulation with isoproterenol and electrical pacing appeared to approximately double the pS2808 density uniformly across the nanodomain. (K-iii) In MCT-RV, a gradient of pS2808 density is seen from the edge of the nanodomain inward, allowing a subdomain of potentiated RyRs to be maintained at the center of the nanodomain (circle). Scale bars: (A) 200 nm, (B–G) 50 nm.
We visually compared the localizations of RyRs which were phosphorylated at Ser2808 within near-surface nanodomains of control and MCT-RV cells (Figure 4D,E, respectively) to find that only a subset was phosphorylated. As a point of comparison, we examined the pSer2808 labeling at the near surface nanodomains in control cells which were stimulated with electrical pacing and the β-adrenoceptor agonist isoproterenol, which is known to evoke acute intrinsic hyperphosphorylation of the RyRs.57 The densities of pSer2808 labeling in these RyR clusters were noticeably higher (Supplementary Figure S9). In cell interior nanodomains (Figure 4F,G), the density of pSer2808 labeling appeared visually comparable for control and MCT-RV cells. Given that RyR clusters were smaller in MCT-RV, the visually identifiable proportion of phosphorylated RyRs was higher in these cells. Histograms of the proportion of phosphorylated RyRs in each cluster (containing ≥4 RyRs; Pphos) between control (purple; Figure 4H) and MCT-RV (blue) cells were comparable (mean ∼0.31 for control and ∼0.30 for MCT-RV) and consisted of similar modes (∼0.24 and ∼0.28, respectively). Few clusters featured either no or complete phosphorylation. A similar distribution was observed for cell interior nanodomains of control cells (purple in Figure 4I; mean ∼0.27 and mode ∼0.16); however, the distribution for Pphos in MCT-RV cells was right-shifted (blue; mean ∼0.45 and mode ∼0.42). This shift in Pphos closely mimicked the shape of the equivalent histogram analyses performed on near-surface and interior RyR clusters of control cells (insets of Figure 4H,I) which were stimulated with isoproterenol and electrical pacing (means ∼0.42 and ∼0.57, respectively).
To investigate whether the likelihood of RyR phosphorylation was dependent on the location within the nanodomain, we plotted the density of pSer2808 puncta against the distance from the edge of the RyR cluster. A near-uniform density was observed in regions ≥50 nm inside of the cluster-boundary of control cells (purple; Figure 4J). In a simulation, we observed that such a uniform density profile is consistent with a random uniform probability of RyR phosphorylation throughout the cluster (Supplementary Figure S10). This density profile appeared to be uniformly amplified by a factor of ∼2 when control cells were stimulated as above (gray; Figure 4J). However, in MCT-RV cells, the pSer2808 puncta density showed a distinct gradient extending from the cluster boundary to the center. This density-distance analysis suggested that healthy cells, when under higher β-stimulation, allow a uniform increase in RyR phosphorylation at Ser2808 throughout each nanodomain (Figure 4K-i and -ii). Despite nanodomains being smaller and/or fragmented, they typically foster a higher density of phosphorylated RyR at the center of the clusters in MCT-RV cells (Figure 4K-iii).
Simulation of Nanodomain Calcium Dynamics
Given the positions of individual RyRs and their respective phosphorylation state, we investigated the likely spatiotemporal patterns of Ca2+ release in these nanodomains determined purely from these structural considerations. Two spatially discretized models based on the RyR and pSer2808 data of flat, approximately median-sized near-surface nanodomains were constructed: one from control (Figure 5A) and the other from MCT-RV (Figure 5B). The centroid positions of each RyR (circles Figure 5C,D) and the phosphorylation identity (yellow for phosphorylated) were marked in each example. Ten independent simulations of the temporal pattern of the local cytoplasmic [Ca2+] in the nanodomain cleft in Ca2+ signals (known as ‘Ca2+ sparks’) were performed for control (Figure 5E) and MCT-RV (Figure 5F) examples. The control cluster consistently gave rise to sparks with amplitudes of 30–80 μM [Ca2+]. By comparison, the MCT-RV nanodomains featured Ca2+ release which lacked the temporal synchronization and development into the full temporal profile of sparks (also see Supplementary Movie). The simulated image series of [Ca2+] in the control (Figure 5G) and MCT-RV (Figure 5H) nanodomains allowed visual examination of likely spatiotemporal pattern of Ca2+ release at resolutions of 10 nm and 0.1 ms. In control nanodomains, we often observed Ca2+ release which was seeded by individual or subgroups of phosphorylated RyRs in the earliest phases of the spark (i.e. at ∼10–20 ms; Figure 5G-ii). These events appeared to cumulatively recruit their nearest neighbors (Figure 5G iii-vi) via CICR, typifying the “triggered saltatory” mechanism of initiating Ca2+ sparks hypothesized previously.18 A similar triggering was observed in MCT-RV clusters (Figure 5H-ii); however, the recruitment of their neighboring RyRs was neither cumulative nor complete. Dephosphorylation of the RyRs in the MCT-RV example, while maintaining all other parameters, showed complete failure of the nanodomain to release Ca2+ (Supplementary Movie). This suggested that RyR hyperphosphorylation at Ser2808 could serve to offset the loss of the nanodomain’s excitability during RyR rearrangement.
Figure 5.

Simulating geometrically realistic spatiotemporal patterns of Ca2+ release based on 10× EExM data. Shown are examples of RyR (gray) and pS2808 (purple) labeling in approximately median-sized nanodomains of control (A) and MCT-RV cells (B). The centroids of the receptors were localized (circles), and receptors identified as phosphorylated were marked (yellow circles) for the control (C) and MCT-RV (D) clusters. A series of local Ca2+ sparks, overplotted as the time course of the local change in cytoplasmic Ca2+ concentration (shown in μM), illustrates how the control (E) geometry facilitates more consistent and pronounced Ca2+ release events compared to that in the MCT-RV cluster (F). Spatiotemporal visualization of the cytoplasmic Ca2+ (color scale shown in μM) at a spatial resolution of 10 nm in the early phases of the Ca2+ sparks illustrates progressive recruitment of RyRs, more readily among those phosphorylated at S2808, in the control nanodomain (G) compared to MCT-RV. The latter shows recruitment of RyR openings, however, often failed to achieve cluster-wide activation in the first 20 ms of the spark. Scale bars: 50 nm. Time stamps in (G and H) shown in ms.
Discussion
10× EExM has allowed us to map the 3D positions of dyadic RyRs in nanodomains deep within the interiors of cardiomyocytes with a resolution which matches that offered by DNA-PAINT for imaging 2D nanodomains at the cell periphery.20 This advancement is enabled by a superior in-plane and axial resolution over existing benchmarks such as 3D dSTORM data published recently.22 With this, we have observed both acute and chronic changes in the phosphorylation of the RyRs within the nanodomain and molecular-scale repositioning of RyRs coinciding with the RV failure pathology.
ExM as a Quantitative True-Molecular-Scale Imaging Technique
Our implementation of 10× EExM examined the components of Ca2+ nanodomains, particularly in the cell interior. We have demonstrated its superior resolution compared to established imaging techniques such as dSTORM and 4× EExM (Figure 1B,C) by mapping 3D cytoskeletal components in cardiomyocyte interiors. With near-surface imaging of both α-actinin lattices and RyR clusters, we showed that the resolution achieved by 10× EExM almost matches that of DNA-PAINT.20 A notable gain in combining the X10 microscopy protocol of Truckenbrodt et al.42 with 3D Airyscan is a distinct improvement in the axial resolution to ∼35 nm. Simulations of the 10× EExM imaging process using two models of curved nanodomains—one of a large RyR cluster and the other of a cluster with a narrower radius of curvature (Supplementary Figure S5)—have confirmed that the 10× EExM imaging protocol is particularly suited for imaging RyRs (spaced ∼40 nm apart) within curved nanodomains. They revealed that the detection accuracy of single RyRs was better where the working resolution was ∼15 nm in-plane and ∼35 axially. Accuracy of aligning fine structures in the image with multichannel 10× EExM is further subject to chromatic alignment of the imaging channels. With the Airyscan imaging system that we used, we found that this alignment error was 10–13% of the effective resolution achieved (see Estimation of the Point Spread Function and Effective Resolution section for details).
As an imaging protocol, 10× EExM was difficult to master initially due a drop in the RyR labeling intensity equivalent to the cube of the expansion factor. However, four key features of our experimental design permitted the single-RyR level of sensitivity to be achieved during the imaging. First, the digestion and removal of the cell material in ExM renders the RI of the sample homogeneous, as described before.37 While dSTORM experiments for mapping RyR23,24 used an 80% glycerol mounting medium to reduce the intracellular RI inhomogeneities, no active clearing approach such as this was adopted. Second, the clearing also led to a near-complete removal of the autofluorescence arising from the intrinsic contractile proteins, mitochondrial NADPH, and flavin co-enzymes, which occupy >40% of the cardiomyocytes’ cytoplasmic volume (discussed by Larcher et al.).58 For EExM, this enhanced the detection of low marker densities substantially. This was a clear advantage over our previous DNA-PAINT and dSTORM experiments18,20 where marker localization accuracy was limited considerably by cellular autofluorescence.59 Third, our selection of bleach-resistant fluorophores, Alexa488 and Janelia549 in particular, also allowed greater consistency in retention and detection of RyRs during the highly oxidative gelation step of ExM42 and Airyscan imaging, respectively. Finally, the choice of the Airyscan method over confocal microscopy for imaging the sample further enhanced the effective resolution improvement and greater photon collection efficiency enabled by its sensitive array detector and the absence of a physical pinhole.60
To validate 10× EExM as an accurate tool for measuring intracellular structures, we performed two key tests to confirm that (i) the expansion of the cellular structures was isotropic and (ii) the effect of gel-to-gel variations in expansion factor (∼ 7–10.5) on the standard deviation of the measurements was minimal (Supplementary Figure S4). There are no ideal calibration standards (e.g., DNA origamis which would expand in proportion to the structures of interest) for these tests currently; however, we used the periodicity of the myocytes’ sarcomeres and fixed-width of z-discs as intrinsic reporters of local gel expansion. The standard deviation of <10% was well in the range of the biological variations of these structures observed previously,61 providing us with assurance on the above requirements. The similar measurements in the cluster size (Figure 2) NND and 3ND (Supplementary Figure S7) between DNA-PAINT and 10× EExM provided additional confirmation that the spatial calibrations of the expansion factor and the isotropy of expansion translate well to the scale of RyR clustering (∼ 30–80 nm). As a super-resolution approach for examining and quantifying structures with 3D complexity, therefore, 10× EExM presents a useful and highly reproducible technique. A comparison with respect to the ease of use of 10× EExM versus high-end localization microscopy methods does not allow a clear-cut conclusion. The 5 day sample preparation protocol and an as yet unproven suitability for examining tissue, particularly heart tissue containing high densities of collagen,42 are still limiting factors that can reduce the practicality of 10× EExM for some experimental settings. On the other hand, the purely chemical approach of EExM is likely more accessible to cell biologists who often have limited expertise in instrumentation development and the intricacies of fluorophore photochemistry, as argued previously.62
3D and Chemical Topography of Nanodomains
By applying 10× EExM to image RyR labeling in rat ventricular myocytes, we observed puncta, whose identities as single RyRs have been confirmed both visually (benchmarking against DNA-PAINT; Figure 2A)20 and quantitatively (by cluster sizes and RyR NND and 3ND measurements; Supplementary Figure S7). The RyR arrays patterns observed with both methods are irregular and, therefore, consistent with the noncrystalline cluster self-assembly which we proposed previously based on near-surface DNA-PAINT data (see Supplementary Figure S2B of refs (20 and 63)). This direct approach to RyR counting made the in situ analysis of RyR clustering more straightforward than indirect protocols (based on event density22,24 or frequency20 calibrations) used in dSTORM and DNA-PAINT experiments previously. The superior 3D data have allowed clear visualization of the nonplanar topology of interior nanodomains (Figure 2D and Supplementary Figure S5). These data have allowed optical comparisons between the fine structure of near-surface and interior nanodomains. Overcoming the uncertainties in RyR counting with techniques like dSTORM,18,23 the 10× EExM data suggest that the two types of nanodomains are similar in size and receptor organization pattern (confirmed by measurements of NND).
An advantage of the multicolor capability of ExM is the ability to map the phosphorylation state of the individual RyRs in situ. The anti-pSer2808 is a well-established antibody and has been used extensively to characterize physiological64,65 and pathological66 RyR phosphorylation. In our analysis, we observed that ∼30% of the RyRs are phosphorylated at Ser2808 at the basal (unstimulated) state. Direct comparisons of the cluster-specific Pphos between near-surface and interior nanodomains provided both visual and quantitative evidence that basal level phosphorylation at Ser2808 is comparable between the two cellular regions. It is well characterized that Ser2808 can be modified primarily by protein kinase A (PKA) but also by calmodulin kinase II (CAMKII),67 commonly under the control of intracellular signaling cascades downstream of β-adrenergic stimulation, to enhance the RyR open probability. The local densities of pSer2808 measured throughout the near-surface nanodomains in control myocytes (Figure 4J) may therefore reflect a basal “tuning” to maintain near-uniform local excitability of RyRs throughout the nanodomain. In an additional iterative simulation which artificially inverted the RyR phosphorylation pattern (supplementary Figure S11), we observed a higher-than-normal likelihood of the nanodomain to ignite full-blown Ca2+ sparks, originating from the locations of pSer2808. This suggested that the nanodomain’s basal pSer2808 tuning may encourage a uniform profile of RyR excitability throughout the nanodomain, potentially as a counter against geometrically determined heterogeneity, proposed in previous models.21 We note that the excitability of individual RyRs is functionally regulated by multiple phosphorylation sites on each of the four RyR subunits, physical interactions with regulators such as FK506 binding protein 12.668 and junctophilin-269 on their cytoplasmic domains. Mapping all of these regulators simultaneously may not be achievable given the bulky size (and potential steric competition) of current probes; however, this would enable a more comprehensive simulation of the intrananodomain heterogeneities in RyR excitability.
We recognize that the in situPphos values estimated for larger nanodomains through our imaging data (Figure 4H,I; ∼30% for control myocytes) are lower than that reported by Western Blot analysis of denatured cardiac homogenates using the same antibody (∼69%).70 The differences in the measurements are not clear, but likely to be methodological, either relating to potentially incomplete labeling of pSer2808 in situ or an unaccounted selectivity to phosphorylated RyRs in the membrane fractionation methods used in in vitro studies.70 We note that the proportional increase in Pphos (by ∼50%) that we see under β-adrenergic stimulation (with electrical pacing in the presence of isoproterenol) closely reproduces the in vitro measurements on pSer2808 made by Huke and Bers.65
Modification and Redistribution of Ryanodine Receptors in Physiology and Pathology
Ser2808 is only one of a handful of PKA and CAMKII targets on the RyR,67 and it is possible that there is little/no relationship between RyR self-assembly pattern and pSer2808. In our experiments, control cells undergoing stimulation showed uniform phosphorylation of RyR throughout the nanodomain, and there was no detectable difference in phosphorylation pattern between near-surface and interior nanodomains. RyR phosphorylation patterns appear different in MCT-RV cells. Interior nanodomains preferentially show hyperphosphorylation at the basal state. The higher systemic β-adrenergic stimulation experienced by these animals71 and the chronic (over a few weeks) upregulation of PKA expression in MCT-RV cells72 could well explain this observation. The exclusivity of this phenotype to interior nanodomains however contrasts clearly with the acute effects of β-adrenergic stimulation observed in control cells. This may simply reflect the broad changes in β-adrenergic intracellular signaling mechanisms which accompany the disease.72 Despite the preservation of Pphos in peripheral nanodomains, a redistribution of pSer2808 is seen (Figure 4J,K). This may be the likely result of an unravelling nanodomain membrane structure which can allow (i) a spatial redistribution of the AKAP scaffold proteins which harbor PKA within the nanodomain,73 (ii) increased access of phosphatases to the peripheral regions of the nanodomain, (iii) or both.
In pathology, we observe a remodelling of the nanodomains which feature a less closely packed RyR-RyR pattern with increased inter-receptor distances, both near the cell surface and in interior regions. This change appeared to be most prominent in the cardiomyocytes of the RV. NND and 3ND measurements in the LV (column 4 of Supplementary Figure S7), by comparison, more closely resembled the distributions in the control cells. The curvatures and the extended shapes of the RyR cluster in the cell interior were less apparent. The superior resolution of 10× EExM has provided a molecular-scale view of the RyR cluster fragmentation, first reported very recently74 using STED and dSTORM at resolutions of 40–80 nm. The 10× EExM data presented in Figure 3 therefore report the smallest spatial scale at which pathological remodelling of heart has been observed to date.
Simulation of Nanoscale Signaling Based on Super-Resolution Data
The simulation (Figure 5) developed from 10× EExM utilizes experimentally determined positions and biochemical states of individual RyRs to interpret intracellular Ca2+ signaling. In Figure 5E,F, we demonstrate its utility, particularly in examining one of the nanodomains’ fundamental features–the ability to produce fast and reproducible Ca2+ sparks. Direct comparisons of the control RyR arrangement with the disease phenotype relative to the Ca2+ revealed the diminishing coupling between adjacent RyRs as they are dispersed. This manifested in both failed Ca2+ sparks as well as unsynchronized opening of individual RyRs within the nanodomain (Figure 5H). While the diminished amplitude of the sparks may be explained by the smaller number of receptors in the diseased nanodomains, the higher variability in the likelihood of evoking a full Ca2+ spark is striking (Figure 5F). Failed Ca2+ sparks in this spatial scale are unlikely to be detectable with the current state-of-the-art Ca2+ imaging techniques which have to contend with a number of limitations which associate contemporary Ca2+ imaging methods. These limitations include high resting cytoplasmic [Ca2+], diffusion of Ca2+ indicator dye, as well as Ca2+, poor photon efficiency particularly when using a pinhole-based detector and the diffraction limited resolution of the imaging system.75 Spatially accurate models such as this are therefore a useful approach to studying the functional phenotype of nanodomain signaling in disease.
Limitations
Our application of 10× EExM has come with a few noteworthy limitations. While we characterized the isotropy of expansion and reliability of the expansion factors estimated in the scales >70 nm, we were limited by the lack of a known calibrant in the range of 10–30 nm. Cross-validation of RyR NND measurements against DNA-PAINT was a useful workaround; however, a DNA origami (which, at present, has been proposed in a preprint manuscript)76 or similar spatial calibration standard which reports the gel expansion in this spatial scale is needed. For now, we find assurance in our approach which is similar to the analyses performed by others using features of intrinsic ultrastructures (e.g., the circularity of the cross-section of centrioles).52 The single RyR positions detected from the 3D 10× EExM data were likely to be limited by two factors. The poorer axial resolution which, as we demonstrate in the 3D simulation, could lead to errors or loss of detection in ∼5% of the receptors, particularly in regions where RyRs are aligned orthogonal to the image-plane (Supplementary Figure S5). Achieving ∼10× expansion is an important determinant because we show in the same simulation that approximately halving of this effective resolution leads to ∼30% drop in the detection accuracy. Second, the binding efficiency of the antibody probes is likely to be limiting. While RyR counts within clusters were comparable to those achieved with DNA-PAINT (and two different Abs) previously,20 there may be steric limitations in the Abs’ access to the targets. The RyR and/or pSer2808 positions may be subject to offsets due to the size of the Ab markers relative to the effective resolution of the data, hence we required a distance-based likelihood criterion for determining the identity of the RyRs carrying the detected phosphorylation. Probes that are more compact but compatible with the pro-ExM chemistry should be examined in the future as a solution to this.
We have used flat, near-surface nanodomains in our simulations to exploit the certainty of the 2D geometry of cytoplasmic Ca2+ diffusion. One of the main limitations in the 10× EExM images of RyR and pSer2808 is the lack of an independent marker of the curvature of the t-tubular membranes62 which determines the 3D diffusion of Ca2+ both within and beyond the nanodomain. This is a limitation particularly in MCT-RV cells where the t-tubular system broadly undergoes remodelling,77 and there is visual evidence in the 10× EExM data of diminishing curvature of the nanodomain. Future investigations therefore would benefit from establishing reliable plasmalemmal and SR markers compatible with ExM. Furthermore, the simulations utilized a general description of RyR dynamics and phenomenological implementation of phosphorylation. Future detailed modeling studies would benefit from biophysically detailed species- and disease-specific formulations of these model components; results presented occurred within parameter ranges which illustrated the differences between control and MCT, but do not necessarily translate to those which may manifest physiological behavior. These simulation results should therefore be considered as a demonstration of the potential for the structural data attained to be used in future mechanistic studies rather than rigorous analysis of the effect of MCT remodelling on cardiac CICR. Nevertheless, these data indicate an important role for nanodomain structure in regulating EC coupling, illustrating that the number of RyRs, their spatial distribution, and both the proportion and distribution of phosphorylation sites can all have an impact on local Ca2+ dynamics and inducibility of CICR; all these factors are observed to be remodelled in MCT and potentially other disease models.
Conclusions
The enhancement to the ExM approach has allowed us to map nanoscale features located deeper within cells at a resolution of ∼15 nm. Exploiting this capability, we have resolved individual RyRs forming 3D complex clusters within nanodomains in the cell interior. Detection of nanometer-scale changes in position and likelihood of site-specific phosphorylation has allowed us to simulate geometrically realistic spatiotemporal profiles of cytoplasmic Ca2+ signals which are encoded by these nanodomains.
Methods
Cell Preparation and Animal Models
Live myocytes were isolated from hearts freshly dissected from Wistar rats, euthanized according to a protocol approved by the UK Home Office. Pulmonary arterial hypertension (PAH) was induced in 5-week old adult rats with a single intraperitoneal injection of monocrotaline (MCT; Sigma-Aldrich), as detailed previously.78 RV-failure was typically observed between days 21 and 28, when the animals were euthanized (references to these cell samples are notated as “MCT-RV” in the text). Isolated right ventricular cells were adhered to coverslips as described before20 and fixed in 2% paraformaldehyde (Sigma) for immunocytochemistry. For examining the effects of stimulating RyR phosphorylation, living cells were simultaneously stimulated with electrical field stimuli at 1 Hz and 100 nM Isoproterenol (Sigma) dissolved in Tyrode’s solution prior to fixation. See Supporting Information experimental procedures for more a detailed protocol. Data presented in this manuscript included cells isolated from four control animals and five MCT-treated animals. Sample numbers stated in the text refer to cell numbers considered in each analysis.
EExM
ExM was performed on fixed and stained cell samples either according to the proExM protocol for 4× expansion34 or X10 microscopy protocol.42 Immunofluorescent labeling protocol is detailed in the Supporting Information. pSer2808 was labeled with anti-RyR2-pSer2808 IgG (A010-30AP, Badrilla Ltd., UK). Among the secondary Abs that we tested, we chose goat antimouse and goat antirabbit conjugates of AlexaFluor488 and JaneliaFluor549 for their strongest photostability during the ExM protocol and shorter wavelength of emission, conducive for superior imaging resolution. The labeling patterns seen with these Abs were also visually confirmed by ExM samples labeled with longer-wavelength secondary Ab (conjugates of Atto 647-N). Samples were subjected to confocal or Airyscan imaging on an inverted LSM880 with Airyscan (Carl Zeiss, Jena) used with a Plan-Apochromat 63× 1.4 NA objective with a working distance of 0.19 mm. Image sampling was set to <40 nm/px in x–y plane and 100 nm in z. The true spatial scale of the images was calculated by multiplying the voxel sampling by the linear expansion factor of the given gel (estimated as detailed in Supplementary Figure S3E). All distances reported in the Results and Discussion sections from ExM data are based on these rescaled distances.
DNA-PAINT and dSTORM of immuno-labeled cells were performed as detailed in previous reports,20,24 on a custom-built Nikon TE2000 TIRF microscope equipped with a 60× 1.49 NA TIRF objective and a 671 nm diode laser (Viasho, China). For dSTORM, the beam angle was inclined to achieve HiLo illumination,55 focused deeper into the sample. Primary DNA-PAINT and dSTORM data were recorded with an Andor Zyla-5.5 sCMOS camera (Andor, Belfast) using the freely available Python Microscopy Environment (PyME) software (www.python-microscopy.org). PYME was used for localizing the fluorophore positions and rendering the point data of dSTORM and DNA-PAINT experiments onto 5 and 1 nm pixel grids for further spatial analyses.
See Supporting Information experimental procedures for more details on imaging protocols.
Spatial Analysis of EExM and DNA-PAINT Data
Segmentation
Local ensembles of punctate RyR densities were segmented into cluster regions on a binary mask using a protocol used previously for segmenting DNA-PAINT images.20,24 These masks were used for the distance-based analysis (Figure 4J) of labeling density using a Euclidean distance transform of the cluster area as detailed before.79
Detection
Punctate labeling densities which were typical for RyR and pSer2808 Ab labeling in EExM and DNA-PAINT data were analyzed by their spatial distribution. A detection algorithm, similar to that used previously,20 was implemented in Python to record the 3D coordinates of the centroid of each punctum; each punctum was approximated as a single RyR, as before.20 For cell surface (2D) DNA-PAINT and 10× EExM data, the detection was 2D. A 3D implementation of this was used for mapping RyR and pSer2808 puncta in the cell interiors. Please see Supporting Information experimental procedures for a detailed protocol. To measure the longitudinal separation between the double peaks of α-actinin, the subpixel centroids of each intensity peak were calculated using a Gaussian line profile fitting procedure detailed previously.61,80 The average distance between the two centroids was calculated for each line (as plotted in Figure 1D).
Estimation of the Point Spread Function and Effective Resolution
The PSF for the confocal deconvolution was estimated by acquiring and averaging between 3D confocal z-stacks of 100 nm yellow-green or red-orange Fluospheres (ThermoFisher) immobilized inside a 2% agarose gel at a depth of ∼20 μm from the coverslip. To estimate the effective PSF of Airyscan postprocessing 3D image data, 20 nm yellow-green Fluosphere beads were imaged within a 2% agarose gel and subjected to the same analysis. The effective PSF in EExM experiments was estimated by scaling the Airyscan PSF by the expansion factor of the specific sample. Line intensity profiles both in-plane and axial dimensions of the EExM PSF volumes (e.g., Supplementary Figure S4B) were fitted with a 1D Gaussian function, and the full-width at half-maximum (fwhm) was taken as a close approximation of the resolution. Variations in this fwhm were observed by taking line profiles from beads embedded within the agarose gel at depths of 5, 30, and 47 μm (overlaid in Supplementary Figure S4B). These reported in-plane fwhm’s of 13.3, 14.8, and 17.3 nm and axial fwhm’s of 33.4, 39, and 46 nm at these depths, respectively.
The estimated PSF is subject to two principal uncertainties. These include the error in localizing the centroids of the bead images prior to averaging (with bead alignment based on a least-squares approach, we estimate that this associates a broadening of the estimated PSF < 1.5%). Further, potential shift variances in the PSF when sampling beads in different locations of the sample would have led to a broader PSF. In the bead data acquired, we did not observe major differences in the bead images from different regions (laterally) within the imaging field which could not be explained by noise or subpixel-scale drift of the beads.
The chromatic alignment of the AlexaFluor488 (green) and JaneliaFluor549 (red) emission channels was important for accurately localizing the pSer2808 signatures of RyRs. We assessed this alignment by acquiring the Airyscan images of Hydro-gel embedded 100 nm TetraSpeck microspheres (Thermo) in the emission channels under the sample imaging parameters (see Supplementary Figure S12). Based on this analysis, the spatial error between the channels was estimated to be ∼10%, subject to the errors in localizing the beads which apply to PSF estimation.
Simulation of Confocal, dSTORM, DNA-PAINT, and EExM Images
3D models of the z-disc (Supplementary Figures S2 and S3) and the curved nanodomains (supplementary Figure S5) were convolved with a PSF that approximated the resolution achieved by each technique. Confocal and Airyscan PSFs were estimated by imaging 100 nm microspheres in the same imaging conditions as the samples. For 4× and 10× EExM, the Airyscan PSF was upscaled as detailed above. dSTORM and DNA-PAINT simulations used the image simulator of PyME detailed previously.20
pSer2808 Reassignment
The 3D coordinates of the RyR and pSer2808 labeling puncta were detected independently (in their separate channels of the two-color data). The RyR puncta which contained a pSer2808 punctum was marked as phosphorylated. Only one RyR was assigned to one pSer2808 punctum.
See Supporting Information experimental procedures for more details on analysis protocols.
Simulation of the Spatiotemporal Ca2+ Dynamics
To simulate Ca2+ dynamics, selected nanodomains were discretized at a resolution of 10 nm and embedded in a 2D grid. Ca2+ dynamics within this grid are described by the regular 2D reaction-diffusion equation. L-type Ca2+ channel flux was represented by an idealized triangular waveform, and RyR dynamics, controlling intracellular calcium release, were described through a simple 2-state stochastic model. The original in vitro study examining the effects of RyR gating kinetics following nonspecific phosphorylation of RyRs by Protein Kinase A reported an increase in the RyR open probability by up to a factor of 3.5.54 To capture an approximate effect of Ser2808-specific phosphorylation in our simulation, the phosphorylated state of the RyR channel was modeled as an increase in the Ca2+ sensitivity of the RyRs (by a factor 1.5–3); phosphorylation sites were given either by the imaging data or assigned randomly according the spatial distributions. Simulations were performed over a range of parameters, 10 simulations per condition, with the behavior of the different geometries and with/without phosphorylation compared under consistent parameter conditions. Equations and parameters are presented in the Supporting Information.
Acknowledgments
The authors acknowledge the Wellcome Trust (Seed Award 207684/Z/17/Z awarded to I.J.), the Medical Research Council (doctoral studentship awarded to I.J. and Strategic Skills Research Fellowship awarded to M.A.C.) for funding. Also acknowledged, is Dr. S. Boxall of the University of Leeds Bioimaging Facility for technical assistance and Dr. S. Truckenbrodt and Prof. D. Steele for insightful discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.8b08742.
Author Contributions
I.J., E.W., and J.C. conceived experiments. T.M.S., M.H., R.N., Y.H., and L.H. performed experiments. T.M.S. made primary experimental observations. E.P., K.K.N., Z.Y., and H.K. provided materials toward primary experiments. I.J., D.B., and C.S. wrote the custom code for image analysis and DNA-PAINT/dSTORM image simulation. I.J. and T.M.S. performed experimental data analysis. I.J., C.S., and M.A.C. made the primary interpretation of results. M.A.C. modeled Ca2+ signaling. I.J. modeled image data. Supervision provided by I.J., A.J.S., J.S., J.C., and C.S.
The authors declare no competing financial interest.
Supplementary Material
References
- Scriven D. R.; Asghari P.; Moore E. D. Microarchitecture of the Dyad. Cardiovasc. Res. 2013, 98, 169–76. 10.1093/cvr/cvt025. [DOI] [PubMed] [Google Scholar]
- Pritchard H. A. T.; Pires P. W.; Yamasaki E.; Thakore P.; Earley S. Nanoscale Remodeling of Ryanodine Receptor Cluster Size Underlies Cerebral Microvascular Dysfunction in Duchenne Muscular Dystrophy. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E9745–E9752. 10.1073/pnas.1804593115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayasinghe I. D.; Munro M.; Baddeley D.; Launikonis B. S.; Soeller C. Observation of the Molecular Organization of Calcium Release Sites in Fast- and Slow-Twitch Skeletal Muscle with Nanoscale Imaging. J. R. Soc., Interface 2014, 11, 20140570. 10.1098/rsif.2014.0570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johenning F. W.; Theis A. K.; Pannasch U.; Ruckl M.; Rudiger S.; Schmitz D. Ryanodine Receptor Activation Induces Long-Term Plasticity of Spine Calcium Dynamics. PLoS Biol. 2015, 13, e1002181 10.1371/journal.pbio.1002181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang K.; Zheng H.; Qin X.; Zhang C.; Yang D.; Wang X.; Wu C.; Zhou Z.; Cheng H. Ca2+ Sparks and Secretion in Dorsal Root Ganglion Neurons. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 12259–64. 10.1073/pnas.0408494102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J. D.; Kuang S.; Misler S.; Polonsky K. S. Ryanodine Receptors in Human Pancreatic Beta Cells: Localization and Effects on Insulin Secretion. FASEB J. 2004, 18, 878–80. 10.1096/fj.03-1280fje. [DOI] [PubMed] [Google Scholar]
- Munro M. L.; Shen X.; Ward M.; Ruygrok P. N.; Crossman D. J.; Soeller C. Highly Variable Contractile Performance Correlates with Myocyte Content in Trabeculae from Failing Human Hearts. Sci. Rep. 2018, 8, 2957. 10.1038/s41598-018-21199-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macquaide N.; Tuan H. T.; Hotta J.; Sempels W.; Lenaerts I.; Holemans P.; Hofkens J.; Jafri M. S.; Willems R.; Sipido K. R. Ryanodine Receptor Cluster Fragmentation and Redistribution in Persistent Atrial Fibrillation Enhance Calcium Release. Cardiovasc. Res. 2015, 108, 387–98. 10.1093/cvr/cvv231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinali C.; Bennett H.; Davenport J. B.; Trafford A. W.; Kitmitto A. Three-Dimensional Reconstruction of Cardiac Sarcoplasmic Reticulum Reveals a Continuous Network Linking Transverse-Tubules: This Organization Is Perturbed in Heart Failure. Circ. Res. 2013, 113, 1219–30. 10.1161/CIRCRESAHA.113.301348. [DOI] [PubMed] [Google Scholar]
- Sun X. H.; Protasi F.; Takahashi M.; Takeshima H.; Ferguson D. G.; Franzini-Armstrong C. Molecular Architecture of Membranes Involved in Excitation-Contraction Coupling of Cardiac Muscle. J. Cell Biol. 1995, 129, 659–71. 10.1083/jcb.129.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzini-Armstrong C.; Protasi F.; Ramesh V. Shape, Size, and Distribution of Ca(2+) Release Units and Couplons in Skeletal and Cardiac Muscles. Biophys. J. 1999, 77, 1528–39. 10.1016/S0006-3495(99)77000-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzini-Armstrong C. The Relationship between Form and Function Throughout the History of Excitation–Contraction Coupling. J. Gen. Physiol. 2018, 150, 189–210. 10.1085/jgp.201711889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asghari P.; Scriven D. R.; Sanatani S.; Gandhi S. K.; Campbell A. I.; Moore E. D. Nonuniform and Variable Arrangements of Ryanodine Receptors within Mammalian Ventricular Couplons. Circ. Res. 2014, 115, 252–62. 10.1161/CIRCRESAHA.115.303897. [DOI] [PubMed] [Google Scholar]
- Asghari P.; Schulson M.; Scriven D. R.; Martens G.; Moore E. D. Axial Tubules of Rat Ventricular Myocytes Form Multiple Junctions with the Sarcoplasmic Reticulum. Biophys. J. 2009, 96, 4651–60. 10.1016/j.bpj.2009.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T.; Martone M. E.; Yu Z.; Thor A.; Doi M.; Holst M. J.; Ellisman M. H.; Hoshijima M. Three-Dimensional Electron Microscopy Reveals New Details of Membrane Systems for Ca2+ Signaling in the Heart. J. Cell Sci. 2009, 122, 1005–13. 10.1242/jcs.028175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell S. W.; Wichmann J. Breaking the Diffraction Resolution Limit by Stimulated Emission: Stimulated-Emission-Depletion Fluorescence Microscopy. Opt. Lett. 1994, 19, 780–2. 10.1364/OL.19.000780. [DOI] [PubMed] [Google Scholar]
- Heilemann M.; van de Linde S.; Schuttpelz M.; Kasper R.; Seefeldt B.; Mukherjee A.; Tinnefeld P.; Sauer M. Subdiffraction-Resolution Fluorescence Imaging with Conventional Fluorescent Probes. Angew. Chem., Int. Ed. 2008, 47, 6172–6. 10.1002/anie.200802376. [DOI] [PubMed] [Google Scholar]
- Baddeley D.; Jayasinghe I. D.; Lam L.; Rossberger S.; Cannell M. B.; Soeller C. Optical Single-Channel Resolution Imaging of the Ryanodine Receptor Distribution in Rat Cardiac Myocytes. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 22275–80. 10.1073/pnas.0908971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandenburg S.; Pawlowitz J.; Fakuade F. E.; Kownatzki-Danger D.; Kohl T.; Mitronova G. Y.; Scardigli M.; Neef J.; Schmidt C.; Wiedmann F.; Pavone F. S.; Sacconi L.; Kutschka I.; Sossalla S.; Moser T.; Voigt N.; Lehnart S. E. Axial Tubule Junctions Activate Atrial Ca2+ Release across Species. Front. Physiol. 2018, 9, 1227. 10.3389/fphys.2018.01227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayasinghe I.; Clowsley A. H.; Lin R.; Lutz T.; Harrison C.; Green E.; Baddeley D.; Di Michele L.; Soeller C. True Molecular Scale Visualization of Variable Clustering Properties of Ryanodine Receptors. Cell Rep. 2018, 22, 557–567. 10.1016/j.celrep.2017.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker M. A.; Williams G. S. B.; Kohl T.; Lehnart S. E.; Jafri M. S.; Greenstein J. L.; Lederer W. J.; Winslow R. L. Superresolution Modeling of Calcium Release in the Heart. Biophys. J. 2014, 107, 3018–3029. 10.1016/j.bpj.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X.; van den Brink J.; Hou Y.; Colli D.; Le C.; Kolstad T. R.; MacQuaide N.; Carlson C. R.; Kekenes-Huskey P. M.; Edwards A. G.; Soeller C.; Louch W. E. 3d Dstorm Imaging Reveals Novel Detail of Ryanodine Receptor Localization in Rat Cardiac Myocytes. J. Physiol. 2019, 597, 399–418. 10.1113/JP277360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y.; Jayasinghe I.; Crossman D. J.; Baddeley D.; Soeller C. Nanoscale Analysis of Ryanodine Receptor Clusters in Dyadic Couplings of Rat Cardiac Myocytes. J. Mol. Cell. Cardiol. 2015, 80, 45–55. 10.1016/j.yjmcc.2014.12.013. [DOI] [PubMed] [Google Scholar]
- Munro M. L.; Jayasinghe I. D.; Wang Q.; Quick A.; Wang W.; Baddeley D.; Wehrens X. H.; Soeller C. Junctophilin-2 in the Nanoscale Organisation and Functional Signalling of Ryanodine Receptor Clusters in Cardiomyocytes. J. Cell Sci. 2016, 129, 4388–4398. 10.1242/jcs.196873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B.; Wang W.; Bates M.; Zhuang X. Three-Dimensional Super-Resolution Imaging by Stochastic Optical Reconstruction Microscopy. Science 2008, 319, 810–3. 10.1126/science.1153529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivier N.; Keller D.; Gönczy P.; Manley S. Resolution Doubling in 3d-Storm Imaging through Improved Buffers. PLoS One 2013, 8, e69004 10.1371/journal.pone.0069004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K.; Zhong G.; Zhuang X. Actin, Spectrin, and Associated Proteins Form a Periodic Cytoskeletal Structure in Axons. Science 2013, 339, 452–6. 10.1126/science.1232251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T. T.; Chong W. M.; Wang W. J.; Mazo G.; Tanos B.; Chen Z.; Tran T. M. N.; Chen Y. D.; Weng R. R.; Huang C. E.; Jane W. N.; Tsou M. B.; Liao J. C. Super-Resolution Architecture of Mammalian Centriole Distal Appendages Reveals Distinct Blade and Matrix Functional Components. Nat. Commun. 2018, 9, 2023. 10.1038/s41467-018-04469-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolin F. P.; Preuss L. E.; Taylor R. C.; Ference R. J. Refractive Index of Some Mammalian Tissues Using a Fiber Optic Cladding Method. Appl. Opt. 1989, 28, 2297–2303. 10.1364/AO.28.002297. [DOI] [PubMed] [Google Scholar]
- Tehrani K. F.; Xu J.; Zhang Y.; Shen P.; Kner P. Adaptive Optics Stochastic Optical Reconstruction Microscopy (Ao-Storm) Using a Genetic Algorithm. Opt. Express 2015, 23, 13677–92. 10.1364/OE.23.013677. [DOI] [PubMed] [Google Scholar]
- Schueder F.; Lara-Gutierrez J.; Beliveau B. J.; Saka S. K.; Sasaki H. M.; Woehrstein J. B.; Strauss M. T.; Grabmayr H.; Yin P.; Jungmann R. Multiplexed 3d Super-Resolution Imaging of Whole Cells Using Spinning Disk Confocal Microscopy and DNA-Paint. Nat. Commun. 2017, 8, 2090. 10.1038/s41467-017-02028-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavsson A. K.; Petrov P. N.; Lee M. Y.; Shechtman Y.; Moerner W. E. 3d Single-Molecule Super-Resolution Microscopy with a Tilted Light Sheet. Nat. Commun. 2018, 9, 123. 10.1038/s41467-017-02563-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F.; Tillberg P. W.; Boyden E. S. Optical Imaging. Science 2015, 347, 543–8. 10.1126/science.1260088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillberg P. W.; Chen F.; Piatkevich K. D.; Zhao Y.; Yu C. C.; English B. P.; Gao L.; Martorell A.; Suk H. J.; Yoshida F.; DeGennaro E. M.; Roossien D. H.; Gong G.; Seneviratne U.; Tannenbaum S. R.; Desimone R.; Cai D.; Boyden E. S. Protein-Retention Expansion Microscopy of Cells and Tissues Labeled Using Standard Fluorescent Proteins and Antibodies. Nat. Biotechnol. 2016, 34, 987–92. 10.1038/nbt.3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chozinski T. J.; Halpern A. R.; Okawa H.; Kim H. J.; Tremel G. J.; Wong R. O.; Vaughan J. C. Expansion Microscopy with Conventional Antibodies and Fluorescent Proteins. Nat. Methods 2016, 13, 485–8. 10.1038/nmeth.3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cang H.; Tong Z.; Beuzer P.; Ye Q.; Axelrod J.; Hong Z. Ex-Storm: Expansion Single Molecule Nanoscopy. bioRxiv 2016, 049403. [Google Scholar]
- Gao M.; Maraspini R.; Beutel O.; Zehtabian A.; Eickholt B.; Honigmann A.; Ewers H. Expansion Stimulated Emission Depletion Microscopy (Exsted). ACS Nano 2018, 12, 4178–4185. 10.1021/acsnano.8b00776. [DOI] [PubMed] [Google Scholar]
- Halpern A. R.; Alas G. C. M.; Chozinski T. J.; Paredez A. R.; Vaughan J. C. Hybrid Structured Illumination Expansion Microscopy Reveals Microbial Cytoskeleton Organization. ACS Nano 2017, 11, 12677–12686. 10.1021/acsnano.7b07200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R.; Chen X.; Lin Z.; Wang Y.; Sun Y. Expansion Enhanced Nanoscopy. Nanoscale 2018, 10, 17552–17556. 10.1039/C8NR04267E. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Yu Z.; Cahoon C. K.; Parmely T.; Thomas N.; Unruh J. R.; Slaughter B. D.; Hawley R. S. Combined Expansion Microscopy with Structured Illumination Microscopy for Analyzing Protein Complexes. Nat. Protoc. 2018, 13, 1869–1895. 10.1038/s41596-018-0023-8. [DOI] [PubMed] [Google Scholar]
- Cahoon C. K.; Yu Z.; Wang Y.; Guo F.; Unruh J. R.; Slaughter B. D.; Hawley R. S. Superresolution Expansion Microscopy Reveals the Three-Dimensional Organization of the Drosophila Synaptonemal Complex. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E6857–e6866. 10.1073/pnas.1705623114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truckenbrodt S.; Maidorn M.; Crzan D.; Wildhagen H.; Kabatas S.; Rizzoli S. O. X10 Expansion Microscopy Enables 25-Nm Resolution on Conventional Microscopes. EMBO Rep. 2018, 19, e45836. 10.15252/embr.201845836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J. B.; Chen F.; Yoon Y. G.; Jung E. E.; Babcock H.; Kang J. S.; Asano S.; Suk H. J.; Pak N.; Tillberg P. W.; Wassie A. T.; Cai D.; Boyden E. S. Iterative Expansion Microscopy. Nat. Methods 2017, 14, 593–599. 10.1038/nmeth.4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. S.; Chang J. B.; Alvarez M. M.; Trujillo-de Santiago G.; Aleman J.; Batzaya B.; Krishnadoss V.; Ramanujam A. A.; Kazemzadeh-Narbat M.; Chen F.; Tillberg P. W.; Dokmeci M. R.; Boyden E. S.; Khademhosseini A. Hybrid Microscopy: Enabling Inexpensive High-Performance Imaging through Combined Physical and Optical Magnifications. Sci. Rep. 2016, 6, 22691. 10.1038/srep22691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F.; Wassie A. T.; Cote A. J.; Sinha A.; Alon S.; Asano S.; Daugharthy E. R.; Chang J. B.; Marblestone A.; Church G. M.; Raj A.; Boyden E. S. Nanoscale Imaging of Rna with Expansion Microscopy. Nat. Methods 2016, 13, 679–84. 10.1038/nmeth.3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Bucur O.; Irshad H.; Chen F.; Weins A.; Stancu A. L.; Oh E. Y.; DiStasio M.; Torous V.; Glass B.; Stillman I. E.; Schnitt S. J.; Beck A. H.; Boyden E. S. Nanoscale Imaging of Clinical Specimens Using Pathology-Optimized Expansion Microscopy. Nat. Biotechnol. 2017, 35, 757–764. 10.1038/nbt.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R.; Asano S. M.; Upadhyayula S.; Pisarev I.; Milkie D. E.; Liu T.-L.; Singh V.; Graves A.; Huynh G. H.; Zhao Y.; Bogovic J.; Colonell J.; Ott C. M.; Zugates C.; Tappan S.; Rodriguez A.; Mosaliganti K. R.; Megason S. G.; Lippincott-Schwartz J.; Hantman A.; et al. Cortical Column and Whole Brain Imaging of Neural Circuits with Molecular Contrast and Nanoscale Resolution. bioRxiv 2018, 374140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang I. E.; Lapan S. W.; Scimone M. L.; Clandinin T. R.; Reddien P. W. Hedgehog Signaling Regulates Gene Expression in Planarian Glia. eLife 2016, 5, e16996 10.7554/eLife.16996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosca T. J.; Luginbuhl D. J.; Wang I. E.; Luo L. Presynaptic Lrp4 Promotes Synapse Number and Function of Excitatory Cns Neurons. eLife 2017, 6, e27347 10.7554/eLife.27347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freifeld L.; Odstrcil I.; Forster D.; Ramirez A.; Gagnon J. A.; Randlett O.; Costa E. K.; Asano S.; Celiker O. T.; Gao R.; Martin-Alarcon D. A.; Reginato P.; Dick C.; Chen L.; Schoppik D.; Engert F.; Baier H.; Boyden E. S. Expansion Microscopy of Zebrafish for Neuroscience and Developmental Biology Studies. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E10799–E10808. 10.1073/pnas.1706281114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang N.; Kim H. J.; Chozinski T. J.; Azpurua J. E.; Eaton B. A.; Vaughan J. C.; Parrish J. Z. Superresolution Imaging of Drosophila Tissues Using Expansion Microscopy. Mol. Biol. Cell 2018, 29, 1413–1421. 10.1091/mbc.E17-10-0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambarotto D.; Zwettler F. U.; Le Guennec M.; Schmidt-Cernohorska M.; Fortun D.; Borgers S.; Heine J.; Schloetel J.-G.; Reuss M.; Unser M.; Boyden E. S.; Sauer M.; Hamel V.; Guichard P. Imaging Cellular Ultrastructures Using Expansion Microscopy (U-Exm). Nat. Methods 2019, 16, 71–74. 10.1038/s41592-018-0238-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesce L.; Cozzolino M.; Lanzanò L.; Diaspro A.; Bianchini P. Enigma at the Nanoscale: Can the Npc Act as an Intrinsic Reporter for Isotropic Expansion Microscopy?. bioRxiv 2018, 449702. [Google Scholar]
- Marx S. O.; Reiken S.; Hisamatsu Y.; Jayaraman T.; Burkhoff D.; Rosemblit N.; Marks A. R. Pka Phosphorylation Dissociates Fkbp12.6 from the Calcium Release Channel (Ryanodine Receptor): Defective Regulation in Failing Hearts. Cell 2000, 101, 365–76. 10.1016/S0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- Tokunaga M.; Imamoto N.; Sakata-Sogawa K. Highly Inclined Thin Illumination Enables Clear Single-Molecule Imaging in Cells. Nat. Methods 2008, 5, 159–61. 10.1038/nmeth1171. [DOI] [PubMed] [Google Scholar]
- Burgoyne T.; Morris E. P.; Luther P. K. Three-Dimensional Structure of Vertebrate Muscle Z-Band: The Small-Square Lattice Z-Band in Rat Cardiac Muscle. J. Mol. Biol. 2015, 427, 3527–3537. 10.1016/j.jmb.2015.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wypijewski K. J.; Tinti M.; Chen W.; Lamont D.; Ashford M. L. J.; Calaghan S. C.; Fuller W. Identification of Caveolar Resident Proteins in Ventricular Myocytes Using a Quantitative Proteomic Approach: Dynamic Changes in Caveolar Composition Following Adrenoceptor Activation. Mol. Cell. Proteomics 2015, 14, 596–608. 10.1074/mcp.M114.038570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larcher V.; Kunderfranco P.; Vacchiano M.; Carullo P.; Erreni M.; Salamon I.; Colombo F. S.; Lugli E.; Mazzola M.; Anselmo A.; Condorelli G. An Autofluorescence-Based Method for the Isolation of Highly Purified Ventricular Cardiomyocytes. Cardiovasc. Res. 2018, 114, 409–416. 10.1093/cvr/cvx239. [DOI] [PubMed] [Google Scholar]
- Mortensen K. I.; Churchman L. S.; Spudich J. A.; Flyvbjerg H. Optimized Localization Analysis for Single-Molecule Tracking and Super-Resolution Microscopy. Nat. Methods 2010, 7, 377–81. 10.1038/nmeth.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff J. The Airyscan Detector from Zeiss: Confocal Imaging with Improved Signal-to-Noise Ratio and Super-Resolution. Nat. Methods 2015, 12, 1205. 10.1038/nmeth.f.388. [DOI] [Google Scholar]
- Jayasinghe I. D.; Crossman D. J.; Soeller C.; Cannell M. B. A New Twist in Cardiac Muscle: Dislocated and Helicoid Arrangements of Myofibrillar Z-Disks in Mammalian Ventricular Myocytes. J. Mol. Cell. Cardiol. 2010, 48, 964–71. 10.1016/j.yjmcc.2009.12.012. [DOI] [PubMed] [Google Scholar]
- Jayasinghe I.; Clowsley A. H.; de Langen O.; Sali S. S.; Crossman D. J.; Soeller C. Shining New Light on the Structural Determinants of Cardiac Couplon Function: Insights from Ten Years of Nanoscale Microscopy. Front. Physiol. 2018, 9, 1472. 10.3389/fphys.2018.01472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungmann R.; Avendano M. S.; Woehrstein J. B.; Dai M.; Shih W. M.; Yin P. Multiplexed 3d Cellular Super-Resolution Imaging with DNA-Paint and Exchange-Paint. Nat. Methods 2014, 11, 313–8. 10.1038/nmeth.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bovo E.; Huke S.; Blatter L. A.; Zima A. V. The Effect of Pka-Mediated Phosphorylation of Ryanodine Receptor on Sr Ca(2+) Leak in Ventricular Myocytes. J. Mol. Cell. Cardiol. 2017, 104, 9–16. 10.1016/j.yjmcc.2017.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huke S.; Bers D. M. Ryanodine Receptor Phosphorylation at Serine 2030, 2808 and 2814 in Rat Cardiomyocytes. Biochem. Biophys. Res. Commun. 2008, 376, 80–5. 10.1016/j.bbrc.2008.08.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrens X. H.; Lehnart S. E.; Reiken S.; Vest J. A.; Wronska A.; Marks A. R. Ryanodine Receptor/Calcium Release Channel Pka Phosphorylation: A Critical Mediator of Heart Failure Progression. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 511–8. 10.1073/pnas.0510113103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.; Valdivia H. H.; Wehrens X. H.; Anderson M. E. A Single Protein Kinase a or Calmodulin Kinase Ii Site Does Not Control the Cardiac Pacemaker Ca2+ Clock. Circ.: Arrhythmia Electrophysiol. 2016, 9, e003180 10.1161/CIRCEP.115.003180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T.; Cornea R. L.; Huke S.; Camors E.; Yang Y.; Picht E.; Fruen B. R.; Bers D. M. Kinetics of Fkbp12.6 Binding to Ryanodine Receptors in Permeabilized Cardiac Myocytes and Effects on Ca Sparks. Circ. Res. 2010, 106, 1743–52. 10.1161/CIRCRESAHA.110.219816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oort R. J.; Garbino A.; Wang W.; Dixit S. S.; Landstrom A. P.; Gaur N.; De Almeida A. C.; Skapura D. G.; Rudy Y.; Burns A. R.; Ackerman M. J.; Wehrens X. H. Disrupted Junctional Membrane Complexes and Hyperactive Ryanodine Receptors after Acute Junctophilin Knockdown in Mice. Circulation 2011, 123, 979–88. 10.1161/CIRCULATIONAHA.110.006437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Imtiaz M. S.; Beard N. A.; Dulhunty A. F.; Thorne R.; vanHelden D. F.; Laver D. R. Ss-Adrenergic Stimulation Increases Ryr2 Activity Via Intracellular Ca2+ and Mg2+ Regulation. PLoS One 2013, 8, e58334 10.1371/journal.pone.0058334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyfarth T.; Gerbershagen H. P.; Giessler C.; Leineweber K.; Heinroth-Hoffmann I.; Ponicke K.; Brodde O. E. The Cardiac Beta-Adrenoceptor-G-Protein(S)-Adenylyl Cyclase System in Monocrotaline-Treated Rats. J. Mol. Cell. Cardiol. 2000, 32, 2315–26. 10.1006/jmcc.2000.1262. [DOI] [PubMed] [Google Scholar]
- Sun F.; Lu Z.; Zhang Y.; Geng S.; Xu M.; Xu L.; Huang Y.; Zhuang P.; Zhang Y. Stagedependent Changes of Β2adrenergic Receptor Signaling in Right Ventricular Remodeling in Monocrotalineinduced Pulmonary Arterial Hypertension. Int. J. Mol. Med. 2018, 41, 2493–2504. 10.3892/ijmm.2018.3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Li J.; Drum B. M.; Chen Y.; Yin H.; Guo X.; Luckey S. W.; Gilbert M. L.; McKnight G. S.; Scott J. D.; Santana L. F.; Liu Q. Loss of Akap150 Promotes Pathological Remodelling and Heart Failure Propensity by Disrupting Calcium Cycling and Contractile Reserve. Cardiovasc. Res. 2017, 113, 147–159. 10.1093/cvr/cvw221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolstad T. R.; van den Brink J.; MacQuaide N.; Lunde P. K.; Frisk M.; Aronsen J. M.; Norden E. S.; Cataliotti A.; Sjaastad I.; Sejersted O. M.; Edwards A. G.; Lines G. T.; Louch W. E. Ryanodine Receptor Dispersion Disrupts Ca(2+) Release in Failing Cardiac Myocytes. eLife 2018, 7, e39427. 10.7554/eLife.39427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratusevich V. R.; Balke C. W. Factors Shaping the Confocal Image of the Calcium Spark in Cardiac Muscle Cells. Biophys. J. 1996, 71, 2942–57. 10.1016/S0006-3495(96)79525-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheible M. B.; Tinnefeld P. Quantifying Expansion Microscopy with DNA Origami Expansion Nanorulers. bioRxiv 2018, 265405. [Google Scholar]
- Fowler E. D.; Drinkhill M. J.; Norman R.; Pervolaraki E.; Stones R.; Steer E.; Benoist D.; Steele D. S.; Calaghan S. C.; White E. Beta1-Adrenoceptor Antagonist, Metoprolol Attenuates Cardiac Myocyte Ca(2+) Handling Dysfunction in Rats with Pulmonary Artery Hypertension. J. Mol. Cell. Cardiol. 2018, 120, 74–83. 10.1016/j.yjmcc.2018.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler E. D.; Benoist D.; Drinkhill M. J.; Stones R.; Helmes M.; Wüst R. C. I.; Stienen G. J. M.; Steele D. S.; White E. Decreased Creatine Kinase Is Linked to Diastolic Dysfunction in Rats with Right Heart Failure Induced by Pulmonary Artery Hypertension. J. Mol. Cell. Cardiol. 2015, 86, 1–8. 10.1016/j.yjmcc.2015.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayasinghe I. D.; Cannell M. B.; Soeller C. Organization of Ryanodine Receptors, Transverse Tubules, and Sodium-Calcium Exchanger in Rat Myocytes. Biophys. J. 2009, 97, 2664–73. 10.1016/j.bpj.2009.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeller C.; Jayasinghe I. D.; Li P.; Holden A. V.; Cannell M. B. Three-Dimensional High-Resolution Imaging of Cardiac Proteins to Construct Models of Intracellular Ca2+ Signalling in Rat Ventricular Myocytes. Exp Physiol 2009, 94, 496–508. 10.1113/expphysiol.2008.043976. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.