

Abstract
This is the first of two invited articles reviewing the development of nucleoside-analogue antiviral drugs, written for a target audience of virologists and other non-chemists, as well as chemists who may not be familiar with the field. Rather than providing a simple chronological account, we have examined and attempted to explain the thought processes, advances in synthetic chemistry and lessons learned from antiviral testing that led to a few molecules being moved forward to eventual approval for human therapies, while others were discarded. The present paper focuses on early, relatively simplistic changes made to the nucleoside scaffold, beginning with modifications of the nucleoside sugars of Ara-C and other arabinose-derived nucleoside analogues in the 1960's. A future paper will review more recent developments, focusing especially on more complex modifications, particularly those involving multiple changes to the nucleoside scaffold. We hope that these articles will help virologists and others outside the field of medicinal chemistry to understand why certain drugs were successfully developed, while the majority of candidate compounds encountered barriers due to low-yielding synthetic routes, toxicity or other problems that led to their abandonment.
Keywords: Nucleoside, History, Modification, Antiviral, Anticancer, Analogue
Highlights
-
•
This is the first of two invited articles reviewing the development of nucleoside-analogue antiviral drugs.
-
•
It is written for a target audience of virologists and other non-chemists, and for chemists unfamiliar with the field.
-
•
Numerous modifications have been made to the nucleoside scaffold in order to impart therapeutic benefits.
-
•
Nucleoside modifications led to the development of potent antivirals such as acyclovir, entecavir, and tenofovir.
-
•
We examine thought processes, progress in synthetic chemistry and results of antiviral testing that led to approved drugs.
1. Nucleosides and nucleotides
Nucleoside and nucleotide analogues have a long and rich history in the field of medicinal chemistry (Perigaud et al., 1992, De Clercq, 2002, De Clercq, 2005a, De Clercq, 2004, Field and De Clercq, 2004, De Clercq, 2010, De Clercq, 2012). The naturally occurring nucleosides represent a unique starting point for drug design due to their involvement in numerous critical biological processes as well as the fact that they serve as essential building blocks for both DNA and RNA synthesis. Because of this, modifications to their structure can be designed and/or refined, based on the key interactions identified in the binding site of target enzymes. Currently there are more than 30 nucleoside/tide analogues on the market approved for use in treating viruses, cancers, parasites, as well as bacterial and fungal infections, with many more currently in clinical and preclinical trials. (Perigaud et al., 1992, De Clercq, 2004, Field and De Clercq, 2004, Jordheim et al., 2013, Niu and Tan, 2015, De Clercq and Li, 2016, Lapponi et al., 2016, Maffioli et al., 2017, Wataya et al., 1984, Isono, 1988, Andriole, 1999).
Nucleosides are composed of a sugar moiety and nucleobase whereas nucleotides are nucleosides that also contain at least one phosphate (or phosphate-like) group. While there are numerous naturally occurring nucleosides found in nature, the five most common in DNA and RNA and other biological processes, include adenosine, guanosine, cytidine, thymidine, and uridine (Fig. 1 ). As a result, even small changes to their structure can have profound effects. Ideally, nucleoside/tide analogues mimic the structure of a natural nucleoside such that they are recognized by cellular or viral enzymes, however due to modifications to their structure, lead to disruption and/or termination of replication or other biological processes (De Clercq, 2005a, De Clercq, 2005b, De Clercq, 2013, De Clercq and Neyts, 2009). Since enzyme recognition is dependent on many factors, including shape, electronics, and hydrogen bonding interactions, it is possible to design an analogue that can have activity against a particular biological target, particularly if the mechanism of action is known and information about the binding site is available.
Fig. 1.

Natural occurring DNA and RNA nucleoside building blocks.
When contemplating potential modifications to the nucleoside scaffold, several different sites can be considered: the sugar moiety, the aromatic heterocyclic base, the glycosidic bond that connects the sugar to the heterocyclic base, and/or the phosphate group of the nucleotide (Fig. 2 ). (Perigaud et al., 1992, Jordheim et al., 2013) Modifications can be made by simply adding a substituent or group to the heterocyclic base or sugar, by replacing an atom in either group, by moving an atom to a different position, or a combination of these approaches (Perigaud et al., 1992, Jordheim et al., 2013). Similar modifications can also be made to the phosphate groups in nucleotide analogues (Roy et al., 2016, De Clercq, 2007a, De Clercq, 2007b, Pertusati et al., 2012). Even the position of the glycosidic bond can be shifted, for example, to another carbon, however, the latter has traditionally been a less common modification (Agrofoglio et al., 1994, Marquez and Lim, 1986). These modifications can also be done in combination, thereby allowing for a large amount of diversity in nucleoside structure and function.
Fig. 2.

Sites for potential modifications in nucleoside/tide drug design including changes to the heterocyclic base, the glycosidic bond, the sugar moiety, and the phosphate group.
2. Modifications to the sugar scaffold
Some of the early examples of nucleoside analogues featured modifications to the sugar. These analogues greatly increased our knowledge of normal nucleoside interactions, but also how these interactions could be exploited for medicinal purposes. These modifications include adding or removing substituents on the furanose sugar ring, changing the ring size, or even removing the furanose oxygen to create an entirely new class of nucleosides.
2.1. 2′-OH modifications
Some of the first nucleosides discovered to have medicinal properties were the arabinose or “Ara” analogues, where the conformation of the hydroxyl group at the C2′ position is inverted, or “up” rather than “down” as is found in ribose nucleosides (Fig. 3 ). (Bergmann and Feeney, 1950, Bergmann and Feeney, 1951, Bergmann and Burke, 1955) The first two Ara nucleosides were natural products and included spongothymidine and spongouridine. Both were isolated from the Caribbean sponge Tethya crypta in the early 1950's (Bergmann and Feeney, 1950, Bergmann and Feeney, 1951, Sipkema et al., 2005). While neither spongothymidine nor spongouridine ever became useful drugs, their discovery led to the synthesis of various other arabinose derived nucleoside analogues such as spongoadenosine (Ara-A) and spongocytidine (Ara-C) both of which were synthesized in the 1960's (Fig. 3). (Bergmann and Burke, 1955, Sipkema et al., 2005, Bauer, 1985, Hamann et al., 2007, Walwick et al., 1959, Lee et al., 1960a)
Fig. 3.

Examples of “Ara” nucleosides where the 2′-OH is in the “up” configuration found in arabinose sugars rather than the “down” configuration typical of ribose nucleosides.
Ara-C, or Cytarabine, was approved for clinical use in 1969 and is considered an essential medicine by the World Health Organization (WHO, 2017) due to its potent activity against many cancers including, non-Hodgkin's Lymphoma, and, most importantly, against many leukemias such as myeloid leukemia, acute lymphatic leukemia, and chronic myelogenous leukemia (Reese and Schiller, 2013, Schilsky et al., 1987, Bodey et al., 1969, Hiddemann, 1991, Momparler, 1974, Rudnick et al., 1979, Herzig et al., 1983). While Ara-C is still currently used primarily for anticancer treatment, its use in antiviral therapy was also explored early on. Unfortunately, there were adverse effects observed for high dose Ara-C during anti-HSV treatments, including myelotoxicity and gastrointestinal toxicities, thus Ara-C was never pursued further as an antiviral drug (Stentoft, 1990, Lauter et al., 1974, Hwang et al., 1985). Ara-A, or Vidarabine, was first approved by the FDA in 1976 and has been used against several viruses including Herpes Simplex Virus 1 and 2 (HSV-1 and HSV-2), as well as Herpes Zoster Virus, which afflicts many AIDS patients (Field and De Clercq, 2004, Bauer, 1985, Hamann et al., 2007, De Clercq, 1982, Suzuki et al., 2006, Lee et al., 1960b). Vidarabine is no longer used due to toxicity issues as well as the discovery of more potent and safer compounds such as Acyclovir, which today is widely prescribed for HSV (Hamann et al., 2007).
In the 70's and 80's, researchers became aware of the unique properties that fluorine imparts to nucleoside analogues (Liu et al., 2008, Pankiewicz and Watanabe, 1993, Pankiewicz, 2000). Fluorine is often used as an isosteric replacement since it is similar in size to a hydrogen, but is also similar in electronegativity to the hydroxyl group found in ribose nucleosides (Liu et al., 2008). Fluorine has found extensive use in both sugar and base modifications (the latter will be discussed later). For example, early studies revealed that fluorine greatly influenced the conformation of the sugar, also known as “sugar pucker” (Saenger, 1984). Because fluorine is the most electronegative element, its presence “locks” the sugar into a specific conformation, which in turn, is one of the factors that affects recognition by different enzymes (Ikeda et al., 1998, Wojtowicz-Rajchel, 2012). For example, some enzymes such as DNA polymerases and reverse transcriptases prefer a “north” conformation, also known as C2′-exo/C3′-endo, while kinases generally prefer a “south” or C2′-endo/C3′-exo conformation (Fig. 4 ) (Saenger, 1984).
Fig. 4.

Standard furanose ring puckering found in ribose and 2′-deoxyribose nucleosides.
Furthermore, it was found that the presence of fluorine served to increase the stability of neighboring bonds (Liu et al., 2008, Bohm et al., 2004, Kirk, 2006, Park et al., 2001a). This observation also led to the discovery that the presence of a fluorine at the 2′-position of the sugar decreased the nucleoside's susceptibility to enzymatic cleavage of the glycosidic bond – a biological process that can inactivate nucleoside drugs (Wojtowicz-Rajchel, 2012, Park et al., 2001a, Gudmundsson et al., 2000). As a result of these observations, fluorine has become a common modification in drug design.
In one of the first examples exploring the effects of fluorine on the sugar, Watanabe et al. synthesized a series of nucleosides with a fluorine at the 2′-position, including both the “up” and “down” analogues, given the activity previously noted by the Ara nucleosides (Fig. 5 ). (Liu et al., 2008, Pankiewicz and Watanabe, 1993, Pankiewicz, 2000) The “up” 2′-F proved to be more active than the “down” analogue, however neither analogue proved to be particularly potent and both were fairly toxic (Pankiewicz and Watanabe, 1993, Pankiewicz, 2000). Some years later, Eli Lilly developed the first example of a nucleoside with a geminal substituents – 2′-deoxy-2′,2′-difluorocytidine, which became known as Gemcitabine (Gemzar) (Hertel et al., 1990, de Sousa Cavalcante and Monteiro, 2014, Brown et al., 2014, Brown et al., 2015). This analogue was a groundbreaking discovery - the installation of two identical substituents (other than H) on the same carbon had not previously been tried before in nucleosides (or any molecule for that matter), and many questioned what the result of two fluorines would be on the overall conformation of the sugar. Logic would suggest that the two opposing dipoles would cancel each other out, leading to no change to the sugar pucker, however it was later shown that the two fluorines pulled the ring carbon “out” of the plane, rather than a north or south confirmation. More importantly, the potent activity of Gemcitabine against a number of cancers was striking (Hertel et al., 1990, de Sousa Cavalcante and Monteiro, 2014, Huang et al., 1991, Oettle, 2014, Hernández et al., 2001). Gemcitabine is still used today against breast, ovarian, pancreatic, bladder, and non-small cell lung cancers (de Sousa Cavalcante and Monteiro, 2014, Oettle, 2014, Bergman et al., 2011, Slusarczyk et al., 2014, Bastiancich et al., 2017).
Fig. 5.

Examples of 2′-F nucleoside analogues where the fluorine can be in the “down” or “up” configuration, or in both the “up” and “down” configuration as in Gemcitabine.
2.2. 3′-OH modifications
Due to the initial success with the 2′-modified nucleoside analogues, scientists also explored modifications at the 3′ carbon. Some of the first 3′ modified nucleosides include 3′-methyl analogues such as 3′-C-methyluridine and 3′-C-methylcytidine (Fig. 6 ). (Mikhailov et al., 1983) From these initial SAR studies, it was found that 3′-C-methyladenosine served as a potent anticancer agent against numerous human leukemia and carcinoma cell lines, with IC50 values of ∼18 μM (Franchetti et al., 2005, Cappellacci et al., 2006). Further analysis found that shifting the methyl from the 3′ position of 3′-C-methyladenosine to another position on the sugar ring was associated with a decrease in activity, thus highlighting the importance of this moiety (Cappellacci et al., 2006). Similarly, this study found that the adenosine nucleobase was the most active analogue against human myelogenous leukemia K562 cells and human colon carcinoma HT-29 and CaCo-2 cell lines, with no antiproliferative activity found with the other nucleobases (Cappellacci et al., 2006). To further test the ability of these 3′-C-methyl analogues as potential therapeutics, the Osolodkin group studied 3′-C-methyluridine and cytidine against Tickborne encephalitis virus, however, none of these analogues demonstrated potent antiviral activity (Orlov et al., 2017). Due to the lack of antiviral activity, as well as the development of more potent anticancer agents, these analogues have not been extensively pursued any further.
Fig. 6.

Examples of 3′-methyl nucleoside analogues where the methyl at the 3′ carbon of the sugar ring is in the “up” configuration and the 3′-OH is in the “down” configuration.
2.3. 2′- and 3′-OH modifications
Since it was known that nucleoside analogues are incorporated into the growing nucleic acid chain via the 3′-OH of the template chain and the 5′-position of the incoming nucleotide, researchers speculated that if a nucleotide was missing the 3′-OH, or an alternative functional group was present at this position, then no further incorporation should occur and extension of the growing DNA chain would be halted (De Clercq and Neyts, 2009, Deval et al., 2007, Deval, 2009). This hypothesis led many researchers to pursue these types of modifications, thereby introducing new class of nucleoside analogues designated as “chain terminators”, and these analogues signaled the beginning of a new era in nucleoside drug design.
The first examples of the chain terminator approach were the 2′,3′-dideoxy nucleosides dideoxycytidine (ddC, Zalcitabine), and dideoxyinosine (ddI, didanosine), both FDA approved nucleoside reverse transcriptase inhibitors against the Human Immunodeficiency Virus (HIV) (Fig. 7 ). (De Clercq, 2012, Veal et al., 1995, Mitsuya and Broder, 1986, Horwitz et al., 1967, Plunkett and Cohen, 1975) These analogues were quickly followed by 2′-deoxy,3′-azidothymidine (AZT, Zidovudine), which demonstrated increased activity against HIV compared to the previous dideoxy analogues with an IC50 of 0.03 μM as compared to 0.049 μM (ddI) and 0.6 μM (ddC) (Smith et al., 2008, Hostetler et al., 1994, Shirasaka et al., 1995, Horwitz et al., 1964). Interestingly, all three compounds were originally pursued as anticancer agents, however, during the 1980's when HIV quickly rose to the forefront of emerging diseases, researchers found that all three were quite effective against HIV (Martin et al., 2010). In addition, the dideoxynucleosides were also used in Sanger sequencing methods in the mid 1970's as a rapid method to determine DNA sequences with DNA polymerases (Sanger and Coulson, 1975, Sanger et al., 1977).
Fig. 7.

Examples of 2′,3′-dideoxy nucleosides that act as obligate chain terminators due to the lack of a 3′-OH for the incoming nucleotide to couple to, thus chain elongation is terminated.
Unfortunately, a number of problems arose with AZT, ddI, and ddC. For example, it was shown that ddI was acid-labile, thus large buffered tablets were necessary to neutralize stomach pH and ensure effective delivery of the compound (Martin et al., 2010). This led to unwanted side effects including pancreatitis and peripheral neuropathy, as well as overall poor patient compliance (Martin et al., 2010). Fortunately, a few years after its initial approval, an enteric coated small capsule form of ddI was developed, which greatly increased patient compliance as well as decreased the previously described side effects (Martin et al., 2010). Similarly, ddC was also associated with numerous adverse effects; the most serious of which was painful peripheral neuropathy that occurred in all patients treated with ddC for longer than 6 weeks (Martin et al., 2010). For this reason, as well as its poor efficacy, ddC is no longer widely used. AZT also suffered from various side effects and problems with toxicity, but most seriously, the development of viral resistance (Boyer et al., 2006, Kerr and Anderson, 1997, Richman et al., 1994). The most common mechanism for the development of resistance is the mutation of one or more residues in the reverse transcriptase binding site (Larder and Kemp, 1989, Huang et al., 1998, Meyer et al., 1999, Gu et al., 1994, Zhang et al., 1994). This leads to the drug either not binding at all, or at the very least, less effective binding.
By 1992, the problem of resistance was well-documented for all three therapies, thus researchers considered combination therapy, i.e. use of two nucleosides rather than the standard monotherapy (Richman et al., 1994, Larder and Kemp, 1989, Meng et al., 1992, Larder et al., 1993, St Clair et al., 1991, Collier et al., 1993). Researchers rationalized that if you could target the replication pathway of HIV with two different drugs, then it would increase the chances of stopping the development of resistance since shutting down one step in a pathway by two different mechanisms, or two different steps, or even two different drugs to target the same step, would greatly reduce the chances of mutant strains gaining dominance (Richman et al., 1994, Meng et al., 1992, Larder et al., 1993, Collier et al., 1993). Moreover, if one drug worked against the mutant strains for the other drug or vice versa, then this would exponentially increase the chances of being more effective (Meng et al., 1992, Larder et al., 1993, Collier et al., 1993, Mascolini, 1997). While today combination therapy is considered essential, in those days there was concern that this would lead to the development of even more serious mutations or resistance to the entire class of drugs. Given there were only a limited amount of drugs available for use, the fear was that this would lead to a lack of drugs left to try (Mascolini, 1997, Mascolini, 2010).
Because of the rapid emergence of resistance against AZT and ddC, the field focused on exploring additional modifications at the 2′ and 3′ sugar positions (Boyer et al., 2006, De Clercq, 2009a, De Clercq, 2009b, Sarafianos et al., 2004, Liotta and Painter, 2016). As a result, efforts to find new and better chain terminators were vigorously pursued. This ultimately led to the discovery of numerous analogues still used today including Stavudine (d4T), Lamivudine (3TC), and Emtricitabine (FTC) (Fig. 8 ). (Martin et al., 2010, Baba et al., 1987, Soudeyns et al., 1991, Schinazi et al., 1992a, Horwitz et al., 1966, Saag, 2006) Like ddI and ddC, Stavudine also lacks the 2′ and 3′-OH moieties, but also introduced unsaturation to the sugar moiety via a double bond between the C2′ and C3′ carbons, which in turn, made the sugar nearly planar due to the increased rigidity of the molecule (Choi et al., 2003). Interestingly, both Lamivudine and Emtricitabine are also examples of “L” nucleosides, the mirror image enantiomers of the naturally occurring “D” nucleosides which will be discussed later in this review. They also feature a sulfur atom instead of a 3′ carbon in the sugar ring (Liotta and Painter, 2016, Vasconcelos et al., 2008). Since the natural nucleosides are “D” nucleosides, it had long been thought that their unnatural enantiomers (non-superimposable mirror images) would not be recognized in biological systems. This proved not to be the case and both were ultimately approved for the use against HIV and Hepatitis B Virus (HBV) after demonstrating low levels of toxicity and diminished side effects compared to the aforementioned chain terminators, including AZT (De Clercq, 2012, Liotta and Painter, 2016, Vasconcelos et al., 2008, Doong et al., 1991).
Fig. 8.

Examples of second generation 2′,3′-dideoxy obligate nucleoside chain terminators.
While Lamivudine and Emtricitabine featured an oxathiolane sugar, researchers found that they could also replace the sulfur atom with an oxygen atom to yield a dioxolane sugar. This led to the development of anti-HIV analogues such as (+)-(2′R,4′R)-dioxolane cytidine and the corresponding (−)-(2′R,4′R)-dioxolane guanosine (Fig. 8). (Kim et al., 1992a, Kim et al., 1993) Like the oxathiolane sugar analogues, the dioxolane analogues are chain terminators due to the lack of a 3′-OH group. These structurally unique deoxynucleosides demonstrated potent anti-HIV activity, with EC50 values of 0.016 μM110 and 0.003 μM111 in PBM cells respectively, with the cytidine analogue displaying greater cytotoxicity than the guanosine analogue (Kim et al., 1992a, Kim et al., 1993). Further studies found that the l-enantiomer of the dioxolane cytidine analogue, Troxacitabine (BCH 4556), demonstrated activity against various cancers including renal cell carcinoma, leukemia, and prostate (Fig. 8). (Kadhim et al., 1997, Grove et al., 1995, Giles et al., 2001, Lee et al., 2006, Grove and Cheng, 1996, Siu et al., 1998, Rabbani et al., 1998) Furthermore, Troxacitabine also demonstrated potent activity against HBV and HIV, thus pointing to the importance of these analogues (Kim et al., 1992b).
2.4. Carbocyclic nucleosides
The use of an atom replacement in the sugar moiety as exemplified by Lamivudine and Emtricitabine were not the first examples of this type of modification. In 1966 an unusual nucleoside was synthesized by Shealy and Clayton that featured a cyclopentyl ring in place of the furanose sugar moiety (Fig. 9 ). (Shealy and Clayton, 1966) This analogue, later named Aristeromycin (Ari), was subsequently isolated from Streptomyces citrocolour and found to have potent antiviral properties against viruses such as measles, parainfluenza, vaccinia virus, and vesicular stomatitis (De Clercq, 1985, De Clercq et al., 1989, Rawal et al., 2016). Similarly, a closely related structural analogue was first isolated from Ampullariella regularis in 1981 (Hayashi et al., 1981), and later synthesized and named Neplanocin A (NpcA). Similar to Ari, NpcA also featured a cyclopentyl ring in place of the furanose sugar, however, in NcpA the cyclopentyl ring possesses a double bond between the C4′ and C6′ carbons (Fig. 9). (Rawal et al., 2016, Hayashi et al., 1980, Hayashi et al., 1981) Both NpcA and Ari were highly active, however, they were also quite toxic, as their triphosphate forms closely mimicked ATP, thus leading to misincorporation and disruption of many important biological systems (De Clercq, 1985, De Clercq et al., 1989, Rawal et al., 2016, Hayashi et al., 1980, Wolfe and Borchardt, 1991).
Fig. 9.

Early carbocyclic nucleosides that feature a C-C-N glycosidic bond that is more stable compared to the corresponding hemiacetal bond found in the natural sugar nucleosides.
Although the toxicity of the two kept them from being used medicinally, the carbocyclic scaffold was still quite attractive to researchers. Carbocyclic nucleosides possess several advantages such as increased lipophillicity and cell permeability, but most importantly, the glycosidic bond was no longer an unstable hemiaminal ether affected by glycoside hydrolases, thus also led to increased stability (Agrofoglio et al., 1994, Marquez and Lim, 1986, Stoeckler et al., 1980, Rodríguez and Comin, 2003, Marquez, 1996). Unfortunately, most of the carbocyclic nucleosides, with only a handful of exceptions, were less active than their corresponding ribose nucleosides (Marquez and Lim, 1986, Marquez, 1996, Marquez et al., 1996).
When comparing the sugar puckering of natural nucleosides and carbocyclic nucleosides such as thymidine and carba-thymidine, it was found that the loss of the furanose oxygen in the carbocyclic nucleosides not only significantly decreased stereoelectronic effects, it also eliminated the anomeric effect as well as decreased important gauche interactions between the furan oxygen and the 3′ hydroxyl groups (Marquez, 1996, Marquez et al., 1996, Boyer et al., 2009, Mahler et al., 2012). As mentioned previously, these interactions typically influence the sugars into either a North or South conformation, or more commonly, an equilibrium of the two (Marquez, 1996, Marquez et al., 1996). The loss of these interactions in the carbocyclic analogues resulted in a 1′-exo sugar confirmation, which differs greatly from the standard 2′-exo/3′-endo (North) or 2′-endo/3′-exo (South) conformations, and thus could explain the decreased activity seen with the carbocyclic derivatives (Marquez, 1996, Marquez et al., 1996, Boyer et al., 2009, Kálmán et al., 1989). Recent studies, however, have shown that alterations such as truncation of the CH2OH group or addition of an endocyclic double bond to the carbocyclic scaffold (such as that found in Entecavir) could increase the interaction between the 2′ and/or 3′-hydroxyl groups and the nucleoside pharmacophore, thus potentially increasing their biological activity (Marquez, 1996, Marquez et al., 1996). This led to a number of groups pursuing novel carbocyclic analogues to see if this increased stability would indeed improve their overall antiviral properties.
2.5. Second generation carbocyclic nucleosides
As Neplanocin A and Aristeromycin were unable to be used as drugs due to the toxicity of their triphosphate forms, focus turned to the design of carbocyclic analogues that could not be phosphorylated at the 5′ position. One pivotal example was 5′-deoxy-Ari, where replacement of the CH2OH group with a simple methyl group led to a dramatic increase in activity against both vaccinia virus as well as vesicular stomatitis virus (Fig. 10 ). (Marquez, 1996, Siddiqi et al., 1992) Another approach that proved advantageous was the development of the 5′-nor analogues by Schneller et al., where the 5′-methylene group was removed resulting in the direct connection of the 5′-OH to the cyclopentyl ring (Patil et al., 1992, Das and Schneller, 2014, Seley et al., 1997a, Seley et al., 1997b, Seley et al., 1997c, Seley et al., 1997d, Seley et al., 1998, Hegde et al., 1999, Hegde et al., 2000, Barnard et al., 2001). Surprisingly, despite the presence of a hydroxyl group, phosphorylation did not occur since the shortened bond did not position the hydroxyl group correctly such that it was recognized by cellular and viral kinases (Patil et al., 1992). Taking it one step further, Borchardt, Schneller, Seley-Radtke and others completely removed the 5′-hydroxymethylene group leading to the truncated Ari and NpcA analogues (Hasobe et al., 1987, Zimmermann et al., 2014, Seley et al., 1997e). These truncations proved extremely fruitful in that they retained similar activity to the Ari and NpcA analogues, however, the associated toxicity was not observed (Zimmermann et al., 2013, Zimmermann et al., 2014, Seley et al., 1997e).
Fig. 10.

Examples of second generation carbocyclic nucleosides with the 5′-OH or 4′-substituent removed to generate "truncated" nucleosides.
One of the most important findings for the carbocyclic analogues was that they proved to be some of the most potent inhibitors against S-adenosyl-l-homocysteine hydrolase (SAHase) – an enzyme key to many biological methylations (Marquez and Lim, 1986, De Clercq et al., 1989, Wolfe and Borchardt, 1991, Marquez, 1996, Wang et al., 2011). SAHase was not the only enzymatic target available for the carbocyclic analogues, as many carbocyclic compounds were also found to be effective polymerase inhibitors (Price et al., 1992, Matyugina et al., 2012). One example is (−)-Carbovir, which resembles the d4T sugar structure in that there is a double bond between the 2′ and 3′ carbons, however, (−)-Carbovir possesses a cyclopentyl ring instead of the furanose ring seen in d4T as well as a guanine nucleobase instead of the thymine nucleobase found in d4T (Fig. 11 ). (Vince et al., 1988, Carter et al., 1990) This was highly strategic in that the unsaturation of the cyclopentyl ring increased the rigidity of the analogue, and the lack of the furanose oxygen lead to increased stabilization of the glycosidic bond of (−)-Carbovir (Vince et al., 1988, Carter et al., 1990, Parker et al., 1993). While (−)-Carbovir has demonstrated its ability to be a potent anti-HIV therapeutic, cytotoxicity, low solubility, and poor oral bioavailability have limited its use in clinical applications (Yeom et al., 1989). In order to alleviate some of these issues, further studies with (−)-Carbovir led to the development of Abacavir, a carbocyclic analogue with a unique modification on the exocyclic amine group at the 6 position of the nucleobase (Fig. 11). (Daluge et al., 1997) The cyclopropyl group acts as a prodrug moiety and, once Abacavir is anabolized to Abacavir 5′-monophosphate, the cyclopropyl group undergoes deamination in vivo to yield (−)-Carbovir 5′-monophosphate (Daluge et al., 1997, Faletto et al., 1997, Tisdale et al., 1997, Yuen et al., 2008). From here, (−)-Carbovir 5′-monophpshate is converted into the 5′-diphosphate and subsequently to the 5′-triphosphate by various cellular kinases to give the bioactive (−)-Carbovir 5′-triphosphate (Vince et al., 1988, Daluge et al., 1997, Faletto et al., 1997). Since Abacavir is converted into the active form of (−)-Carbovir in vivo, it also acts as a potent HIV-1 therapeutic. Abacavir is typically administered with other nucleoside analogues as part of combination therapy, and has demonstrated the ability to overcome some of the bioavailability limitations associated with (−)-Carbovir (Daluge et al., 1997, Tisdale et al., 1997, Harrigan et al., 2000).
Fig. 11.

Structure of the 2′,3′-dideoxy obligate chain terminator Carbovir, a nucleoside that features unsaturation in the sugar ring.
One of the most important examples of the carbocyclic nucleosides is Entecavir, which possesses a 3′-OH group but introduced a highly unique exocyclic double bond in place of the furanose oxygen (Fig. 12 ). (Bisacchi et al., 1997, Wilber et al., 2011) Entecavir is an FDA approved drug for treating both HIV and HBV (Bisacchi et al., 1997, Wilber et al., 2011, Tang et al., 2013). Interestingly, the presence of the exocyclic double bond is critical to the increased binding affinity, as it fits into a hydrophobic pocket of the HBV polymerase, increasing the efficacy of Entecavir against HBV (Tang et al., 2013, Rawal et al., 2015). Initially however, this was not part of the rationale of the overall design - the exocyclic double bond was merely chosen to increase rigidity of the nucleoside and prevent the sugar ring flipping between the North and South conformations, as well as to maximize the distance and orientation between the nucleobase and the 5′-hydroxyl (Tang et al., 2013, Langley et al., 2007).
Fig. 12.
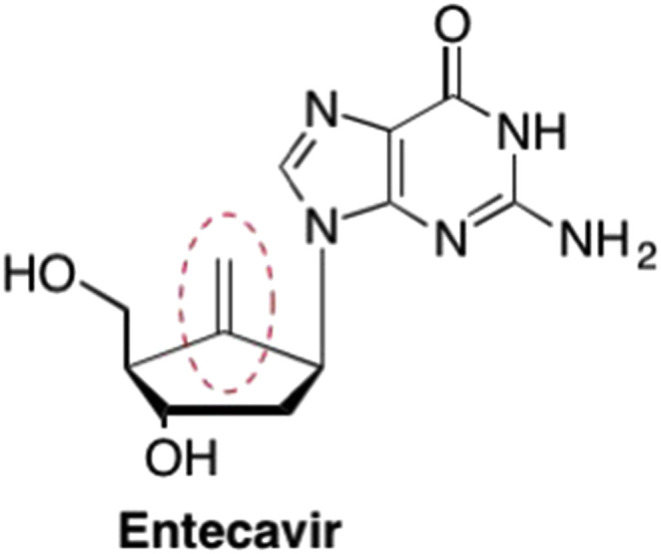
Structure of the 2′-deoxy non-obligate chain terminator Entecavir that features a unique exocyclic double bond which leads to unfavorable steric interactions resulting in chain termination despite the presence of a 3′-OH.
Interestingly, Entecavir is one of the rare examples of a rationally (although some might consider it irrationally!) designed nucleoside (Fig. 13 ). In 1992, another carbocyclic nucleoside, Lobucavir, was garnering much attention, due to its potent antiviral activity against HBV and HSV (Marquez, 1996, Hayashi et al., 1990, Norbeck et al., 1990). Lobucavir possesses a cyclobutyl “sugar”, i.e. four-membered ring, and resembles the naturally occurring Oxetanocin G, which was isolated from bacteria in 1986 and later demonstrated antiviral activity against HIV (Fig. 14 ). (Marquez, 1996)
Fig. 13.

The spatial and structural similarities between Lobucavir and Entecavir that provided the rationale for Entecavir's unique design.
Fig. 14.

Examples of nucleosides with four-membered rings including the corresponding carbocyclic (“carba”) analogues.
At the time, researchers at Bristol Myers Squibb, including the lead chemist on the project Robert Zahler, were looking to design something that would occupy the same space as Lobucavir, a unique carbocyclic cyclobutyl nucleoside structurally similar to the naturally occurring Oxetanocins, which also possess a four member sugar ring (Bisacchi et al., 1997, Hayashi et al., 1990). More specifically, they wanted the nucleoside to mimic the orientation of the nucleobase, as well as the distance between the 5′-OH and the guanine base (Fig. 13). (Wilber et al., 2011) Zahler felt strongly that these two factors were the key to Lobucavir's potent activity. Since computational chemistry was only in its early stages, chemists relied on physical models, thus Zahler rationalized that it didn't matter what was in the middle, only that it kept the base and 5′-OH group positioned in the correct distance and the right spatial orientation (Wilber et al., 2011). More specifically, knowing that a sugar ring was prone to flipping conformations, which could subsequently abolish the activity, he knew it would be important to hold the sugar ring rigid enough such that this flip did not occur (Wilber et al., 2011, Tang et al., 2013, Langley et al., 2007). In addition, since ring flipping would impact the orientation of the heterocyclic base into either an anti or syn conformation, which would also impact recognition, mimicking the specific distance between the 5′-OH and the heterocyclic base could potentially be achieved by holding the base in the preferred conformation. Zahler tried a number of approaches but ultimately conceived of a cyclopentyl ring wherein an exocyclic double bond replaced the furanose oxygen (Bisacchi et al., 1997, Wilber et al., 2011, Tang et al., 2013). Not only would this rigidify the ring enough to avoid the ring flip, he also predicted this would hold the nucleobase and OH group in the correct orientation and distance (Bisacchi et al., 1997, Wilber et al., 2011). As it turned out, the overlap with Lobucavir was quite remarkable and Entecavir still represents one of the best and most widely prescribed anti-HBV drugs on the market (Langley et al., 2007). Notably, it has also been shown to be particularly effective against HIV/HBV co-infected patients (McMahon et al., 2007, Pessôa et al., 2008).
2.6. Alternative ring size nucleosides
As introduced briefly above, the naturally occurring oxetanocins possess a four-membered sugar ring. Oxetanocin A is an adenosine mimic that possessed a 4-membered sugar ring and was found to be very active against HIV as well as HSV-1 and HSV-2 (Marquez, 1996, Groaz et al., 2013). Oxetanocin G is the corresponding guanosine mimic which proved effective against HIV (Fig. 14). (Marquez, 1996) Interestingly, the carbocyclic derivatives of both Oxetanocin A (carba-oxetanocin A) and Oxetanocin G (carba-oxetanocin G; Lobucavir) also displayed high antiviral activity against numerous viruses, including HSV-1, HSV-2, Human Cytomegalovirus (HCMV), and Varicella Zoster Virus (Fig. 14). (Marquez, 1996, Hayashi et al., 1990, Norbeck et al., 1990, De Clercq et al., 2001, Sekiyama et al., 1998)
From these initial studies and the great success of Entecavir, as well as the oxetanocins and Lobucavir, it was clear that ring size could play an important role in recognition and activity. One such ring size were the cyclopropyl nucleosides, which introduced extreme rigidity to the sugar scaffold however exhibited only moderate antiviral activity against HSV and HIV (Sekiyama et al., 1998, Ashton et al., 1988, Qiu et al., 1998, Aihong and Joon, 2007). Later Zemlicka et al. developed the Z-isomers of 2-hydroxymethylcyclopropylidenemethyl purines and pyrimidines (Synadenol and Synguanol) that demonstrated potent activity against HCMV (IC50 = 1–2.1 μM and 0.04–2.1 μM) and Epstein-Barr virus (IC50 = 0.2μM and 0.3μM) (Fig. 15 ). (Qiu et al., 1998, Baldanti et al., 2002)
Fig. 15.

Examples of cyclopropyl nucleoside analogues that feature three-membered sugar rings.
In contrast, Herdewijn et al. developed numerous six-membered ring nucleosides including the cyclohexenyl and hexose nucleoside analogues shown in Fig. 16 (Wang et al., 2000). The cyclohexenyl analogues were pursued for two primary reasons – cyclohexene is a bioisostere of a saturated furanose ring, and the cyclohexenyl analogues are more resistant to glycoside hydrolysis due to the change in the glycosidic bond to a hemiaminal ether as previously discussed (Wang et al., 2000, Barral et al., 2005, Herdewijn and De Clercq, 2001, Wang and Herdewijn, 1999). Studies demonstrated that the enantiomers of one particular cyclohexenyl nucleoside, cyclohexenyl guanine, exhibited potent and selective activity against HSV-1, HSV-2, VZV, and HCMV (Wang et al., 2000, Herdewijn and De Clercq, 2001, Wang and Herdewijn, 1999). Unfortunately, due to tedious synthetic procedures, these compounds did not lead to any clinical candidates, however, they did serve to increase our understanding of other aspects of drug design and biological activity.
Fig. 16.

Examples of nucleoside analogues that feature a six-membered ring instead of the typical five-membered ring found in natural nucleosides.
2.7. Replacement of oxygen in the furanose ring by heteroatoms
Given the potent activity exhibited by the carbocyclic nucleosides, researchers next turned to replacing the furanose oxygen with other atoms to see the potential impact on biological activity. One such replacement included that of the isosteric sulfur atom to yield thionucleosides, which have a similar benefit to carbocyclic nucleosides in that they are also unaffected by glycoside hydrolases (Fig. 17 ). (Messini et al., 1999, Elzagheid et al., 1999, Crnugelj et al., 2002) Sulfur is related to oxygen in several ways, including a similar electronic configuration and the same number of valence electrons, however, sulfur is less electronegative and larger in size than oxygen. In contrast, sulfur does not participate in hydrogen bonding like oxygen, thus this substitution results in the loss of important interactions in enzyme binding pockets (Paulsen, 1966). While numerous analogues were synthesized by Secrist et al. (Messini et al., 1999, Elzagheid et al., 1999) and others (Paulsen, 1966, Inoue et al., 2005, Crnugelj et al., 2002) none led to a marketable drug.
Fig. 17.

Examples of non-furanose sugars where the oxygen is replaced with either an isosteric sulfur atom or a selenium atom.
Many years later, Jeong et al. developed the analogous selenonucleosides, however, none showed meaningful anticancer or antiviral activity, most likely due to lack of recognition by cellular kinases, as they were not phosphorylated (Fig. 17). (Sahu et al., 2014, Yu et al., 2013) Extension of the methylene group at the 5′ position of these compounds however, resulted in potent anti-HSV-1 activity with EC50 values as low as 2.3 μM since these analogues were able to be phosphorylated by cellular kinases (Sahu et al., 2014, Yu et al., 2013).
Another modification that proved to be highly successful is the use of the nitrogen-containing imino sugar found in the immucillins (Fig. 18 ) developed by Schramm et al. (Kicska et al., 2001, Schramm and Tyler, 2003), however those will be discussed below in the C-nucleoside section of this review.
Fig. 18.
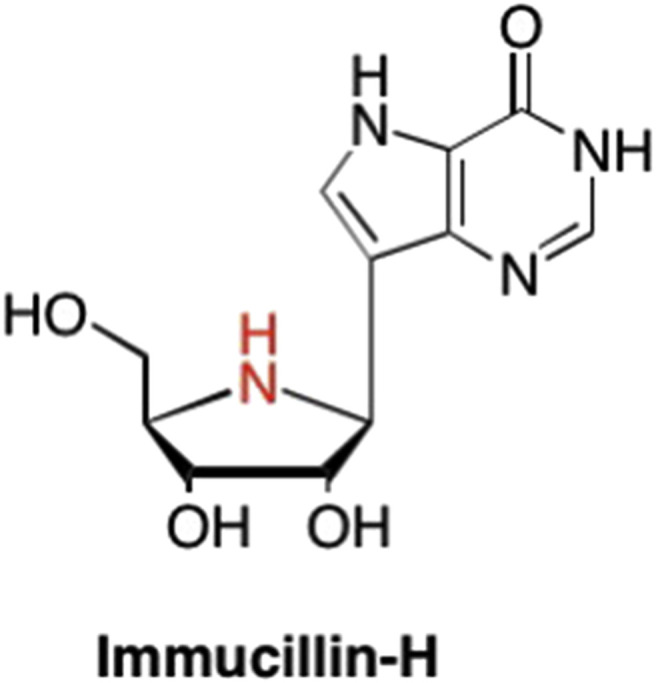
Structure of the imino sugar modified nucleoside Immucillin-H, where the furanose oxygen is replaced with a nitrogen.
2.8. Acyclic nucleosides
While 2′ and 3′-OH modified analogues such as AZT, ddI, and ddC were effective antiviral analogues, researchers concurrently speculated as to just how much of the sugar scaffold they could remove and still retain the activity of the nucleoside. Studies found that not only could the 2′ and 3′-OH moieties be removed, but the 2′ and 3′-carbons could also be excised to yield what was dubbed an acyclic nucleoside (Fig. 19 ). (Deval, 2009, De Clercq et al., 2001, Sekiyama et al., 1998, King, 1988, Elion, 1993, Kimberlin, 2001, Freeman and Gardiner, 1996)
Fig. 19.

Comparison of the structure of guanosine with the acyclic nucleosides Ganciclovir, which is missing a 2′ carbon and corresponding 2′-OH, Acyclovir, which is missing both the 2′ and 3′ hydroxyl groups, and Penciclovir, the analogous carbocyclic version of Ganciclovir.
One of the most medically important acyclic nucleosides is Acyclovir, an acyclic guanosine mimic that has displayed high specificity and activity towards HSV-1 and HSV-2 (Fig. 19). (King, 1988, Elion, 1993, Kimberlin, 2001, Freeman and Gardiner, 1996, Reardon and Spector, 1989, McMahon et al., 2008) This high specificity and activity is due to many factors. For example, Acyclovir is more readily recognized and phosphorylated by viral thymidine kinases than the corresponding cellular thymidine kinases (King, 1988, Reardon and Spector, 1989) at a rate of >3000 fold higher, thus leading to higher concentrations of monophosphorylated Acyclovir in infected cells as opposed to uninfected cells (Elion, 1993, McMahon et al., 2008). In addition, the triphosphorylated Acyclovir is recognized and incorporated 100 fold more efficiently by the viral DNA polymerase as compared to the cellular polymerase (King, 1988, Reardon and Spector, 1989). It has also been shown that Acyclovir's inherent flexibility allows for optimized interactions in target enzyme binding sites (Alvarez-Ros and Palafox, 2014, Tuttle and Krenitsky, 1984, Wesołowski et al., 1987).
Unfortunately, like most nucleoside analogues, Acyclovir suffers from poor water solubility and thus a low oral bioavailability (de Miranda and Blum, 1983, Elion et al., 1977). One way to alleviate this issue is to add prodrug moieties to the nucleoside analogues to increase lipophillicity and ultimately increase the delivery of the active analogue to the active site. In the case of Acyclovir, various amino acid esters of Acyclovir were synthesized including the valine ester prodrug of Acyclovir, (Valacyclovir), which served to increase oral bioavailability 3-to-5 fold in humans (Fig. 20 ). (Colla et al., 1983, Bras et al., 2001, Weller et al., 1993, Steingrimsdottir et al., 2000, Perry and Faulds, 1996, Soul-Lawton et al., 1995, Smiley et al., 1996) This increase in bioavailability also resulted in an improved efficacy against herpesviruses such as varicella-zoster virus and cytomegalovirus infections, that had previously shown less susceptibility to Acyclovir (Weller et al., 1993, Perry and Faulds, 1996, Soul-Lawton et al., 1995, Smiley et al., 1996, Burnette et al., 1995, Beutner, 1995). Moreover, Valacyclovir could be administered in the form of eye drops to treat herpetic keratitis, in contrast to Acyclovir, which had to be administered as an ointment (Maudgal et al., 1984). The increased absorption of Valacyclovir is attributed to the facilitated transport of Valacyclovir across the intestinal membrane by human oligopeptide transporter 1 (hPepT1), as compared to simple diffusion utilized by Acyclovir (de Vrueh et al., 1998, Balimane et al., 1998). Valacyclovir has also been utilized for treatment against HSV-1 and HSV-2 (Perry and Faulds, 1996, Smiley et al., 1996, Beutner, 1995).
Fig. 20.

Examples of the prodrug analogues of Acyclovir, Ganciclovir, and Pencyclovir that increase lipophillicity and subsequent bioavailability.
The initial discovery of Acyclovir led many researchers to pursue additional nucleoside analogues utilizing the acyclic sugar scaffold. One example is Ganciclovir, also an acyclic guanosine mimic, however, unlike Acyclovir, Ganciclovir retains the 3′-hydroxyl group (Fig. 19). (Kimberlin, 2001, Freeman and Gardiner, 1996, Hewlett et al., 2004, Fan-Havard et al., 1989) Ganciclovir is most commonly used to treat HCMV in immunocompromised patients, and, like Acyclovir, HCMV infected cells have a higher affinity for Ganciclovir than non-infected cells (Kimberlin, 2001, Fan-Havard et al., 1989). Unfortunately, since HCMV treatment is usually long term, instances of resistance to Ganciclovir have been reported (Kimberlin, 2001, Hewlett et al., 2004, Komatsu et al., 2014). Like Acyclovir, Gancyclovir also demonstrates low bioavailability which can be increased with the addition of a valine ester to yield the prodrug Valganciclovir (Fig. 20). (Curran and Noble, 2001, Martin et al., 1987) Valganciclovir is currently utilized for both treatment of CMV retinitis in patients with HIV as well as a prophylactic treatment for the prevention of CMV in transplant patients (Reischig et al., 2008, Paya et al., 2004, Martin et al., 2002, Piketty et al., 2000, De Clercq and Field, 2006, Chamberlain et al., 2008).
Another example is Penciclovir, a carbocyclic guanosine analogue similar in structure to both Acyclovir and Ganciclovir, however, Penciclovir has a methylene group instead of the oxygen in the acyclic sugar moiety (Fig. 19). (Boyd et al., 1987, Boyd et al., 1993, Harnden and Jarvest, 1985) Like Acyclovir, Penciclovir is also a potent therapeutic against both HSV-1 and HSV-2, however, surprisingly Penciclovir demonstrated a lower bioavailability than Acyclovir, thus researchers attempted to find a suitable prodrug (De Clercq and Field, 2006, Harnden and Jarvest, 1985, Vere Hodge et al., 1989). These studies ultimately led to the development of Famciclovir, a prodrug analogue that utilizes a typical acetate prodrug for the hydroxyl moieties, but also features the removal of the carbonyl group on the guanosine nucleobase (Fig. 20). (Vere Hodge et al., 1989, Harnden et al., 1989) After enzymatic oxidation in vivo of the nucleobase to add the carbonyl to the purine ring and hydrolysis of the acetate groups, Famciclovir yields Penciclovir (Vere Hodge et al., 1989, Harnden et al., 1989, Vere Hodge, 1993). Due to the increased bioavailability associated with Famciclovir, this analogue has been utilized for years as a treatment against both HSV-1 and HSV-2 as well as against herpes zoster infections as it can be administered less frequently and at lower doses as compared to Acyclovir treatment (De Clercq and Field, 2006, Degreef and Group, 1994, Tyring et al., 1995, Sacks et al., 1996, Mertz et al., 1997).
2.9. Acyclic nucleoside phosphonates
During DNA/RNA replication, nucleosides are phosphorylated by various host cell kinases into their active triphosphate form, then are taken up by polymerases and incorporated into the growing chain (De Clercq and Neyts, 2009, Deval, 2009). The same process is required for modified nucleosides, however, one of the major limitations to utilizing nucleosides as drugs is the specificity of the kinases involved in the various phosphorylation steps. These steps then become rate limiting for the overall conversion to the active triphosphate form. Since nucleoside analogues are not always recognized efficiently and thus may initially appear inactive, it became important to design analogues that could overcome this issue (De Clercq, 2007a, De Clercq, 2007b, Pertusati et al., 2012, De Clercq and Holý, 2005, De Clercq et al., 2005). In that regard, researchers attempted to utilize monophosphate analogues, however, the phosphate group renders the nucleotides extremely polar, therefore making it difficult for the compounds to cross the cellular membrane. Moreover, this also increases the susceptibility of these compounds towards degradation by cellular enzymes, particularly phosphatases, thus was not a viable solution (Pertusati et al., 2012, De Clercq and Holý, 2005, Macchi et al., 2015).
In the late 1980's, Antonín Hóly and his team circumvented this issue by adding a phosphonate, rather than a phosphate, group to the acyclic nucleoside scaffold. This not only increased the stability of the phosphate bond, but also successfully delivered a monophosphate mimic into the cell, thereby overcoming that first rate-limiting phosphorylation step, as well as overcoming issues with delivery (De Clercq, 2007a, De Clercq, 2007b, De Clercq and Holý, 2005, Macchi et al., 2015, Hóly and Rosenberg, 1987, Holý, 2006). The corresponding acyclic nucleoside phosphonates (ANPs) were associated with prolonged antiviral activity due to the fact that these compounds were no longer susceptible to cellular phosphatases that would cleave the phosphorester bond, reverting it back to the parent nucleoside (De Clercq, 2007a, De Clercq, 2007b, Pertusati et al., 2012, Macchi et al., 2015, Engel, 1977). The addition of the phosphonate moiety also increased the scope of the antiviral activities as seen with ANPs such as Cidofovir, Adefovir, and Tenofovir (Fig. 21 ). (De Clercq, 2003, De Clercq, 2007a, De Clercq, 2007b)
Fig. 21.

Examples of acyclic nucleoside phosphonates such as Cidofovir, Adefovir, and Tenofovir, where the phosphate group is replaced by the more stable phosphonate group.
Cidofovir (S-HPMPC) (Fig. 21) is a cytosine derivative that retains the 2′-carbon and corresponding 2′-hydroxyl group and has demonstrated activity against most DNA viruses including: polyoma-, papilloma-, adeno-, pox-virus, HSV-1, HSV-2, VZV, HCMV, and Epstein-Barr virus (De Clercq, 2003, De Clercq, 2007a, De Clercq, 2007b, De Clercq, 2013b, De Clercq and Holý, 2005, Lalezari et al., 1997). While the primary use of Cidofovir is for the treatment of HCMV retinitis in AIDS patients, Cidofovir is currently one of the only licensed antiviral drugs that has been stockpiled for the possible use as short term prophylaxis against smallpox in the event of a biological weapons attack (De Clercq, 2003, De Clercq, 2007a, De Clercq, 2007b, De Clercq and Holý, 2005, Holý, 2006). Unfortunately Cidofovir, like almost all of the acyclic nucleoside phosphonates, is associated with severe side effects, including nephrotoxicity, impaired renal function, and other side effects due to the presence of the phosphonate moiety. Thus, Cidofovir contains a black box warning, which limits its use to short term therapies (De Clercq, 2003, Lalezari et al., 1997, Smee et al., 2002).
Another acyclic nucleoside phosphonate that initially garnered much attention was Adefovir (R-PMEA, Fig. 21), an adenosine analogue that was first utilized in the treatment of HIV, however, the high dosage needed was associated with toxicity and numerous adverse side effects (De Clercq, 2005b, De Clercq, 2007a, De Clercq and Holý, 2005, Holý, 2006, Hadziyannis et al., 2003, Dando and Plosker, 2003, Angus et al., 2003). Adefovir is still used in some countries, however at a much lower dosage, as it was subsequently proven to be a very effective and inexpensive treatment for chronic Hepatitis B infections (De Clercq, 2003, De Clercq, 2005b, De Clercq, 2007a, De Clercq, 2007b, De Clercq and Holý, 2005, Holý, 2006).
In contrast, one of the most widely used phosphonate nucleoside drugs is Tenofovir (R-PMPA). Tenofovir (Fig. 22 ) is an adenosine analogue that is utilized to treat HIV as well as chronic Hepatitis B infections (De Clercq, 2003, De Clercq, 2005b, De Clercq, 2007a, De Clercq, 2007b, De Clercq, 2009a, De Clercq, 2009b, De Clercq and Holý, 2005, Holý, 2006). Unfortunately, like most acyclic nucleoside phosphonates, Tenofovir is associated with a very low bioavailability, however, this was greatly increased with the addition of a prodrug moiety (De Clercq, 2003, Fung et al., 2002, Porche, 2002, Chapman et al., 2003). Tenofovir disoproxyl fumarate (TDF) was initially marketed in 2001, and demonstrated a higher potency and greater efficacy than the parent Tenofovir (Fig. 22). (Fung et al., 2002, Porche, 2002, Chapman et al., 2003) Most recently however, Tenofovir Alafenamide (TAF) has largely replaced TDF in HIV treatments, primarily due to the significant difference in dosage – only 30 mgs vs 300 mgs, as well as greatly increased levels of the nucleotide inside the virally infected cell (De Clercq, 2016a, Ray et al., 2016). Recently growing concerns over increasing incidents of nephrotoxicity and other renal issues related to TDF have arisen, similar other phosphonates, although these side effects took years to arise (Rawal et al., 2015, De Clercq, 2016a, Ray et al., 2016, Birkus et al., 2015). Importantly, almost no reports of resistance against TAF have been reported, thereby rendering TAF a much more effective and safe drug (De Clercq, 2016a, Ray et al., 2016, Stray et al., 2017).
Fig. 22.

Structure of the acyclic nucleoside phosphonate Tenofovir and its prodrug analogues Tenofovir disoproxyl fumarate and Tenofovir alafenamide.
Not just limited to antiviral uses, several other acyclic nucleoside phosphonates, including the 9-(3-hydroxy-2-phosphonylmethoxypropyl) derivatives, have demonstrated potent activity against different members of the Trypanosoma genus that cause lethal African sleeping sickness (Fig. 21). (Kaminsky et al., 1994, Kaminsky et al., 1996) The adenosine analogues were the most effective of the 9-(3-hydroxy-2-phosphonylmethoxypropyl) derivatives, especially the S enantiomer, which demonstrated potent activity against T.b. rhodesiense with an EC50 of 0.028 μg/mL (Kaminsky et al., 1994, Kaminsky et al., 1996). These potent analogues are still under investigation as potential therapeutics and have yet to reach clinical trials.
3. Modifications to the nucleobase
Concurrently to the aforementioned approaches to modify the sugar moiety, numerous modifications were also being made to both the purine and the pyrimidine heterocyclic bases of the nucleosides. Analogous to various interactions involved in recognition and binding for the sugar of the nucleoside, enzyme-ligand recognition and binding also involves many different types of interactions with the base. Thus changing the sterics, electronics, or hydrogen bonding interactions can also significantly impact recognition, and subsequently, biological activity (Jordheim et al., 2013, De Clercq and Neyts, 2009, Deval, 2009). Modifications to the heterobase have primarily focused on adding substituents onto the base, removing or adding atoms to the rings themselves, or changing positions of the atoms. These types of changes have served to create many new classes of nucleoside analogues over the years.
3.1. Modifications at the 5 position of pyrimidines
Although various modifications can be made to the pyrimidine ring, one of the most common early modifications in nucleoside drug design involved adding substituents to the C5 position of the pyrimidine ring. This modification can potentially alter the sterics, the electronic environment, and even the hydrogen bonding interactions between the enzyme binding site and the nucleoside analogue (De Clercq, 2010, Prusoff, 1959, Kaufman and Heidelberger, 1964, Whitley, 1996). One early nucleoside that utilized this modification was Idoxuridine, a 2′-deoxyuridine analogue with an iodine at the C5 position on the uridine ring (Fig. 23 ). (De Clercq, 2010, De Clercq, 2013b, Prusoff, 1959) Idoxuridine was one of the first examples of a pyrimidine analogue and was originally developed as an anticancer agent, however, subsequent studies later determined that treatment with Idoxuridine was also associated with profound antiviral activity, most notably against DNA viruses such as HSV (Prusoff, 1959). Other noteworthy C5 modified analogues that were developed early on included 5-trifluorothymidine (Trifluridine), which was primarily used for ocular herpes infections, and 5-bromovinyl deoxyuridine (Brivudine), which was utilized for treating VZV (Fig. 23). (De Clercq, 2010, De Clercq, 2013b, Bauer, 1985, Kaufman and Heidelberger, 1964, Whitley, 1996, De Clercq et al., 1979)
Fig. 23.

Examples of typical heterocyclic modifications at the 5-position of the pyrimidine moiety.
As mentioned previously, use of a fluorine in the sugars of the nucleosides was known to be advantageous. This also proved true for the nucleobases. The anticancer drug 5-fluororuracil (5-FU), and the corresponding 2-deoxyribonucleoside floxuridine are important examples of a bioisosteric replacement of fluorine for hydrogen (Fig. 23). In a series of biologically mediated reactions, 5-FU is first converted to a nucleoside and then to its monophosphate, which subsequently becomes part of a ternary complex between thymidylate synthase and its cofactor 5,10-methylenetetrahydrofolate (Longley et al., 2003, Santi et al., 1987). Because the hydrogen normally present at C5 is subsequently abstracted by thymidylate synthase, thereby causing the complex to disassemble, which ultimately leads to thymidine (and tetrahydrofolate), replacement of the hydrogen with the fluorine results in a dead-end complex that cannot come apart and therefore does not lead to the production of thymidine (Longley et al., 2003, Santi et al., 1987). Since this process is a critical source of thymidine, and cancer cells need elevated levels of thymidine to replicate, use of 5-FU is highly effective (Longley et al., 2003, Santi et al., 1987). Notably, addition of fluorine at the C5 position also renders cytosine nucleosides stable to deaminases, enzymes that are known to inactivate many nucleosides with amine groups on their bases, further highlighting the importance of this particular structural modification (Secrist et al., 1985, Montgomery et al., 1983, Park et al., 2001b).
Another commonly pursued modification at the C5 position involved addition of various alkyl groups, which were systematically pursued by systematically increasing the length of the alkyl chain one methylene group (Fig. 24 ). These types of homologous modifications were pursued for two main reasons – one, to increase the drug's lipophillicity to aid in delivery, and two, to potentially increase the fit of the drug into a hydrophobic pocket in an enzyme binding site (Silverman and Holladay, 2014, Dohme et al., 1926). Initial studies revealed that while increasing the chain length often led to an increase in activity, at a certain point this trend reversed and the nucleoside lost activity (Dohme et al., 1926). This is most likely due to the nucleoside becoming too lipophilic, which can lead to aggregation and accumulation in fatty tissues (Silverman and Holladay, 2014, Dohme et al., 1926). While these modifications helped increase our knowledge of the parameters of enzyme binding sites and the importance of lipophilicity, most of these analogues proved to be clinically relevant, and led to the oft quoted phrase, “methyl, ethyl, propyl, butyl, futile”!
Fig. 24.

Example of a homologous series of modifications that increase lipophillicity up to a certain threshold.
These original C5 alkyl modifications led to the discovery of other long chain substituted analogues such as the bicyclic pyrimidine nucleoside analogues (BCNAs) originally discovered by Balzarini and McGuigan (Fig. 25 ). (Balzarini and McGuigan, 2002, McGuigan et al., 1999) Through structure activity relationship studies, even more potent BCNAs were discovered with high specificity towards VZV treatment (Balzarini and McGuigan, 2002). Analogues CF-1368 and CF-1743 were especially potent, with EC50 values of 0.027 ± 0.013 and 0.0002 ± 0.00017 μM compared to Brivudine and Acyclovir (EC50 = 0.009 ± 0.004 μM and 3.4 ± 0.67 μM respectively), two analogues already used in the clinic to treat VZV infections (Balzarini and McGuigan, 2002, Andrei et al., 2005). Not only were these analogues extremely potent against VZV, they also demonstrated low cytotoxicity at levels up to 50 μM (Balzarini and McGuigan, 2002, McGuigan et al., 1999). As with the C5 alkyl study, increased branching of the BCNAs was associated with a decrease in antiviral activity, with the optimum length proving to be C8-C10 (McGuigan et al., 1999, Luoni et al., 2005).
Fig. 25.

Examples of bicyclic pyrimidine nucleoside analogues (BCNAs) with potent anti-VZV activity.
3.2. Nitrogen substitutions and rearrangements
As mentioned earlier, adding substituents was not the only type of modification that was routinely tried in an effort to find activity or temper side effects. Other common modifications included removing or relocating various atoms from the nucleobase. For example, removal of a nitrogen from the purine or pyrimidine ring system (referred to as a “deaza” analogues) could be utilized to explore the effect of hydrogen bonding interactions in enzyme binding sites. Given that removal or addition of a strong hydrogen bond donor or acceptor affects reactivity as well as properties at other positions on the bases, this approach can have a profound affect. One important example is 3-deaza-deoxyguanosine, where the N3 is replaced by a CH (Fig. 26 ). (Mian and Khwaja, 1983, Liu et al., 2001) Not only did this modification result in broad spectrum antiviral activity against both DNA and RNA viruses, potent antitumor properties against leukemia L1210 and P388 cell lines were also observed (Mian and Khwaja, 1983, Revankar et al., 1984). Other medically relevant deaza analogues are 7-deazaadenosine analogues, such as Tubercidin, where the N7 was replaced by a CH group (Fig. 26). (Olsen et al., 2004, Acs et al., 1964, Itoh et al., 1972) Tubercidin was originally isolated from the bacteria Streptomyces tubercidicus, and has demonstrated potent activity as an antibiotic against Streptococcus faecalis (Olsen et al., 2004, Acs et al., 1964, Bloch et al., 1967). While many early deaza compounds were purines, others similarly used a deaza approach in pyrimidine analogues, such as 3-deazauridine, where the N3 of the pyrimidine ring was replaced by a CH (Fig. 26). (Ebrahimi et al., 2001, Patching et al., 2005) While the 3-deazauridine analogues did not display meaningful biological activity, these analogues often helped to identify the specific requirements of the various amino acids involved in binding within enzyme binding sites (Patching et al., 2005).
Fig. 26.

Examples of “deaza” modifications to the heterobase scaffold where a nitrogen is removed from either the purine or the pyrimidine ring.
Another common nitrogen modification was the addition of a nitrogen to the purine ring system (referred to as “aza” analogues). This type of modification can introduce new hydrogen bonding donors or acceptors, and depending upon their position in the ring system, can direct the reactivity of the aromatic ring itself towards nucleophilic and electrophilic substitutions. One example of this type of modification is present in 5-azacytidine, an antibiotic analogue originally isolated from Streptoverticillium ladakanus (Fig. 27 ). (Winkley and Robins, 1970) The addition of the extra nitrogen also endowed 5-azacytidine with profound anticancer properties, specifically against acute myelogenous leukemia and MCF07 human breast cancers, as well as in the treatment of myelodysplastic syndrome (Winkley and Robins, 1970, Chang et al., 2014, Gryn et al., 2002, Raj and Mufti, 2006).
Fig. 27.

Examples of “aza” modifications to the heterobase scaffold where a nitrogen is added to either the purine or pyrimidine ring.
While nitrogens can be removed or added to the pyrimidine or purine scaffold, nitrogens can also be repositioned in the ring, thus retaining the same number of nitrogens as the parent nucleoside, but also introducing alternative hydrogen bond donors or acceptors to that can therefore interact with new areas of the enzyme binding site. This is especially true for the adenosine mimic 8-aza-7-deazapyrazole pyrimidine (Fig. 27). Due to the antiparasitic activity associated with this analogue, numerous researchers subsequently attempted to add various substituents to the pyrazole moiety in order to increase the activity, however, to date this approach has not proven particularly fruitful (Petrie et al., 1985, Bhat et al., 1981, Montgomery et al., 1974, Rideout et al., 1982).
3.3. Expanded purine nucleobases
Since changing the size of the sugar ring led to clinically relevant results for some nucleoside analogues, researchers also pursued increasing the size of the nucleobase. In that regard, Nelson Leonard was an early pioneer in the development of base modified nucleosides. He first introduced the nucleoside field to this concept with his benzyl-expanded purines including lin-benzoadenosine, as well as the corresponding proximal and distal analogues (Fig. 28 ). (Leonard et al., 1975, Leonard et al., 1976, Leonard et al., 1978, Leonard, 1982) These analogues were originally developed in order to probe adenosine metabolizing enzyme binding sites to determine the upper limits of enzyme-substrate recognition (Leonard et al., 1978, Leonard, 1982). Through numerous studies, it was determined that this modification served to not only increase the aromaticity and polarizability of the heterocyclic base, but also increased π-π stacking, an important element of recognition in enzyme binding sites (Leonard et al., 1975, Leonard et al., 1976, Lee and Kool, 2006, Lynch et al., 2006, Krueger et al., 2007). Unfortunately, while some of these compounds demonstrated interesting biological activity, none proved to be potent enough to continue on to the clinic.
Fig. 28.

Structures of the “benzene-expanded” purine nucleosides originally developed by Leonard, in which there is a benzene spacer ring separating the imidazole and the pyrimidine components of the purine nucleobase scaffold.
Many years later, Seley-Radtke took Leonard's shape modifications one step further, and replaced Leonard's benzene spacer ring with various five-membered heteroaromatic rings, most notably, a series of thieno-expanded purines (Fig. 29 ). The tricyclic thieno analogues were associated with numerous benefits while still retaining the essential recognition elements of the parent nucleoside, including increased aromaticity and polarizability of the base (Seley-Radtke et al., 2008, Zhang et al., 2008, Wauchope et al., 2012, Chen et al., 2015). These analogues are also smaller than Leonard's benzyl spacer derivatives, thus can base pair more readily (Seley-Radtke et al., 2008, Zhang et al., 2008, Wauchope et al., 2012). These shape modified nucleosides showed some interesting biological activity in several areas including HCV and cancer (Wallace et al., 2004, Seley et al., 2000), as well as to serve as probes in various enzyme binding sites (Quirk and Seley, 2005), however, the synthesis of these analogues was excessively tedious and plagued with overall unacceptably low yields, thus have not been pursued extensively since the initial studies.
Fig. 29.

Structures of the “thieno-expanded” purine nucleosides developed by Seley-Radtke based on Leonard's original benzyl-expanded derivatives, where a 5 membered thiophene ring separates the imidazole and pyrimidine components of the nucleobase scaffold.
4. Other structural modifications
While numerous analogues with either a sugar or a nucleobase modification were employed, researchers have of course, not been limited to one or two modifications per analogue, and, as such, many analogues feature multiple modifications to the nucleobase and/or sugar, including double modifications to the same position on the sugar, as well as to the glycosidic bond. Although the more complex nucleoside/tide analogues will be reviewed more extensively in the second paper of this series, there are several additional classes of nucleosides that will be briefly covered below as they fall into some of the categories already discussed above.
4.1. C-nucleosides
As mentioned previously, one of the limiting factors in the development of nucleoside analogues is stability of the glycosidic bond to cleavage by various glycoside hydrolases. While some researchers opted to remove the furanose oxygen and create carbocyclic analogues to overcome issues with the glycosidic bond instability, others attempted to remove the N3 nitrogen in the purine ring to create C-nucleosides (Fig. 30 ). (De Clercq, 2016b, Stambaský et al., 2009, Temburnikar and Seley-Radtke, 2018)
Fig. 30.

Comparison of the structure of adenosine and the corresponding C-adenosine analogue where the glycosidic bond is removed due to removal of the N1 nitrogen in the purine ring.
One of the first C-nucleosides discovered was pseudouridine, a naturally derived nucleoside analogue, where the pyrimidine ring is attached to the ribose moiety at C5 instead of C6 (Fig. 31 ). (De Clercq, 2016b, Stambaský et al., 2009, Srinivasan and Borek, 1964, Charette and Gray, 2000) Pseudouridine is most commonly found in both transfer RNA and ribosomal RNA and is the most abundant naturally occurring C-nucleoside (Stambaský et al., 2009, Charette and Gray, 2000). While no significant biological activity has been associated with pseudouridine, its discovery aided in the discovery and identification of other medically relevant C-nucleosides (Stambaský et al., 2009). One such analogue is Showdomycin, which is a naturally occurring 5-membered uridine mimic first isolated in 1964 from Streptomyces showdonesis by Nishimura et al. (Fig. 31). (Nishimura et al., 1964) Showdomycin proved to be an effective antibiotic against several Gram-positive and Gram-negative bacteria, particularly against Streptococcus haemolyticus and Streptococcus pyogenes (Roy-Burman et al., 1968, Darnall et al., 1967). Furthermore, this analogue has also demonstrated potent antitumor activity against Ehrlich mouse ascites as well as HeLa cells in vitro (Stambaský et al., 2009, Roy-Burman et al., 1968, Darnall et al., 1967).
Fig. 31.

Examples of early naturally occurring C-nucleosides Pseudouridine and Showdomycin.
Other important examples of C-nucleosides include the aforementioned Immucillin-H (Forodesine) and BCX-4430 (Fig. 32 ). (Warren et al., 2014, Bergeron-Brlek et al., 2015, Korycka et al., 2007) While both Immucillin-H and BCX-4430 feature an imino sugar, where the furanose oxygen has been replaced with a secondary amine, not all C-nucleosides feature this type of sugar (De Clercq, 2016b, Stambaský et al., 2009, Korycka et al., 2007, Boutureira et al., 2013). The addition of the NH in the imino sugar imparts a significant benefit over corresponding carbocyclic sugars in that the nitrogen is a hydrogen bond donor (whereas the furanose oxygen was simply a hydrogen bond acceptor), thus expanding the potential interactions of imino sugars in enzyme binding sites (Schramm and Tyler, 2003, Bergeron-Brlek et al., 2015). This benefit is especially seen in Immucillin-H, a transition state analogue that inhibits purine nucleoside phosphorylase (PNP), which in turn inhibits the growth of malignant T cells in human leukemia (Kicska et al., 2001, Korycka et al., 2007, Bantia et al., 2001). Similarly, BCX-4430 has demonstrated the ability to be a broad spectrum antiviral agent, with micromolar activity against numerous viruses including Ebola, Zika, Yellow Fever, and Middle East Respiratory Syndrome Coronavirus (MERS-CoV), just to name a few (Warren et al., 2014, Julander et al., 2014, Julander et al., 2017, Taylor et al., 2016, Eyer et al., 2017).
Fig. 32.

Structure of C-nucleosides with an imino sugar.
4.2. Iso-nucleosides
Other nucleosides that feature an altered glycosidic bond are the iso-nucleosides, which were also developed by Nelson Leonard and are represented by Iso-adenosine (Iso-A), wherein the adenine base is connected to the sugar at N3 instead of N9 (Fig. 33 ). (Leonard and Laursen, 1963, Leonard and Laursen, 1965, Kumar et al., 1988, Leonard et al., 1986) This modification was associated with various biological activities, most notably against lymphoblastic leukemia cells in vitro (Gerzon et al., 1966). Unfortunately, Iso-A is not particularly stable since it is hyperconjugated and has a tendency to rearrange in an effort to restore the aromaticity, thus resulting in re-forming adenosine (Seley et al., 2003). Furthermore, Iso-A was also unstable due to the presence of a glycosidic bond, rendering the analogue susceptible to cleavage by both acids, bases, and glycosidic hydrolases (Seley et al., 2003). This led to further investigation by Seley-Radtke et al., wherein a carbocyclic Iso-Neplanocin A was synthesized (Fig. 33). (Seley et al., 2003) This analogue overcame the instability of the glycosidic bond, with the N3 product preferably formed (Seley et al., 2003). Unfortunately none of these analogues exhibited any clinically useful levels of antiviral or anticancer activity.
Fig. 33.

Examples of Iso-adenosine analogues where the sugar moiety is now attached to the N3 instead of the N7 as in natural nucleosides. Iso-analogues can also be formed with attachment of the nucleobase at the 2′-carbon instead of the 1′-carbon.
More recent examples of iso-nucleosides were introduced by Nair et al., and featured the glycosidic bond relocated to C2′ on the sugar (Fig. 33). (Nair and Nuesca, 1992, Nair et al., 1995, Nair and Jahnke, 1995) These analogues proved to exhibit much greater stability in acidic conditions compared to other dideoxy analogues, where the half-life of C2′-iso-adenosine was greater than 16 days at pH 1 compared to a half-life of dideoxyadenosine of 0.5 h at pH 3 (Nair and Nuesca, 1992, Nair et al., 1995, Nair and Jahnke, 1995). Moreover, this analogue demonstrated potent micromolar anti-HIV activity in vitro with no apparent cytotoxicity (Nair and Nuesca, 1992, Nair et al., 1995, Nair and Jahnke, 1995). While these analogues possessed potent activity against HIV integrase, they have not been extensively pursued for other purposes (Nair et al., 1995, Nair and Jahnke, 1995).
4.3. L-nucleosides
Most of the examples of modified nucleosides discussed thus far were initially synthesized as racemic mixtures of the D and L nucleosides, however, in the 1980s it became apparent that, in general, only one of the enantiomers in a racemic mixture was responsible for the activity. In addition, it was also found that in some cases the l-enantiomer could prove to be the cause of toxicity, or to interfere with or lower the activity of the more active enantiomer, although this wasn't always true. For example in some cases, the l-enantiomer showed different activities or even synergistic activities. Indeed, some L nucleosides have exhibited quite potent antiviral activities when the corresponding D nucleosides did not, particularly against HBV (Gumina et al., 2001, Gumina et al., 2007).
The most notable examples of biologically relevant L nucleosides were FTC and 3TC, identified earlier in Fig. 6 and again below in Fig. 34 . Resolution of the two enantiomers of (±)-BCH-189 to give the l-enantiomer (dubbed 3TC) proved to quite important, as 3TC was found to be exponentially more active against both HIV-1 and HBV (EC50 0.002 μM and 0.01 μM) compared to its D-counterpart (EC50 0.2 μM and 0.5 μM) (Schinazi et al., 1992a, Schinazi et al., 1992b, Gumina et al., 2001, Jarvis and Faulds, 1999). Moreover, not only was 3TC more potent than the d-enantiomer or the racemic mixture (±)-BCH-189, 3TC also displayed less toxicity, likely due to the fact that it was quite resistant to deamination (Gumina et al., 2001, Chang et al., 1992). The potent activity against both HIV-1 and HBV also makes 3TC one of the leading treatments of coinfections of hepatitis B and HIV in patients (Gumina et al., 2001, Mathé and Gosselin, 2006).
Fig. 34.

Examples of the l-enantiomer of nucleoside analogues that were assumed to be inactive since naturally occurring nucleosides are the D-enantiomers. Later it was shown that the L-nucleosides exhibit potent biological activity against a variety of pathogens.
Another L-nucleoside that garnered much attention early on was telbivudine (L-dT) for its potent effect in the treatment of chronic Hepatitis B infections (Fig. 34). (Han, 2005, Kim et al., 2006) L-dT is the l-enantiomer of naturally occurring thymidine, and once transformed into the active triphosphate form, is incorporated by viral DNA polymerase and induces chain termination (Han, 2005, Kim et al., 2006). In comparison to other nucleoside analogues used to treat HBV infections such as 3TC, treatment with L-dT elicits higher therapeutic and biochemical responses in patients (Lai et al., 2005, Lai et al., 2007). Moreover, L-dT is specific for HBV polymerase, and is not recognized by human polymerases, making treatment with L-dT safe and effective (Kim et al., 2006, Semizarov et al., 1997, Bryant et al., 2001a).
Another early L-nucleoside was 1-(2-deoxy-2-fluoro-β-L-arabino-furanosyl)-5-methyluracil (L-FMAU), a thymidine analogue that displayed potent antiviral activity against HBV (EC50 = 0.1 μM in Hep-G2/2.2.15 cells) with no toxicity reported up to 100 μM in vitro (Fig. 34). (Chu et al., 1995, Du et al., 1999) Initially there was hope that L-FMAU could be utilized in treatment of chronic HBV infections, however ultimately more potent drugs were discovered and it was no longer used clinically (Chu et al., 1995, Du et al., 1999, Wright et al., 1995, Peek et al., 2001). Another example of a promising L-nucleoside was L-7-deaza-5′-noraristeromycin, which not only featured an L-nucleoside, but also utilized the 5′-nor carbocyclic sugar scaffold as well as a deazapurine heterobase (Fig. 34). (Seley et al., 1997a, Seley et al., 1998, Siddiqi et al., 1995) Initial studies found potent activity for this analogue against several pathogens including HBV and four strains of African trypanosomes, one of which is known to cause east African sleeping sickness (Seley et al., 1997a, Seley et al., 1998). Finally, Elvucitabine, or L-d4FC, was a cytidine dideoxynucleoside reverse transcriptase inhibitor that also demonstrated micromolar activity against HBV (Fig. 34). (Mathé and Gosselin, 2006, Bryant et al., 2001a, Bryant et al., 2001b, Li et al., 1998) The potent activities demonstrated by some of these analogues, particularly against HBV, has led to continued interest in pursuing L-nucleosides.
5. Conclusions
Nucleoside analogues continue to play an essential role in the treatment of diseases, thus the pursuit of new nucleoside analogues is critical to solving global health issues such as emerging and reemerging infectious diseases and other pathogens. The examples covered in this review are by no means meant to be exhaustive. It would be impossible to show all of the various nucleoside modifications that have been synthesized in each of these broad categories, however, it was our intention to provide a flavor of the historical progression of nucleoside development and the various types of structural modifications that have been pursued to date. As the field has progressed and new information has become available about nucleoside structure, enzyme recognition, and biological activity, new and more complex modifications have been pursued, including multiple modifications to the same scaffold. Those and other types of approaches will be reviewed in the second paper in this series.
References
- Acs G., Reich E., Mori M. Biological and biochemical properties of the analogue antibiotic tubercidin. Proc. Natl. Acad. Sci. U. S. A. 1964;52:493–501. doi: 10.1073/pnas.52.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrofoglio L., Suhas E., Farese A., Condom R., Challand S.R., Earl R.A., Guedj R. Synthesis of carbocyclic nucleosides. Tetrahedron. 1994;50:10611–10670. [Google Scholar]
- Aihong K., Joon H.H. Synthesis and antiviral activity of C-fluoro-branched cyclopropyl nucleosides. Eur. J. Med. Chem. 2007;42:487–493. doi: 10.1016/j.ejmech.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Alvarez-Ros M.C., Palafox M.A. Conformational analysis, molecular structure and solid state simulation of the antiviral drug acyclovir (zovirax) using density functional theory methods. Pharm. Basel. 2014;7:695–722. doi: 10.3390/ph7060695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrei G., Sienaert R., McGuigan C., De Clercq E., Balzarini J., Snoeck R. Susceptibilities of several clinical varicella-zoster virus (VZV) isolates and drug-resistant VZV strains to bicyclic furano pyrimidine nucleosides. Antimicrob. Agents Chemother. 2005;49:1081–1086. doi: 10.1128/AAC.49.3.1081-1086.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andriole V.T. Current and future antifungal therapy: new targets for antifungal agents. J Antimicrob. Chemother. 1999;44:151–162. doi: 10.1093/jac/44.2.151. [DOI] [PubMed] [Google Scholar]
- Angus P., Vaughan R., Xiong S., Yang H., Delaney W., Gibbs C., Brosgart C., Colledge D., Edwards R., Ayres A., Bartholomeusz A., Locarnini S. Resistance to adefovir dipivoxil therapy associated with the selection of a novel mutation in the HBV polymerase. Gastroenterology. 2003;125:292–297. doi: 10.1016/s0016-5085(03)00939-9. [DOI] [PubMed] [Google Scholar]
- Ashton W.T., Meurer L.C., Cantone C.L., Field K.A., Hannah J., Karkas J.D., Liou R., Patel G.F., Perry H.C. Synthesis and antiherpetic activity of (+/-)-9- (Z)-2-(hydroxymethyl)cyclopropyl methyl guanine and related compounds. J. Med. Chem. 1988;31:2304–2315. doi: 10.1021/jm00120a010. [DOI] [PubMed] [Google Scholar]
- Baba M., Pauwels R., Herdewijn P., De Clercq E., Desmyter J., Vandeputte M. Both 2',3'-dideoxythymidine and its 2',3'-unsaturated derivative (2',3'-dideoxythymidinene) are potent and selective inhibitors of human immunodeficiency virus replication in vitro. Biochem. Biophys. Res. Commun. 1987;142:128–134. doi: 10.1016/0006-291x(87)90460-8. [DOI] [PubMed] [Google Scholar]
- Baldanti F., Sarasini A., Drach J.C., Zemlicka J., Gerna G. Z-isomers of 2-hydroxymethylcyclopropylidenemethyl adenine (synadenol) and guanine (synguanol) are active against ganciclovir- and foscarnet-resistant human cytomegalovirus UL97 mutants. Antivir. Res. 2002;56:273–278. doi: 10.1016/s0166-3542(02)00129-8. [DOI] [PubMed] [Google Scholar]
- Balimane P.V., Tamai I., Guo A., Nakanishi T., Kitada H., Leibach F.H., Tsuji A., Sinko P.J. Direct evidence for peptide transporter (PepT1)-mediated uptake of a nonpeptide prodrug, valacyclovir. Biochem. Biophys. Res. Commun. 1998;250:246–251. doi: 10.1006/bbrc.1998.9298. [DOI] [PubMed] [Google Scholar]
- Balzarini J., McGuigan C. Bicyclic pyrimidine nucleoside analogues (BCNAs) as highly selective and potent inhibitors of varicella-zoster virus replication. J. Antimicrob. Chemother. 2002;50:5–9. doi: 10.1093/jac/dkf037. [DOI] [PubMed] [Google Scholar]
- Bantia S., Miller P.J., Parker C.D., Ananth S.L., Horn L.L., Kilpatrick J.M., Morris P.E., Hutchison T.L., Montgomery J.A., Sandhu J.S. Purine nucleoside phosphorylase inhibitor BCX-1777 (Immucillin-H)–a novel potent and orally active immunosuppressive agent. Int. Immunopharmacol. 2001;1:1199–1210. doi: 10.1016/s1567-5769(01)00056-x. [DOI] [PubMed] [Google Scholar]
- Barnard D.L., Stowell V.D., Seley K.L., Hegde V.R., Das S.R., Rajappan V.P., Schneller S.W., Smee D.F., Sidwell R.W. Inhibition of measles virus replication by 5'-nor carbocyclic adenosine analogues. Antivir. Chem. Chemother. 2001;12:241–250. doi: 10.1177/095632020101200405. [DOI] [PubMed] [Google Scholar]
- Barral K., Courcambeck J., Pèpe G., Balzarini J., Neyts J., De Clercq E., Camplo M. Synthesis and antiviral evaluation of cis-substituted cyclohexenyl and cyclohexanyl nucleosides. J. Med. Chem. 2005;48:450–456. doi: 10.1021/jm0493966. [DOI] [PubMed] [Google Scholar]
- Bastiancich C., Bastiat G., Lagarce F. Gemcitabine and glioblastoma: challenges and current perspectives. Drug Discov. Today. 2017;2:416–423. doi: 10.1016/j.drudis.2017.10.010. [DOI] [PubMed] [Google Scholar]
- Bauer D.J. A history of the discovery and clinical application of antiviral drugs. Br. Med. Bull. 1985;41:309–314. doi: 10.1093/oxfordjournals.bmb.a072069. [DOI] [PubMed] [Google Scholar]
- Bergeron-Brlek M., Meanwell M., Britton R. Direct synthesis of imino-C-nucleoside analogues and other biologically active iminosugars. Nat. Commun. 2015;6:6903. doi: 10.1038/ncomms7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman A.M., Adema A.D., Balzarini J., Bruheim S., Fichtner I., Noordhuis P., Fodstad O., Myhren F., Sandvold M.L., Hendriks H.R., Peters G.J. Antiproliferative activity, mechanism of action and oral antitumor activity of CP-4126, a fatty acid derivative of gemcitabine, in in vitro and in vivo tumor models. Invest. New Drugs. 2011;29:456–466. doi: 10.1007/s10637-009-9377-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann W., Burke D.C. Contributions to the study of marine products. Xxxix. The nucleosides of sponges. Iii.1 spongothymidine and Spongouridine2. J. Org. Chem. 1955;20:1501–1507. [Google Scholar]
- Bergmann W., Feeney R.J. The isolation of a new thymine pentoside from Sponges1. J. Am. Chem. Soc. 1950;72:2809–2810. [Google Scholar]
- Bergmann W., Feeney R.J. Contributions to the study of marine products. Xxxii. The nucleosides of sponges. I.1. J. Org. Chem. 1951;16:981–987. [Google Scholar]
- Beutner K.R. Valacyclovir: a review of its antiviral activity, pharmacokinetic properties, and clinical efficacy. Antivir. Res. 1995;28:281–290. doi: 10.1016/0166-3542(95)00066-6. [DOI] [PubMed] [Google Scholar]
- Bhat G.A., Montero J.L., Panzica R.P., Wotring L.L., Townsend L.B. Pyrazolopyrimidine nucleosides. 12. Synthesis and biological activity of certain pyrazolo[3,4-d]pyrimidine nucleosides related to adenosine. J. Med. Chem. 1981;24:1165–1172. doi: 10.1021/jm00142a009. [DOI] [PubMed] [Google Scholar]
- Birkus G., Bam R.A., Willkom M., Frey C.R., Tsai L., Stray K.M., Yant S.R., Cihlar T. Intracellular activation of tenofovir alafenamide and the effect of viral and host protease inhibitors. Antimicrob. Agents Chemother. 2015;60:316–322. doi: 10.1128/AAC.01834-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisacchi G.S., Chao S.T., Bachard C., Daris J.P., Innamio S., Jacobs G.A., Kocy O., Lapointe P., Martel A., Merchant Z., Slusarchyk W.A., Sundeen J.E., Young M.G., Colonno R., Zahler R. BMS-200475, a novel carbocyclic 2′-deoxyguanosine analog with potent and selective anti-hepatitis B virus activity in vitro. Bioorg. Med. Chem. Lett. 1997;7:127–132. [Google Scholar]
- Bloch A., Leonard R.J., Nichol C.A. On the mode of action of 7-deaza-adenosine (tubercidin) Biochim. Biophys. Acta. 1967;138:10–25. doi: 10.1016/0005-2787(67)90581-3. [DOI] [PubMed] [Google Scholar]
- Bodey G.P., Freireich E.J., Monto R.W., Hewlett J.S. Cytosine arabinoside (NSC-63878) therapy for acute leukemia in adults. Cancer Chemother. Rep. 1969;53:59–66. [PubMed] [Google Scholar]
- Bohm H., Banner D., Bendels S., Kansy M., Kuhn B., Muller K., Obst-Sander U., Stahl M. Fluorine in medicinal chemistry. Chembiochem. 2004;5:637–643. doi: 10.1002/cbic.200301023. [DOI] [PubMed] [Google Scholar]
- Boutureira O., Matheu M.I., Díaz Y., Castillón S. Synthesis of C-Nucleosides. In: Merino P., editor. Chemical Synthesis of Nucleoside Analogues. 2013. [DOI] [Google Scholar]
- Boyd M.R., Bacon T.H., Sutton D., Cole M. Antiherpesvirus activity of 9-(4-hydroxy-3-hydroxy-methylbut-1-yl)guanine (BRL 39123) in cell culture. Antimicrob. Agents Chemother. 1987;31:1238–1242. doi: 10.1128/aac.31.8.1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd M.R., Safrin S., Kern E.R. Penciclovir: a review of its spectrum of activity, selectivity, and cross-resistance pattern. Antivir. Chem. Chemother. 1993;4:3–11. [Google Scholar]
- Boyer P.L., Sarafianos S.G., Clark P.K., Arnold E., Hughes S.H. Why do HIV-1 and HIV-2 use different pathways to develop AZT resistance? PLoS Pathog. 2006;2:e10. doi: 10.1371/journal.ppat.0020010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer P.L., Vu B.C., Ambrose Z., Julias J.G., Warnecke S., Liao C., Meier C., Marquez V.E., Hughes S.H. The nucleoside analogue D-carba T blocks HIV-1 reverse transcription. J. Med. Chem. 2009;52:5356–5364. doi: 10.1021/jm801176e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bras A.P., Sitar D.S., Aoki F.Y. Comparative bioavailability of acyclovir from oral valacyclovir and acyclovir in patients treated for recurrent genital herpes simplex virus infection. Can. J. Clin. Pharmacol. 2001;8:207–211. [PubMed] [Google Scholar]
- Brown K., Dixey M., Weymouth-Wilson A., Linclau B. The synthesis of gemcitabine. Carbohydr. Res. 2014;387:59–73. doi: 10.1016/j.carres.2014.01.024. [DOI] [PubMed] [Google Scholar]
- Brown K., Weymouth-Wilson A., Linclau B. A linear synthesis of gemcitabine. Carbohydr. Res. 2015;406:71–75. doi: 10.1016/j.carres.2015.01.001. [DOI] [PubMed] [Google Scholar]
- Bryant M.L., Bridges E.G., Placidi L., Faraj A., Loi A.G., Pierra C., Dukhan D., Gosselin G., Imbach J.L., Hernandez B., Juodawlkis A., Tennant B., Korba B., Cote P., Marion P., Cretton-Scott E., Schinazi R.F., Sommadossi J.P. Antiviral L-nucleosides specific for hepatitis B virus infection. Antimicrob. Agents Chemother. 2001;45:229–235. doi: 10.1128/AAC.45.1.229-235.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant M.L., Bridges E.G., Placidi L., Faraj A., Loi A.G., Pierra C., Dukhan D., Gosselin G., Imbach J.L., Hernandez B., Juodawlkis A., Tennant B., Korba B., Cote P., Cretton-Scott E., Schinazi R.F., Sommadossi J.P. Anti-HBV specific beta-L-2'-deoxynucleosides. Nucleosides Nucleotides Nucleic Acids. 2001;20:597–607. doi: 10.1081/NCN-100002336. [DOI] [PubMed] [Google Scholar]
- Burnette T.C., Harrington J.A., Reardon J.E., Merrill B.M., de Miranda P. Purification and characterization of a rat liver enzyme that hydrolyzes valaciclovir, the L-valyl ester prodrug of acyclovir. J. Biol. Chem. 1995;270:15827–15831. doi: 10.1074/jbc.270.26.15827. [DOI] [PubMed] [Google Scholar]
- Cappellacci L., Franchetti P., Petrelli R., Riccioni S., Vita P.N., Jayaram H., Grifantini M. Purine and pyrimidine nucleoside analogs of 3'-C-methyladenosine as antitumor agents. Collect Czechoslov Chem. Comm. 2006;71:1088–1098. [Google Scholar]
- Carter S.G., Kessler J.A., Rankin C.D. Activities of (-)-carbovir and 3'-azido-3'-deoxythymidine against human immunodeficiency virus in vitro. Antimicrob. Agents Chemother. 1990;34:1297–1300. doi: 10.1128/aac.34.6.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain C.E., Penzak S.R., Alfaro R.M., Wesley R., Daniels C.E., Hale D., Kirk A.D., Mannon R.B. Pharmacokinetics of low and maintenance dose valganciclovir in kidney transplant recipients. Am. J. Transpl. 2008;8:1297–1302. doi: 10.1111/j.1600-6143.2008.02220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C.N., Doong S.L., Zhou J.H., Beach J.W., Jeong L.S., Chu C.K., Tsai C.H., Cheng Y.C., Liotta D., Schinazi R. Deoxycytidine deaminase-resistant stereoisomer is the active form of (+/-)-2',3'-dideoxy-3'-thiacytidine in the inhibition of hepatitis B virus replication. J. Biol. Chem. 1992;267:13938–13942. [PubMed] [Google Scholar]
- Chang H.W., Wang H.C., Chen C.Y., Hung T.W., Hou M.F., Yuan S.S., Huang C.J., Tseng C.N. 5-azacytidine induces anoikis, inhibits mammosphere formation and reduces metalloproteinase 9 activity in MCF-7 human breast cancer cells. Molecules. 2014;19:3149–3159. doi: 10.3390/molecules19033149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman T.M., McGavin J.K., Noble S. Tenofovir disoproxil fumarate. Drugs. 2003;63:1597–1608. doi: 10.2165/00003495-200363150-00006. [DOI] [PubMed] [Google Scholar]
- Charette M., Gray M.W. Pseudouridine in RNA: what, where, how, and why. IUBMB Life. 2000;49:341–351. doi: 10.1080/152165400410182. [DOI] [PubMed] [Google Scholar]
- Chen Z., Ku T.C., Seley-Radtke K.L. Thiophene-expanded guanosine analogues of Gemcitabine. Bioorg. Med. Chem. Lett. 2015;25:4274–4276. doi: 10.1016/j.bmcl.2015.07.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y., George C., Comin M.J., Barchi J.J., Kim H.S., Jacobson K.A., Balzarini J., Mitsuya H., Boyer P.L., Hughes S.H., Marquez V.E. A conformationally locked analogue of the anti-HIV agent stavudine. An important correlation between pseudorotation and maximum amplitude. J. Med. Chem. 2003;46:3292–3299. doi: 10.1021/jm030116g. [DOI] [PubMed] [Google Scholar]
- Chu C.K., Ma T., Shanmuganathan K., Wang C., Xiang Y., Pai S.B., Yao G.Q., Sommadossi J.P., Cheng Y.C. Use of 2'-fluoro-5-methyl-beta-L-arabinofuranosyluracil as a novel antiviral agent for hepatitis B virus and Epstein-Barr virus. Antimicrob. Agents Chemother. 1995;39:979–981. doi: 10.1128/aac.39.4.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colla L., De Clercq E., Busson R., Vanderhaeghe H. Synthesis and antiviral activity of water-soluble esters of acyclovir [9-[(2-hydroxyethoxy)methyl]guanine] J. Med. Chem. 1983;26:602–604. doi: 10.1021/jm00358a029. [DOI] [PubMed] [Google Scholar]
- Collier A.C., Coombs R.W., Fischl M.A., Skolnik P.R., Northfelt D., Boutin P., Hooper C.J., Kaplan L.D., Volberding P.A., Davis L.G., Henrard D.R., Weller S., Corey L. Combination therapy with zidovudine and didanosine compared with zidovudine alone in HIV-1 infection. Ann. Intern Med. 1993;119:786–793. doi: 10.7326/0003-4819-119-8-199310150-00003. [DOI] [PubMed] [Google Scholar]
- Crnugelj M., Dukhan D., Barascut J.-L., Imbachb J.-L., Plavec J. How SCN anomeric effects and energetic preference across [SCCO] fragments steer conformational equilibria in 4'-thionucleosides. 1 H NMR and ab initio MO study. J. Chem. Soc. Perkin Trans. 2 Phys. Org. Chem. 2002;2:255–262. [Google Scholar]
- Curran M., Noble S. Valganciclovir. Drugs. 2001;61 doi: 10.2165/00003495-200161080-00013. 1145-50 ; discussion 1151–1152. [DOI] [PubMed] [Google Scholar]
- Daluge S.M., Good S.S., Faletto M.B., Miller W.H., St Clair M.H., Boone L.R., Tisdale M., Parry N.R., Reardon J.E., Dornsife R.E., Averett D.R., Krenitsky T.A. 1592U89, a novel carbocyclic nucleoside analog with potent, selective anti-human immunodeficiency virus activity. Antimicrob. Agents Chemother. 1997;41:1082–1093. doi: 10.1128/aac.41.5.1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dando T., Plosker G. Adefovir dipivoxil: a review of its use in chronic hepatitis B. Drugs. 2003;63:2215–2234. doi: 10.2165/00003495-200363200-00007. [DOI] [PubMed] [Google Scholar]
- Darnall K.R., Townsend L.B., Robins R.K. The structure of showdomycin, a novel carbon-linked nucleoside antibiotic related to uridine. Proc. Natl. Acad. Sci. U. S. A. 1967;57:548–553. doi: 10.1073/pnas.57.3.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S.R., Schneller S.W. The 5'-nor aristeromycin analogues of 5'-deoxy-5'-methylthioadenosine and 5'-deoxy-5'-thiophenyladenosine. Nucleosides Nucleotides Nucleic Acids. 2014;33:668–677. doi: 10.1080/15257770.2014.917671. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Specific targets for antiviral drugs. Biochem. J. 1982;205:1–13. doi: 10.1042/bj2050001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E. Antiviral and antimetabolic activities of neplanocins. Antimicrob. Agents Chemother. 1985;28:84–89. doi: 10.1128/aac.28.1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E. Strategies in the design of antiviral drugs. Nat. Rev. Drug Discov. 2002;1:13–25. doi: 10.1038/nrd703. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Clinical potential of the acyclic nucleoside phosphonates cidofovir, adefovir, and tenofovir in treatment of DNA virus and retrovirus infections. Clin. Microbiol. Rev. 2003;16:569–596. doi: 10.1128/CMR.16.4.569-596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E. Antivirals and antiviral strategies. Nat. Rev. Microbiol. 2004;2:704–720. doi: 10.1038/nrmicro975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E. Recent highlights in the development of new antiviral drugs. Curr. Opin. Microbiol. 2005;8:552–560. doi: 10.1016/j.mib.2005.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E. Antiviral drug discovery and development: where chemistry meets with biomedicine. Antivir. Res. 2005;67:56–75. doi: 10.1016/j.antiviral.2005.05.001. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Acyclic nucleoside phosphonates: past, present and future. Bridging chemistry to HIV, HBV, HCV, HPV, adeno-, herpes-, and poxvirus infections: the phosphonate bridge. Biochem. Pharmacol. 2007;73:911–922. doi: 10.1016/j.bcp.2006.09.014. [DOI] [PubMed] [Google Scholar]
- De Clercq E. The acyclic nucleoside phosphonates from inception to clinical use: historical perspective. Antivir. Res. 2007;75:1–13. doi: 10.1016/j.antiviral.2006.10.006. [DOI] [PubMed] [Google Scholar]
- De Clercq E. The history of antiretrovirals: key discoveries over the past 25 years. Rev. Med. Virol. 2009;19:287–299. doi: 10.1002/rmv.624. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV. Int. J. Antimicrob. Agents. 2009;33:307–320. doi: 10.1016/j.ijantimicag.2008.10.010. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Historical perspectives in the development of antiviral agents against poxviruses. Viruses. 2010;2:1322–1339. doi: 10.3390/v2061322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E. Milestones in the discovery of antiviral agents: nucleosides and nucleotides. Acta Pharm. Sin. B. 2012;2:535–548. [Google Scholar]
- De Clercq E. Antiviral agents. Adv. Pharmacol. 2013;67:317–345. [Google Scholar]
- De Clercq E. Selective anti-herpesvirus agents. Antivir. Chem. Chemother. 2013;23:93–101. doi: 10.3851/IMP2533. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Tenofovir alafenamide (TAF) as the successor of tenofovir disoproxil fumarate (TDF) Biochem. Pharmacol. 2016;119:1–7. doi: 10.1016/j.bcp.2016.04.015. [DOI] [PubMed] [Google Scholar]
- De Clercq E. C-nucleosides to Be revisited. J. Med. Chem. 2016;59:2301–2311. doi: 10.1021/acs.jmedchem.5b01157. [DOI] [PubMed] [Google Scholar]
- De Clercq E., Field H.J. Antiviral prodrugs - the development of successful prodrug strategies for antiviral chemotherapy. Br. J. Pharmacol. 2006;147:1–11. doi: 10.1038/sj.bjp.0706446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E., Holý A. Acyclic nucleoside phosphonates: a key class of antiviral drugs. Nat. Rev. Drug Discov. 2005;4:928–940. doi: 10.1038/nrd1877. [DOI] [PubMed] [Google Scholar]
- De Clercq E., Li G. Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev. 2016;29:695–747. doi: 10.1128/CMR.00102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E., Neyts J. Antiviral agents acting as DNA or RNA chain terminators. Handb. Exp. Pharmacol. 2009:53–84. doi: 10.1007/978-3-540-79086-0_3. [DOI] [PubMed] [Google Scholar]
- De Clercq E., Descamps J., De Somer P., Barr P.J., Jones A.S., Walker R.T. (E)-5-(2-Bromovinyl)-2'-deoxyuridine: a potent and selective anti-herpes agent. Proc. Natl. Acad. Sci. U. S. A. 1979;76:2947–2951. doi: 10.1073/pnas.76.6.2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E., Cools M., Balzarini J., Marquez V.E., Borcherding D.R., Borchardt R.T., Drach J.C., Kitaoka S., Konno T. Broad-spectrum antiviral activities of neplanocin A, 3-deazaneplanocin A, and their 5'-nor derivatives. Antimicrob. Agents Chemother. 1989;33:1291–1297. doi: 10.1128/aac.33.8.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E., Andrei G., Snoeck R., De Bolle L., Naesens L., Degrève B., Balzarini J., Zhang Y., Schols D., Leyssen P., Ying C., Neyts J. Acyclic/carbocyclic guanosine analogues as anti-herpesvirus agents. Nucleosides Nucleotides Nucleic Acids. 2001;20:271–285. doi: 10.1081/NCN-100002298. [DOI] [PubMed] [Google Scholar]
- De Clercq E., Andrei G., Balzarini J., Leyssen P., Naesens L., Neyts J., Pannecouque C., Snoeck R., Ying C., Hockova D., Holy A. Antiviral potential of a new generation of acyclic nucleoside phosphonates, the 6-[2-(phosphonomethoxy)alkoxy]-2,4-diaminopyrimidines. Nucleosides Nucleotides Nucleic Acids. 2005;24:331–341. doi: 10.1081/ncn-200059772. [DOI] [PubMed] [Google Scholar]
- de Miranda P., Blum M.R. Pharmacokinetics of acyclovir after intravenous and oral administration. J. Antimicrob. Chemother. 1983;12(Suppl. B):29–37. doi: 10.1093/jac/12.suppl_b.29. [DOI] [PubMed] [Google Scholar]
- de Sousa Cavalcante L., Monteiro G. Gemcitabine: metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. Eur. J. Pharmacol. 2014;741:8–16. doi: 10.1016/j.ejphar.2014.07.041. [DOI] [PubMed] [Google Scholar]
- de Vrueh R.L., Smith P.L., Lee C.P. Transport of L-valine-acyclovir via the oligopeptide transporter in the human intestinal cell line, Caco-2. J. Pharmacol. Exp. Ther. 1998;286:1166–1170. [PubMed] [Google Scholar]
- Degreef H., Group F.H.Z.C.S. Famciclovir, a new oral antiherpes drug: results of the first controlled clinical study demonstrating its efficacy and safety in the treatment of uncomplicated herpes zoster in immunocompetent patients. Int. J. Antimicrob. Agents. 1994;4:241–246. doi: 10.1016/0924-8579(94)90024-8. [DOI] [PubMed] [Google Scholar]
- Deval J. Antimicrobial strategies: inhibition of viral polymerases by 3'-hydroxyl nucleosides. Drugs. 2009;69:151–166. doi: 10.2165/00003495-200969020-00002. [DOI] [PubMed] [Google Scholar]
- Deval J., Powdrill M.H., D'Abramo C.M., Cellai L., Götte M. Pyrophosphorolytic excision of nonobligate chain terminators by hepatitis C virus NS5B polymerase. Antimicrob. Agents Chemother. 2007;51:2920–2928. doi: 10.1128/AAC.00186-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohme A.R.L., Cox E.H., Miller E. The preparation of the acyl and alkyl derivatives of resorcinol. J. Am. Chem. Soc. 1926;48:1688–1693. [Google Scholar]
- Doong S.L., Tsai C.H., Schinazi R.F., Liotta D.C., Cheng Y.C. Inhibition of the replication of hepatitis B virus in vitro by 2',3'-dideoxy-3'-thiacytidine and related analogues. Proc. Natl. Acad. Sci. U. S. A. 1991;88:8495–8499. doi: 10.1073/pnas.88.19.8495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J., Choi Y., Lee K., Chun B.K., Hong J.H., Chu C.K. A practical synthesis of L-FMAU from L-arabinose. Nucleosides Nucleotides. 1999;18:187–195. doi: 10.1080/15257779908043066. [DOI] [PubMed] [Google Scholar]
- Ebrahimi M., Rossi P., Rogers C., Harbison G.S. Dependence of 13C NMR chemical shifts on conformations of rna nucleosides and nucleotides. J. Magn. Reson. 2001;150:1–9. doi: 10.1006/jmre.2001.2314. [DOI] [PubMed] [Google Scholar]
- Elion G.B. Acyclovir: discovery, mechanism of action, and selectivity. J. Med. Virol. 1993;(Suppl. 1):2–6. doi: 10.1002/jmv.1890410503. [DOI] [PubMed] [Google Scholar]
- Elion G.B., Furman P.A., Fyfe J.A., de Miranda P., Beauchamp L., Schaeffer H.J. Selectivity of action of an antiherpetic agent, 9-(2-hydroxyethoxymethyl) guanine. Proc. Natl. Acad. Sci. U. S. A. 1977;74:5716–5720. doi: 10.1073/pnas.74.12.5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elzagheid M.I., Oivanen M., Walker R.T., Secrist J.A., III Kinetics for the acid-catalyzed hydrolysis of purine and cytosine 2′-Deoxy-4′-thionucleosides. Nucleosides Nucleotides. 1999;18:181–186. [Google Scholar]
- Engel R. Phosphonates as analogues of natural phosphates. Chem. Rev. 1977;77:349–367. [Google Scholar]
- Eyer L., Zouharová D., Širmarová J., Fojtíková M., Štefánik M., Haviernik J., Nencka R., de Clercq E., Růžek D. Antiviral activity of the adenosine analogue BCX4430 against West Nile virus and tick-borne flaviviruses. Antivir. Res. 2017;142:63–67. doi: 10.1016/j.antiviral.2017.03.012. [DOI] [PubMed] [Google Scholar]
- Faletto M.B., Miller W.H., Garvey E.P., St Clair M.H., Daluge S.M., Good S.S. Unique intracellular activation of the potent anti-human immunodeficiency virus agent 1592U89. Antimicrob. Agents Chemother. 1997;41:1099–1107. doi: 10.1128/aac.41.5.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan-Havard P., Nahata M.C., Brady M.T. Ganciclovir–a review of pharmacology, therapeutic efficacy and potential use for treatment of congenital cytomegalovirus infections. J. Clin. Pharm. Ther. 1989;14:329–340. doi: 10.1111/j.1365-2710.1989.tb00256.x. [DOI] [PubMed] [Google Scholar]
- Field H.J., De Clercq E. Antiviral drugs - a short history of their discovery and development. Microbiol. Today. 2004;31:58–61. [Google Scholar]
- Franchetti P., Cappellacci L., Pasqualini M., Petrelli R., Vita P., Jayaram H.N., Horvath Z., Szekeres T., Grifantini M. Antitumor activity of C-methyl-beta-D-ribofuranosyladenine nucleoside ribonucleotide reductase inhibitors. J. Med. Chem. 2005;48:4983–4989. doi: 10.1021/jm048944c. [DOI] [PubMed] [Google Scholar]
- Freeman S., Gardiner J.M. Acyclic nucleosides as antiviral compounds. Mol. Biotechnol. 1996;5:125–137. doi: 10.1007/BF02789061. [DOI] [PubMed] [Google Scholar]
- Fung H.B., Stone E.A., Piacenti F.J. Tenofovir disoproxil fumarate: a nucleotide reverse transcriptase inhibitor for the treatment of HIV infection. Clin. Ther. 2002;24:1515–1548. doi: 10.1016/s0149-2918(02)80058-3. [DOI] [PubMed] [Google Scholar]
- Gerzon K., Johnson I.S., Boder G.B., Cline J.C., Simpson P.J., Speth C., Leonard N.J., Laursen R.A. Biological activities of 3-isoadenosine. Biochim. Biophys. Acta. 1966;119:445–461. doi: 10.1016/0005-2787(66)90120-1. [DOI] [PubMed] [Google Scholar]
- Giles F.J., Cortes J.E., Baker S.D., Thomas D.A., O'Brien S., Smith T.L., Beran M., Bivins C., Jolivet J., Kantarjian H.M. Troxacitabine, a novel dioxolane nucleoside analog, has activity in patients with advanced leukemia. J. Clin. Oncol. 2001;19:762–771. doi: 10.1200/JCO.2001.19.3.762. [DOI] [PubMed] [Google Scholar]
- Groaz E., Herdewijn P. Uncommon three-, four-, and six-membered nucleosides. In: Merino P., editor. Chemical Synthesis of Nucleoside Analogues. John Wiley & Sons, Inc.; Hoboken, NJ, USA: 2013. pp. 605–654. [Google Scholar]
- Grove K.L., Cheng Y.C. Uptake and metabolism of the new anticancer compound beta-L-(-)-dioxolane-cytidine in human prostate carcinoma DU-145 cells. Cancer Res. 1996;56:4187–4191. [PubMed] [Google Scholar]
- Grove K.L., Guo X., Liu S.H., Gao Z., Chu C.K., Cheng Y.C. Anticancer activity of beta-L-dioxolane-cytidine, a novel nucleoside analogue with the unnatural L configuration. Cancer Res. 1995;55:3008–3011. [PubMed] [Google Scholar]
- Gryn J., Zeigler Z.R., Shadduck R.K., Lister J., Raymond J.M., Sbeitan I., Srodes C., Meisner D., Evans C. Treatment of myelodysplastic syndromes with 5-azacytidine. Leuk. Res. 2002;26:893–897. doi: 10.1016/s0145-2126(02)00028-0. [DOI] [PubMed] [Google Scholar]
- Gu Z., Gao Q., Fang H., Salomon H., Parniak M.A., Goldberg E., Cameron J., Wainberg M.A. Identification of a mutation at codon 65 in the IKKK motif of reverse transcriptase that encodes human immunodeficiency virus resistance to 2',3'-dideoxycytidine and 2',3'-dideoxy-3'-thiacytidine. Antimicrob. Agents Chemother. 1994;38:275–281. doi: 10.1128/aac.38.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudmundsson K.S., Freeman G.A., Drach J.C., Townsend L.B. Synthesis of fluorosugar analogues of 2,5,6-trichloro-1-(beta-D-ribofuranosyl)benzimidazole as antivirals with potentially increased glycosidic bond stability. J. Med. Chem. 2000;43:2473–2478. doi: 10.1021/jm990219s. [DOI] [PubMed] [Google Scholar]
- Gumina G., Song G.Y., Chu C.K. L-Nucleosides as chemotherapeutic agents. FEMS Microbiol. Lett. 2001;202:9–15. doi: 10.1111/j.1574-6968.2001.tb10773.x. [DOI] [PubMed] [Google Scholar]
- Gumina G., Chong Y., Chu C.K. L-nucleosides as chemotherapeutic agents. In: Peters G.J., editor. Deoxynucleoside Analogs in Cancer Therapy. Humana Press; Totowa, NJ: 2007. pp. 173–198. [Google Scholar]
- Hadziyannis S.J., Tassopoulos N.C., Heathcote E.J., Chang T.T., Kitis G., Rizzetto M., Marcellin P., Lim S.G., Goodman Z., Wulfsohn M.S., Xiong S., Fry J., Brosgart C.L., Group A.D.S. Adefovir dipivoxil for the treatment of hepatitis B e antigen-negative chronic hepatitis B. N. Engl. J. Med. 2003;348:800–807. doi: 10.1056/NEJMoa021812. [DOI] [PubMed] [Google Scholar]
- Hamann M., Hill R., Roggo S. Marine natural products. Key advances to the practical application of this resource in drug development. Chimia. 2007;61:313–321. [Google Scholar]
- Han S.H. Telbivudine: a new nucleoside analogue for the treatment of chronic hepatitis B. Expert Opin. Investig. Drugs. 2005;14:511–519. doi: 10.1517/13543784.14.4.511. [DOI] [PubMed] [Google Scholar]
- Harnden M.R., Jarvest R.L. An improved synthesis of the antiviral acyclonucleoside 9-(4-hydroxy-3-hydroxymethylbut-1-yl)guanine. Tetrahedron Lett. 1985;26:4265–4268. [Google Scholar]
- Harnden M.R., Jarvest R.L., Boyd M.R., Sutton D., Vere Hodge R.A. Prodrugs of the selective antiherpesvirus agent 9-[4-hydroxy-3-(hydroxymethyl)but-1-yl]guanine (BRL 39123) with improved gastrointestinal absorption properties. J. Med. Chem. 1989;32:1738–1743. doi: 10.1021/jm00128a012. [DOI] [PubMed] [Google Scholar]
- Harrigan P.R., Stone C., Griffin P., Nájera I., Bloor S., Kemp S., Tisdale M., Larder B. Resistance profile of the human immunodeficiency virus type 1 reverse transcriptase inhibitor abacavir (1592U89) after monotherapy and combination therapy. CNA2001 Investigative Group. J. Infect. Dis. 2000;181:912–920. doi: 10.1086/315317. [DOI] [PubMed] [Google Scholar]
- Hasobe M., McKee J.G., Borcherding D.R., Borchardt R.T. 9-(trans-2',trans-3'-Dihydroxycyclopent-4'-enyl)-adenine and -3-deazaadenine: analogs of neplanocin A which retain potent antiviral activity but exhibit reduced cytotoxicity. Antimicrob. Agents Chemother. 1987;31:1849–1851. doi: 10.1128/aac.31.11.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M., Yaginuma S., Muto N., Tsujino M. Structures of neplanocins, new antitumor antibiotics. Nucleic Acids Symp. Ser. 1980:s65–s67. [PubMed] [Google Scholar]
- Hayashi M., Yaginuma S., Yoshioka H., Nakatsu K. Studies on neplanocin A, new antitumor antibiotic. II. Structure determination. J. Antibiot. Tokyo. 1981;34:675–680. doi: 10.7164/antibiotics.34.675. [DOI] [PubMed] [Google Scholar]
- Hayashi S., Norbeck D.W., Rosenbrook W., Fine R.L., Matsukura M., Plattner J.J., Broder S., Mitsuya H. Cyclobut-A and cyclobut-G, carbocyclic oxetanocin analogs that inhibit the replication of human immunodeficiency virus in T cells and monocytes and macrophages in vitro. Antimicrob. Agents Chemother. 1990;34:287–294. doi: 10.1128/aac.34.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde V.R., Seley K.L., Chen X., Schneller S.W. The synthesis of carbocyclic 5′-nor thymidine and an isomer as oligonucleotide monomers. Nucleosides Nucleotides. 1999;18:1905–1910. [Google Scholar]
- Hegde V.R., Seley K.L., Schneller S.W. Carbocyclic 5'-noruridine. Nucleosides Nucleotides Nucleic Acids. 2000;19:269–273. doi: 10.1080/15257770008033008. [DOI] [PubMed] [Google Scholar]
- Herdewijn P., De Clercq E. The cyclohexene ring as bioisostere of a furanose ring: synthesis and antiviral activity of cyclohexenyl nucleosides. Bioorg. Med. Chem. Lett. 2001;11:1591–1597. doi: 10.1016/s0960-894x(01)00270-0. [DOI] [PubMed] [Google Scholar]
- Hernández P., Olivera P., Dueñas-Gonzalez A., Pérez-Pastenes M.A., Zárate A., Maldonado V., Meléndez-Zajgla J. Gemcitabine activity in cervical cancer cell lines. Cancer Chemother. Pharmacol. 2001;48:488–492. doi: 10.1007/s002800100370. [DOI] [PubMed] [Google Scholar]
- Hertel L.W., Boder G.B., Kroin J.S., Rinzel S.M., Poore G.A., Todd G.C., Grindey G.B. Evaluation of the antitumor activity of gemcitabine (2',2'-difluoro-2'-deoxycytidine) Cancer Res. 1990;50:4417–4422. [PubMed] [Google Scholar]
- Herzig R.H., Wolff S.N., Lazarus H.M., Phillips G.L., Karanes C., Herzig G.P. High-dose cytosine arabinoside therapy for refractory leukemia. Blood. 1983;62:361–369. [PubMed] [Google Scholar]
- Hewlett G., Hallenberger S., Rübsamen-Waigmann H. Antivirals against DNA viruses (hepatitis B and the herpes viruses) Curr. Opin. Pharmacol. 2004;4:453–464. doi: 10.1016/j.coph.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Hiddemann W. Cytosine arabinoside in the treatment of acute myeloid leukemia: the role and place of high-dose regimens. Ann. Hematol. 1991;62:119–128. doi: 10.1007/BF01702925. [DOI] [PubMed] [Google Scholar]
- Holý A. Antiviral acyclic nucleoside phosphonates structure activity studies. Antivir. Res. 2006;71:248–253. doi: 10.1016/j.antiviral.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Hóly A., Rosenberg I. Acyclic nucleotide analogs .3. Synthesis of 9-(2-Phosphonylmethoxyethyl)Adenine and related-compounds. Collect Czechoslov Chem. Comm. 1987;52:2801–2809. [Google Scholar]
- Horwitz J.P., Chua J., Noel M. Nucleosides. V. The monomesylates of 1-(2'-Deoxy-β-D-lyxofuranosyl)thymine. J. Org. Chem. 1964;29:2076–2078. [Google Scholar]
- Horwitz J.R., Chua J., Da Rooge M.A., Noel M., Klundt I.L. Nucleosides. IX. The formation of 2',2'-unsaturated pyrimidine nucleosides via a novel beta-elimination reaction. J. Org. Chem. 1966;31:205–211. doi: 10.1021/jo01339a045. [DOI] [PubMed] [Google Scholar]
- Horwitz J.P., Chua J., Noel M., Donatti J.T. Nucleosides. XI. 2',3'-dideoxycytidine. J. Org. Chem. 1967;32:817–818. doi: 10.1021/jo01278a070. [DOI] [PubMed] [Google Scholar]
- Hostetler K.Y., Richman D.D., Sridhar C.N., Felgner P.L., Felgner J., Ricci J., Gardner M.F., Selleseth D.W., Ellis M.N. Phosphatidylazidothymidine and phosphatidyl-ddC: assessment of uptake in mouse lymphoid tissues and antiviral activities in human immunodeficiency virus-infected cells and in Rauscher leukemia virus-infected mice. Antimicrob. Agents Chemother. 1994;38:2792–2797. doi: 10.1128/aac.38.12.2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P., Chubb S., Hertel L.W., Grindey G.B., Plunkett W. Action of 2',2'-difluorodeoxycytidine on DNA synthesis. Cancer Res. 1991;51:6110–6117. [PubMed] [Google Scholar]
- Huang H., Chopra R., Verdine G.L., Harrison S.C. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science. 1998;282:1669–1675. doi: 10.1126/science.282.5394.1669. [DOI] [PubMed] [Google Scholar]
- Hwang T.L., Yung W.K., Estey E.H., Fields W.S. Central nervous system toxicity with high-dose Ara-C. Neurology. 1985;35:1475–1479. doi: 10.1212/wnl.35.10.1475. [DOI] [PubMed] [Google Scholar]
- Ikeda H., Fernandez R., Wilk A., Barchi J.J., Huang X., Marquez V.E. The effect of two antipodal fluorine-induced sugar puckers on the conformation and stability of the Dickerson-Drew dodecamer duplex [d(CGCGAATTCGCG)]2. Nucleic Acids Res. 1998;26:2237–2244. doi: 10.1093/nar/26.9.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue N., Kaga D., Minakawa N., Matsuda A. Practical synthesis of 2'-deoxy-4'-thioribonucleosides: substrates for the synthesis of 4'-thioDNA. J. Org. Chem. 2005;70:8597–8600. doi: 10.1021/jo051248f. [DOI] [PubMed] [Google Scholar]
- Isono K. Nucleoside antibiotics: structure, biological activity, and biosynthesis. J. Antibiot. Tokyo. 1988;41:1711–1739. doi: 10.7164/antibiotics.41.1711. [DOI] [PubMed] [Google Scholar]
- Itoh T., Kitano S., Mizuno Y. Synthetic studies of potential antimetabolites. XIII. Synthesis of 7-amino-3-β-d-ribofuranosyl-3H-imidazo 4,5-b pyridine (1-deazaadenosine) and related nucleosides. J. Heterocycl. Chem. 1972;9:465–470. [Google Scholar]
- Jarvis B., Faulds D. Lamivudine. A review of its therapeutic potential in chronic hepatitis B. Drugs. 1999;58:101–141. doi: 10.2165/00003495-199958010-00015. [DOI] [PubMed] [Google Scholar]
- Jordheim L.P., Durantel D., Zoulim F., Dumontet C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013;12:447–464. doi: 10.1038/nrd4010. [DOI] [PubMed] [Google Scholar]
- Julander J.G., Bantia S., Taubenheim B.R., Minning D.M., Kotian P., Morrey J.D., Smee D.F., Sheridan W.P., Babu Y.S. BCX4430, a novel nucleoside analog, effectively treats yellow fever in a Hamster model. Antimicrob. Agents Chemother. 2014;58:6607–6614. doi: 10.1128/AAC.03368-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julander J.G., Siddharthan V., Evans J., Taylor R., Tolbert K., Apuli C., Stewart J., Collins P., Gebre M., Neilson S., Van Wettere A., Lee Y.M., Sheridan W.P., Morrey J.D., Babu Y.S. Efficacy of the broad-spectrum antiviral compound BCX4430 against Zika virus in cell culture and in a mouse model. Antivir. Res. 2017;137:14–22. doi: 10.1016/j.antiviral.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadhim S.A., Bowlin T.L., Waud W.R., Angers E.G., Bibeau L., DeMuys J.M., Bednarski K., Cimpoia A., Attardo G. Potent antitumor activity of a novel nucleoside analogue, BCH-4556 (beta-L-dioxolane-cytidine), in human renal cell carcinoma xenograft tumor models. Cancer Res. 1997;57:4803–4810. [PubMed] [Google Scholar]
- Kálmán A., Yoritsánszky T., Béres J., Sági G. Crystal and molecular structure of (+)-Carba-Thymidine, C11 H10 N2 O6. Nucleosides Nucleotides. 1989;9:235–243. [Google Scholar]
- Kaminsky R., Zweygarth E., De Clercq E. Antitrypanosomal activity of phosphonylmethoxyalkylpurines. J. Parasitol. 1994;80:1026–1030. [PubMed] [Google Scholar]
- Kaminsky R., Schmid C., Grether Y., Holý A., DeClercq E., Naesens L., Brun R. (S)-9-(3-hydroxy-2-phosphonylmethoxypropyl)adenine [(S)-HPMPA]: a purine analogue with trypanocidal activity in vitro and in vivo. Trop. Med. Int. Health. 1996;1:255–263. doi: 10.1111/j.1365-3156.1996.tb00036.x. [DOI] [PubMed] [Google Scholar]
- Kaufman H.E., Heidelberger C. Therapeutic antiviral action of 5-trifluoromethyl-2'-deoxyuridine in herpes simplex keratitis. Science. 1964;145:585–586. doi: 10.1126/science.145.3632.585. [DOI] [PubMed] [Google Scholar]
- Kerr S.G., Anderson K.S. Pre-steady-state kinetic characterization of wild type and 3'-azido-3'-deoxythymidine (AZT) resistant human immunodeficiency virus type 1 reverse transcriptase: implication of RNA directed DNA polymerization in the mechanism of AZT resistance. Biochemistry. 1997;36:14064–14070. doi: 10.1021/bi9713862. [DOI] [PubMed] [Google Scholar]
- Kicska G.A., Long L., Hörig H., Fairchild C., Tyler P.C., Furneaux R.H., Schramm V.L., Kaufman H.L. Immucillin H, a powerful transition-state analog inhibitor of purine nucleoside phosphorylase, selectively inhibits human T lymphocytes. Proc. Natl. Acad. Sci. U. S. A. 2001;98:4593–4598. doi: 10.1073/pnas.071050798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.O., Ahn S.K., Alves A.J., Beach J.W., Jeong L.S., Choi B.G., Van Roey P., Schinazi R.F., Chu C.K. Asymmetric synthesis of 1,3-dioxolane-pyrimidine nucleosides and their anti-HIV activity. J. Med. Chem. 1992;35:1987–1995. doi: 10.1021/jm00089a007. [DOI] [PubMed] [Google Scholar]
- Kim H.O., Shanmuganathan K., Alves A.J., Jeong L.S., Beach J.W., Schinazi R.F., Chang C.N., Cheng Y.C., Chu C.K. Potent anti-HIV and anti-HBV activities of (−)-L-β-dioxolane-C and (+)-L-β-dioxolane-T and their asymmetric syntheses. Tetrahedron Lett. 1992;33:6899–6902. [Google Scholar]
- Kim H.O., Schinazi R.F., Nampalli S., Shanmuganathan K., Cannon D.L., Alves A.J., Jeong L.S., Beach J.W., Chu C.K. 1,3-dioxolanylpurine nucleosides (2R,4R) and (2R,4S) with selective anti-HIV-1 activity in human lymphocytes. J. Med. Chem. 1993;36:30–37. doi: 10.1021/jm00053a004. [DOI] [PubMed] [Google Scholar]
- Kim J.W., Park S.H., Louie S.G. Telbivudine: a novel nucleoside analog for chronic hepatitis B. Ann. Pharmacother. 2006;40:472–478. doi: 10.1345/aph.1G027. [DOI] [PubMed] [Google Scholar]
- Kimberlin D.W. Acyclovir derivatives and other new antiviral agents. Semin. Pediatr. Infect. Dis. 2001;12:224–234. [Google Scholar]
- King D.H. History, pharmacokinetics, and pharmacology of acyclovir. J. Am. Acad. Dermatol. 1988;18:176–179. doi: 10.1016/s0190-9622(88)70022-5. [DOI] [PubMed] [Google Scholar]
- Kirk K. Fluorine in medicinal chemistry: recent therapeutic applications of fluorinated small molecules. J. Fluor. Chem. 2006;127:1013–1029. [Google Scholar]
- Komatsu T.E., Pikis A., Naeger L.K., Harrington P.R. Resistance of human cytomegalovirus to ganciclovir/valganciclovir: a comprehensive review of putative resistance pathways. Antivir. Res. 2014;101:12–25. doi: 10.1016/j.antiviral.2013.10.011. [DOI] [PubMed] [Google Scholar]
- Korycka A., Błoński J.Z., Robak T. Forodesine (BCX-1777, Immucillin H)–a new purine nucleoside analogue: mechanism of action and potential clinical application. Mini Rev. Med. Chem. 2007;7:976–983. doi: 10.2174/138955707781662636. [DOI] [PubMed] [Google Scholar]
- Krueger A.T., Lu H., Lee A.H., Kool E.T. Synthesis and properties of size-expanded DNAs: toward designed, functional genetic systems. Acc. Chem. Res. 2007;40:141–150. doi: 10.1021/ar068200o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Wilson S.R., Leonard N.J. Structure of 3-isoadenosine. Acta Crystallogr. C. 1988;44(Pt 3):508–510. doi: 10.1107/s0108270187011144. [DOI] [PubMed] [Google Scholar]
- Lai C.L., Leung N., Teo E.K., Tong M., Wong F., Hann H.W., Han S., Poynard T., Myers M., Chao G., Lloyd D., Brown N.A., Group T.P.I.I. A 1-year trial of telbivudine, lamivudine, and the combination in patients with hepatitis B e antigen-positive chronic hepatitis B. Gastroenterology. 2005;129:528–536. doi: 10.1016/j.gastro.2005.05.053. [DOI] [PubMed] [Google Scholar]
- Lai C.L., Gane E., Liaw Y.F., Hsu C.W., Thongsawat S., Wang Y., Chen Y., Heathcote E.J., Rasenack J., Bzowej N., Naoumov N.V., Di Bisceglie A.M., Zeuzem S., Moon Y.M., Goodman Z., Chao G., Constance B.F., Brown N.A., Group G.S. Telbivudine versus lamivudine in patients with chronic hepatitis B. N. Engl. J. Med. 2007;357:2576–2588. doi: 10.1056/NEJMoa066422. [DOI] [PubMed] [Google Scholar]
- Lalezari J.P., Stagg R.J., Kuppermann B.D., Holland G.N., Kramer F., Ives D.V., Youle M., Robinson M.R., Drew W.L., Jaffe H.S. Intravenous cidofovir for peripheral cytomegalovirus retinitis in patients with AIDS. A randomized, controlled trial. Ann. Intern Med. 1997;126:257–263. doi: 10.7326/0003-4819-126-4-199702150-00001. [DOI] [PubMed] [Google Scholar]
- Langley D.R., Walsh A.W., Baldick C.J., Eggers B.J., Rose R.E., Levine S.M., Kapur A.J., Colonno R.J., Tenney D.J. Inhibition of hepatitis B virus polymerase by entecavir. J. Virol. 2007;81:3992–4001. doi: 10.1128/JVI.02395-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapponi M.J., Rivero C.W., Zinni M.A., Britos C.N., Trelles J.A. New developments in nucleoside analogues biosynthesis: a review. J. Mol. Catal. B Enzym. 2016;133:218–233. [Google Scholar]
- Larder B.A., Kemp S.D. Multiple mutations in HIV-1 reverse transcriptase confer high-level resistance to zidovudine (AZT) Science. 1989;246:1155–1158. doi: 10.1126/science.2479983. [DOI] [PubMed] [Google Scholar]
- Larder B.A., Kellam P., Kemp S.D. Convergent combination therapy can select viable multidrug-resistant HIV-1 in vitro. Nature. 1993;365:451–453. doi: 10.1038/365451a0. [DOI] [PubMed] [Google Scholar]
- Lauter C.B., Bailey E.J., Lerner A.M. Assessment of cytosine arabinoside as an antiviral agent in humans. Antimicrob. Agents Chemother. 1974;6:598–602. doi: 10.1128/aac.6.5.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A.H., Kool E.T. Exploring the limits of DNA size: naphtho-homologated DNA bases and pairs. J. Am. Chem. Soc. 2006;128:9219–9230. doi: 10.1021/ja0619004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W.W., Benitez A., Goodman L., Baker B.R. Potential anticancer agents. XL. Synthesis of the banomer of 9-(d-arabinofuranosyl)-adenine. J. Am. Chem. Soc. 1960;82:2648–2649. [Google Scholar]
- Lee W.W., Benitez A., Goodman L., Baker B.R. Potential anticancer Agents.1 Xl. Synthesis of the β-anomer of 9-(d-arabinofuranosyl)-adenine. J. Am. Chem. Soc. 1960;82:2648–2649. [Google Scholar]
- Lee C.K., Rowinsky E.K., Li J., Giles F., Moore M.J., Hidalgo M., Capparelli E., Jolivet J., Baker S.D. Population pharmacokinetics of troxacitabine, a novel dioxolane nucleoside analogue. Clin. Cancer Res. 2006;12:2158–2165. doi: 10.1158/1078-0432.CCR-05-2249. [DOI] [PubMed] [Google Scholar]
- Leonard N.J. Dimensional probes of enzyme-coenzyme binding-sites. Acc. Chem. Res. 1982;15:128–135. [Google Scholar]
- Leonard N.J., Laursen R.A. The synthesis of 3-β-D-Ribofuranosyladenine. J. Am. Chem. Soc. 1963;85:2026–2028. [Google Scholar]
- Leonard N.J., Laursen R.A. Synthesis of 3-β-D-Ribofuranosyladenine and (3-β-d-ribofuranosyladenine)-5'-phosphate*. Biochemistry. 1965;4:354–365. [Google Scholar]
- Leonard N.L., Morrice A.G., Sprecker M.A. Linear benzoadenine. A stretched-out analog of adenine. J. Org. Chem. 1975;40:356–363. doi: 10.1021/jo00891a021. [DOI] [PubMed] [Google Scholar]
- Leonard N.J., Sprecker M.A., Morrice A.G. Defined dimensional changes in enzyme substrates and cofactors. Synthesis of lin-benzoadenosine and enzymatic evaluation of derivatives of the benzopurines. J. Am. Chem. Soc. 1976;98:3987–3994. doi: 10.1021/ja00429a040. [DOI] [PubMed] [Google Scholar]
- Leonard N.J., Scopes D.I., VanDerLijn P., Barrio J.R. Dimensional probes of the enzyme binding sites of adenine nucleotides. Biological effects of widening the adenine ring by 2.4 A. Biochemistry. 1978;17:3677–3685. doi: 10.1021/bi00611a001. [DOI] [PubMed] [Google Scholar]
- Leonard N.J., Cruickshank K.A., Groziak M.P., Clauson G.L., Devadas B. Relatives of Watson-Crick DNA, RNA cross sections. Ann. N. Y. Acad. Sci. 1986;471:255–265. doi: 10.1111/j.1749-6632.1986.tb48041.x. [DOI] [PubMed] [Google Scholar]
- Li X., Carmichael E., Feng M., King I., Doyle T.W., Chen S.H. Bis-S-acyl-2-thioethyl (SATE)-bearing monophosphate prodrug of beta-L-FD4C as potent anti-HBV agent. Bioorg. Med. Chem. Lett. 1998;8:57–62. doi: 10.1016/s0960-894x(97)10178-0. [DOI] [PubMed] [Google Scholar]
- Liotta D.C., Painter G.R. Discovery and development of the anti-human immunodeficiency virus drug, emtricitabine (emtriva, FTC) Acc. Chem. Res. 2016 doi: 10.1021/acs.accounts.6b00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.C., Luo M.Z., Mozdziesz D.E., Lin T.S., Dutschman G.E., Gullen E.A., Cheng Y.C., Sartorelli A.C. Synthesis of halogen-substituted 3-deazaadenosine and 3-deazaguanosine analogues as potential antitumor/antiviral agents. Nucleosides Nucleotides Nucleic Acids. 2001;20:1975–2000. doi: 10.1081/NCN-100108327. [DOI] [PubMed] [Google Scholar]
- Liu P., Sharon A., Chu C.K. Fluorinated nucleosides: synthesis and biological implication. J. Fluor Chem. 2008;129:743–766. doi: 10.1016/j.jfluchem.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longley D.B., Harkin D.P., Johnston P.G. 5-fluorouracil: mechanisms of action and clinical strategies. Nat. Rev. Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- Luoni G., McGuigan C., Andrei G., Snoeck R., De Clercq E., Balzarini J. Bicyclic nucleoside inhibitors of Varicella-Zoster virus: the effect of branching in the p-alkylphenyl side chain. Bioorg. Med. Chem. Lett. 2005;15:3791–3796. doi: 10.1016/j.bmcl.2005.05.071. [DOI] [PubMed] [Google Scholar]
- Lynch S.R., Liu H., Gao J., Kool E.T. Toward a designed, functioning genetic system with expanded-size base pairs: solution structure of the eight-base xDNA double helix. J. Am. Chem. Soc. 2006;128:14704–14711. doi: 10.1021/ja065606n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macchi B., Romeo G., Chiacchio U., Frezza C., Giofrè S.V., Marino-Merlo F., Mastino A. Phosphonated nucleoside analogues as antiviral agents. In: Diederich W.E., Steuber H., editors. Therapy of Viral Infections. Springer Berlin Heidelberg; Berlin, Heidelberg: 2015. pp. 53–91. [Google Scholar]
- Maffioli S.I., Zhang Y., Degen D., Carzaniga T., Del Gatto G., Serina S., Monciardini P., Mazzetti C., Guglierame P., Candiani G., Chiriac A.I., Facchetti G., Kaltofen P., Sahl H.G., Dehò G., Donadio S., Ebright R.H. Antibacterial nucleoside-analog inhibitor of bacterial RNA polymerase. Cell. 2017;169:1240–1248. doi: 10.1016/j.cell.2017.05.042. e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahler M., Reichardt B., Hartjen P., van Lunzen J., Meier C. Stereoselective synthesis of D- and L-carbocyclic nucleosides by enzymatically catalyzed kinetic resolution. Chemistry. 2012;18:11046–11062. doi: 10.1002/chem.201200733. [DOI] [PubMed] [Google Scholar]
- Marquez V.E. Carbocyclic nucleosides. Adv. Antivir. Drug Des. 1996;2:89–146. [Google Scholar]
- Marquez V.E., Lim M.I. Carbocyclic nucleosides. Med. Res. Rev. 1986;6:1–40. doi: 10.1002/med.2610060102. [DOI] [PubMed] [Google Scholar]
- Marquez V.E., Siddiqui M.A., Ezzitouni A., Russ P., Wang J., Wagner R.W., Matteucci M.D. Nucleosides with a twist. Can fixed forms of sugar ring pucker influence biological activity in nucleosides and oligonucleotides? J. Med. Chem. 1996;39:3739–3747. doi: 10.1021/jm960306+. [DOI] [PubMed] [Google Scholar]
- Martin J.C., Tippie M.A., McGee D.P., Verheyden J.P. Synthesis and antiviral activity of various esters of 9-[(1,3-dihydroxy-2-propoxy)methyl]guanine. J. Pharm. Sci. 1987;76:180–184. doi: 10.1002/jps.2600760221. [DOI] [PubMed] [Google Scholar]
- Martin D.F., Sierra-Madero J., Walmsley S., Wolitz R.A., Macey K., Georgiou P., Robinson C.A., Stempien M.J., Group V.S. A controlled trial of valganciclovir as induction therapy for cytomegalovirus retinitis. N. Engl. J. Med. 2002;346:1119–1126. doi: 10.1056/NEJMoa011759. [DOI] [PubMed] [Google Scholar]
- Martin J.C., Hitchcock M.J., De Clercq E., Prusoff W.H. Early nucleoside reverse transcriptase inhibitors for the treatment of HIV: a brief history of stavudine (D4T) and its comparison with other dideoxynucleosides. Antivir. Res. 2010;85:34–38. doi: 10.1016/j.antiviral.2009.10.006. [DOI] [PubMed] [Google Scholar]
- Mascolini M. Looking past two drugs, looking past three? J. Int. Assoc. Physicians AIDS Care. 1997;3:9–15. 18-22, 46. [PubMed] [Google Scholar]
- Mascolini M. 2010. Immunologic Markers to Evaluate Immune-based Therapies in HIV Disease Meeting Report. [Google Scholar]
- Mathé C., Gosselin G. L-nucleoside enantiomers as antivirals drugs: a mini-review. Antivir. Res. 2006;71:276–281. doi: 10.1016/j.antiviral.2006.04.017. [DOI] [PubMed] [Google Scholar]
- Matyugina E.S., Khandazhinskaya A.P., Kochetkov S.N. Carbocyclic nucleoside analogues: classification, target enzymes, mechanisms of action and synthesis. Russ. Chem. Rev. 2012;81:729–746. [Google Scholar]
- Maudgal P.C., De Clercq K., Descamps J., Missotten L. Topical treatment of experimental herpes simplex keratouveitis with 2'-O-glycylacyclovir. A water-soluble ester of acyclovir. Arch. Ophthalmol. 1984;102:140–142. doi: 10.1001/archopht.1984.01040030118049. [DOI] [PubMed] [Google Scholar]
- McGuigan C., Yarnold C.J., Jones G., Velázquez S., Barucki H., Brancale A., Andrei G., Snoeck R., De Clercq E., Balzarini J. Potent and selective inhibition of varicella-zoster virus (VZV) by nucleoside analogues with an unusual bicyclic base. J. Med. Chem. 1999;42:4479–4484. doi: 10.1021/jm990346o. [DOI] [PubMed] [Google Scholar]
- McMahon M.A., Jilek B.L., Brennan T.P., Shen L., Zhou Y., Wind-Rotolo M., Xing S., Bhat S., Hale B., Hegarty R., Chong C.R., Liu J.O., Siliciano R.F., Thio C.L. The HBV drug entecavir - effects on HIV-1 replication and resistance. N. Engl. J. Med. 2007;356:2614–2621. doi: 10.1056/NEJMoa067710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon M.A., Siliciano J.D., Lai J., Liu J.O., Stivers J.T., Siliciano R.F., Kohli R.M. The antiherpetic drug acyclovir inhibits HIV replication and selects the V75I reverse transcriptase multidrug resistance mutation. J. Biol. Chem. 2008;283:31289–31293. doi: 10.1074/jbc.C800188200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng T., Fischl M.A., Boota A.M., Spector S.A., Bennett D., Bassiakos Y., Lai S.H., Wright B., Richman D.D. Combination therapy with zidovudine and dideoxycytidine in patients with advanced human immunodeficiency virus infection: a phase i/ii study. Ann. Intern Med. 1992;116:13–20. doi: 10.7326/0003-4819-116-1-13. [DOI] [PubMed] [Google Scholar]
- Mertz G.J., Loveless M.O., Levin M.J., Kraus S.J., Fowler S.L., Goade D., Tyring S.K. Oral famciclovir for suppression of recurrent genital herpes simplex virus infection in women. A multicenter, double-blind, placebo-controlled trial. Collaborative Famciclovir Genital Herpes Research Group. Arch. Intern Med. 1997;157:343–349. [PubMed] [Google Scholar]
- Messini L., Tiwari K.N., Montgomery J.A., Secrist J.A. Synthesis and biological activity of 4'-thio-2'-deoxy purine nucleosides. Nucleosides Nucleotides. 1999;18:683–685. doi: 10.1080/15257779908041540. [DOI] [PubMed] [Google Scholar]
- Meyer P.R., Matsuura S.E., Mian A.M., So A.G., Scott W.A. A mechanism of AZT resistance: an increase in nucleotide-dependent primer unblocking by mutant HIV-1 reverse transcriptase. Mol. Cell. 1999;4:35–43. doi: 10.1016/s1097-2765(00)80185-9. [DOI] [PubMed] [Google Scholar]
- Mian A.M., Khwaja T.A. Synthesis and antitumor activity of 2-deoxyribofuranosides of 3-deazaguanine. J. Med. Chem. 1983;26:286–291. doi: 10.1021/jm00356a034. [DOI] [PubMed] [Google Scholar]
- Mikhailov S.N., Beigelman L.N., Gurskaya G.V., Padyukova N.S., Yakovlev G.I., Karpeisky M.Y. Synthesis and properties of 3′-C-methylnucleosides and their phosphoric esters. Carb Res. 1983;124:75–96. [Google Scholar]
- Mitsuya H., Broder S. Inhibition of the in vitro infectivity and cytopathic effect of human T-lymphotrophic virus type III/lymphadenopathy-associated virus (HTLV-III/LAV) by 2',3'-dideoxynucleosides. Proc. Natl. Acad. Sci. U. S. A. 1986;83:1911–1915. doi: 10.1073/pnas.83.6.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momparler R.L. A model for the chemotherapy of acute leukemia with 1-beta-D-arabinofuranosylcytosine. Cancer Res. 1974;34:1775–1787. [PubMed] [Google Scholar]
- Montgomery J.A., Shortnacy A.T., Thomas H.J. Analogs of 5'-deoxy-5'-(methylthio)adenosine. J. Med. Chem. 1974;17:1197–1207. doi: 10.1021/jm00257a014. [DOI] [PubMed] [Google Scholar]
- Montgomery J.A., Shortnacy A.T., Secrist J.A. Synthesis and biological evaluation of 2-fluoro-8-azaadenosine and related compounds. J. Med. Chem. 1983;26:1483–1489. doi: 10.1021/jm00364a023. [DOI] [PubMed] [Google Scholar]
- Nair V., Jahnke T.S. Antiviral activities of isometric dideoxynucleosides of D- and L-related stereochemistry. Antimicrob. Agents Chemother. 1995;39:1017–1029. doi: 10.1128/aac.39.5.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair V., Nuesca Z.M. Isodideoxynucleosides: a conceptually new class of nucleoside antiviral agents. J. Am. Chem. Soc. 1992;114:7951–7953. [Google Scholar]
- Nair V., St Clair M.H., Reardon J.E., Krasny H.C., Hazen R.J., Paff M.T., Boone L.R., Tisdale M., Najera I., Dornsife R.E. Antiviral, metabolic, and pharmacokinetic properties of the isomeric dideoxynucleoside 4(S)-(6-amino-9H-purin-9-yl)tetrahydro-2(S)-furanmethanol. Antimicrob. Agents Chemother. 1995;39:1993–1999. doi: 10.1128/aac.39.9.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura H., Mayama M., Komatsu Y., Kato H., Shimaoka N., Tanaka Y. Showdomycin, a new antibiotic from a Streptomyces SP. J. Antibiot. Tokyo. 1964;17:148–155. [PubMed] [Google Scholar]
- Niu G., Tan H. Nucleoside antibiotics: biosynthesis, regulation, and biotechnology. Trends Microbiol. 2015;23:110–119. doi: 10.1016/j.tim.2014.10.007. [DOI] [PubMed] [Google Scholar]
- Norbeck D.W., Kern E., Hayashi S., Rosenbrook W., Sham H., Herrin T., Plattner J.J., Erickson J., Clement J., Swanson R. Cyclobut-A and cyclobut-G: broad-spectrum antiviral agents with potential utility for the therapy of AIDS. J. Med. Chem. 1990;33:1281–1285. doi: 10.1021/jm00167a002. [DOI] [PubMed] [Google Scholar]
- Oettle H. Progress in the knowledge and treatment of advanced pancreatic cancer: from benchside to bedside. Cancer Treat. Rev. 2014;40:1039–1047. doi: 10.1016/j.ctrv.2014.07.003. [DOI] [PubMed] [Google Scholar]
- Olsen D.B., Eldrup A.B., Bartholomew L., Bhat B., Bosserman M.R., Ceccacci A., Colwell L.F., Fay J.F., Flores O.A., Getty K.L., Grobler J.A., LaFemina R.L., Markel E.J., Migliaccio G., Prhavc M., Stahlhut M.W., Tomassini J.E., MacCoss M., Hazuda D.J., Carroll S.S. A 7-deaza-adenosine analog is a potent and selective inhibitor of hepatitis C virus replication with excellent pharmacokinetic properties. Antimicrob. Agents Chemother. 2004;48:3944–3953. doi: 10.1128/AAC.48.10.3944-3953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlov A.A., Drenichev M.S., Oslovsky V.E., Kurochkin N.N., Solyev P.N., Kozlovskaya L.I., Palyulin V.A., Karganova G.G., Mikhailov S.N., Osolodkin D.I. New tools in nucleoside toolbox of tick-borne encephalitis virus reproduction inhibitors. Bioorg. Med. Chem. Lett. 2017;27:1267–1273. doi: 10.1016/j.bmcl.2017.01.040. [DOI] [PubMed] [Google Scholar]
- Pankiewicz K.W. Fluorinated nucleosides. Carbohydr. Res. 2000;327:87–105. doi: 10.1016/s0008-6215(00)00089-6. [DOI] [PubMed] [Google Scholar]
- Pankiewicz K.W., Watanabe K.A. vol. 64. Elsevier; Amsterdam, PAYS-BAS: 1993. (Synthesis of 2'-β-fluoro-substituted Nucleosides by a Direct Approach). [Google Scholar]
- Park B., Kitteringham N., O'Neill P. Metabolism of fluorine-containing drugs. Annu. Rev. Pharmacol. Toxicol. 2001;41:443–470. doi: 10.1146/annurev.pharmtox.41.1.443. [DOI] [PubMed] [Google Scholar]
- Park B., Kitteringham N., O'Neill P. Metabolism of fluorine-containing drugs. Annu. Rev. Pharmacol. Toxicol. 2001;41:443–470. doi: 10.1146/annurev.pharmtox.41.1.443. [DOI] [PubMed] [Google Scholar]
- Parker W.B., Shaddix S.C., Bowdon B.J., Rose L.M., Vince R., Shannon W.M., Bennett L.L. Metabolism of carbovir, a potent inhibitor of human immunodeficiency virus type 1, and its effects on cellular metabolism. Antimicrob. Agents Chemother. 1993;37:1004–1009. doi: 10.1128/aac.37.5.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patching S.G., Baldwin S.A., Baldwin A.D., Young J.D., Gallagher M.P., Henderson P.J., Herbert R.B. The nucleoside transport proteins, NupC and NupG, from Escherichia coli: specific structural motifs necessary for the binding of ligands. Org. Biomol. Chem. 2005;3:462–470. doi: 10.1039/b414739a. [DOI] [PubMed] [Google Scholar]
- Patil S.D., Schneller S.W., Hosoya M., Snoeck R., Graciela A., Balzarini J., De Clercq E. The synthesis and antiviral properties of (.+-.)-5'-noraristeromycin and related purine carbocyclic nucleosides. A new lead for anti-human cytomegalovirus agent design. J. Med. Chem. 1992;35:3372–3377. doi: 10.1021/jm00096a012. [DOI] [PubMed] [Google Scholar]
- Paulsen H. Carbohydrates containing nitrogen or sulfur in the “hemiacetal” ring. Angew. Chem. Int. Ed. 1966;5:495–510. [Google Scholar]
- Paya C., Humar A., Dominguez E., Washburn K., Blumberg E., Alexander B., Freeman R., Heaton N., Pescovitz M.D., Group V.S.O.T.S. Efficacy and safety of valganciclovir vs. oral ganciclovir for prevention of cytomegalovirus disease in solid organ transplant recipients. Am. J. Transpl. 2004;4:611–620. doi: 10.1111/j.1600-6143.2004.00382.x. [DOI] [PubMed] [Google Scholar]
- Peek S.F., Cote P.J., Jacob J.R., Toshkov I.A., Hornbuckle W.E., Baldwin B.H., Wells F.V., Chu C.K., Gerin J.L., Tennant B.C., Korba B.E. Antiviral activity of clevudine [L-FMAU, (1-(2-fluoro-5-methyl-beta, L-arabinofuranosyl) uracil)] against woodchuck hepatitis virus replication and gene expression in chronically infected woodchucks (Marmota monax) Hepatology. 2001;33:254–266. doi: 10.1053/jhep.2001.20899. [DOI] [PubMed] [Google Scholar]
- Perigaud C., Gosselin G., Imbach J.L. Nucleoside analogues as chemotherapeutic agents: a review. Nucleoside Nucleotides. 1992;11:903–945. [Google Scholar]
- Perry C.M., Faulds D. Valaciclovir. A review of its antiviral activity, pharmacokinetic properties and therapeutic efficacy in herpesvirus infections. Drugs. 1996;52:754–772. doi: 10.2165/00003495-199652050-00009. [DOI] [PubMed] [Google Scholar]
- Pertusati F., Serpi M., McGuigan C. Medicinal chemistry of nucleoside phosphonate prodrugs for antiviral therapy. Antivir. Chem. Chemother. 2012;22:181–203. doi: 10.3851/IMP2012. [DOI] [PubMed] [Google Scholar]
- Pessôa M.G., Gazzard B., Huang A.K., Brandão-Mello C.E., Cassetti I., Mendes-Corrêa M.C., Soriano V., Phiri P., Hall A., Brett-Smith H. Efficacy and safety of entecavir for chronic HBV in HIV/HBV coinfected patients receiving lamivudine as part of antiretroviral therapy. AIDS. 2008;22:1779–1787. doi: 10.1097/QAD.0b013e32830b3ab5. [DOI] [PubMed] [Google Scholar]
- Petrie C.R., Cottam H.B., McKernan P.A., Robins R.K., Revankar G.R. Synthesis and biological activity of 6-azacadeguomycin and certain 3,4,6-trisubstituted pyrazolo[3,4-d]pyrimidine ribonucleosides. J. Med. Chem. 1985;28:1010–1016. doi: 10.1021/jm00146a007. [DOI] [PubMed] [Google Scholar]
- Piketty C., Bardin C., Gilquin J., Gairard A., Kazatchkine M.D., Chast F. Monitoring plasma levels of ganciclovir in AIDS patients receiving oral ganciclovir as maintenance therapy for CMV retinitis. Clin. Microbiol. Infect. 2000;6:117–120. doi: 10.1046/j.1469-0691.2000.00014.x. [DOI] [PubMed] [Google Scholar]
- Plunkett W., Cohen S.S. Two approaches that increase the activity of analogs of adenine nucleosides in animal cells. Cancer Res. 1975;35:1547–1554. [PubMed] [Google Scholar]
- Porche D.J. Tenofovir disoproxil fumarate (Viread) J. Assoc. Nurses AIDS Care. 2002;13:100–102. doi: 10.1177/10529002013003007. [DOI] [PubMed] [Google Scholar]
- Price P.M., Banerjee R., Jeffrey A.M., Acs G. The mechanism of inhibition of hepatitis B virus replication by the carbocyclic analog of 2'-deoxyguanosine. Hepatology. 1992;16:8–12. doi: 10.1002/hep.1840160103. [DOI] [PubMed] [Google Scholar]
- Prusoff W.H. Synthesis and biological activities of iododeoxyuridine, an analog of thymidine. Biochim. Biophys. Acta. 1959;32:295–296. doi: 10.1016/0006-3002(59)90597-9. [DOI] [PubMed] [Google Scholar]
- Qiu Y.L., Ksebati M.B., Ptak R.G., Fan B.Y., Breitenbach J.M., Lin J.S., Cheng Y.C., Kern E.R., Drach J.C., Zemlicka J. (Z)- and (E)-2-((hydroxymethyl)cyclopropylidene)methyladenine and -guanine. New nucleoside analogues with a broad-spectrum antiviral activity. J. Med. Chem. 1998;41:10–23. doi: 10.1021/jm9705723. [DOI] [PubMed] [Google Scholar]
- Quirk S., Seley K.L. Substrate discrimination by the human GTP fucose pyrophosphorylase. Biochemistry. 2005;44:10854–10863. doi: 10.1021/bi0503605. [DOI] [PubMed] [Google Scholar]
- Rabbani S.A., Harakidas P., Bowlin T., Attardo G. Effect of nucleoside analogue BCH-4556 on prostate cancer growth and metastases in vitro and in vivo. Cancer Res. 1998;58:3461–3465. [PubMed] [Google Scholar]
- Raj K., Mufti G.J. Azacytidine (Vidaza(R)) in the treatment of myelodysplastic syndromes. Ther. Clin. Risk Manag. 2006;2:377–388. doi: 10.2147/tcrm.2006.2.4.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawal R.K., Konreddy A.K., Chu C.K. Mechanism of adefovir, tenofovir and entecavir resistance: molecular modeling studies of how a novel anti-HBV agent (FMCA) can overcome the drug resistance. Curr. Med. Chem. 2015;22:3922–3932. doi: 10.2174/0929867322666150904144802. [DOI] [PubMed] [Google Scholar]
- Rawal R.K., Bariwal J., Singh V. Chemistry and bioactivities of aristeromycins: an overview. Curr. Top. Med. Chem. 2016;16:3258–3273. doi: 10.2174/1568026616666160506145300. [DOI] [PubMed] [Google Scholar]
- Ray A.S., Fordyce M.W., Hitchcock M.J. Tenofovir alafenamide: a novel prodrug of tenofovir for the treatment of Human Immunodeficiency Virus. Antivir. Res. 2016;125:63–70. doi: 10.1016/j.antiviral.2015.11.009. [DOI] [PubMed] [Google Scholar]
- Reardon J.E., Spector T. Herpes simplex virus type 1 DNA polymerase. Mechanism of inhibition by acyclovir triphosphate. J. Biol. Chem. 1989;264:7405–7411. [PubMed] [Google Scholar]
- Reese N.D., Schiller G.J. High-dose cytarabine (HD araC) in the treatment of leukemias: a review. Curr. Hematol. Malig. Rep. 2013;8:141–148. doi: 10.1007/s11899-013-0156-3. [DOI] [PubMed] [Google Scholar]
- Reischig T., Jindra P., Hes O., Svecová M., Klaboch J., Treska V. Valacyclovir prophylaxis versus preemptive valganciclovir therapy to prevent cytomegalovirus disease after renal transplantation. Am. J. Transpl. 2008;8:69–77. doi: 10.1111/j.1600-6143.2007.02031.x. [DOI] [PubMed] [Google Scholar]
- Revankar G.R., Gupta P.K., Adams A.D., Dalley N.K., McKernan P.A., Cook P.D., Canonico P.G., Robins R.K. Synthesis and antiviral/antitumor activities of certain 3-deazaguanine nucleosides and nucleotides. J. Med. Chem. 1984;27:1389–1396. doi: 10.1021/jm00377a002. [DOI] [PubMed] [Google Scholar]
- Richman D.D., Meng T.C., Spector S.A., Fischl M.A., Resnick L., Lai S. Resistance to AZT and ddC during long-term combination therapy in patients with advanced infection with human immunodeficiency virus. J. Acquir Immune Defic. Syndr. 1994;7:135–138. [PubMed] [Google Scholar]
- Rideout J.L., Krenitsky T.A., Koszalka G.W., Cohn N.K., Chao E.Y., Elion G.B., Latter V.S., Williams R.B. Pyrazolo[3,4-d]pyrimidine ribonucleosides as anticoccidials. 2. Synthesis and activity of some nucleosides of 4-(alkylamino)-1H-pyrazolo[3, 4-d]pyrimidines. J. Med. Chem. 1982;25:1040–1044. doi: 10.1021/jm00351a007. [DOI] [PubMed] [Google Scholar]
- Rodríguez J.B., Comin M.J. New progresses in the enantioselective synthesis and biological properties of carbocyclic nucleosides. Mini Rev. Med. Chem. 2003;3:95–114. doi: 10.2174/1389557033405331. [DOI] [PubMed] [Google Scholar]
- Roy B., Depaix A., Périgaud C., Peyrottes S. Recent trends in nucleotide synthesis. Chem. Rev. 2016;116:7854–7897. doi: 10.1021/acs.chemrev.6b00174. [DOI] [PubMed] [Google Scholar]
- Roy-Burman S., Roy-Burman P., Visser D.W. Showdomycin, a new nucleoside antibiotic. Cancer Res. 1968;28:1605–1610. [PubMed] [Google Scholar]
- Rudnick S.A., Cadman E.C., Capizzi R.L., Skeel R.T., Bertino J.R., McIntosh S. High dose cytosine arabinoside (HDARAC) in refractory acute leukemia. Cancer. 1979;44:1189–1193. doi: 10.1002/1097-0142(197910)44:4<1189::aid-cncr2820440404>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Saag M.S. Emtricitabine, a new antiretroviral agent with activity against HIV and hepatitis B virus. Clin. Infect. Dis. 2006;42:126–131. doi: 10.1086/498348. [DOI] [PubMed] [Google Scholar]
- Sacks S.L., Aoki F.Y., Diaz-Mitoma F., Sellors J., Shafran S.D. Patient-initiated, twice-daily oral famciclovir for early recurrent genital herpes. A randomized, double-blind multicenter trial. Canadian Famciclovir Study Group. JAMA. 1996;276:44–49. [PubMed] [Google Scholar]
- Saenger W. Defining terms for the nucleic acids. In: Cantor C.R., editor. Principles of Nucleic Acid Structure. Springer-Verlag New York Inc; 1984. pp. 9–28. [Google Scholar]
- Sahu P.K., Kim G., Yu J., Ahn J.Y., Song J., Choi Y., Jin X., Kim J.H., Lee S.K., Park S., Jeong L.S. Stereoselective synthesis of 4'-selenonucleosides via seleno-Michael reaction as potent antiviral agents. Org. Lett. 2014;16:5796–5799. doi: 10.1021/ol502899b. [DOI] [PubMed] [Google Scholar]
- Sanger F., Coulson A.R. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J. Mol. Biol. 1975;94:441–448. doi: 10.1016/0022-2836(75)90213-2. [DOI] [PubMed] [Google Scholar]
- Sanger F., Nicklen S., Coulson A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U. S. A. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santi D.V., McHenry C.S., Raines R.T., Ivanetich K.M. Kinetics and thermodynamics of the interaction of 5-fluoro-2'-deoxyuridylate with thymidylate synthase. Biochemistry. 1987;26:8606–8613. doi: 10.1021/bi00400a017. [DOI] [PubMed] [Google Scholar]
- Sarafianos S.G., Das K., Hughes S.H., Arnold E. Taking aim at a moving target: designing drugs to inhibit drug-resistant HIV-1 reverse transcriptases. Curr. Opin. Struct. Biol. 2004;14:716–730. doi: 10.1016/j.sbi.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Schilsky R.L., Williams S.F., Ultmann J.E., Watson S. Sequential hydroxyurea-cytarabine chemotherapy for refractory non-Hodgkin's lymphoma. J. Clin. Oncol. 1987;5:419–425. doi: 10.1200/JCO.1987.5.3.419. [DOI] [PubMed] [Google Scholar]
- Schinazi R.F., McMillan A., Cannon D., Mathis R., Lloyd R.M., Peck A., Sommadossi J.P., St Clair M., Wilson J., Furman P.A. Selective inhibition of human immunodeficiency viruses by racemates and enantiomers of cis-5-fluoro-1-[2-(hydroxymethyl)-1,3-oxathiolan-5-yl]cytosine. Antimicrob. Agents Chemother. 1992;36:2423–2431. doi: 10.1128/aac.36.11.2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinazi R.F., Chu C.K., Peck A., McMillan A., Mathis R., Cannon D., Jeong L.S., Beach J.W., Choi W.B., Yeola S. Activities of the four optical isomers of 2',3'-dideoxy-3'-thiacytidine (BCH-189) against human immunodeficiency virus type 1 in human lymphocytes. Antimicrob. Agents Chemother. 1992;36:672–676. doi: 10.1128/aac.36.3.672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm V.L., Tyler P.C. Imino-sugar-based nucleosides. Curr. Top. Med. Chem. 2003;3:525–540. doi: 10.2174/1568026033452465. [DOI] [PubMed] [Google Scholar]
- Secrist J.A., Shortnacy A.T., Montgomery J.A. 2-Fluoroformycin and 2-aminoformycin. Synthesis and biological activity. J. Med. Chem. 1985;28:1740–1742. doi: 10.1021/jm00149a033. [DOI] [PubMed] [Google Scholar]
- Sekiyama T., Hatsuya S., Tanaka Y., Uchiyama M., Ono N., Iwayama S., Oikawa M., Suzuki K., Okunishi M., Tsuji T. Synthesis and antiviral activity of novel acyclic nucleosides: discovery of a cyclopropyl nucleoside with potent inhibitory activity against herpesviruses. J. Med. Chem. 1998;41:1284–1298. doi: 10.1021/jm9705869. [DOI] [PubMed] [Google Scholar]
- Seley K.L., Schneller S.W., Rattendi D., Bacchi C.J. (+)-7-Deaza-5‘-noraristeromycin as an anti-trypanosomal agent. J. Med. Chem. 1997;40:622–624. doi: 10.1021/jm9605039. [DOI] [PubMed] [Google Scholar]
- Seley K.L., Schneller S.W., Rattendi D., Lane S., Bacchi C.J. Synthesis and antitrypanosomal activities of a series of 7-deaza-5'-noraristeromycin derivatives with variations in the cyclopentyl ring substituents. Antimicrob. Agents Chemother. 1997;41:1658–1661. doi: 10.1128/aac.41.8.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seley K.L., Schneller S.W., De Clercq E. A methylated derivative of 5‘-noraristeromycin. J. Org. Chem. 1997;62:5645–5646. [Google Scholar]
- Seley K.L., Schneller S.W., Korba B. A 5′-noraristeromycin enantiomer with activity towards hepatitis B virus. Nucleosides Nucleotides. 1997;16:2095–2099. [Google Scholar]
- Seley K.L., Schneller S.W., Rattendi D., Lane S., Bacchi C.J. Synthesis and anti-trypanosomal activity of various 8-aza-7-deaza-5'noraristeromycin derivatives. J. Med. Chem. 1997;40:625–629. doi: 10.1021/jm9606148. [DOI] [PubMed] [Google Scholar]
- Seley K.L., Schneller S.W., De Clercq E., Rattendi D., Lane S., Bacchi C.J., Korba B. The importance of the 4′-hydroxyl hydrogen for the anti-trypanosomal and antiviral properties of (+)-5′-Noraristeromycin and two 7-deaza analogues. Bioorg. Med. Chem. 1998;6:797–801. doi: 10.1016/s0968-0896(98)00036-4. [DOI] [PubMed] [Google Scholar]
- Seley K.L., Januszczyk P., Hagos A., Zhang L., Dransfield D.T. Synthesis and antitumor activity of thieno-separated tricyclic purines. J. Med. Chem. 2000;43:4877–4883. doi: 10.1021/jm000326i. [DOI] [PubMed] [Google Scholar]
- Seley K.L., Mosley S.L., Zeng F. Carbocyclic isoadenosine analogues of neplanocin A. Org. Lett. 2003;5:4401–4403. doi: 10.1021/ol035696q. [DOI] [PubMed] [Google Scholar]
- Seley-Radtke K.L., Zhang Z., Wauchope O.R., Zimmermann S.C., Ivanov A., Korba B. Hetero-expanded purine nucleosides. Design, synthesis and preliminary biological activity. Nucleic Acids Symp. Ser. Oxf. 2008:635–636. doi: 10.1093/nass/nrn321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semizarov D.G., Arzumanov A.A., Dyatkina N.B., Meyer A., Vichier-Guerre S., Gosselin G., Rayner B., Imbach J.L., Krayevsky A.A. Stereoisomers of deoxynucleoside 5'-triphosphates as substrates for template-dependent and -independent DNA polymerases. J. Biol. Chem. 1997;272:9556–9560. doi: 10.1074/jbc.272.14.9556. [DOI] [PubMed] [Google Scholar]
- Shealy Y.F., Clayton J.D. 9- β-DL-2α,3α-Dihydroxy-4β-(hydroxymethyl)- cyclopentyl adenine, the Carbocyclic Analog of Adenosine1,2. J. Am. Chem. Soc. 1966;88:3885–3887. [Google Scholar]
- Shirasaka T., Chokekijchai S., Yamada A., Gosselin G., Imbach J.L., Mitsuya H. Comparative analysis of anti-human immunodeficiency virus type 1 activities of dideoxynucleoside analogs in resting and activated peripheral blood mononuclear cells. Antimicrob. Agents Chemother. 1995;39:2555–2559. doi: 10.1128/aac.39.11.2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqi S.M., Schneller S.W., Ikeda S., Snoeck R., Andrei G., Balzarini J., De Clercq E. S-Adenosyl-L-homocysteine hydrolase inhibitors as anti-viral agents: 5′-deoxyaristeromycin. Nucleosides Nucleotides. 1992;12:185–198. [Google Scholar]
- Siddiqi S.M., Chen X., Rao J., Schneller S.W., Ikeda S., Snoeck R., Andrei G., Balzarini J., De Clercq E. 3-deaza- and 7-deaza-5'-noraristeromycin and their antiviral properties. J. Med. Chem. 1995;38:1035–1038. doi: 10.1021/jm00006a023. [DOI] [PubMed] [Google Scholar]
- Silverman R.B., Holladay M.W. The Organic Chemistry of Drug Design and Drug Action. third ed. Elsevier; 2014. Lead discovery and lead modification; pp. 19–122. [Google Scholar]
- Sipkema D., Franssen M.C., Osinga R., Tramper J., Wijffels R.H. Marine sponges as pharmacy. Mar. Biotechnol. (NY) 2005;7:142–162. doi: 10.1007/s10126-004-0405-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu L.L., Attardo G., Izbicka E., Lawrence R., Cerna C., Gomez L., Davidson K., Finkle C., Marsolais C., Rowinsky E.K., Von Hoff D.D. Activity of (-)-2'-deoxy-3'-oxacytidine (BCH-4556) against human tumor colony-forming units. Ann. Oncol. 1998;9:885–891. doi: 10.1023/a:1008387019062. [DOI] [PubMed] [Google Scholar]
- Slusarczyk M., Lopez M.H., Balzarini J., Mason M., Jiang W.G., Blagden S., Thompson E., Ghazaly E., McGuigan C. Application of ProTide technology to gemcitabine: a successful approach to overcome the key cancer resistance mechanisms leads to a new agent (NUC-1031) in clinical development. J. Med. Chem. 2014;57:1531–1542. doi: 10.1021/jm401853a. [DOI] [PubMed] [Google Scholar]
- Smee D.F., Sidwell R.W., Kefauver D., Bray M., Huggins J.W. Characterization of wild-type and cidofovir-resistant strains of camelpox, cowpox, monkeypox, and vaccinia viruses. Antimicrob. Agents Chemother. 2002;46:1329–1335. doi: 10.1128/AAC.46.5.1329-1335.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiley M.L., Murray A., de Miranda P. Valacyclovir HCl (Valtrex): an acyclovir prodrug with improved pharmacokinetics and better efficacy for treatment of zoster. Adv. Exp. Med. Biol. 1996;394:33–39. doi: 10.1007/978-1-4757-9209-6_4. [DOI] [PubMed] [Google Scholar]
- Smith R.A., Gottlieb G.S., Anderson D.J., Pyrak C.L., Preston B.D. Human immunodeficiency virus types 1 and 2 exhibit comparable sensitivities to Zidovudine and other nucleoside analog inhibitors in vitro. Antimicrob. Agents Chemother. 2008;52:329–332. doi: 10.1128/AAC.01004-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soudeyns H., Yao X.I., Gao Q., Belleau B., Kraus J.L., Nguyen-Ba N., Spira B., Wainberg M.A. Anti-human immunodeficiency virus type 1 activity and in vitro toxicity of 2'-deoxy-3'-thiacytidine (BCH-189), a novel heterocyclic nucleoside analog. Antimicrob. Agents Chemother. 1991;35:1386–1390. doi: 10.1128/aac.35.7.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soul-Lawton J., Seaber E., On N., Wootton R., Rolan P., Posner J. Absolute bioavailability and metabolic disposition of valaciclovir, the L-valyl ester of acyclovir, following oral administration to humans. Antimicrob. Agents Chemother. 1995;39:2759–2764. doi: 10.1128/aac.39.12.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan P.R., Borek E. Enzymatic alteration of nucleic acid structure. Science. 1964;145:548–553. doi: 10.1126/science.145.3632.548. [DOI] [PubMed] [Google Scholar]
- St Clair M.H., Martin J.L., Tudor-Williams G., Bach M.C., Vavro C.L., King D.M., Kellam P., Kemp S.D., Larder B.A. Resistance to ddI and sensitivity to AZT induced by a mutation in HIV-1 reverse transcriptase. Science. 1991;253:1557–1559. doi: 10.1126/science.1716788. [DOI] [PubMed] [Google Scholar]
- Stambaský J., Hocek M., Kocovský P. C-nucleosides: synthetic strategies and biological applications. Chem. Rev. 2009;109:6729–6764. doi: 10.1021/cr9002165. [DOI] [PubMed] [Google Scholar]
- Steingrimsdottir H., Gruber A., Palm C., Grimfors G., Kalin M., Eksborg S. Bioavailability of aciclovir after oral administration of aciclovir and its prodrug valaciclovir to patients with leukopenia after chemotherapy. Antimicrob. Agents Chemother. 2000;44:207–209. doi: 10.1128/aac.44.1.207-209.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stentoft J. The toxicity of cytarabine. Drug Saf. 1990;5:7–27. doi: 10.2165/00002018-199005010-00003. [DOI] [PubMed] [Google Scholar]
- Stoeckler J.D., Cambor C., Parks R.E. Human erythrocytic purine nucleoside phosphorylase: reaction with sugar-modified nucleoside substrates. Biochem. 1980;19:102–107. doi: 10.1021/bi00542a016. [DOI] [PubMed] [Google Scholar]
- Stray K.M., Park Y., Babusis D., Callebaut C., Cihlar T., Ray A.S., Perron M. Tenofovir alafenamide (TAF) does not deplete mitochondrial DNA in human T-cell lines at intracellular concentrations exceeding clinically relevant drug exposures. Antivir. Res. 2017;140:116–120. doi: 10.1016/j.antiviral.2017.01.014. [DOI] [PubMed] [Google Scholar]
- Suzuki M., Okuda T., Shiraki K. Synergistic antiviral activity of acyclovir and vidarabine against herpes simplex virus types 1 and 2 and varicella-zoster virus. Antivir. Res. 2006;72:157–161. doi: 10.1016/j.antiviral.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Tang H., Griffin J., Innaimo S., Lehman-Mckeeman L., Llamoso C. The discovery and development of a potent antiviral drug, entecavir, for the treatment of chronic hepatitis B. J. Clin. Transl. Hepatol. 2013;1:51–58. doi: 10.14218/JCTH.2013.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor R., Kotian P., Warren T., Panchal R., Bavari S., Julander J., Dobo S., Rose A., El-Kattan Y., Taubenheim B., Babu Y., Sheridan W.P. BCX4430-A broad-spectrum antiviral adenosine nucleoside analog under development for the treatment of Ebola virus disease. J. Infect. Public Health. 2016;9:220–226. doi: 10.1016/j.jiph.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temburnikar K., Seley-Radtke K.L. Recent advances in synthetic approaches to C-Nucleosides. Beilstein J. Org. Chem. 2018;14:772–785. doi: 10.3762/bjoc.14.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisdale M., Alnadaf T., Cousens D. Combination of mutations in human immunodeficiency virus type 1 reverse transcriptase required for resistance to the carbocyclic nucleoside 1592U89. Antimicrob. Agents Chemother. 1997;41:1094–1098. doi: 10.1128/aac.41.5.1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttle J.V., Krenitsky T.A. Effects of acyclovir and its metabolites on purine nucleoside phosphorylase. J. Biol. Chem. 1984;259:4065–4069. [PubMed] [Google Scholar]
- Tyring S., Barbarash R.A., Nahlik J.E., Cunningham A., Marley J., Heng M., Jones T., Rea T., Boon R., Saltzman R. Famciclovir for the treatment of acute herpes zoster: effects on acute disease and postherpetic neuralgia. A randomized, double-blind, placebo-controlled trial. Collaborative Famciclovir Herpes Zoster Study Group. Ann. Intern Med. 1995;123:89–96. doi: 10.7326/0003-4819-123-2-199507150-00002. [DOI] [PubMed] [Google Scholar]
- Vasconcelos T., Ferreira M., Goncalves R., da Silva E., de Souza M. Lamivudine, an important drug in aids treatment. J. Sulfur Chem. 2008;29:559–571. [Google Scholar]
- Veal G.J., Barry M.G., Back D.J. Zalcitabine (DDC) phosphorylation and drug-interactions. Antivir. Chem. Chemother. 1995;6:379–384. [Google Scholar]
- Vere Hodge R.A. Famciclovir and penciclovir. The mode of action of famciclovir including its conversion to penciclovir. Antivir. Chem. Chemother. 1993;4:67–84. [Google Scholar]
- Vere Hodge R.A., Sutton D., Boyd M.R., Harnden M.R., Jarvest R.L. Selection of an oral prodrug (BRL 42810; famciclovir) for the antiherpesvirus agent BRL 39123 [9-(4-hydroxy-3-hydroxymethylbut-l-yl)guanine; penciclovir] Antimicrob. Agents Chemother. 1989;33:1765–1773. doi: 10.1128/aac.33.10.1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vince R., Hua M., Brownell J., Daluge S., Lee F.C., Shannon W.M., Lavelle G.C., Qualls J., Weislow O.S., Kiser R. Potent and selective activity of a new carbocyclic nucleoside analog (carbovir: NSC 614846) against human immunodeficiency virus in vitro. Biochem. Biophys. Res. Commun. 1988;156:1046–1053. doi: 10.1016/s0006-291x(88)80950-1. [DOI] [PubMed] [Google Scholar]
- Wallace L.J., Candlish D., Hagos A., Seley K.L., de Koning H.P. Selective transport of a new class of purine antimetabolites by the protozoan parasite Trypanosoma brucei. Nucleosides Nucleotides Nucleic Acids. 2004;23:1441–1444. doi: 10.1081/NCN-200027660. [DOI] [PubMed] [Google Scholar]
- Walwick E.R., Roberts W.K., Dekker C.A. Cyclisation during the phosphorylation of uridine and cytidine bypolyphosphoric acid-A new route to the O-2, 2′-cyclonucleosides. Proc. Chem. Soc. Lond. 1959;3:84. [Google Scholar]
- Wang J., Herdewijn P. Enantioselective synthesis and conformational study of cyclohexene carbocyclic nucleosides. J. Org. Chem. 1999;64:7820–7827. [Google Scholar]
- Wang J., Froeyen M., Hendrix C., Andrei G., Snoeck R., De Clercq E., Herdewijn P. The cyclohexene ring system as a furanose mimic: synthesis and antiviral activity of both enantiomers of cyclohexenylguanine. J. Med. Chem. 2000;43:736–745. doi: 10.1021/jm991171l. [DOI] [PubMed] [Google Scholar]
- Wang J., Rawal R.K., Chu C.K. Recent advances in carbocyclic nucleosides: synthesis and biological activity. In: Zhang L.H., Xi Z., Chattopadhyaya J., editors. Medicinal Chemistry of Nucleic Acids. John Wiley & Sons, Inc.; Hoboken, NJ, USA: 2011. pp. 1–100. [Google Scholar]
- Warren T.K., Wells J., Panchal R.G., Stuthman K.S., Garza N.L., Van Tongeren S.A., Dong L., Retterer C.J., Eaton B.P., Pegoraro G., Honnold S., Bantia S., Kotian P., Chen X., Taubenheim B.R., Welch L.S., Minning D.M., Babu Y.S., Sheridan W.P., Bavari S. Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430. Nature. 2014;508:402–405. doi: 10.1038/nature13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wataya Y., Hiraoka O., Sonobe Y., Yoshioka A., Matsuda A., Miyasaka T., Saneyoshi M., Ueda T. Anti-parasite activity of nucleoside analogues in Leishmania tropica promastigotes. Nucleic Acids Symp. Ser. 1984:69–71. [PubMed] [Google Scholar]
- Wauchope O.R., Johnson C., Krishnamoorthy P., Andrei G., Snoeck R., Balzarini J., Seley-Radtke K.L. Synthesis and biological evaluation of a series of thieno-expanded tricyclic purine 2'-deoxy nucleoside analogues. Bioorg. Med. Chem. 2012;20:3009–3015. doi: 10.1016/j.bmc.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller S., Blum M.R., Doucette M., Burnette T., Cederberg D.M., de Miranda P., Smiley M.L. Pharmacokinetics of the acyclovir pro-drug valaciclovir after escalating single- and multiple-dose administration to normal volunteers. Clin. Pharmacol. Ther. 1993;54:595–605. doi: 10.1038/clpt.1993.196. [DOI] [PubMed] [Google Scholar]
- Wesołowski T.A., Godzik A., Geller M. Calculations of the conformational properties of acyclonucleosides. Part I. Stable conformations of acyclovir. Acta Biochim. Pol. 1987;34:111–122. [PubMed] [Google Scholar]
- Whitley R.J. The past as prelude to the future: history, status, and future of antiviral drugs. Ann. Pharmacother. 1996;30:967–971. doi: 10.1177/106002809603000911. [DOI] [PubMed] [Google Scholar]
- WHO Model List of Essential Medicines. http://www.who.int/medicines/publications/essentialmedicines/20th_EML2017_FINAL_amendedAug2017.pdf?ua=1.
- Wilber R., Kreter B., Bifano M., Danetz S., Lehman-McKeeman L., Tenney D.J., Meanwell N., Zahler R., Brett-Smith H. Discovery and development of entecavir. In: Kazmierski W.M., editor. Antiviral Drugs: from Basic Discovery through Clinical Trials. John Wiley & Sons, Inc.; Hoboken, NJ, USA: 2011. pp. 401–416. [Google Scholar]
- Winkley M.W., Robins R.K. Direct glycosylation of 1,3,5-triazinones. A new approach to the synthesis of the nucleoside antibiotic 5-azacytidine (4-amino-1-beta-D-ribofuranosyl-1,3,5-triazin-2-one) and related derivatives. J. Org. Chem. 1970;35:491–495. doi: 10.1021/jo00827a045. [DOI] [PubMed] [Google Scholar]
- Wojtowicz-Rajchel H. Synthesis and applications of fluorinated nucleoside analogues. J. Fluor. Chem. 2012;143:11–48. [Google Scholar]
- Wolfe M.S., Borchardt R.T. S-adenosyl-L-homocysteine hydrolase as a target for antiviral chemotherapy. J. Med. Chem. 1991;34:1521–1530. doi: 10.1021/jm00109a001. [DOI] [PubMed] [Google Scholar]
- Wright J.D., Ma T., Chu C.K., Boudinot F.D. Pharmacokinetics of 1-(2-deoxy-2-fluoro-beta-L-arabinofuranosyl)-5-methyluracil (L-FMAU) in rats. Pharm. Res. 1995;12:1350–1353. doi: 10.1023/a:1016234009624. [DOI] [PubMed] [Google Scholar]
- Yeom Y.H., Remmel R.P., Huang S.H., Hua M., Vince R., Zimmerman C.L. Pharmacokinetics and bioavailability of carbovir, a carbocyclic nucleoside active against human immunodeficiency virus, in rats. Antimicrob. Agents Chemother. 1989;33:171–175. doi: 10.1128/aac.33.2.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J., Kim J.H., Lee H.W., Alexander V., Ahn H.C., Choi W.J., Choi J., Jeong L.S. New RNA purine building blocks, 4'-selenopurine nucleosides: first synthesis and unusual mixture of sugar puckerings. Chemistry. 2013;19:5528–5532. doi: 10.1002/chem.201300741. [DOI] [PubMed] [Google Scholar]
- Yuen G.J., Weller S., Pakes G.E. A review of the pharmacokinetics of abacavir. Clin. Pharmacokinet. 2008;47:351–371. doi: 10.2165/00003088-200847060-00001. [DOI] [PubMed] [Google Scholar]
- Zhang D., Caliendo A.M., Eron J.J., DeVore K.M., Kaplan J.C., Hirsch M.S., D'Aquila R.T. Resistance to 2',3'-dideoxycytidine conferred by a mutation in codon 65 of the human immunodeficiency virus type 1 reverse transcriptase. Antimicrob. Agents Chemother. 1994;38:282–287. doi: 10.1128/aac.38.2.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Wauchope O.R., Seley-Radtke K.L. Mechanistic studies in the synthesis of a series of thieno-expanded xanthosine and guanosine nucleosides. Tetrahedron. 2008;64:10791–10797. doi: 10.1016/j.tet.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann S.C., Sadler J.M., O'Daniel P.I., Kim N.T., Seley-Radtke K.L. “Reverse” carbocyclic fleximers: synthesis of a new class of adenosine deaminase inhibitors. Nucleosides Nucleotides Nucleic Acids. 2013;32:137–154. doi: 10.1080/15257770.2013.771187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann S.C., O'Neill E., Ebiloma G.U., Wallace L.J., De Koning H.P., Seley-Radtke K.L. Design and synthesis of a series of truncated neplanocin fleximers. Molecules. 2014;19:21200–21214. doi: 10.3390/molecules191221200. [DOI] [PMC free article] [PubMed] [Google Scholar]