

Abstract
The rising incidence of cholangiocarcinoma (CCA) coupled with a low 5‐year survival rate that remains below 10% delineates the urgent need for more effective treatment strategies. Although several recent studies provided detailed information on the genetic landscape of this fatal malignancy, versatile model systems to functionally dissect the immediate clinical relevance of the identified genetic alterations are still missing. To enhance our understanding of CCA pathophysiology and facilitate rapid functional annotation of putative CCA driver and tumor maintenance genes, we developed a tractable murine CCA model by combining the cyclization recombination (Cre)‐lox system, RNA interference, and clustered regularly interspaced short palindromic repeats/CRISPR associated protein 9 (CRISPR/Cas9) technology with liver organoids, followed by subsequent transplantation into immunocompetent, syngeneic mice. Histologically, resulting tumors displayed cytokeratin 19–positive ductal structures surrounded by a desmoplastic stroma—hallmark features of human CCAs. Despite their initial biliary phenotype in vitro, organoids retained the plasticity to induce a broader differentiation spectrum of primary liver cancers following transplantation into recipient mice, depending on their genetic context. Thus, the organoid system combines the advantage of using nontransformed, premalignant cells to recapitulate liver tumorigenesis as a multistep process, with the advantage of a reproducible and expandable cell culture system that abrogates the need for recurrent isolations of primary cells. Conclusion: Genetically modified liver organoids are able to transform into histologically accurate CCAs. Depending on the oncogenic context, they are also able to give rise to liver cancers that show features of hepatocellular carcinomas. The model can be used to functionally explore candidate cancer genes of primary liver cancers in immunocompetent animals and evaluate novel treatment regimens.
Abbreviations
- 2D
2‐dimensional
- 3D
3‐dimensional
- CCA
cholangiocarcinoma
- CK7
cytokeratin 7
- CK19
cytokeratin 19
- Cre
cyclization recombination
- CRISPR/Cas9
clustered regularly interspaced short palindromic repeats/CRISPR associated protein 9
- EpCAM
epithelial cell adhesion molecule
- GFP
green fluorescent protein
- HCC
hepatocellular carcinoma
- Kras
Kirsten rat sarcoma oncogene
- lsl
lox‐stop‐lox
- Myc
myelocytomatosis oncogene
- p44/42 MAPK
p44/42 mitogen‐activated protein kinase
- Pten
phosphatase and tensin homolog
- sgRNA
single‐guide RNA
- shRNA
short hairpin RNA
- sq
subcutaneous
- WT
wild type
Three‐dimensional (3D) organoid cultures have been derived successfully from a variety of organ systems and cell types, such as intestinal cells, cerebral tissues, and, more recently, liver cells.1 These culture systems exhibit commitment to the respective tissue of origin and faithfully retain their genetic profiles, even after long‐term expansion.2 Although the exact localization of the liver organoid–initiating cell remains elusive, the organoids cultured under conditions described by Huch et al. likely originate from epithelial cell adhesion molecule (EpCAM+) biliary epithelial cells and not from hepatocytes, the most abundant cell type in the liver.2, 3 Consequently, these cells readily express biliary markers in vitro. Seminal work recently demonstrated that, apart from normal liver tissue, organoids can be established from human liver cancer specimens, and organoids derived from hepatocellular carcinoma (HCC), cholangiocarcinoma (CCA), or mixed subtypes are able to maintain the genetic and histological hallmarks of the initial tumor specimen in vitro.4
CCA is a dismal malignancy that is highly refractory to current chemotherapeutic regimens and targeted therapies. At the time of diagnosis, most patients present with advanced disease not amenable to surgical resection.5 Histologically, CCA commonly exhibits a ductal growth pattern with an abundant fibrous stroma.
Despite an increasing wealth of sequencing data from human tumor specimens and the identification of recurrent genetic alterations, as well as key prognostic transcriptome signatures, detailed understanding of actionable molecular alterations remains elusive.6, 7, 8 Here, we describe how primary murine liver organoids can be used to study CCA in vivo and in vitro and aid in the translational annotation of the CCA mutagenome.
Materials and Methods
Additional information, including antibodies, sample preparations, introduction of short hairpin RNAs (shRNAs) and single‐guide RNAs (sgRNAs) into organoids, bioinformatic analyses, and statistical procedures, is provided in the Supporting Information.
Animal Experiments
All experiments involving mice were performed according to animal protocols approved by local authorities (the Lower Saxony State Office for Consumer Protection and Food Safety). Animals were maintained under standard housing conditions with a 12‐hour day–night cycle and access to food and water ad libitum. All interventions were performed during the day cycle. Recipient mice (C57BL/6J and NSG [NOD.Cg‐PrkdcscidIl2rgtm1Wjl/SzJZtm], 5‐8 weeks old) were obtained from the local animal facility (Hannover Medical School, Germany). Kirsten rat sarcoma oncogene Krastm4Tyj (KraslslG12D)9 mice were a gift from Dieter Saur (Munich, Germany), and Krastm4Tyj‐Trp53tm1Brn (KraslslG12D/wt; p53lox/lox)10 mice were a gift from Florian Kühnel (Hannover, Germany). C57BL/6J mice were used as recipients for organoids derived from C57BL/6J or syngeneic KraslslG12D mice. Organoids derived from KraslslG12D/wt; p53lox/lox animals (mixed background) were implanted into NSG mice.
Isolation of Murine Liver Organoids
Murine organoids were isolated from adult C57BL/6J mice, KraslslG12D mice, or KraslslG12D/wt; p53lox/lox mice according to published protocols.11 Briefly, murine livers were minced and enzymatically digested in Earle’s Balanced Salt Solution (EBSS; Thermo Fisher, Waltham, MA) containing 2.5 mg/mL Collagenase Type IV (Sigma‐Aldrich, St. Louis, MO) and 0.1 mg/mL DNase I (Sigma‐Aldrich) for 20 to 40 minutes with repeated pipetting and passed through a 70‐µm cell strainer. After additional washes, cells were spun at 300g, resuspended in 100% Growth Factor Reduced Matrigel (Corning, NY), and plated (two 50‐µL droplets per 24‐well). After solidification, Matrigel droplets were overlaid with 500‐µL murine liver organoid media according to published protocols.11 Alternatively, residual fragments retained in the cell strainer after collagenase digest were plated in Matrigel. For passaging, organoids were mechanically disrupted by repeated pipetting using a P200 pipette tip, followed by a 5‐minute to 8‐minute enzymatic digestion in TrypLE Express solution (Thermo Fisher).
Tumor Cell Isolation
Tumors were minced and enzymatically digested in a shaking incubator with Collagenase IV 1 mg/mL (Sigma‐Aldrich) in EBSS (Thermo Fisher) for 30 minutes at 37°C. Cells were washed, spun at 300g, and resuspended either in Matrigel and overlaid with organoid expansion medium for organoid culture or resuspended and plated on tissue culture dishes in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% penicillin‐streptomycin for 2‐dimensional (2D) culture.
Cell Viability Assay
KraslslG12D/wt; p53lox/lox and KrasG12D/wt; p53∆/∆ organoids were seeded in 100‐µL 10% Matrigel per 96‐well. Two, 24, and 48 hours after seeding, the organoids were lysed and CellTiter‐Glo 3D Luminescent Cell Viability Assay (Promega, Madison, WI) was performed according to the manufacturer’s protocol.
For inhibitor treatment, KrasG12D/wt; p53∆/∆; LMP_shRenilla.713 tumor cell lines established as 2D or 3D cultures from primary, organoid‐derived tumors were plated at 1,000 cells (2D cell line) and 10,000 cells (3D cell line) per 96‐well and treated with selumetinib (3.36 µM; MedChem Express, Monmouth, NJ), NVP‐BKM120 (0.25 µM; MedChem Express), or a combination of both inhibitors for 24 hours and 48 hours. At the indicated time points, cells were lysed using CellTiter‐Glo Luminescent Cell Viability Assay or CellTiter‐Glo 3D Luminescent Cell Viability Assay, and luminescence was acquired on a Glomax Multi Detection System (Promega, Madison, WI).
Flow Cytometry
Single cell suspensions from murine livers or organoids were prepared and incubated with primary antibody (1:100 dilution) for 30 minutes at 4°C (Allophycocyanin‐EpCAM; Thermo Fisher; Cat. #17‐5791‐80). Flow cytometry was performed on a FacsCanto (BD Biosciences, San Jose, CA).
Results
Expression of Oncogenic Kras in Combination With p53 Loss in Transplanted Liver Organoids Gives Rise to CCA in Recipient Mice
We previously developed a murine model for CCA based on the transplantation of fetal liver cells.12 Because fetal liver cells cannot be propagated long term in vitro, the model required the continuous maintenance of multi‐allelic colonies, timed matings, and isolation of primary cells. We aimed to evaluate the use of liver organoids to create a CCA model that eliminates the need for repetitive fetal liver cell isolations while maintaining the advantage of using nontransformed, premalignant cells to recapitulate tumorigenesis as a multistep process.
The Kras oncogene and the tumor suppressor Tp53 are among the most frequently mutated genes in CCA.13, 14 Hence, we isolated organoids from adult KraslslG12D/wt; p53lox/lox mice and cultured them according to published protocols.11 As anticipated, we could detect expression of cytokeratin 19 (CK19), cytokeratin 7 (CK7), sex determining region Y (SRY)‐box 9, and EpCAM, in line with a biliary phenotype of liver organoids in vitro (Fig. 1 A,B; Supporting Fig. S1). Interestingly, when we slightly modified the isolation procedure by plating the fragments that remained after collagenase digestion instead of plating liver cell suspensions, organoids budded abundantly from the fragments, leading to an accelerated establishment of stable organoid cultures (Supporting Fig. S2).
Figure 1.
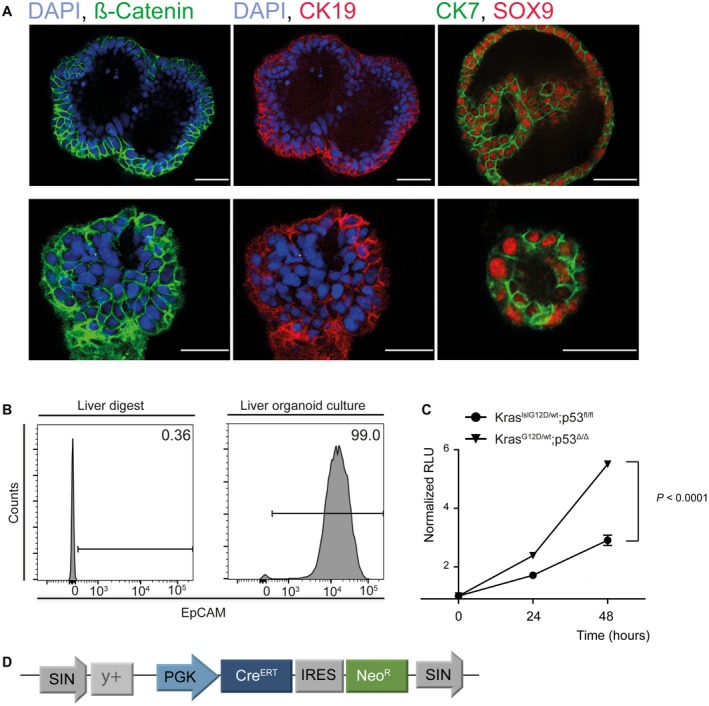
Murine liver organoids express markers of biliary differentiation. (A) Immunofluorescence staining of organoids reveals expression of biliary markers such as CK7, CK19, and SOX9. Scale bars correspond to 50 µm. (B) The adult murine liver contains only a minor fraction of EpCAM‐positive cells as determined by flow cytometry (left histogram), whereas nearly all liver‐derived organoids are EpCAM‐positive (right histogram). (C) Activation of a latent KrasG12D and loss of p53 lead to increased proliferation as assessed by cell viability assay. (D) Schematic of the retroviral vector pSIN‐PGK‐CreERT‐IRES‐NeomycinResistance that was used to activate the latent Kras allele and delete p53 following tamoxifen treatment. Abbreviations: DAPI, 4′,6‐diamidino‐2‐phenylindole; RLU, relative light units; and SOX9, sex determining region Y (SRY)‐box 9.
To activate the latent mutant Kras allele and excise the p53 allele, we transduced the organoids with a retroviral vector containing a tamoxifen regulatable Cre‐recombinase and a neomycin resistance cassette (Fig. 1D), and confirmed excision of the respective lox sites by polymerase chain reaction (PCR) (Supporting Fig. S3). Following activation of the latent alleles, proliferation of the organoids increased without any overt morphological alterations (Fig. 1C and Supporting Fig. S3B).
Next, we aimed to determine whether transplantation of the genetically modified organoids leads to tumor development in recipient mice (Fig. 2A). Following subcutaneous (sq) injection, tumors developed with 100% penetrance and were harvested after 8 weeks. Consistent with activation of mutant Kras and loss of p53, Cre‐recombined organoids before transplantation as well as organoids derived from resulting tumors (tumoroids) exhibited increased phosphorylated p44/42 mitogen‐activated protein kinase (p‐p44/42 MAPK) levels and loss of p21 (cyclin‐dependent kinase inhibitor 1A) expression (Fig. 2C). Histological examination uniformly revealed gland‐forming adenocarcinomas with CK19‐positive tumor cells surrounded by an abundant stromal reaction, compatible with moderately differentiated CCA (Fig. 2B).
Figure 2.
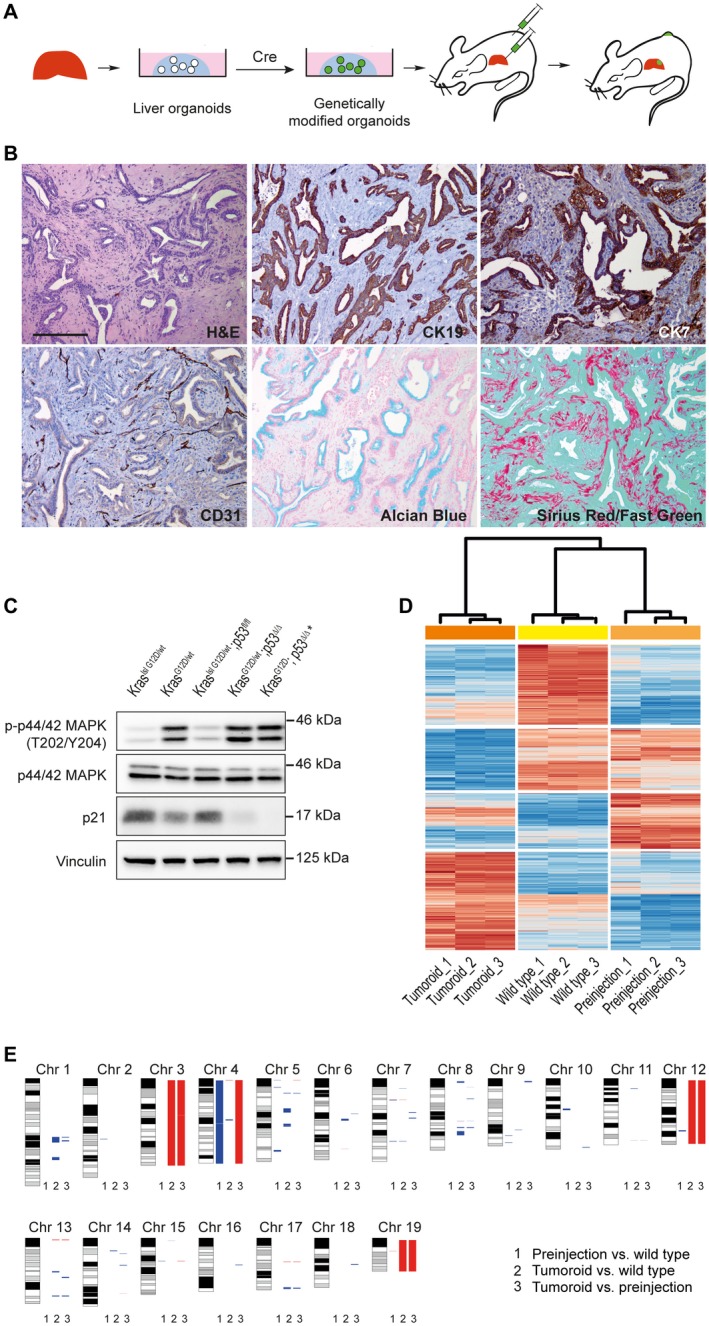
Genetically modified liver organoids give rise to CCA in vivo. (A) Technical outline: Murine organoids lead to tumor formation in recipient mice following Cre‐mediated activation of mutant Kras and loss of the tumor suppressor p53. (B) Tumors exhibit glandular differentiation and stromal desmoplasia, resembling human CCA. CK7 and CK19 expression in tumor cells confirm ductal differentiation. Alcian blue staining indicates elevated production of mucins. Cluster of differentiation 31 stains blood vessels, and sirius red/fast green confirms the presence of collagen fibers. Scale bars correspond to 200 µm. (C) Immunoblotting shows p‐p44/42 MAPK up‐regulation in mutant Kras expressing organoids and p21 down‐regulation following loss of p53. (Genotypes of organoids before Cre activation and after Cre activation are indicated.) Asterisk marks tumor‐derived cell line. (D) Expression profiles of KraslslG12D/wt; p53lox/lox (wild type) organoids, KrasG12D/wt; p53Δ/Δ (pre‐injection) organoids, and organoids derived from tumors of transplanted KrasG12D/wt; p53Δ/Δ organoids (tumoroids). Unsupervised cluster analyses were performed based on differentially expressed genes using Euclidean distance. (E) Array comparative genomic hybridization analysis reveals acquisition of copy number alterations after genetic modification of organoids and during in vivo tumorigenesis. Red, deletion; blue, amplification. Lane 1: KrasG12D/wt; p53Δ/Δ pre‐injection versus KraslslG12D/wt; p53lox/lox organoids (WT). Lane 2: KrasG12D/wt; p53Δ/Δ tumoroids versus KraslslG12D/wt; p53lox/lox organoids (WT). Lane 3: KrasG12D/wt; p53Δ/Δ tumoroids versus KrasG12D/wt; p53Δ/Δ organoids (pre‐injection). Abbreviations: aCGH, array comparative genomic hybridization; CD31, cluster of differentiation 31; Chr, chromosome; CNAs, copy number alterations; H&E, hematoxylin and eosin; lsl, lox‐stop‐lox; p21, cyclin‐dependent kinase inhibitor 1A; and p‐p44/42 MAPK, phosphorylated p44/42 mitogen‐activated protein kinase.
Organoids Alter Their Genetic and Molecular Profiles During Neoplastic Transformation
To delineate whether liver organoids acquire additional genetic alterations following genetic modification during in vitro culture and in vivo tumorigenesis, we performed genomic copy number analysis of KrasG12D/wt; p53∆/∆ organoids before injection into recipient mice and of organoids derived from the resulting tumor in comparison to the parental wild type (WT) organoids. In vitro activation of mutant Kras and loss of p53 led to multiple genomic deletions and amplifications in pre‐injection organoids and even more so after in vivo tumorigenesis in the isolated tumoroids (Fig. 2E). Several of these alterations correspond to regions with oncogenic potential altered in human CCA patient samples (Supporting Table S1).
Next, we performed whole transcriptome analysis to assess transcriptome profiles of WT organoids and Cre‐activated pre‐injection organoids as well as tumoroids. Unsupervised cluster analyses revealed that WT organoids and Cre‐activated pre‐injection organoids shared similar expression profiles, whereas tumoroids exhibited a markedly distinct expression profile (Fig. 2D). Gene set enrichment analysis (GSEA) demonstrated that during the progression from wild type to pre‐injection cells, gene sets associated with cell cycle regulation, E2F and myelocytomatosis oncogene (MYC) target genes were activated, in line with increased proliferation of the genetically modified organoids observed in vitro (Fig. 1C). Tumoroids isolated from murine CCAs exhibited activation of several gene sets associated with inflammation and paracrine signaling when compared with pre‐injection organoids, which might be of importance during tumor progression (Supporting Fig. S4).
Liver Organoids for Preclinical Drug Testing In Vivo and In Vitro
Therapeutic approaches are limited in patients with CCA, and first‐line chemotherapy includes gemcitabine‐based treatment protocols. Therefore, we tested the effect of gemcitabine on tumors arising in our murine CCA model. Similar to human patients, gemcitabine treatment led to a moderate survival benefit (median overall survival gemcitabine versus vehicle: 42 days versus 32 days; Supporting Fig. S5), but not to complete tumor regression, suggesting that the model is suitable for preclinical drug testing in an immunocompetent in vivo situation.
Apart from more complex in vivo model systems, tumor cell lines are an indispensable tool to study the molecular pharmacology and explore the efficacy of anticancer drugs in a preclinical setting.
To assess the scalability of the system for drug testing, we first tested whether the system is suitable to generate cancer‐derived cell lines that are serially transplantable, while maintaining the histological features of the primary malignancy. Tumors derived from sq‐injected KrasG12D/wt; p53∆/∆ organoids were collagenase‐digested, and cells were cultured and passaged either under standard 2D cell culture conditions in serum‐containing media or kept as 3D tumoroid cultures over 14 days (Fig. 3A). Both 2D and 3D cell lines gave rise to predominantly moderately differentiated CCAs with similar relative stromal content (CK19‐negative area) following sq transplantation (Fig. 3A‐C). Thus, the organoid model can be used to generate well‐expandable, genetically defined 2D and 3D tumor cell lines that maintain the histology of the primary tumor following retransplantation. Next, we interrogated whether in vitro susceptibility toward targeted therapies depends on the culture condition. Combinatorial treatment with mitogen‐activated protein kinase kinase (MAPKK) and phosphatidylinositol 3‐kinase (PI3K) inhibitors has been suggested for Kras mutant cancers15, 16 and CCA.17 Genetically engineered tumor‐derived cell lines established in parallel as 2D versus 3D cultures from the same Kras mutant murine CCA were treated with a MAPKK inhibitor (selumetinib) and a PI3K inhibitor (BKM‐120), alone and in combination. Independent of the culture conditions, both 2D and 3D cultures exhibited a comparable response to targeted therapy in vitro, as assessed by luminescence assay (Fig. 3D), indicating that, although organoids can be conveniently used to engineer genetically defined primary tumors, 2D cell lines derived from these tumors are a viable tool for in vitro drug studies. Cell cycle analysis revealed that 24‐hour treatment of the 2D cell lines with the respective drugs caused an accumulation of cells in the gap 0 phase/gap 1 phase (G0/G1 phase) and a concomitant S‐phase reduction (Fig. 3D, lower panel). Cell lines derived from non‐Kras mutant tumors did not exhibit comparable growth inhibition (Supporting Fig. S8C).
Figure 3.
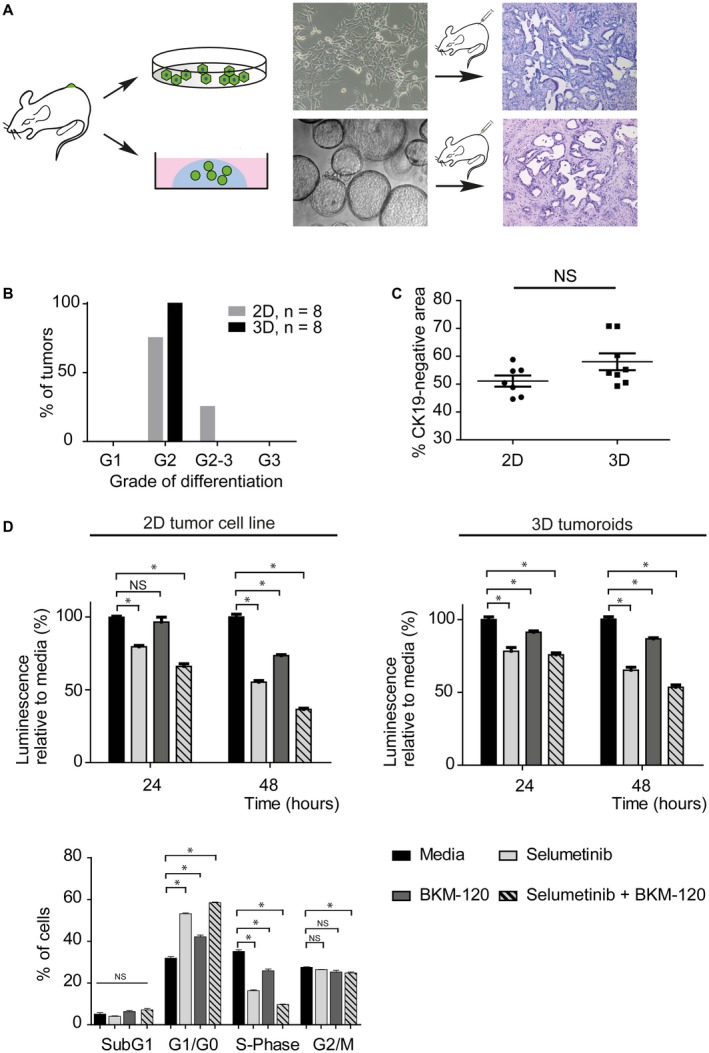
Organoid‐derived tumors are serially transplantable as 2D or 3D cell lines. (A) Genetically defined tumor cell lines established as 2D or 3D (tumoroid) cultures from primary, organoid‐derived tumors maintain the histology of the parental tumor following retransplantation. (B) Differentiation grading of tumors derived from murine‐reinjected 2D cell lines and tumoroids. (C) Stromal content of 2D and 3D cell line–derived tumors does not differ significantly. The CK19‐negative area was used as a surrogate for stromal content. (D) The efficiency of targeted therapy does not depend on the culture conditions. Both 2D and 3D cultured tumor cell lines show a comparable response to targeted therapies in vitro. Cell‐cycle fluorescence‐activated cell sorting reveals the accumulation of cells in the G0/G1 phase and a concomitant S‐phase reduction following drug treatment in the 2D cell line. Abbreviations: FACS, fluorescence‐activated cell sorting; G1, grade 1; G2, grade 2; G3, grade 3; and NS, not significant.
Liver Organoids Can Be Used to Probe Candidate Cancer Genes In Vivo
The advent of next‐generation sequencing technologies led to the identification of the mutational landscape of human CCA. However, the relevance of individual genetic alterations for initiation and maintenance of the tumors is still unknown. To probe whether our model is suitable for the functional characterization of candidate tumor suppressor genes, we retrovirally introduced an shRNA directed against the tumor suppressor phosphatase and tensin homolog (shPten)18 or a nontargeting control (shRenilla) into KrasG12D/wt; p53∆/∆ recombined organoids. Pten knockdown activated thymoma viral proto‐oncogene (Akt) signaling (Fig. 4C) and markedly accelerated tumor growth compared with control‐transduced cells (Fig. 4A). Histologically, tumors from both groups were indistinguishable and not altered by loss of Pten (Fig. 4B,D). This experiment highlights the ease with which candidate cancer genes can be probed by RNA interference–mediated knockdown. Of note, a comparable transgenic approach would have required breeding six different transgenic alleles (e.g., Albumin‐Cre; KraslslG12D/wt; p53lox/lox; Ptenlox/lox).
Figure 4.
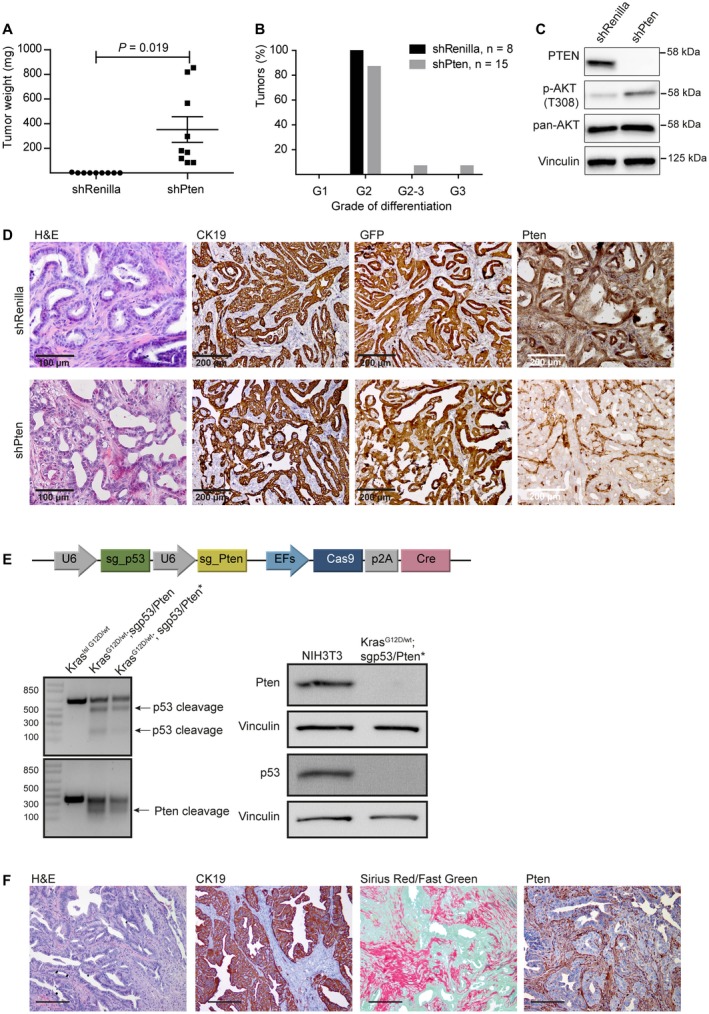
In vivo validation of liver cancer genes in the organoid model. (A) Murine organoids can be used to validate candidate cancer drivers in cholangiocarcinogenesis. KrasG12D; p53Δ/Δ organoids were transduced with retroviruses expressing GFP‐coupled shRenilla (control) or shPten (LMP_shRenilla or LMP_shPten) and transplanted into recipient mice. Loss of Pten significantly accelerates tumor development over controls, as assessed by tumor weight. (B) Genetically modified organoids give rise to predominantly moderately differentiated CCAs. (C) Immunoblotting confirms loss of Pten in shPten‐expressing tumor cell lines and up‐regulation of downstream mediators (phosphorylated Akt). (D) Immunohistochemistry shows loss of Pten only in CK19 and GFP‐positive tumor cells, but not in the recipient‐derived surrounding stroma. (E) Schematic of the sgp53/sgPten‐CC vector that facilitates efficient genome editing in the CCA model using CRISPR/Cas9 technology. KraslslG12D liver organoids were transfected with the sgp53/sgPten‐CC vector that simultaneously encodes for sgRNAs directed against the tumor suppressors p53 and Pten, the Cas9 enzyme, and Cre recombinase. Efficient cleavage of p53 and Pten by the sgp53/sgPten‐CC vector in murine pre‐injection organoids and tumoroids is detected by T7 endonuclease assays (left), and immunoblotting confirms loss of Pten‐protein and p53‐protein expression in a tumor‐derived CCA cell line (right; protein extracts from NIH3T3 cells serve as a control). Asterisks mark tumor‐derived cell lines. (F) Immunohistochemistry on CRISPR/Cas9‐generated tumors reveals hallmark features of CCA histology as described. Scale bar corresponds to 200 µm. Abbreviations: AKT, thymoma viral proto‐oncogene; G1, grade 1; G2, grade 2; G3, grade 3; and H&E, hematoxylin and eosin.
The shRNAs were expressed from a retroviral backbone (LMP) co‐encoding for a fluorescent reporter gene (green fluorescent protein [GFP]) that, in addition to testing candidate cancer drivers, allows tracing of cells derived from the transplanted organoids in the recipient animal. The stroma cells were GFP‐negative, indicating that stroma cells were exclusively derived from the recipient host and not from epithelial‐to‐mesenchymal transition of tumor cells (Fig. 4D). Stromal content did not differ significantly in tumors with and without loss of PTEN (Supporting Fig. S6).
To address whether the prominent stromal reaction is a direct consequence of injecting the Kras mutant organoids into the subcutaneous microenvironment or whether recruitment of stromal cells is rather an intrinsic feature of the transplanted organoids, we orthotopically injected organoids into the liver. Despite the profoundly different injection sites, tumors developing within the liver were histologically comparable to tumors that formed from sq‐injected organoids. In both settings, the organoid‐derived, Kras mutant tumor cells were able to recruit stromal cells and induce a desmoplastic reaction (Supporting Fig. S7A,B).
In contrast to RNA interference, CRISPR/Cas9 technology facilitates genome editing and allows for complete loss of function or the generation of genomic deletions. To introduce genetic perturbations into liver organoids using CRISPR/Cas9, we transfected a plasmid co‐encoding for a Cre‐recombinase, Cas9 protein, and sgRNAs directed against the tumor suppressors Pten and p53 into KraslslG12D/wt organoids (sgp53/sgPten‐CC)19 (Fig. 4E, top panel). Cre‐mediated recombination was confirmed by PCR and efficient target cleavage by T7 endonuclease assays (Fig. 4E, left panel). Similar to their shRNA‐transgenic counterparts, these organoids gave rise to CK19‐positive, moderately differentiated murine CCAs (Fig. 4F). Isolated tumoroids showed complete loss of p53 and Pten expression (Fig. 4E, right panel). In summary, liver organoids can be genetically modified with either RNA interference or CRISPR/Cas9 technology to interrogate gene function in vivo, and fluorescent marker proteins facilitate the tracing of transplanted cells in the recipient animal.
Organoids Expressing a Biliary Marker Phenotype Are Able to Give Rise to Tumors Resembling HCC Following Malignant Transformation
Next, we assessed whether the intrinsic biliary marker constellation expressed by the organoids in vitro translates into the development of tumors of biliary differentiation or whether the organoids retain the plasticity to give rise to a broader differentiation spectrum. We transduced organoids derived from WT C57/Bl6 livers with retroviruses encoding for murine myelocytomatosis oncogene (Myc) and co‐expressing a red fluorescent reporter (mCherry), an shRNA directed against p53 coupled to a GFP reporter, and transfected an sgRNA targeting Apc (adenomatous polyposis coli) (Fig. 5A,B and Supporting Fig. S8A). Intriguingly, substituting Kras with Myc as an oncogenic driver resulted in tumors of a different and distinct differentiation: Unlike the duct‐forming, stroma‐rich adenocarcinomas that arise in the context of mutant Kras, Myc‐overexpressing tumors display a compact, solid growth pattern of tumor cell nests lacking prominent desmoplasia. The tumor cells stained negative for CK19 and exhibited more prominent nucleoli, overall more resembling HCC than CCA (Fig. 5C).
Figure 5.
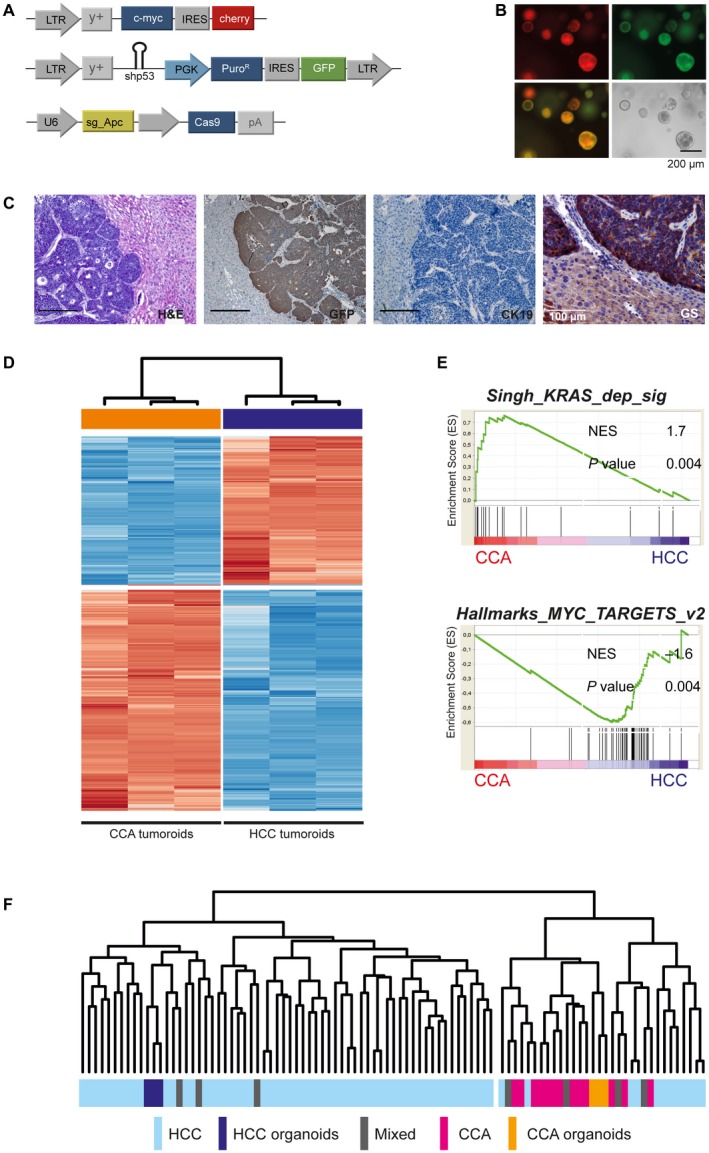
Liver organoids retain their plasticity to differentiate into tumors resembling HCC. (A) Liver organoids derived from WT C57Bl/6J mice were stably transduced with retroviruses expressing Myc coupled with a red fluorescent protein (mCherry), a GFP‐coupled shRNA directed against p53, and transiently transfected with pX459_sgAPC. (B) Stable integration of both retroviral vectors is indicated by expression of fluorescent markers, GFP and mCherry. (C) Myc‐driven liver tumors develop at a latency of about 40 days and are histologically distinct from Kras‐driven murine CCAs. Tumor cells stain negative for CK19 and strongly express glutamine synthetase. Immunohistochemistry confirms GFP positivity. Scale bars represent 200 µm unless otherwise indicated. (D) Unsupervised cluster analyses based on differentially expressed genes between HCC and CCA tumoroids based on Euclidean distance. (E) Gene‐set enrichment analyses of key gene sets activated in CCA and HCC tumoroids. (F) Integrative analyses of a publicly available cohort of 70 human HCCs, 17 CCAs, and 7 liver cancers of mixed HCC/CCA histology, with Myc HCC‐like and KrasG12D/wt CCA tumoroids. Integration and cross‐species comparisons were performed as described by Lee et al.26 Unsupervised cluster analyses confirm that the HCC and CCA tumoroids share gene‐expression profiles with authentic human cancers. Abbreviations: Mixed, liver cancers of mixed HCC/CCA histology; and NES, normalized enrichment score.
Comparative analyses of the CCA tumoroids and the HCC‐like tumoroids by RNA sequencing identified 4,723 differentially expressed genes. Unsupervised clustering confirmed the distinct gene expression profile of Myc overexpressing and Kras mutant tumoroids (Fig. 5D). Consistently, GSEA (Fig. 5E) confirmed that Kras‐mutated tumoroids were predominantly driven by activation of Kras‐dependent gene sets, whereas HCC‐like tumoroids showed an activation of MYC target genes, overall matching the differences in histology and thus validating the approach presented here. Similar to what we observed in CCA organoids (Fig. 2D), Myc‐overexpressing pre‐injection organoids and WT C57Bl/6J organoids, but not tumoroids, clustered together (Supporting Fig. S8B). Moreover, comparative analyses and cross‐species integration of the tumoroid expression signatures with a previously published data set of 70 human HCCs, 13 CCAs, and 7 liver cancers of mixed HCC/CCA histology20 confirmed that both the HCC‐like and CCA‐like tumoroids closely reflected the transcriptome profile of respective authentic human cancers (Fig. 5F).
These results indicate that, despite their initial biliary phenotype, the genetic profile of the liver organoids significantly contributes to the morphology of resulting tumors in vivo.
Discussion
CCA is the second‐most common primary liver cancer. A rising incidence and pronounced resistance to current treatment regimens emphasize the need for a better pathophysiological understanding, which is pivotal for the development of novel treatment approaches. Multiple studies have investigated the mutational landscape of CCAs.13, 14 To functionally address the role of potential cancer drivers during tumor development and maintenance, as well as to identify effective drug targets, histologically accurate and genetically flexible in vivo systems are needed. Organoid cultures offer unique opportunities to model CCA. Unlike other primary culture systems, such as fetal liver cells, organoids can be serially passaged in an untransformed state, cryoconserved, infinitely expanded, and genetically modified according to specific objectives of individual investigations. Importantly, organoid cultures offer the opportunity to minimize cost and time required for continuous animal husbandry and/or repeated isolations of primary cells, thus significantly accelerating the translation of scientific hypotheses into experimental approaches.
We demonstrate that genetic modifications can be rapidly introduced into liver organoid cultures by RNA interference or CRISPR/Cas9 technology. Co‐introduction of antibiotic resistance genes facilitates the in vitro selection of efficiently targeted cell populations before transplantation. This feature is especially convenient when studying genetic alterations that do not convey a selective advantage to the emerging tumor and might otherwise be outgrown by nonmodified clones. Furthermore, compared with traditional transgenic models, this model can incorporate genetic alterations in multiple genes simultaneously, offering the genetic flexibility to model highly complex genetic interactions across diverse genomic loci.
Our work demonstrates that, in the context of mutant Kras, liver organoids derived from adult murine livers can give rise to tumors histologically matching CCAs with 100% penetrance when injected orthotopically or sq in syngeneic animals. Unlike in transgenic approaches, in which poorly differentiated sarcomatoid tumors or overlaps to tumors of hepatocellular differentiation are frequently observed,21 more than 90% of CCAs are moderately differentiated CCAs (G2). Given the decisive importance of the tumor stroma in tumor development and therapy response, the observed distinct stromal reaction in the genetically modified organoid model closely recapitulates this hallmark feature of human CCAs. Therefore, the model can be highly instrumental to study tumor–stroma interactions in the emerging field of stroma biology.
Liver tumorigenesis is considered a multistep process. Using expression profiling and array comparative genomic hybridization, we provide initial evidence for how the molecular profile of organoids undergoes gradual genetic changes, reflecting the key transforming events from cells that are WT or harbor latent cancer alleles, toward a “premalignant” in vitro activated phenotype following introduction of defined genetic alterations, until reaching the fully transformed state of malignant tumoroids.
Indeed, transcriptome alterations are most pronounced in tumoroids, likely due to the outgrowth of highly malignant clones under selective pressure, in combination with the acquisition of additional lesions during in vivo tumorigenesis. Similarly, the acquisition of pro‐oncogenic copy number alterations can be detected during the progression from in vitro modified organoids to tumoroids. Our study highlights that the de novo introduction of driver oncogenes in combination with tumor suppressor gene loss is capable of introducing specific genomic changes, thus significantly altering the genetic landscape of liver organoids.
The murine liver organoids complement the human counterpart by enabling scientific work in genetically induced tumors that allow, among others, the study of vulnerabilities in the presence of defined combinations of genetic alterations that occur in large fractions of patients with CCA. In addition, considering the significant advances made in the field of tumor immunology, transplantable murine organoids allow us to study CCA in an immunocompetent and syngeneic environment. An additional benefit of our model is the simplicity with which the transition between in vitro and in vivo work can be accomplished. Retransplantation of both 2D tumor‐derived cell lines and tumoroids gives rise to cancers that recapitulate the histology of the primary tumors, which indicates that the tumor cell lines retain crucial features of corresponding parental tumors. In addition, 2D and 3D tumor‐derived cell lines exhibit comparable susceptibility to targeted therapies. These results suggest that these cell lines are a competent and viable tool for functional in vitro studies, such as drug screening.
The cellular origin of liver cancer remains unresolved. Indeed, in experimental animal models, hepatocytes, as well as bipotent progenitor cells, can give rise to CCA and HCC in a context‐dependent and genotype‐dependent fashion.20, 22, 23, 24, 25 Liver organoid cultures are most likely initiated from cells that reside within the biliary epithelial compartment, as suggested by the expression of biliary markers and the inability of mature hepatocytes to contribute to organoid cultures under the described conditions. Despite their biliary phenotype, we were able to show that, in direct dependence on the oncogenic driver, organoids retain their plasticity and can be transformed into tumors that show a markedly different histology than CCA and more closely resemble HCC. Importantly, these tumors also exhibit expression profiles that cluster with human HCCs. Therefore, our genetically modified liver organoids can recapitulate the full morphological and molecular spectrum of primary human liver cancer.
In summary, we have shown that, depending on the genetic context, organoids can give rise to aggressive liver cancers that exhibit characteristics of human CCA as well as HCCs in immunocompetent mice. These tumors not only histologically resemble their human counterparts but also show similar expression profiles. Thus, organoid‐based mouse models can serve as a scalable system to facilitate the rapid interrogation of putative cancer genes and vulnerabilities using RNA interference and CRISPR/Cas9 technology.
Supporting information
Acknowledgments
We thank Sandra Rohrmoser and Meriame Nassiri for their expert technical assistance. Craig Dorrell (OHSU, Portland, OR) has generously shared his protocols and his expertise in murine organoid cultures. Johannes Zuber (IMP, Vienna) kindly provided the PCIN (pSIN‐PGK‐CreERT‐IRES‐NeomycinResistance) vector. We also thank Dieter Saur (Munich, Germany) for the KraslslG12D mice and Florian Kühnel (Hannover, Germany) for the KraslslG12D/wt; p53lox/lox mice.
Potential conflict of interest: Dr. Dow consults for and owns stock in Mirimus Inc.
Supported by the German Cancer Aid Deutsche Krebshilfe (111757 to M.S.); the Else Kröner‐Fresenius Foundation (2015_A225 to A.S.); Hannover Medical School intramural grant HiLF (to A.S.); the Fritz‐Thyssen grant (10.15.1.025MN to S.M. and A.V.); and the SFB/TR 209 Liver Cancer grant (to A.V., A.S., M.S., and P.S.).
Contributor Information
Arndt Vogel, Email: vogel.arndt@mh-hannover.de.
Michael Saborowski, Email: saborowski.michael@mh-hannover.de.
References
Author names in bold designate shared co‐first authorship.
- 1. Clevers H. Modeling development and disease with organoids. Cell 2016;165:1586‐1597. [DOI] [PubMed] [Google Scholar]
- 2. Huch M, Gehart H, van Boxtel R, Hamer K, Blokzijl F, Verstegen MM, et al. Long‐term culture of genome‐stable bipotent stem cells from adult human liver. Cell 2015;160:299‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li B, Dorrell C, Canaday PS, Pelz C, Haft A, Finegold M, et al. Adult mouse liver contains two distinct populations of cholangiocytes. Stem Cell Rep 2017;9:478‐489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Broutier L, Mastrogiovanni G, Verstegen MM, Francies HE, Gavarro LM, Bradshaw CR, et al. Human primary liver cancer‐derived organoid cultures for disease modeling and drug screening. Nat Med 2017;23:1424‐1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schweitzer N, Fischer M, Kirstein MM, Berhane S, Kottas M, Sinn M, et al. Risk estimation for biliary tract cancer: development and validation of a prognostic score. Liver Int 2017;37:1852‐1860. [DOI] [PubMed] [Google Scholar]
- 6. Farshidfar F, Zheng S, Gingras MC, Newton Y, Shih J, Robertson AG, et al. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH‐mutant molecular profiles. Cell Rep 2017;19:2878‐2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chan‐On W, Nairismagi ML, Ong CK, Lim WK, Dima S, Pairojkul C, et al. Exome sequencing identifies distinct mutational patterns in liver fluke‐related and non‐infection‐related bile duct cancers. Nat Genet 2013;45:1474‐1478. [DOI] [PubMed] [Google Scholar]
- 8. Nakamura H, Arai Y, Totoki Y, Shirota T, Elzawahry A, Kato M, et al. Genomic spectra of biliary tract cancer. Nat Genet 2015;47:1003‐1010. [DOI] [PubMed] [Google Scholar]
- 9. Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K‐ras. Genes Dev 2001;15:3243‐3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet 2001;29:418‐425. [DOI] [PubMed] [Google Scholar]
- 11. Broutier L, Andersson‐Rolf A, Hindley CJ, Boj SF, Clevers H, Koo BK, et al. Culture and establishment of self‐renewing human and mouse adult liver and pancreas 3D organoids and their genetic manipulation. Nat Protoc 2016;11:1724‐1743. [DOI] [PubMed] [Google Scholar]
- 12. Saborowski A, Saborowski M, Davare MA, Druker BJ, Klimstra DS, Lowe SW. Mouse model of intrahepatic cholangiocarcinoma validates FIG‐ROS as a potent fusion oncogene and therapeutic target. Proc Natl Acad Sci U S A 2013;110:19513‐19518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wardell CP, Fujita M, Yamada T, Simbolo M, Fassan M, Karlic R, et al. Genomic characterization of biliary tract cancers identifies driver genes and predisposing mutations. J Hepatol 2018;68:959‐969. [DOI] [PubMed] [Google Scholar]
- 14. Jusakul A, Cutcutache I, Yong CH, Lim JQ, Huang MN, Padmanabhan N, et al. Whole‐genome and epigenomic landscapes of etiologically distinct subtypes of cholangiocarcinoma. Cancer Discov 2017;7:1116‐1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med 2008;14:1351‐1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alagesan B, Contino G, Guimaraes AR, Corcoran RB, Deshpande V, Wojtkiewicz GR, et al. Combined MEK and PI3K inhibition in a mouse model of pancreatic cancer. Clin Cancer Res 2015;21:396‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ewald F, Norz D, Grottke A, Hofmann BT, Nashan B, Jucker M. Dual Inhibition of PI3K‐AKT‐mTOR‐ and RAF‐MEK‐ERK‐signaling is synergistic in cholangiocarcinoma and reverses acquired resistance to MEK‐inhibitors. Invest New Drugs 2014;32:1144‐1154. [DOI] [PubMed] [Google Scholar]
- 18. Miething C, Scuoppo C, Bosbach B, Appelmann I, Nakitandwe J, Ma J, et al. PTEN action in leukaemia dictated by the tissue microenvironment. Nature 2014;510:402‐406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. O’Rourke KP, Loizou E, Livshits G, Schatoff EM, Baslan T, Manchado E, et al. Transplantation of engineered organoids enables rapid generation of metastatic mouse models of colorectal cancer. Nat Biotechnol 2017;35:577‐582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Holczbauer A, Factor VM, Andersen JB, Marquardt JU, Kleiner DE, Raggi C, et al. Modeling pathogenesis of primary liver cancer in lineage‐specific mouse cell types. Gastroenterology 2013;145:221‐231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. O’Dell MR, Huang JL, Whitney‐Miller CL, Deshpande V, Rothberg P, Grose V, et al. Kras(G12D) and p53 mutation cause primary intrahepatic cholangiocarcinoma. Cancer Res 2012;72:1557‐1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fan B, Malato Y, Calvisi DF, Naqvi S, Razumilava N, Ribback S, et al. Cholangiocarcinomas can originate from hepatocytes in mice. J Clin Invest 2012;122:2911‐2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sekiya S, Suzuki A. Intrahepatic cholangiocarcinoma can arise from Notch‐mediated conversion of hepatocytes. J Clin Invest 2012;122:3914‐3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guest RV, Boulter L, Kendall TJ, Minnis‐Lyons SE, Walker R, Wigmore SJ, et al. Cell lineage tracing reveals a biliary origin of intrahepatic cholangiocarcinoma. Cancer Res 2014;74:1005‐1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, et al. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell‐specific gene knock‐outs using Cre recombinase. J Biol Chem 1999;274:305‐315. [DOI] [PubMed] [Google Scholar]
- 26. Lee JS, Heo J, Libbrecht L, Chu IS, Kaposi‐Novak P, Calvisi DF, et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nature Med 2006;12:410‐416. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials