

Abstract
Interleukin‐22 (IL‐22) has context‐dependent hepatoprotective or adverse properties in vitro and in animal models. IL‐22 binding protein (IL‐22BP) is a soluble inhibitor of IL‐22 signaling. The role of IL‐22 and IL‐22BP in patients with acute‐on‐chronic liver failure (ACLF) is unclear. Beginning in August 2013, patients with liver cirrhosis with and without ACLF were prospectively enrolled and followed at predefined time points. IL‐22 and IL‐22BP concentrations were quantified and associated with clinical endpoints. The impact of IL‐22BP on hepatocellular IL‐22 signaling was assessed by functional experiments. A total of 139 patients were analyzed, including 45 (32%), 52 (37%), and 42 (30%) patients with compensated/stable decompensated liver cirrhosis, acute decompensation of liver cirrhosis, and ACLF at baseline, respectively. Serum levels of IL‐22 and IL‐22BP were strongly associated with the presence of, or progression to, ACLF (P < 0.001), and with mortality (P < 0.01). Importantly, the mean IL‐22BP levels exceeded IL‐22 levels more than 300‐fold. Furthermore, IL‐22BP/IL‐22 ratios were lowest in patients with adverse outcomes (i.e., ACLF and death). In vitro experiments showed that IL‐22BP at these concentrations inhibits hepatocellular IL‐22 signaling, including the induction of acute‐phase proteins. The capacity of patient serum to induce signal transducer and activator of transcription 3 phosphorylation was substantially higher in the presence of low versus high IL‐22BP/IL‐22 ratios. Conclusion: Our study reveals that high IL‐22 levels and low ratios of IL‐22BP/IL‐22 are associated with ACLF and mortality of patients with cirrhosis. Excessive secretion of IL‐22BP can neutralize IL‐22 in vitro and may prevent—likely in a context‐specific manner—hepatoprotective, but also adverse effects, of IL‐22 in patients with cirrhosis.
Abbreviations
- ACLF
acute‐on‐chronic liver failure
- BP
binding protein
- CLIF‐EASL
Chronic Liver Failure–European Association for the Study of Liver
- CRP
C‐reactive protein
- ELISA
enzyme‐linked immunosorbent assay
- GAPDH
glyceraldehyde 3‐phosphate dehydrogenase
- GGT
gamma‐glutamyltransferase
- IL
interleukin
- INR
international normalized ratio
- LBP
LPS binding protein
- LPS
lipopolysaccharide
- mAb
monoclonal antibody
- mRNA
messenger RNA
- STAT
signal transducer and activator of transcription
- TNF‐α
tumor necrosis factor alpha
Acute‐on‐chronic liver failure (ACLF) is a severe complication of liver cirrhosis characterized by hepatic and extrahepatic organ failures.1 Depending on the severity of ACLF, mortality rates of up to 79% within 90 days have been observed.1 Established precipitating events of ACLF include infections, excessive alcohol consumption, exposure to toxins, and bleeding episodes.1 These events are able to strongly augment the liver cirrhosis–associated systemic inflammatory response, characterized by high levels of proinflammatory and anti‐inflammatory cytokines, switch of monocyte phenotypes, and immune paralysis.2
Interleukin‐22 (IL‐22) is a member of the IL‐10 cytokine family and is generated by, for example, Th17 and Th22 cells, as well as subpopulations of γδ T cells and natural killer cells.3 IL‐22 acts by means of a transmembrane receptor complex consisting of the two subunits IL‐22Rα and IL‐10R2, which are only expressed on nonimmune cells such as hepatocytes, kidney cells, keratinocytes, or epithelial cells of the intestinal or respiratory tract.3 IL‐22 receptor signaling is mediated through Janus kinase (JAK) signal transducer and activator of transcription (STAT) signaling, and phosphorylated STAT3 constitutes the most important transcription factor downstream of the IL‐22 receptor.3 Of note, IL‐22 binding to its transmembrane receptor can be prevented by the IL‐22 binding protein (IL‐22BP), which is a secreted single‐chain receptor for IL‐22 that acts as a bait with significantly higher affinity to IL‐22 than IL‐22Rα.4, 5, 6, 7
IL‐22 signaling promotes epithelial immune responses through induction of epithelial defensins and strengthening of cellular barriers.8 Furthermore, IL‐22 has been shown to promote liver regeneration in vitro as well as in animal models.9 Consequently, IL‐22 is considered a promising therapeutic agent in advanced liver diseases, and intravenous administration of IL‐22 is currently being evaluated in a clinical trial in patients with alcoholic hepatitis (NCT02655510). However, high serum levels of IL‐22 have been associated with adverse outcomes in patients with alcoholic and nonalcoholic liver disease.10, 11 Possible explanations for this phenomenon may include unwanted proinflammatory effects of IL‐22, which can augment Th17 immune responses, as well as insufficient biological activity of IL‐22 due to inhibitory mechanisms such as IL‐22BP.12, 13
In the present study we aim to explore the behavior of this special cytokine and explore the role of its inhibitor IL‐22BP in the progression of liver cirrhosis to ACLF, a clinical scenario in which strategies to augment epithelial barrier functions and promotion of liver regeneration appear to be particularly attractive.
Patients and Methods
Study Population
Since August 2013, consecutive patients admitted to the University Hospital Frankfurt, Germany, with acute decompensation of liver cirrhosis and/or ACLF according to the criteria of the Chronic Liver Failure–European Association for the Study of Liver (CLIF‐EASL) consortium,1 were prospectively enrolled in our liver cirrhosis cohort study. In 2015, the cohort was extended to patients with compensated or stable decompensated liver cirrhosis.
The diagnosis of liver cirrhosis was based on a combination of clinical, laboratory, and imaging findings (ultrasound and transient elastography or shear wave elastography) or, less frequently, liver biopsy. Acute decompensation of liver cirrhosis was defined as the presence of one of the following criteria: new onset/progression of hepatic encephalopathy (HE) graded by West‐Haven criteria,14 gastrointestinal hemorrhage, bacterial infection, and ascites grade 2‐3 (graded according to Moore et al.15). ACLF was diagnosed according to the ACLF criteria proposed by the CLIF‐EASL consortium.1
Exclusion criteria were age below 18 years, pregnancy or breastfeeding, presence of hepatocellular carcinoma (HCC) beyond Milan criteria, presence of infection with human immunodeficiency virus, or therapy with immunosuppressive agents.
All patients provided written informed consent to the study protocol, and the study was approved by the local ethics committee of the University Hospital Frankfurt, Germany.
Clinical Data Collection and Biobanking
Clinical data and biomaterials of patients with acute decompensation of liver cirrhosis or ACLF were collected at baseline, follow‐up days 7 and 28, and follow‐up week 12. Clinical data and biomaterials of patients with compensated or stable decompensation of liver cirrhosis were collected at baseline and at least every 3 months of follow‐up, or at the time of development of acute decompensation or ACLF.
Demographic and clinical characteristics, including age, sex, body mass index, underlying cause of liver cirrhosis, listing for liver transplantation, presence or absence of diabetes/portal vein thrombosis/HCC or cholangiocellular carcinoma, alcohol consumption, duration of abstinence, presence and grade of ascites/HE, gastrointestinal hemorrhage, presence and type of infection, use of renal replacement therapy or catecholamines, PaO2/FiO2 and stage of ACLF, were obtained from clinical databases. Additionally, laboratory parameters were extracted, including peripheral blood cell counts, hemoglobin, C‐reactive protein (CRP), serum sodium, serum potassium, serum calcium, serum chloride, serum phosphate, serum creatinine, serum urea, serum bilirubin, serum alanine aminotransferase, serum aspartate aminotransferase, serum gamma‐glutamyltransferase (GGT), serum alkaline phosphatase, serum ferritin, international normalized ratio (INR), activated partial thromboplastin time, and serum albumin.
Serum and viable peripheral blood mononuclear cells were collected at the indicated baseline and follow‐up time points. All patients who were enrolled in this prospective cohort study until April 2017 were included in the present analysis.
Quantification of Cytokines
Serum concentrations of IL‐22, IL‐22BP, and IL‐17A were quantified using the Human IL‐22 Quantikine ELISA (enzyme‐linked immunosorbent assay) Kit (R & D Systems, Minneapolis, MN), the Human IL‐22BP/IL‐22RA2 ELISA Kit (Lifespan Biosciences, Seattle, WA), and the LEGEND MAX Human IL‐17A ELISA Kit (BioLegend, San Diego, CA), according to manufacturers’ instructions. Samples and standard curve values were measured at 450 nm on an EnVision 2104 Multilabel Plate Reader (PerkinElmer, Waltham, MA). The specificity of the IL‐22 and IL‐22BP ELISA with respect to detection of IL‐22BP and IL‐22 (and vice versa) was assessed (Supporting Table S4). The presence of very high concentrations of IL‐22BP interfered with the IL‐22 ELISA (in line with the data sheets of all available IL‐22 ELISAs) with the consequence that IL‐22 serum concentrations might be slightly underestimated.
Cell Culture
Human hepatoma cell line Huh7 was purchased from the Japanese Collection of Research Bioresources cell bank (JCRB0403; Osaka, Japan), and the human hepatoma cell line HepG2 was purchased from the American Type Culture Collection (HB‐8065; Manassas, VA). The Huh7 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) 1 g/L glucose (Gibco 31885‐023) supplemented with 10% fetal calf serum and 1% penicillin/streptomycin. HepG2 cells were cultured in DMEM 4.5 g/L glucose (Gibco 41965‐039). Recombinant human IL‐22 (Peprotech, Rocky Hill, NJ) and recombinant human IL‐22BP/IL‐22RA2 protein (Sino Biological, Wayne, PA) were used for in vitro assays. Primary human hepatocytes were purchased from ScienCell Research Laboratories (Carlsbad, CA) and cultured in hepatocyte medium on poly‐l‐lysine‐coated plates according to the manufacturer’s instructions. Cell viability was assessed using the WST‐1 Cell Proliferation Reagent from Takara Bio (Kusatsu, Shiga, Japan).
Immunocytochemistry
Immunocytochemistry of the IL‐22 receptor chain IL‐22Rα1 was performed using a primary antibody from Santa Cruz Biotechnology (sc‐134366; Dallas, TX) and an Alexa Fluor 488 goat antimouse immunoglobulin G (A11001; Life Technologies, Carlsbad, CA) as secondary antibody and bisbenzimide H 33342 trihydrochloride (Sigma Aldrich, St. Louis, MO) as nuclear counterstaining on an Olympus FluoView FV1000 microscope (Tokyo, Japan). To this end, human hepatoma cell lines were fixed onto coverslips with 4% paraformaldehyde, permeabilized with 0.25% Triton‐X in phosphate‐buffered saline (PBS), and blocked with 5% goat serum (Sigma Aldrich) in PBS.
Immunoblot Analyses
Immunoblot analyses were performed as described16 using the following antibodies: STAT1 p84/p91 antibody (E‐23; Santa Cruz Biotechnology); Phospho‐STAT1 (Tyr701) rabbit monoclonal antibody (mAb) (#7649; Cell Signaling Technology, Danvers, MA); STAT3 mouse mAb (124H6; Cell Signaling Technology); phospho‐STAT3 (Tyr705) rabbit mAb (D3A7; Cell Signaling Technology); STAT5 antibody (#9363; Cell Signaling Technology); phospho‐STAT5 XP rabbit mAb (D47E7; Cell Signaling Technology); lipopolysaccharide (LPS) binding protein (LBP) (AF870; R & D Systems); and anti‐ß‐actin antibody mouse monoclonal (A1978; Sigma Aldrich).
Quantitative Polymerase Chain Reaction
Quantitative polymerase chain reaction was performed using SYBR green technology as described,16 using the following primers: human GAPDH (glyceraldehyde 3‐phosphate dehydrogenase) forward (GAA GAT GGT GAT GGG ATT TC); human GAPDH reverse (GAA GGT GAA GGT CGG AGT C); human TNF‐α (tumor necrosis factor alpha) forward (GGC AGT CAG ATC ATC TTC TCG AA); human TNF‐α reverse (GAA GGC CTA AGG TCC ACT TGT GT); human LBP forward (TTC GGT CAA CCT CCT GTT GG TTC GGT CAA CCT CCT GTT GG); and human LBP reverse (CGC AAA TCC TGC TCT CCA GT).
Statistical Analyses
Statistical analyses were performed using BiAS, Version 11.06, and Graphpad Prism 5. Group differences were assessed using χ 2 contingency tables or Wilcoxon–Mann‐Whitney U tests, as appropriate. P values less than 0.05 were considered to be statistically significant. Associations of outcomes with continuous or dichotomic variables were assessed in linear and logistic regression models, respectively. After univariate analyses, multivariate analyses were performed for significant associations. Multivariate models were obtained by backward selection, using a P value greater than 0.15 for removal from the model.
Results
Study Population: Baseline Characteristics
A total of 139 patients were included in the present analyses according to the previously defined selection criteria. Of those, 45 (32%), 52 (37%), and 42 (30%) patients had compensated or stable decompensated liver cirrhosis, acute decompensation of liver cirrhosis, and ACLF at baseline, respectively. Of the patients with ACLF, 47.6% were classified as ACLF grade 1, 28.6% as ACLF grade 2, and 23.8% as ACLF grade 3 at baseline. Of the patients without ACLF, 38 (39%) had proven infections, whereas 34 (81%) of patients with ACLF had infections. The most common infections were spontaneous bacterial peritonitis (38% of all infections), urinary tract infections (22% of all infections), pneumonia (22% of all infections), and cellulitis (8% of all infections).
As expected, patients with ACLF had significantly higher serum levels of CRP (2.7 mg/dL versus 5.3 mg/dL, P = 0.001) and peripheral blood leukocytes (8.51/nL versus 11.75/nL, P = 0.016) (Table 1). In addition, hemoglobin values were significantly lower in patients with ACLF compared with patients with decompensated cirrhosis (10.8 g/dL versus 9.3 g/dL, P = 0.006). Baseline characteristics of all patients are given in Table 1.
Table 1.
Baseline Characteristics and Laboratory Results of Included Patients
| Compensated/Stable Decompensated Cirrhosis (n = 45) | Acute Decompensation of Cirrhosis (n = 52) | ACLF (n = 42) | P Value* | |
|---|---|---|---|---|
| Age (years), mean (SD) | 57 (12) | 55 (11) | 55 (10) | 0.9 |
| Male gender, n (%) | 25 (55.3) | 37 (71.2) | 32 (76.2) | 0.1 |
| BMI (kg/m2), mean (SD) | 26.1 (5.5) | 25.2 (5.6) | 27.9 (6.5) (n = 34) | 0.08 |
| Diabetes, n (%) | 6 (13.3) | 10 (19.2) | 14 (33.3) | 0.19 |
| Listed for LTX, n (%) | 19 (42.2) | 7 (13.5) | 13 (31.0) | 0.07 |
| Origin of cirrhosis | ||||
| Alcohol, n (%) | 17 (37.8) | 31 (59.6) | 26 (61.9) | 0.9 |
| HCV/HBV, n (%) | 6 (13.3) | 4 (7.7) | 4 (9.5) | 1.0 |
| NASH, n (%) | 3 (6.7) | 2 (3.8) | 3 (7.1) | 0.8 |
| Other, n (%) | 42.2 | 15 (28.8) | 9 (21.4) | 0.6 |
| Complications of liver cirrhosis, organ failures | ||||
| Portal vein thrombosis, n (%) | 4 (8.9) | 1 (1.9) | 6 (14.3) | 0.06 |
| Gastrointestinal hemorrhage, n (%) | 0 (0) | 10 (19.2) | 7 (16.3) | 0.9 |
| Infection, n (%) | 0 (0) | 38 (73.1) | 34 (81.0) | 0.5 |
| Alcoholic hepatitis, n (%) | 1 (2.2) | 19 (32.1) | 10 (23.8) | 0.3 |
| Ascites | ||||
| Grade 0, n (%) | 32 (71.1) | 14 (27.0) | 7 (16.7) | 0.3 |
| Grade 1, n (%) | 8 (17.8) | 8 (15.4) | 10 (23.8) | 0.4 |
| Grade 2, n (%) | 2 (4.4) | 12 (23.0) | 10 (23.8) | 1.0 |
| Grade 3, n (%) | 3 (6.7) | 18 (34.6) | 15 (35.8) | 1.0 |
| Hepatic encephalopathy | ||||
| Grade 0, n (%) | 25 (55.6) | 44 (84.6) | 18 (51.4) | 0.002 |
| Grade I/II, n (%) | 20 (44.4) | 15.4 | 15 (42.9) | 0.009 |
| Grade III/IV, n (%) | 0 (0) | 0 (0) | 2 (5.7) | 0.3 |
| Kidney failure | ||||
| Creatinine < 1.5mg/dL, n (%) | 44 (97.8) | 44 (84.6) | 11 (26.2) | < 0.001 |
| Creatinine 1.5‐1.9 mg/dL, n (%) | 1 (2.2) | 7 (13.5) | 4 (9.5) | 0.789 |
| Creatinine > 2.0 mg/dL or renal replacement therapy, n (%) | 0 (0) | 1 (1.9) | 27 (64.3) | < 0.001 |
| Circulatory failure, n (%) | 0 (0) | 1 (1.9) | 13 (31.0) | < 0.001 |
| Respiratory failure, n (%) | 0 (0) | 1 (1.9) | 7 (16.7) | 0.008 |
| Admission to intensive care unit, n (%) | 1 (2.2) | 10 (18.9) | 25 (60.0) | < 0.001 |
| Laboratory data | ||||
| Leukocytes (per nL), mean (SD) | 6.60 (4.08) | 8.51 (4.85) | 11.75 (6.60) | 0.02 |
| Hemoglobin (g/dL), mean (SD) | 12.3 (2.5) | 10.8 (2.8) | 9.3 (1.7) | 0.006 |
| Platelets (per nL), mean (SD) | 153 (84) | 140 (103) | 106 (71) | 0.06 |
| CRP (mg/dL), mean (SD) | 1.3 (2.0) (n = 29) | 2.7 (2.9) (n = 48) | 5.3 (4.5) (n = 40) | 0.001 |
| Sodium (mmol/L), mean (SD) | 137 (4) (n = 40) | 135 (5.5) (n = 50) | 129 (20) (n = 38) | 0.1 |
| Potassium (mmol/L), mean (SD) | 4.33 (0.61) (n = 39) | 4.08 (0.69) (n = 50) | 4.10 (2.51) (n = 37) | 0.9 |
| Calcium (mmol/L), mean (SD) | 2.24 (0.21) (n = 41) | 2.06 (0.23) (n = 39) | 2.02 (0.33) (n = 31) | 0.7 |
| Creatinine (mg/dL), mean (SD) | 0.83 (0.25) | 1.04 (0.41) | 2.66 (1.39) | < 0.001 |
| Urea (mg/dL), mean (SD) | 32.67 (15.67) (n = 33) | 41.22 (30.04) (n = 45) | 102.22 (49.22) (n = 36) | < 0.001 |
| Bilirubin (mg/dL), mean (SD) | 2.8 (4.7) (n = 44) | 7.2 (9.2) | 14.9 (13.6) (n = 41) | 0.01 |
| AST (U/L), mean (SD) | 51 (39) (n = 40) | 124 (132) (n = 47) | 139 (138) (n = 36) | 0.8 |
| ALT (U/L), mean (SD) | 33 (22) (n = 44) | 67 (108) | 51 (39) (n = 41) | 0.7 |
| γGT (U/L), mean (SD) | 137 (145) (n = 44) | 163 (181) | 189 (183) (n = 41) | 0.3 |
| AP (U/L), mean (SD) | 130 (92) (n = 37) | 157 (93) (n = 49) | 167 (120) (n = 34) | 0.9 |
| INR, mean (SD) | 1.23 (0.36) (n = 43) | 1.54 (0,45) (n = 49) | 2.16 (0.85) (n = 40) | < 0.001 |
| APTT (seconds), mean (SD) | 33 (7) (n = 32) | 37 (13) (n = 42) | 43 (11) (n = 34) | 0.01 |
| Albumin (g/dL), mean (SD) | 4.0 (0.8) (n = 29) | 2.8 (0.5) (n = 33) | 2.9 (0.6) (n = 27) | 0.7 |
For decompensated cirrhosis versus ACLF. In 7 patients with mechanical ventilation, evaluation of hepatic encephalopathy was not possible. Significant P values are highlighted in bold.
Abbreviations: ALT, alanine aminotransferase; AP, alkaline phosphatase; AST, aspartate aminotransferase; BMI, body mass index; GT, gamma‐glutamyltransferase; and LTX, liver transplantation.
Study Population: Follow‐up Until Day 28
A summary of laboratory data at days 7 and 28 are given in Supporting Tables S1 and S2. Mean serum creatinine (P = 0.00001), bilirubin (P = 0.07), and INR (P = 0.0004) of those patients with ACLF who survived until day 7 were still substantially higher compared with patients with acute decompensation of cirrhosis (Supporting Table S1).
Of note, 6 (6%) and 11 (11%) of 97 patients without ACLF at baseline progressed to ACLF until days 7 and 28 of follow‐up, respectively. Of the patients with ACLF (n = 42) at baseline, 15 (36%) recovered to compensated/decompensated cirrhosis, whereas ACLF persisted (or progressed in stage) in 27 (64%) of patients. Until days 7 and 28 of follow‐up, 7 (17%) and 13 (31%) versus 0 (0%) and 1 (1%) patients with versus without ACLF at baseline died (P < 0.05 each). None of the patients received a liver transplant between baseline and day 28 of follow‐up.
IL‐22 Serum Concentration Is Associated With ACLF, Infections, and Mortality
IL‐22 serum levels were quantified at baseline in all patients with compensated liver cirrhosis, decompensated liver cirrhosis, or ACLF. In addition, serum cytokine levels of 16 healthy volunteers were measured as a reference (mean age 39 years [SD = 12], 9 women and 7 men). Serum levels of IL‐22 were almost identical in patients with compensated/stable decompensated liver cirrhosis compared with healthy controls (not significant), but significantly higher in patients with acute decompensation of liver cirrhosis and highest in patients with ACLF (P = 0.0001) (Fig. 1A). The association remained significant in a multivariate analysis of factors associated with ACLF at baseline (Supporting Table S3). In this regard, an IL‐22 serum concentration of 47.68 pg/mL was identified by receiver operating characteristic analysis as the optimal cutoff to predict the presence of ACLF with good accuracy (area under the curve = 0.78; P < 0.00001) (Fig. 1B). Furthermore, there was a significant association between IL‐22 serum levels and the grade of ACLF (Fig. 1C).
Figure 1.
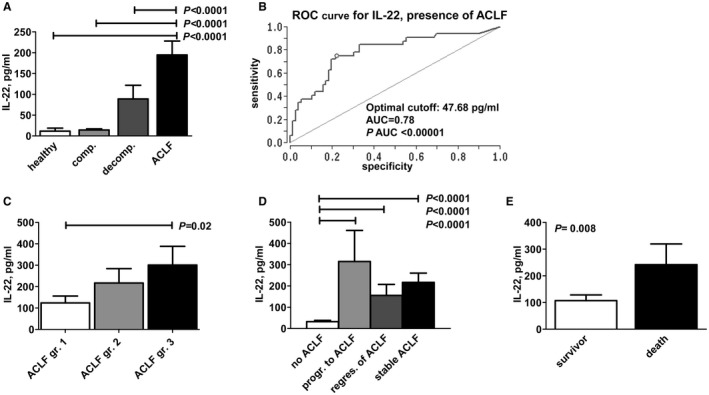
IL‐22 serum concentrations are associated with adverse outcomes of liver cirrhosis. (A) IL‐22 serum concentrations are shown according to the stage of liver cirrhosis. (B) Receiver operating characteristic analysis for IL‐22 serum concentration for the presence of ACLF. An IL‐22 serum concentration of 47.68 pg/mL has been identified as an optimal cutoff to predict ACLF. (C) IL‐22 serum concentration according to the grade of ACLF, as defined by the CLIF‐EASL consortium. (D) Association between serum concentration of IL‐22 and short‐term outcome of patients with liver cirrhosis. IL‐22 concentrations were quantified at baseline and grouped according to the respective outcomes until day 28 of follow‐up as follows: No ACLF: patients without ACLF at baseline and without progression to ACLF until follow‐up; progression (prog.) to ACLF: patients without ACLF at baseline but with progression to ACLF until follow‐up; regression (regr.) of ACLF: patients with ACLF at baseline but with resolution of ACLF until follow‐up; stable ACLF: patients with persistent ACLF from baseline until follow‐up. (E) IL‐22 concentrations were quantified at baseline and compared between patients who died or who survived until day 28 of follow‐up. Abbreviations: AUC, area under the curve; comp., compensated; decomp., decompensated; gr., grade; and ROC, receiver operating characteristic.
We next assessed associations between IL‐22 serum concentration and progression to adverse outcomes during short‐term follow‐up. In patients without ACLF at baseline and without progression to ACLF until day 28 of follow‐up, low IL‐22 serum concentrations at baseline were observed (32.45 pg/mL), whereas patients with stable ACLF from baseline to day 28 showed significantly higher serum concentrations of IL‐22 (216.82 pg/mL; P < 0.0001). In comparison, baseline IL‐22 serum levels were intermediate in patients who recovered from ACLF between baseline and day 28 (155.85 pg/mL), whereas highest baseline IL‐22 serum concentrations were observed in patients who had no ACLF at baseline but progressed to ACLF until day 28 of follow‐up (314 pg/mL; P < 0.0001) (Fig. 1D). Furthermore, IL‐22 serum levels were significantly lower in patients who survived until day 28 (107.25 pg/mL versus 241.94 pg/mL, P = 0.008) (Fig. 1E).
Finally, we explored the relationship between IL‐22 serum levels and infections in patients with or without ACLF, as infections are important precipitating events of ACLF and because infection‐triggered ACLF is burdened with a particularly high mortality.17, 18 Of note, patients without ACLF with proven infections had significantly higher IL‐22 serum concentrations than patients without ACLF and absence of infections (118 versus 15.40 pg/mL; P = 0.004) (Fig. 2). In contrast, IL‐22 serum concentrations did not differ significantly in patients with ACLF with versus without infections (Fig. 2).
Figure 2.
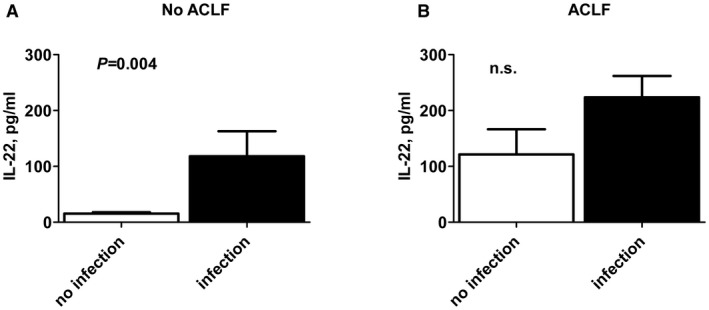
IL‐22 serum concentrations are associated with infections in patients without ACLF. (A) IL‐22 serum concentrations were quantified at baseline in patients without (A) or with (B) ACLF and compared within these groups according to the presence or absence of infection, as indicated.
Collectively, these data indicate that serum concentrations of IL‐22 are significantly associated with adverse outcomes of liver cirrhosis, namely, development of ACLF, death, and infections.
Dissociated Kinetics of IL‐22 and IL‐17A in Patients With Liver Cirrhosis
Because IL‐22 is considered to be a hallmark cytokine of Th17 cell‐mediated immune responses, we also quantified serum levels of IL‐17A, the distinctive cytokine of Th17 responses. In contrast to IL‐22, no relevant differences in IL‐17A serum concentrations between patients with acute decompensation of liver cirrhosis and ACLF were identified (7.03 versus 7.51 pg/mL; not significant), and IL‐17A serum levels were only slightly lower in patients with compensated liver cirrhosis (5.05 pg/mL) or in healthy controls (4.90 pg/mL) (Fig. 3A). Furthermore, no association between IL‐17A serum concentrations and the grade of ACLF, and no relevant difference in IL‐17A serum levels between survivors and nonsurvivors were observed (Fig. 3B,C). Hence, the kinetics of IL‐22 in patients with liver cirrhosis appear to be rather independent from Th17 cell responses.
Figure 3.
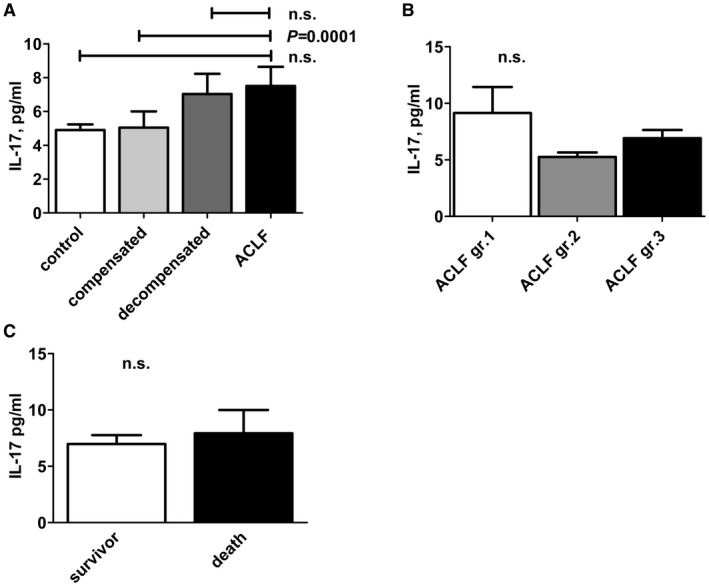
IL‐17A serum concentrations are not associated with ACLF. (A) IL‐17A serum concentrations are shown according to the indicated stage of liver cirrhosis. (B) IL‐17A serum concentrations are shown according to the grade of ACLF, as defined by the CLIF‐EASL consortium. (C) IL‐17A concentrations were quantified at baseline and compared between patients who died or who survived until day 28 of follow‐up. Abbreviations: comp., compensated; decomp., decompensated; and gr., grade.
Excessive Secretion of IL‐22BP in Patients With Liver Cirrhosis
This described association between high serum levels of IL‐22, a cytokine with known hepatoprotective effects, and adverse outcomes of liver cirrhosis may indicate insufficient biological activity of IL‐22 in humans with liver cirrhosis, or context‐specific dominant adverse effects of the proinflammatory functions of IL‐22.
To further explore these possibilities, we assessed the serum concentrations of IL‐22BP, the antagonist of IL‐22‐signaling, in our cohort. Of note, a progressive, significant increase of IL‐22BP serum levels was observed in patients with compensated/stable decompensated liver cirrhosis versus acute decompensation of liver cirrhosis versus ACLF (Fig. 4A). Importantly, mean IL‐22BP levels were exceeding IL‐22 levels more than 300‐fold with respect to the picomolar serum concentration of both proteins (compare Figs. 1 and 4). We therefore calculated ratios of IL‐22BP/IL‐22 serum levels. IL‐22BP/IL‐22 ratios were lowest in patients with ACLF and in patients with acutely decompensated cirrhosis, but significantly higher in patients with compensated/stable decompensated cirrhosis or in healthy controls (P < 0.05) (Fig. 4B). Furthermore, higher IL‐22BP serum levels were observed in patients who died until day 28 (88,256.40 pg/mL versus 58,842.60 pg/mL, P = 0.01), but ratios of IL‐22BP/IL‐22 serum concentrations were numerically (but not significantly) lower in patients who died compared with patients who survived until day 28 (4,148 versus 1,843; P = 0.09) (Fig. 4C,D). In contrast, there was an association between higher ratios of IL‐22BP/IL‐22 serum concentrations and impaired liver synthesis capacity (i.e., high INR and low serum albumin concentration) (Fig. 4E).
Figure 4.
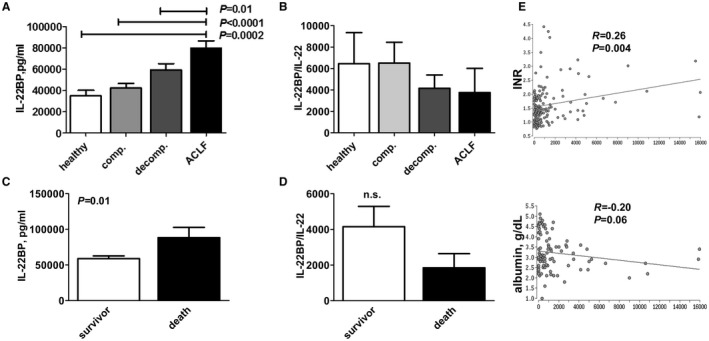
Liver cirrhosis stage‐dependent excess of IL‐22BP. (A) IL‐22BP serum concentrations are shown according to the indicated stage of liver cirrhosis. (B) The ratio of serum concentrations of IL‐22BP divided through serum concentrations of IL‐22 (shown in Fig. 1) were calculated and compared among patients with the indicated stages of liver cirrhosis. Significantly lower ratios of IL‐22BP/IL‐22 are observed in patients with decompensated liver cirrhosis and ACLF compared with patients with compensated liver cirrhosis or healthy controls. (C) IL‐22BP concentrations were quantified at baseline and compared between patients who died or who survived until day 28 of follow‐up. (D) The ratio of serum concentrations of IL‐22BP divided through serum concentrations of IL‐22 were calculated and compared between patients who died or who survived until day 28 of follow‐up. (E) The ratio of IL‐22BP to IL‐22 serum concentration is associated with liver synthesis capacity. The ratio of serum concentrations of IL‐22BP divided through serum concentrations of IL‐22 were calculated in all patients with liver cirrhosis and correlated with INR (left panel) and with serum albumin concentration (right panel). Abbreviations: comp., compensated; decomp., decompensated; and gr., grade.
IL‐22BP Inhibits Hepatocellular IL‐22‐Induced JAK/STAT Signaling
To further assess the possible functional relevance of excessive IL‐22BP secretion in patients with liver cirrhosis, we exposed human hepatoma cells to IL‐22 using concentrations covering the range observed in our patient cohort. As expected, both Huh‐7 and HepG2 cells expressed the IL‐22 receptor (Fig. 5A). Exposure of Huh‐7 or HepG2 cells with 5 ng/mL IL‐22 for 30 minutes resulted in a robust phosphorylation of the key transcription factors of IL‐22 signaling STAT1 and STAT3 (but not of STAT5, not shown) (Fig. 5B). In this regard, STAT3 promotes the well‐described positive effect of IL‐22 on hepatocellular proliferation/survival, whereas STAT1 is rather a proinflammatory transcription factor inducing a large number of proinflammatory mediators and cytokines.12
Figure 5.
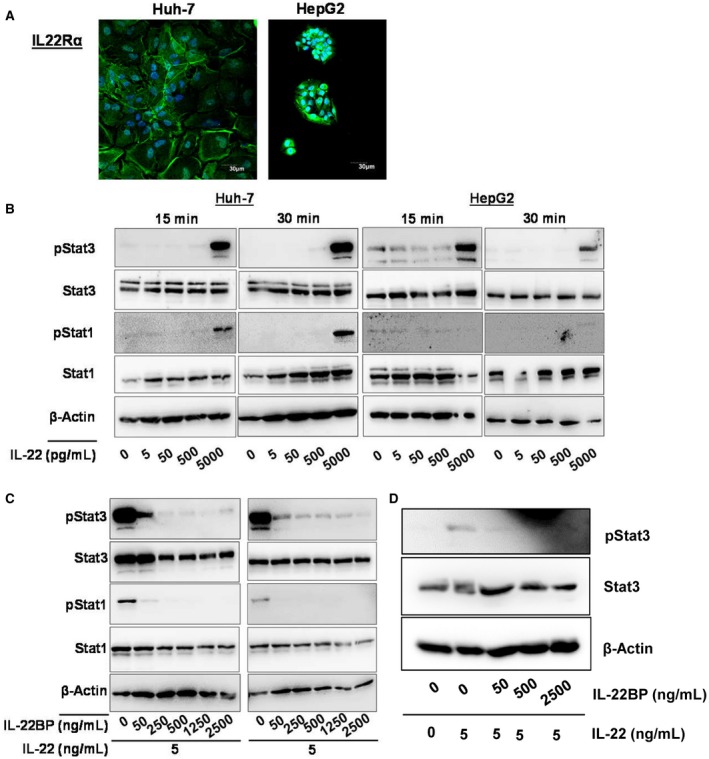
IL‐22BP inhibits hepatocellular IL‐22 signaling. (A) Expression of IL‐22Rα on Huh‐7 and HepG2 cells. IL‐22Rα was labeled with AF488 and nuclei were stained with DAPI (4´,6‐diamidino‐2‐phenylindole). (B) IL‐22 induces Stat1 and Stat3 phosphorylation in Huh‐7 and HepG2 cells. (C) IL‐22BP inhibits IL‐22‐induced Stat1 and Stat3 phosphorylation at physiological dose ranges. (D) IL‐22BP inhibits IL‐22 downstream signaling in primary human hepatocytes. Primary human hepatocytes were stimulated with or without 5 ng/mL of IL‐22 and ascending concentrations of IL‐22BP for 15 minutes, as indicated, and cellular phospho‐Stat3, total Stat3, and β‐actin protein levels were quantified by immunoblot analysis.
Next, we asked whether IL‐22BP at concentrations observed in our cohort is sufficient to suppress hepatocellular IL‐22 signaling. To address this question, human hepatoma cell lines were incubated with IL‐22 together with various concentrations of IL‐22BP. Supplementation of IL‐22BP to IL‐22 in ratios of 10 and 50 or more substantially and completely blocked IL‐22‐induced STAT3 phosphorylation in vitro, respectively (Fig. 5C). This finding could be confirmed in primary human hepatocytes (Fig. 5D).
As described previously, in vivo ratios of IL‐22BP to IL‐22 were even higher. We therefore tested the ability of patient serum with low versus high ratios of IL‐22BP/IL‐22 to induce STAT3 phosphorylation in human hepatoma cell lines. The capacity of patient serum to induce STAT3 phosphorylation was substantially higher for the presence of low versus high IL‐22BP/IL‐22 ratios, and STAT3 phosphorylation was blocked and became a neutralizing antibody against the IL‐22 receptor (Fig. 6).
Figure 6.
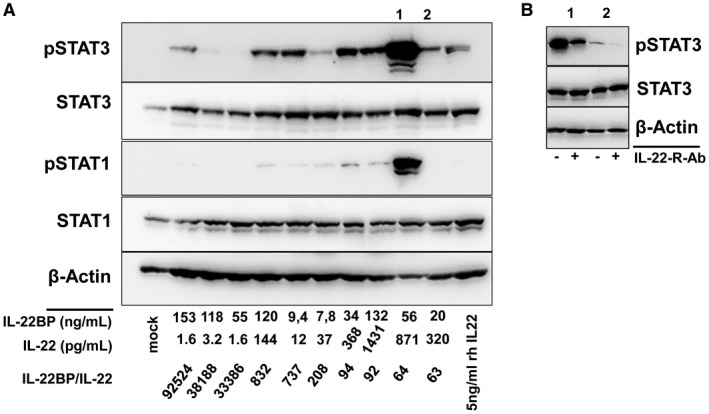
Ability of patient serum to induced hepatocellular IL‐22 signaling depends on the IL‐22BP/IL‐22 ratio. Huh‐7 cells were incubated with representative serum samples of patients with low and high IL‐22BP/IL‐22 ratios for 15 minutes. (A) Western blot analyses for the indicated antigens were performed. IL‐22BP and IL‐22 concentrations as well as IL‐22BP/IL‐22 ratios of the patient samples are shown. (B) Cells were pretreated with or without a neutralizing antibody against the IL‐22 receptor before exposure to patient serum with high IL‐22 activity (1 and 2 from [A]) for 15 minutes.
IL‐22BP Inhibits IL‐22‐Induced Production of Proinflammatory Mediators
To elaborate on the effect of the described inhibition of IL‐22‐induced STAT‐phosphorylation by IL‐22BP in hepatocytes, we assessed the two relevant functions of IL‐22 signaling: promotion of cell viability and induction of proinflammatory mediators. IL‐22 numerically (but not statistically significantly) increased the viability of Huh‐7 cells, an effect that was abolished by IL‐22BP (Fig. 7A). Furthermore, hepatocellular stimulation with IL‐22 resulted in a moderate induction of TNF‐α messenger RNA (mRNA) in Huh‐7 cells (Fig. 7B), as well as a strong induction of LBP mRNA levels (Fig. 7C), an important acute‐phase protein and target gene of IL‐22.20 The induction of LPB by IL‐22 was confirmed on the protein level in HepG2 cells as well as in primary human hepatocytes (Fig. 7D,E). Of note, supplementation of IL‐22BP to IL‐22 in ratios of 10‐500 completely blocked IL‐22‐induced production of LBP in both HepG2 cells and in primary human hepatocytes (Fig. 7E).
Figure 7.
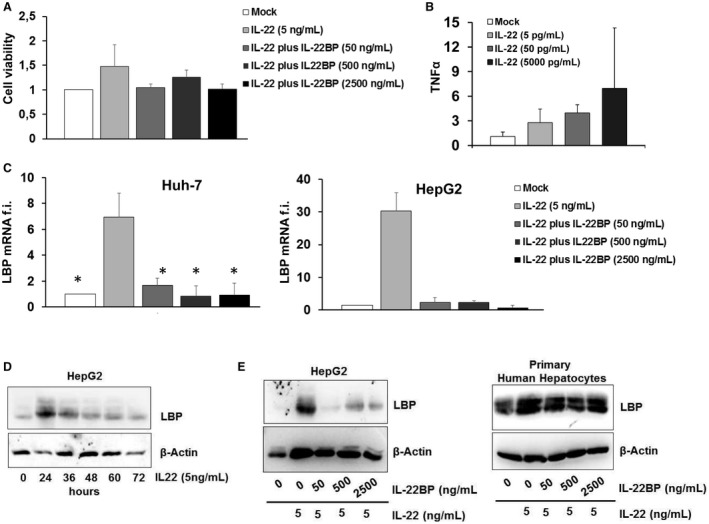
Effect of IL‐22BP on IL‐22‐induced production of proinflammatory mediators and cell viability. (A) Cell viability was assessed in Huh‐7 cells using the WST‐1 assay 6 hours after stimulation with IL‐22 with or without IL‐22BP at the indicated concentrations. (B) Induction of TNF‐α mRNA in Huh‐7.5 cells treated for 6 hours with the indicated concentrations of IL‐22. TNF‐α mRNA levels were quantified by quantitative polymerase chain reaction (PCR) and expressed relative to GAPDH (glyceraldehyde 3‐phosphate dehydrogenase). (C) Huh‐7 cells and HepG2 cells were stimulated with IL‐22 with or without IL‐22BP at the indicated concentrations for 6 hours. mRNA levels of LBP were quantified by quantitative PCR and expressed relative to GAPDH. (D) Quantification of LBP protein levels in HepG2 cells stimulated with IL‐22 for the indicated time points. (E) IL‐22BP abolishes IL‐22‐mediated LBP protein production in HepG2 cells and primary human hepatocytes. Cells were stimulated with 5 ng/mL of IL‐22 and ascending concentrations of IL‐22BP for 24 hours. Protein levels were analyzed by immunoblotting using β‐actin as a control.
Discussion
The present prospective cohort study reveals a strong association between IL‐22 serum levels and adverse outcomes of liver cirrhosis (i.e., ACLF, infections, and death). However, serum concentrations of IL‐22BP, a soluble inhibitor of IL‐22 signaling, exceed IL‐22 concentrations excessively and are capable of inhibiting hepatocellular IL‐22 signaling at these concentrations in vitro. In line, high IL‐22BP/IL‐22 ratios are associated with impaired liver synthesis capacity in vivo but also with an inhibition of IL‐22‐induced production of acute‐phase proteins in primary hepatocytes. Importantly, we observed declining ratios of IL‐22BP/IL‐22 during the progression of liver cirrhosis to ACLF and ultimately death, which may not be sufficient to prevent proinflammatory adverse effects of IL‐22 in these clinical scenarios, which are characterized by deleterious levels of systemic inflammation.
In general, IL‐22 is considered to have beneficial properties in liver diseases. In this regard, a key study has shown that IL‐22 promotes the expansion of liver progenitor/stem cells in mice and in patients with hepatitis B in a STAT3‐dependent manner.9 Furthermore, IL‐22 protects hepatocytes from apoptosis in a miR‐15a/16‐1‐dependent manner in mice.19 In addition, a number of studies have shown convincingly that IL‐22 strengthens the intestinal barrier by targeting intestinal stem cells and epithelial cells and by inducing important intestinal defensins or lectins such as Reg3β.20 These and other studies suggest that IL‐22 could be an attractive agent for the treatment of advanced liver disease. Indeed, IL‐22 application in animal models of concanavalin A–induced hepatitis or alcoholic hepatitis reduced the severity of liver disease.21, 22 In light of these findings, therapeutic application of IL‐22 is currently being evaluated in a controlled clinical trial for the treatment of alcoholic hepatitis (NCT02655510).
However, IL‐22 also has proinflammatory functions that may adversely affect the outcome of patients with severe liver diseases. For example, IL‐22 was shown to promote liver inflammation by recruiting Th17 cells to the liver in humans and mice with HBV infection.23 As shown in our study and in previous studies, IL‐22 can induce hepatocellular production of proinflammatory mediators and acute‐phase proteins.24 In the present study, high IL‐22 serum concentrations in patients with liver cirrhosis were associated with the presence of and progression to ACLF as well as with mortality. The association between IL‐22 and these adverse endpoints in patients with liver cirrhosis might be explained by the proinflammatory properties of IL‐22, which might be insufficiently prevented by IL‐22BP, as ratios of IL‐22BP/IL‐22 are lower in patients with ACLF compared with patients without ACLF. IL‐22BP could therefore play a context‐specific role in advanced liver cirrhosis. One may speculate that excessive secretion of IL‐22BP in patients with compensated and decompensated liver cirrhosis results in nonfunctional IL‐22 signaling in these scenarios and prevents beneficial effects of IL‐22 on liver function and regeneration—an assumption that is in line with the observed association of high IL‐22BP/IL‐22 ratios and impaired liver synthesis capacity. However, as discussed previously, IL‐22BP levels may not be sufficient to prevent adverse proinflammatory functions of IL‐22 in patients with ACLF in whom systemic inflammation is detrimental for short‐term prognosis. This latter notion would be in line with a recently identified protective role of IL‐22BP in a murine model of acute liver failure.12 Collectively, future studies evaluating therapeutic IL‐22 administration may consider that high doses of IL‐22 may be required to overcome the inhibitory functions of IL‐22BP, and that a cautious use of IL‐22 in severely ill patients with organ failures may be warranted.
Our study has a number of limitations. First, we were not able to quantify hepatic IL‐22 and IL‐22BP levels, as liver biopsies are rarely performed in patients with advanced liver disease in our hospital. Furthermore, the number of patients who died during follow‐up was relatively low. Finally, the clinical relevance of IL‐22 as a biomarker for ACLF is probably rather limited in light of excellent prognosis scores of this syndrome. However, the value of our study is rather in exploring the behavior of this special and important cytokine and its inhibitor IL‐22BP during the progression of liver cirrhosis.
In conclusion, our study shows that high IL‐22 levels and low ratios of IL‐22BP/IL‐22 are associated with ACLF and the mortality of patients with cirrhosis. Excessive secretion of IL‐22BP can neutralize IL‐22 in vitro and may prevent—likely in a context‐specific manner—hepatoprotective, but also adverse, effects of IL‐22 in patients with cirrhosis.
Author Contributions
The authors have contributed to the manuscript by planning the study (C.M.L.), collecting the data (K.S., S.R., S.B., C.C., M.M., D.F., A.W., C.M.L.), analyzing and interpreting the data (K.S., S.R., A.W., S.Z., C.W., C.M.L.), and preparing and revising of the manuscript (all authors).
Supporting information
Supported by the Deutsche Forschungsgemeinschaft (LA 2806/2‐1 and LA 2806/5‐1 to C.M.L.).
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Moreau R, Jalan R, Gines P, Pavesi M, Angeli P, Cordoba J, et al. Acute‐on‐chronic liver failure is a distinct syndrome that develops in patients with acute decompensation of cirrhosis. Gastroenterology 2013;144:1426‐1437,e1421‐e1429. [DOI] [PubMed] [Google Scholar]
- 2. Claria J, Stauber RE, Coenraad MJ, Moreau R, Jalan R, Pavesi M, et al. Systemic inflammation in decompensated cirrhosis: characterization and role in acute‐on‐chronic liver failure. Hepatology 2016;64:1249‐1264. [DOI] [PubMed] [Google Scholar]
- 3. Wolk K, Witte E, Witte K, Warszawska K, Sabat R. Biology of interleukin‐22. Semin Immunopathol 2010;32:17‐31. [DOI] [PubMed] [Google Scholar]
- 4. Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, et al. IL‐22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 2012;491:259‐263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kotenko SV, Izotova LS, Mirochnitchenko OV, Esterova E, Dickensheets H, Donnelly RP, et al. Identification, cloning, and characterization of a novel soluble receptor that binds IL‐22 and neutralizes its activity. J Immunol 2001;166:7096‐7103. [DOI] [PubMed] [Google Scholar]
- 6. Pelczar P, Witkowski M, Perez LG, Kempski J, Hammel AG, Brockmann L, et al. A pathogenic role for T cell‐derived IL‐22BP in inflammatory bowel disease. Science 2016;354:358‐362. [DOI] [PubMed] [Google Scholar]
- 7. Xu W, Presnell SR, Parrish‐Novak J, Kindsvogel W, Jaspers S, Chen Z, et al. A soluble class II cytokine receptor, IL‐22RA2, is a naturally occurring IL‐22 antagonist. Proc Natl Acad Sci U S A 2001;98:9511‐9516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JK, et al. A human natural killer cell subset provides an innate source of IL‐22 for mucosal immunity. Nature 2009;457:722‐725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Feng D, Kong X, Weng H, Park O, Wang H, Dooley S, et al. Interleukin‐22 promotes proliferation of liver stem/progenitor cells in mice and patients with chronic hepatitis B virus infection. Gastroenterology 2012;143:188‐198,e187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu Y, Verma VK, Malhi H, Gores GJ, Kamath PS, Sanyal A, et al. Lipopolysaccharide downregulates macrophage‐derived IL‐22 to modulate alcohol‐induced hepatocyte cell death. Am J Physiol Cell Physiol 2017;313:C305‐C313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kronenberger B, Rudloff I, Bachmann M, Brunner F, Kapper L, Filmann N, et al. Interleukin‐22 predicts severity and death in advanced liver cirrhosis: a prospective cohort study. BMC Med 2012;10:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kleinschmidt D, Giannou AD, McGee HM, Kempski J, Steglich B, Huber FJ, et al. A protective function of IL‐22BP in ischemia reperfusion and acetaminophen‐induced liver injury. J Immunol 2017;199:4078‐4090. [DOI] [PubMed] [Google Scholar]
- 13. Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham‐Anderson J, Wu J, et al. Interleukin‐22, a T(H)17 cytokine, mediates IL‐23‐induced dermal inflammation and acanthosis. Nature 2007;445:648‐651. [DOI] [PubMed] [Google Scholar]
- 14. Blei AT, Cordoba J. Practice Parameters Committee of the American College of Gastroenterology. Hepatic encephalopathy. Am J Gastroenterol 2001;96:1968‐1976. [DOI] [PubMed] [Google Scholar]
- 15. Moore KP, Wong F, Gines P, Bernardi M, Ochs A, Salerno F, et al. The management of ascites in cirrhosis: report on the consensus conference of the International Ascites Club. Hepatology 2003;38:258‐266. [DOI] [PubMed] [Google Scholar]
- 16. Lange CM, Gouttenoire J, Duong FH, Morikawa K, Heim MH, Moradpour D. Vitamin D receptor and Jak‐STAT signaling crosstalk results in calcitriol‐mediated increase of hepatocellular response to IFN‐alpha. J Immunol 2014;192:6037‐6044. [DOI] [PubMed] [Google Scholar]
- 17. Fernandez J, Acevedo J, Wiest R, Gustot T, Amoros A, Deulofeu C, et al. Bacterial and fungal infections in acute‐on‐chronic liver failure: prevalence, characteristics and impact on prognosis. Gut 2018;67:1870‐1880. [DOI] [PubMed] [Google Scholar]
- 18. Mucke MM, Rumyantseva T, Mucke VT, Schwarzkopf K, Joshi S, Kempf VAJ, et al. Bacterial infection‐triggered acute‐on‐chronic liver failure is associated with increased mortality. Liver Int 2018;38:645‐653. [DOI] [PubMed] [Google Scholar]
- 19. Lu Z, Liu J, Liu X, Huang E, Yang J, Qian J, et al. MiR‐15a, 16‐1 suppresses AHR‐dependent IL‐22 secretion in CD4(+) T cells and contributes to immune‐mediated organ injury. Hepatology. In press. [DOI] [PubMed] [Google Scholar]
- 20. Dudakov JA, Hanash AM, van den Brink MR. Interleukin‐22: immunobiology and pathology. Annu Rev Immunol 2015;33:747‐785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ki SH, Park O, Zheng M, Morales‐Ibanez O, Kolls JK, Bataller R, et al. Interleukin‐22 treatment ameliorates alcoholic liver injury in a murine model of chronic‐binge ethanol feeding: role of signal transducer and activator of transcription 3. Hepatology 2010;52:1291‐1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Radaeva S, Sun R, Pan HN, Hong F, Gao B. Interleukin 22 (IL‐22) plays a protective role in T cell‐mediated murine hepatitis: IL‐22 is a survival factor for hepatocytes via STAT3 activation. Hepatology 2004;39:1332‐1342. [DOI] [PubMed] [Google Scholar]
- 23. Zhao J, Zhang Z, Luan Y, Zou Z, Sun Y, Li Y, et al. Pathological functions of interleukin‐22 in chronic liver inflammation and fibrosis with hepatitis B virus infection by promoting T helper 17 cell recruitment. Hepatology 2014;59:1331‐1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou Z, Xu MJ, Gao B. Hepatocytes: a key cell type for innate immunity. Cell Mol Immunol 2016;13:301‐ 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials