

Abstract
Primary biliary cholangitis (PBC) is typically associated with elevated serum bile acid levels and pruritus, but pruritus is often refractory to treatment with existing therapies. This phase 2 study assessed the efficacy and safety of maralixibat, a selective, ileal, apical, sodium‐dependent, bile acid transporter inhibitor, in adults with PBC and pruritus. Adults with PBC and pruritus who had received ursodeoxycholic acid (UDCA) for ≥6 months or were intolerant to UDCA were randomized 2:1 to maralixibat (10 or 20 mg/day) or placebo for 13 weeks in combination with UDCA (when tolerated). The primary outcome was change in Adult Itch Reported Outcome (ItchRO™) average weekly sum score (0, no itching; 70, maximum itching) from baseline to week 13/early termination (ET). The study enrolled 66 patients (maralixibat [both doses combined], n = 42; placebo, n = 24). Mean ItchRO™ weekly sum scores decreased from baseline to week 13/ET with maralixibat (–26.5; 95% confidence interval [CI], –31.8, –21.2) and placebo (–23.4; 95% CI, –30.3, –16.4). The difference between groups was not significant (P = 0.48). In the maralixibat and placebo groups, adverse events (AEs) were reported in 97.6% and 70.8% of patients, respectively. Gastrointestinal disorders were the most frequently reported AEs (maralixibat, 78.6%; placebo, 50.0%). Conclusion: Reductions in pruritus did not differ significantly between maralixibat and placebo. However, a large placebo effect may have confounded assessment of pruritus. Lessons learned from this rigorously designed and executed trial are indispensable for understanding how to approach trials assessing pruritus as the primary endpoint and the therapeutic window of bile acid uptake inhibition as a therapeutic strategy in PBC.
Abbreviations
- AE
adverse event
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- C4
7α‐hydroxy‐4‐cholesten‐3‐one
- CI
confidence interval
- ET
early termination
- FGF
fibroblast growth factor
- GGT
gamma‐glutamyl transferase
- IBAT
ileal bile acid transporter
- ItchRO™
Itch Reported Outcome
- ITT
intent‐to‐treat
- LDL‐C
low‐density lipoprotein cholesterol
- LS
least squares
- MOS‐Sleep
Medical Outcomes Study Sleep
- PBC
primary biliary cholangitis
- PBC‐40
primary biliary cholangitis‐specific health‐related quality‐of‐life instrument
- PGTB
Patient Global Therapeutic Benefit
- PIC
Patient Impression of Change
- sBA
serum bile acid
- UDCA
ursodeoxycholic acid
- ULN
upper limit of normal
Primary biliary cholangitis (PBC), formerly known as primary biliary cirrhosis,1 is a rare, chronic, progressive, autoimmune, cholestatic liver disease characterized by damage to intrahepatic bile ducts. Pruritus is a common clinical symptom of PBC, reported to affect 20%‐75% of individuals,2 and can significantly impair patients’ quality of life.3 In particular, severe pruritus impairs sleep quality.4 The etiology of pruritus is thought to be associated with increased serum bile acid (sBA) levels,5 but the relationship is not fully understood. The pathophysiology of pruritus involves multiple contributing factors, including bile acids, autotaxin, histamine, opioidergic tone, serotonin, and substance P.6
Ursodeoxycholic acid (UDCA) is the first‐line pharmacologic intervention for the treatment of patients with PBC7 and has been shown to improve serum biochemical markers of liver disease and transplant‐free survival.8, 9, 10 However, a significant improvement in pruritus is not typically observed.11 Individuals also remain unresponsive to recommended agents for the treatment of cholestatic pruritus, which include cholestyramine, rifampicin, naltrexone, and sertraline.12 New pharmacologic interventions are therefore needed.
Maralixibat chloride (SHP625; formerly LUM001 or lopixibat) is a potent, apical, sodium‐dependent, bile acid transporter competitive inhibitor (here on referred to as an ileal bile acid transporter [IBAT] inhibitor) with minimal systemic absorption.13, 14, 15 In animal models of cholestasis, maralixibat blocked reabsorption of bile acids in the terminal ileum, thereby reducing enterohepatic recirculation to the liver and increasing fecal excretion of bile acids.16 In a rat partial bile duct ligation model of cholestasis, maralixibat reduced elevations in sBA levels, improved liver function, and reduced liver tissue damage compared with control.17 Reduced pruritus has been observed in patients with cholestatic liver disease following partial external biliary diversion surgery or administration of bile acid sequestrants, which also interrupt enterohepatic circulation of bile acids.18, 19, 20 Use of a pharmacologic agent, such as maralixibat, to block reabsorption of bile acids may avoid the complications and disfigurement associated with surgery. Here, we present the results of a placebo‐controlled phase 2 study of the efficacy, safety, and tolerability of maralixibat in adults with PBC and pruritus.
Patients and Methods
ETHICS
This study (ClinicalTrials.gov identifier NCT01904058) was conducted in accordance with the Declaration of Helsinki as well as with applicable national legislation and the International Conference on Harmonisation Harmonised Tripartite Guideline for Good Clinical Practice (E6). All patients gave written informed consent before study enrollment.
STUDY POPULATION
This study enrolled adults aged 18‐80 years who were diagnosed with PBC according to the American Association for the Study of Liver Diseases practice guidelines.11 Patients had to have significant pruritus as evidenced by an average daily score >4.0 on the 10‐point Adult Itch Reported Outcome (ItchRO™) questionnaire (Fig. 1) for 2 consecutive weeks during the screening period and to have been receiving UDCA for at least 6 months (stable dose for at least 3 months before baseline) or have an intolerance to UDCA (no UDCA for at least 3 months before baseline). Changes in UDCA regimen were not permitted during the study. Participants could also continue to receive rifampin, antihistamines, and opiate antagonists during the study if initiated at least 30 days before screening and if the regimen was maintained throughout the study; the same was true for selective serotonin reuptake inhibitors and serotonin–norepinephrine reuptake inhibitors if initiated at least 90 days before screening. Key exclusion criteria were evidence of biliary obstruction or overt malignancy on ultrasound or equivalent imaging in the 12 months before the screening visit, evidence of significant concomitant significant liver diseases, advanced clinical complications of PBC or clinically significant hepatic decompensation, bile acid resin use within 30 days before randomization, and total bilirubin levels of more than twice the upper limit of normal (ULN) or alanine aminotransferase (ALT) or aspartate aminotransferase (AST) levels more than 5‐fold ULN at screening.
Figure 1.
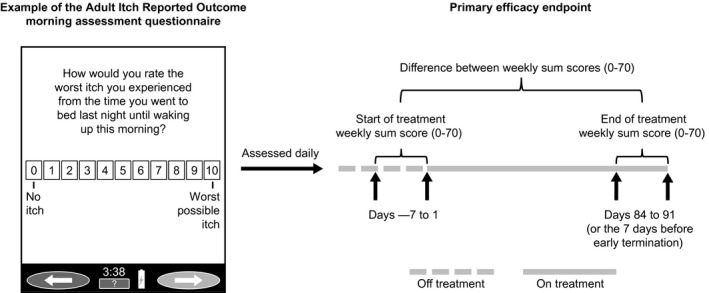
Adult Itch Reported Outcome weekly sum score. Patients scored their itch each morning and evening. Data analysis included only the highest score available each day. Missing daily scores were not imputed. The weekly sum score for a given week was only calculated if daily scores were completed for at least 4 of the 7 days that week (compliant week). Missing daily scores for a compliant week were imputed using the average daily score for that week. If for a given week more than three daily scores were missing, then weekly sum scores from the most recent compliant week were used in a last observation carried forward format.
STUDY DESIGN
The study was conducted between August 2013 and April 2015 at 24 locations, including medical centers, clinics, and hospitals, in the United States, United Kingdom, and Canada and comprised a screening period of up to 5 weeks, a 13‐week treatment period, and a 4‐week safety follow‐up period (Fig. 2). The 13‐week treatment period comprised 2‐4 weeks of dose escalation followed by 9‐11 weeks of stable dose treatment. To allow patients to acclimate to the study drug, maralixibat was initiated at 2.5 mg/day and increased at weekly intervals to 5, 10, or 20 mg/day. Patients who did not complete to week 13 were invited to attend an early termination (ET) visit.
Figure 2.
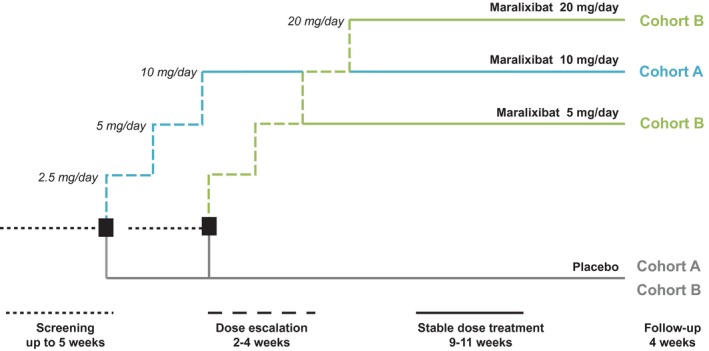
Study design. Dose selection for cohort B was guided by the tolerability of maralixibat in the first 18 patients who completed 4 weeks of treatment in cohort A; the decision was based on the number of patients who lowered their dose or suspended or stopped active treatment owing to gastrointestinal intolerance related to the maralixibat (≥5 patients, cohort B would have received 5 mg maralixibat; <5 patients, cohort B would have received 20 mg maralixibat). Clinic visits were scheduled at baseline (week 0) and weeks 2, 4, 8, and 13. Telephone contact was scheduled at weeks 1, 3, and 17 (follow‐up call).
Two dose cohorts were planned (cohort A, maralixibat 10 mg; cohort B, maralixibat 5 mg or 20 mg). Dose selection for cohort B was guided by tolerance to maralixibat in the first 18 patients to complete 4 weeks of treatment in cohort A. If dosing of active treatment had to be reduced, suspended, or stopped owing to gastrointestinal intolerance in 5 or more of these 18 patients, then patients in cohort B would receive maralixibat 5 mg; otherwise, patients would receive maralixibat 20 mg.
In each dose cohort, patients were randomized 2:1 using an interactive web response system to receive once‐daily oral maralixibat or matching placebo. The sponsor prepared the randomization list. All patients, monitors, study center personnel, and the sponsor were blinded to treatment throughout the study. Further details of the randomization procedure can be found in the Supporting Materials.
PRIMARY EFFICACY OUTCOME
The primary efficacy outcome was change from baseline to study endpoint (week 13/ET) in pruritus as measured by the Adult ItchRO™ weekly sum score (Fig. 1). Patients completed the Adult ItchRO™ electronic diary twice daily (morning and evening); itch severity was rated from 0 (no itching) to 10 (very severe itching). The highest score from the morning and evening assessments was used as the daily score, and weekly sum scores were calculated as the sum of daily scores over the 7 days before each visit.
SECONDARY EFFICACY OUTCOMES
Secondary efficacy outcomes, assessed at weeks 4, 8, 13, and 13/ET, were Adult ItchRO™ weekly sum scores, fasting sBA levels, serum 7α‐hydroxy‐4‐cholesten‐3‐one (C4) levels, alkaline phosphatase (ALP) levels,21 and 5‐D Itch scores. 5‐D Itch is a validated, self‐reported measure of pruritus that includes five domains (duration, degree, direction, disability, and distribution) and gives a total score ranging from 5 (no itching) to 25 (most severe itching).22
EXPLORATORY EFFICACY OUTCOMES
Exploratory efficacy outcomes included changes from baseline to weeks 4, 8, 13, and 13/ET in levels of ALT and AST and gamma‐glutamyl transferase (GGT).21 Changes from baseline for levels of fibroblast growth factor (FGF) 19 and FGF‐21 (potential regulators of bile acid synthesis),23 total and conjugated bilirubin and autotaxin,24 and total cholesterol and low‐density lipoprotein cholesterol (LDL‐C)21 were assessed at weeks 4, 13, and 13/ET.
Changes from baseline in PBC‐40 domain scores were assessed at weeks 4, 8, 13, and 13/ET. PBC‐40 is a validated, patient‐derived, PBC‐specific, health‐related quality‐of‐life measure comprising 40 items, each scored on a scale from 1 (least impact) to 5 (greatest impact) and distributed across six domains (itch, emotional, cognitive, symptoms, social, and fatigue). Individual item scores were summed for each domain, with higher scores indicating poorer quality of life.25
Changes from baseline in Medical Outcomes Study Sleep (MOS‐Sleep) Scale scores were assessed at weeks 4, 13, and 13/ET. The MOS‐Sleep Scale is a self‐reported 12‐item measure evaluating six dimensions of sleep in patients with chronic illness (sleep disturbance, snoring, awaken short of breath or headache, sleep adequacy, somnolence, sleep quantity/optimal sleep indicator). Items were scored, converted (range, 0‐100), and averaged according to the MOS‐Sleep Scale User’s Manual,26 with higher scores indicating greater sleep dysfunction. MOS‐Sleep Scale composite scores were also determined (sleep problems index I [6/12 items] and sleep problems index II [9/12 items]). Individuals were considered to have had optimal sleep if, on average, they slept 7‐8 hours each night during the past 4 weeks.
Self‐Administered Patient Impression of Change (PIC) and Patient Global Therapeutic Benefit (PGTB) were assessed at week 13 and week 13/ET. The PIC scale is a self‐reported assessment of itching that is scored from 1 (much better) to 7 (much worse). Treatment response was predefined as a PIC score ≤3. The PGTB scale is a self‐reported assessment of whether treatment benefit in terms of a reduction in itching outweighs the side effects and is scored from 1 (definitely) to 5 (definitely not). Treatment response was predefined as a PGTB score ≤2.
BIOANALYTICAL METHODOLOGY
Levels of sBA and C4 were determined using validated routine liquid chromatography–mass spectrometry methodology. Levels of FGF‐19, FGF‐21, and autotaxin were determined using commercially available enzyme‐linked immunosorbent assays. See Supporting Materials for further information. All chemistry analytes (ALP, ALT, AST, conjugated bilirubin, GGT, total bilirubin) and lipid panel analytes (LDL‐C and total cholesterol) were analyzed colorimetrically using commercially available tests at a central laboratory (Clinical Reference Laboratory, Lenexa, KS).
SAFETY ASSESSMENTS
Assessment of the safety and tolerability of maralixibat included evaluation of adverse events (AEs) and vital signs. AEs were coded using the Medical Dictionary for Regulatory Activities, version 16.0, and graded as mild, moderate, or severe in the opinion of the site investigator reporting the AE, using the Common Terminology Criteria for AEs, version 4.0. Predefined AEs of special interest included gastrointestinal disorders.
STATISTICAL ANALYSIS
Efficacy was assessed in the intent‐to‐treat (ITT) population, which included all randomized patients. Data for patients receiving placebo in cohorts A and B were pooled and compared with combined data for patients receiving maralixibat 10 mg or 20 mg (maralixibat overall) and with data from each maralixibat dose group.
The study design required randomization of 60 patients (maralixibat, n = 40; placebo, n = 20) to provide 80% power to detect an effect size of 0.780 and 90% power to detect an effect size of 0.903 for the difference between active treatment and placebo in mean change from baseline to week 13/ET in the Adult ItchRO™ weekly sum score (α = 0.05). Because no published information was available, these target effect sizes were based on other itch measures in this study population and on clinical judgment.
Between‐group differences in the primary efficacy outcome were assessed by analysis of covariance, with treatment and stratified ALP level as factors and baseline Adult ItchRO™ weekly sum score as a covariate. Least‐squares (LS) mean changes from baseline with 95% confidence intervals (CIs) were calculated for each treatment group and the group receiving placebo. LS mean differences between maralixibat and placebo with 95% CIs were calculated with pairwise treatment P values.
All secondary and exploratory outcomes involving continuous measures were analyzed using an analysis of covariance model, adjusted for the baseline value for each outcome measure. Exploratory outcomes involving categorical data were analyzed using a Cochran–Mantel−Haenszel test. All P values were considered nominal (unadjusted for multiplicity). Therefore, discussion of the results for secondary and exploratory outcomes is based on whether 95% CIs overlap between the maralixibat and placebo groups.
A hierarchical testing sequence was applied. If the difference for both maralixibat doses combined (maralixibat overall) versus placebo was statistically significant (P < 0.05), then further analyses of maralixibat 20 mg versus placebo and maralixibat 10 mg versus placebo were performed. A P value was considered nominal if the value from the preceding test was ≥0.05.
Correlations between change (absolute and percentage) in Adult ItchRO™ weekly sum score and levels of sBA, C4, or FGF‐19 were explored in post hoc analyses using Spearman correlation statistics. The proportions of patients with a response (reduction in sBA levels of at least 70% and an improvement of at least 2.5 points on the Adult ItchRO™ from baseline to week 13/ET) were calculated in the overall study population (maralixibat and placebo groups combined) in a post hoc subgroup analysis that stratified patients by baseline sBA level (greater than 3 times ULN and 3 times ULN or less). All post hoc statistics were considered descriptive rather than inferential.
Safety outcomes were assessed descriptively in all patients who had received at least one dose of the study drug (safety population; these patients were the same as those in the ITT population); inferential statistics were not performed.
Results
DISPOSITION AND BASELINE CHARACTERISTICS OF PATIENTS
Of the 66 patients randomized, 61 patients (92.4%) completed the 13‐week study (Fig. 3). All randomized patients received at least one dose of the study drug and had at least one postbaseline Adult ItchRO™ assessment.
Figure 3.
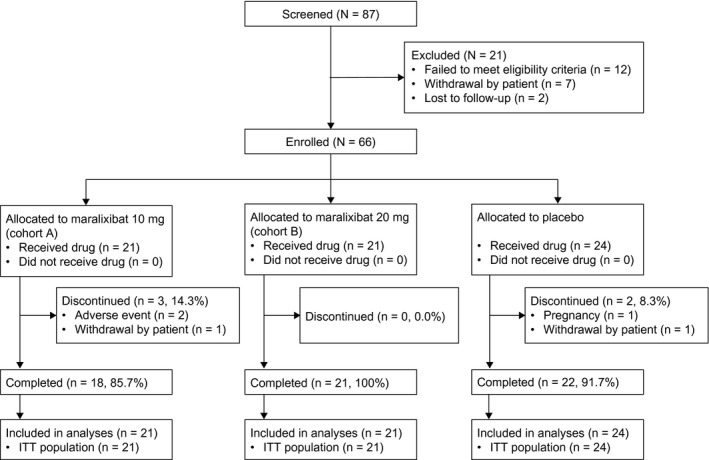
Patient disposition. Percentages are based on the ITT population.
The majority of patients were receiving ongoing treatment with UDCA during the study (maralixibat 10 mg, 100.0%; maralixibat 20 mg, 90.5%; placebo, 79.2%). The numbers of patients receiving stable concomitant therapy for pruritus in the maralixibat overall (n = 42) and placebo (n = 24) cohorts, respectively, were 21 (50.0%) and 9 (37.5%): antihistamines (11 [26.2%]; 5 [20.8%]), analgesics (5 [11.9%]; 3 [12.5%]), lipid‐modifying agents (4 [9.5%]; 1 [4.2%]), psychoanaleptics (2 [4.8%]; 1 [4.2%]), psycholeptics (2 [4.8%]; 0 [0.0%]), and other nervous system drugs (1 [2.4%]; 0 [0.0%]) (Supporting Table S1). Some patients received more than one concomitant medication.
Most baseline characteristics were well matched across the treatment groups (Table 1), although sBA levels (mean ± SD) were numerically lower with maralixibat overall (42.8 ± 69.99 μmol/L) than with placebo (55.8 ± 68.41 μmol/L), and proportionately more patients receiving maralixibat (85.7%) had a liver biopsy consistent with PBC than patients in the placebo group (75.0%).
Table 1.
Baseline Demographics and Disease Characteristics
| Maralixibat 10 mg (n = 21) | Maralixibat 20 mg (n = 21) | Maralixibat Overall (n = 42) | Placebo (n = 24) | |
|---|---|---|---|---|
| Patient demographics | ||||
| Age, years | 54.7 ± 12.74 | 53.5 ± 10.53 | 54.1 ± 11.56 | 52.0 ± 9.32 |
| Female sex, n (%) | 20 (95.2) | 17 (81.0) | 37 (88.1) | 23 (95.8) |
| Disease characteristics | ||||
| Adult ItchRO™ weekly sum score | 48.1 ± 13.36 | 52.1 ± 13.78 | 50.1 ± 13.56 | 51.8 ± 12.14 |
| Serum bile acids, μmol/L | 33.1 ± 30.59 | 52.5 ± 94.39 | 42.8 ± 69.99 | 55.8 ± 68.41 |
| ALP, U/L | 288.2 ± 193.91 | 257.6 ± 190.38 | 272.9 ± 190.43 | 264.9 ± 152.27 |
All values are mean ± SD unless otherwise stated.
EFFICACY OUTCOMES
Pruritus
Adult ItchRO™ weekly sum scores significantly decreased from baseline to week 13/ET in the maralixibat overall group (LS mean change, –26.5; 95% CI, –31.8, –21.2; P < 0.0001) and the placebo group (–23.4; 95% CI, –30.3, –16.4; P < 0.0001) (Fig. 4A; Supporting Table S2). However, the reduction was not significantly different between placebo and maralixibat overall (LS mean difference, –3.1; 95% CI, –11.9, 5.6; P = 0.48), maralixibat 20 mg (–4.0; 95% CI, –14.2, 6.2; P = 0.44), or maralixibat 10 mg (–2.3; 95% CI, –12.6, 8.0; P = 0.66) (Fig. 4C; Supporting Table S2). Reductions from baseline in itch were similar in all groups at weeks 8 and 13 (Fig. 4C; Supporting Table S2). Similar findings were observed when UDCA use was included as a factor in the analysis of covariance model and when only patients with UDCA use were assessed (data not shown).
Figure 4.
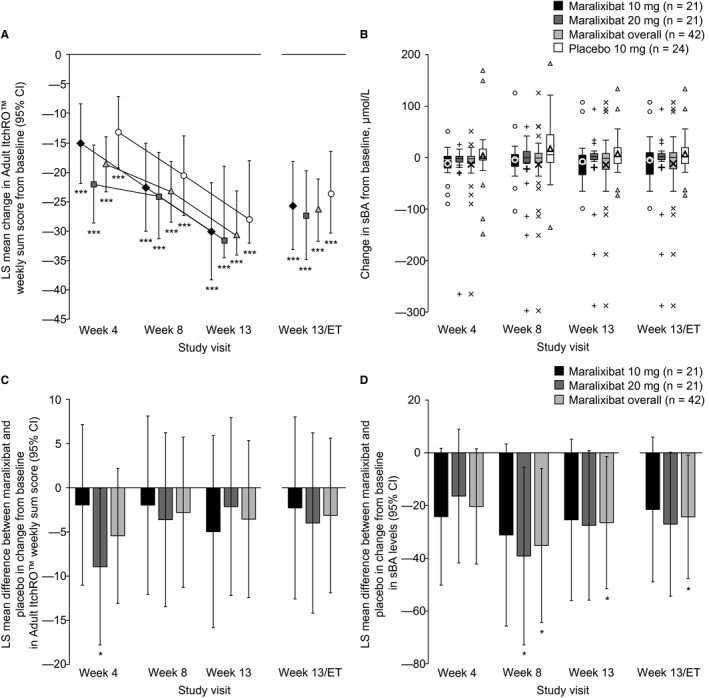
Adult ItchRO™ weekly sum scores and serum bile acids. (A,B) Change from baseline and (C,D) difference in change for maralixibat versus placebo. (A,C) Adult ItchRO™ weekly sum scores and (B,D) sBA levels are presented at each time point. *P < 0.05 (analysis of covariance model); ***P < 0.0001. In panel B, boxes represent the IQR; the band within each box represents the median; the upper and lower error bars, respectively, represent the maximum and minimum data points excluding outliers; small data points represent outliers (individual data points outside 1.5 × IQR); and the bold, single, large data points represent mean change. All statistical comparisons except those for the primary outcome were considered nominal (not adjusted for multiplicity). Abbreviation: IQR, interquartile range.
Scores for the 5‐D Itch measure and the PBC‐40 Itch domain also decreased from baseline to weeks 4, 8, 13, and 13/ET in all groups but did not differ between the maralixibat and placebo groups (Table 2; Supporting Table S2).
Table 2.
Changes in Pruritus, Quality of Life, Sleep, and Patient Perception of Treatment Benefits from Baseline to Week 13/et
| Maralixibat 10 mg (n = 21) | Maralixibat 20 mg (n = 21) | Maralixibat Overall (n = 42) | Placebo (n = 24) | |
|---|---|---|---|---|
| 5‐D Itch score | ||||
| LS mean change from baseline (95% CI) | –6.9 | –6.7 | –6.8 | –6.3 |
| (–8.9, –4.8)‡ | (–8.8, –4.6)‡ | (–8.2, –5.3)‡ | (–8.3, –4.4)‡ | |
| LS mean difference from placebo (95% CI) | –0.5 | –0.4 | –0.5 | N/A |
| (–3.4, 2.3) | (–3.2, 2.5) | (–2.9, 2.0) | ||
| PBC‐40 domain score | ||||
| Itch (3 items) | ||||
| LS mean change from baseline (95% CI) | –4.0 | –4.1 | –4.1 | –3.8 |
| (–5.3, –2.8)‡ | (–5.4, –2.9)‡ | (–5.0, –3.2)‡ | (–5.0, –2.6)‡ | |
| LS mean difference from placebo (95% CI) | –0.3 | –0.4 | –0.3 | N/A |
| (–2.0, 1.5) | (–2.1, 1.4) | (–1.8, 1.2) | ||
| Emotional (3 items) | ||||
| LS mean change from baseline (95% CI) | –1.4 | –1.2 | –1.3 | –0.5 |
| (–2.2, –0.7)‡ | (–2.0, –0.4)† | (–1.9, –0.8)‡ | (–1.3, 0.2) | |
| LS mean difference from placebo (95% CI) | –0.9 | –0.7 | –0.8 | N/A |
| (–2.0, 0.2) | (–1.7, 0.4) | (–1.7, 0.1) | ||
| Cognitive (6 items) | ||||
| LS mean change from baseline (95% CI) | –1.5 | –0.5 | –1.0 | –1.6 |
| (–3.0, –0.1)* | (–2.0, 0.9) | (–2.0, 0.0)* | (–3.0, –0.3)* | |
| LS mean difference from placebo (95% CI) | 0.1 | 1.1 | 0.6 | N/A |
| (–1.8, 2.1) | (–0.8, 3.1) | (–1.1, 2.3) | ||
| Symptoms (7 items) | ||||
| LS mean change from baseline (95% CI) | –0.8 | –0.4 | –0.6 | –1.8 |
| (–2.3, 0.7) | (–1.9, 1.1) | (–1.6, 0.4) | (–3.2, –0.4)* | |
| LS mean difference from placebo (95% CI) | 1.0 | 1.4 | 1.2 | N/A |
| (–1.0, 3.0) | (–0.7, 3.4) | (–0.6, 2.9) | ||
| Social (10 items) | ||||
| LS mean change from baseline (95% CI) | –2.0 | –2.8 | –2.4 | –2.1 |
| (–4.4, 0.4) | (–5.2, –0.3)* | (–4.1, –0.7)† | (–4.5, 0.2) | |
| LS mean difference from placebo (95% CI) | 0.1 | –0.6 | –0.2 | N/A |
| (–3.2, 3.5) | (–4.0, 2.7) | (–3.1, 2.6) | ||
| Fatigue (11 items) | ||||
| LS mean change from baseline (95% CI) | –2.3 | –2.8 | –2.5 | –4.9 |
| (–5.5, 0.9) | (–6.0, 0.4) | (–4.8, –0.3)* | (–7.9, –1.8)† | |
| LS mean difference from placebo (95% CI) | 2.6 | 2.1 | 2.3 | N/A |
| (–1.8, 7.0) | (–2.3, 6.5) | (–1.4, 6.1) | ||
| MOS‐Sleep Scale score | ||||
| Sleep disturbances (4 items) | ||||
| LS mean change from baseline (95% CI) | –9.2 | –9.0 | –9.1 | –15.6 |
| (–18.8, 0.5) | (–18.6, 0.7) | (–15.9, –2.3)* | (–24.8, –6.4)† | |
| LS mean difference from placebo (95% CI) | 6.4 | 6.6 | 6.5 | N/A |
| (–6.9, 19.8) | (–6.7, 19.9) | (–4.9, 18.0) | ||
| Snoring (1 item) | ||||
| LS mean change from baseline (95% CI) | –8.5 | –5.7 | –7.1 | 0.3 |
| (–17.5, 0.6) | (–14.9, 3.6) | (–13.5, –0.6)* | (–8.4, 8.9) | |
| LS mean difference from placebo (95% CI) | –8.7 | –5.9 | –7.3 | N/A |
| (–21.2, 3.7) | (–18.8, 6.9) | (–18.2, 3.5) | ||
| Awaken short of breath or headache (1 item) | ||||
| LS mean change from baseline (95% CI) | –6.3 | –3.0 | –4.6 | –0.9 |
| (–16.4, 3.9) | (–13.2, 7.2) | (–11.8, 2.6) | (–10.5, 8.8) | |
| LS mean difference from placebo (95% CI) | –5.4 | –2.1 | –3.7 | N/A |
| (–19.4, 8.7) | (–16.2, 12.0) | (–15.8, 8.3) | ||
| Sleep adequacy (2 items) | ||||
| LS mean change from baseline (95% CI) | 7.1 | 2.1 | 4.6 | 3.9 |
| (–3.8, 18.0) | (–8.8, 13.0) | (–3.1, 12.3) | (–6.4, 14.2) | |
| LS mean difference from placebo (95% CI) | 3.2 | –1.8 | 0.7 | N/A |
| (–11.8, 18.2) | (–16.8, 13.2) | (–12.1, 13.5) | ||
| Somnolence (3 items) | ||||
| LS mean change from baseline (95% CI) | –5.0 | –10.3 | –7.7 | –11.7 |
| (–14.3, 4.2) | (–19.6, –1.1)* | (–14.2, –1.2)* | (–20.5, –3.0)† | |
| LS mean difference from placebo (95% CI) | 6.7 | 1.4 | 4.1 | N/A |
| (–6.0, 19.4) | (–11.3, 14.1) | (–6.8, 15.0) | ||
| Sleep problems index I (6 items) | ||||
| LS mean change from baseline (95% CI) | –8.3 | –5.7 | –7.0 | –8.8 |
| (–16.0, –0.6)* | (–13.4, 2.0) | (–12.5, –1.5)* | (–16.1, –1.4)* | |
| LS mean difference from placebo (95% CI) | 0.5 | 3.1 | 1.8 | N/A |
| (–10.2, 11.1) | (–7.6, 13.7) | (–7.4, 10.9) | ||
| Sleep problems index II (9 items) | ||||
| LS mean change from baseline (95% CI) | –7.8 | –7.2 | –7.5 | –10.9 |
| (–15.2, –0.4)* | (–14.6, 0.2) | (–12.7, –2.3)† | (–17.9, –3.9)† | |
| LS mean difference from placebo (95% CI) | 3.1 | 3.8 | 3.4 | N/A |
| (–7.1, 13.3) | (–6.5, 13.9) | (–5.3, 12.2) | ||
| Sleep quantity, number of hours (1 item) | ||||
| LS mean change from baseline (95% CI) | 0.1 | 0.5 | 0.3 | 0.3 |
| (–0.4, 0.6) | (0.0, 1.0) | (0.0, 0.6) | (–0.2, 0.7) | |
| LS mean difference from placebo (95% CI) | –0.2 | 0.2 | 0.0 | N/A |
| (–0.8, 0.5) | (–0.5, 0.9) | (–0.6, 0.6) | ||
| Optimal sleep indicator (1 item) | ||||
| Baseline | ||||
| n | 21 | 21 | 42 | 24 |
| Yes, n (%) | 6 (28.6) | 8 (38.1) | 14 (33.3) | 4 (16.7) |
| No, n (%) | 15 (71.4) | 13 (61.9) | 28 (66.7) | 20 (83.3) |
| P value versus placebo | 0.34 | 0.11 | 0.14 | N/A |
| Week 13/ET | ||||
| n | 21 | 21 | 42 | 23 |
| Yes, n (%) | 9 (42.9) | 5 (23.8) | 14 (33.3) | 8 (34.8) |
| No, n (%) | 12 (57.1) | 16 (76.2) | 28 (66.7) | 15 (65.2) |
| P value versus placebo | 0.59 | 0.42 | 0.91 | N/A |
| Patient Impression of Change | ||||
| Week 13/ET | ||||
| n | 21 | 21 | 42 | 22 |
| Responder (score ≤3), n (%) | 19 (90.5) | 17 (81.0) | 36 (85.7) | 15 (68.2) |
| Nonresponder (score >3), n (%) | 2 (9.5) | 4 (19.0) | 6 (14.3) | 7 (31.8) |
| P value versus placebo | 0.07 | 0.39 | 0.10 | N/A |
| Patient Global Therapeutic Benefit | ||||
| Week 13/ET | ||||
| n | 21 | 21 | 42 | 22 |
| Responder (score ≤2), n (%) | 14 (66.7) | 17 (81.0) | 31 (73.8) | 14 (63.6) |
| Nonresponder (score >2), n (%) | 7 (33.3) | 4 (19.0) | 11 (26.2) | 8 (36.4) |
| P value versus placebo | 0.80 | 0.24 | 0.40 | N/A |
P < 0.05; † P < 0.01; ‡ P < 0.001 (analysis of covariance model with treatment group, ALP level [strata] and treatment group by ALP level interaction as factors, and baseline value as a covariate). Categorical data were analyzed using a Cochran–Mantel−Haenszel test. All P values are nominal (not adjusted for multiplicity).
Abbreviation: N/A, not applicable.
Serum Bile Acids and Synthesis Regulation
In the maralixibat groups, fasting sBA levels decreased from baseline to weeks 4, 8, 13, and 13/ET, whereas increases were observed in the placebo group (Fig. 4B; Supporting Table S3). The LS mean reduction in fasting sBA levels from baseline to week 13/ET was numerically greater in the maralixibat 20 mg group (–17.0 µmol/L; 95% CI, –36.9, 2.9) than in the 10 mg group (–11.4 µmol/L; 95% CI, –31.4, 8.5) (Fig. 4B; Supporting Table S3). In the placebo group, the LS mean increase in fasting sBA levels from baseline to week 13/ET was 10.1 µmol/L (95% CI, –8.7, 28.8).
Levels of C4 increased from baseline to week 13/ET with both doses of maralixibat and decreased with placebo (Table 3; Supporting Table S3). LS mean increases in C4 levels from baseline to week 13/ET were 21.5 ng/mL (95% CI, 10.9, 32.1) with maralixibat 10 mg and 5.5 ng/mL (95% CI, –5.2, 16.2) with maralixibat 20 mg compared with a decrease of 2.2 ng/mL (95% CI, –12.5, 8.1) in the placebo group (Table 3; Supporting Table S3).
Table 3.
Changes in Laboratory Parameters from Baseline to Week 13/et
| Maralixibat 10 mg (n = 21) | Maralixibat 20 mg (n = 21) | Maralixibat Overall (n = 42) | Placebo (n = 24) | |
|---|---|---|---|---|
| Bile acid synthesis | ||||
| C4, ng/mL | ||||
| LS mean change from baseline (95% CI) | 21.5 | 5.5 | 13.5 | –2.2 |
| (10.9, 32.1)‡ | (–5.2, 16.2) | (6.0, 21.0)‡ | (–12.5, 8.1) | |
| LS mean difference from placebo (95% CI) | 23.7 | 7.7 | 15.7 | N/A |
| (8.9, 38.5)† | (–7.2, 22.7) | (2.9, 28.5)* | ||
| Potential regulators of bile acid synthesis | ||||
| FGF‐19, pg/mL | ||||
| LS mean change from baseline (95% CI) | 7.1 | –31.8 | –12.3 | –5.8 |
| (–31.1, 45.4) | (–70.9, 7.3) | (–39.3, 14.6) | (–42.5, 30.9) | |
| LS mean difference from placebo (95% CI) | 12.9 | –26.0 | –6.5 | N/A |
| (–39.7, 65.5) | (–80.8, 28.8) | (–52.6, 39.5) | ||
| FGF‐21, pg/mL | ||||
| LS mean change from baseline (95% CI) | –32.2 | 28.9 | –1.7 | 20.8 |
| (–99.7, 35.3) | (–33.6, 91.4) | (–47.1, 43.8) | (–39.3, 80.9) | |
| LS mean difference from placebo (95% CI) | –53.0 | 8.1 | –22.4 | N/A |
| (–143.5, 37.5) | (–78.5, 94.7) | (–97.8, 52.9) | ||
| Cholestasis | ||||
| ALP, U/L | ||||
| LS mean change from baseline (95% CI) | –7.4 | 17.1 | 4.9 | 7.3 |
| (–41.8, 27.0) | (–17.3, 51.5) | (–19.5, 29.2) | (–24.7, 39.3) | |
| LS mean difference from placebo (95% CI) | –14.7 | 9.8 | –2.4 | N/A |
| (–61.7, 32.3) | (–37.2, 56.8) | (–42.6, 37.8) | ||
| Autotaxin, ng/mL | ||||
| LS mean change from baseline (95% CI) | –167.7 | –82.5 | –125.1 | 62.8 |
| (–275.6, –59.8)† | (–188.9, 23.9) | (–201.1, –49.1)† | (–42.5, 168.1) | |
| LS mean difference from placebo (95% CI) | –230.5 | –145.3 | –187.9 | N/A |
| (–381.9, –79.0)† | (–296.1, 5.6) | (–318.8, –56.9)† | ||
| GGT, U/L | ||||
| LS mean change from baseline (95% CI) | 28.6 | 61.9 | 45.3 | 45.0 |
| (–46.5, 103.7) | (–13.1, 137.0) | (–7.7, 98.2) | (–24.6, 114.6) | |
| LS mean difference from placebo (95% CI) | –16.4 | 16.9 | 0.3 | N/A |
| (–118.7, 85.9) | (–85.5, 119.4) | (–87.2, 87.7) | ||
| Total bilirubin, mg/dL | ||||
| LS mean change from baseline (95% CI) | 0.00 | 0.09 | 0.04 | –0.05 |
| (–0.13, 0.13) | (–0.04, 0.21) | (–0.05, 0.13) | (–0.17, 0.07) | |
| LS mean difference from placebo (95% CI) | 0.05 | 0.14 | 0.09 | N/A |
| (–0.12, 0.22) | (–0.04, 0.31) | (–0.06, 0.24) | ||
| Conjugated bilirubin, mg/dL | ||||
| LS mean change from baseline (95% CI) | 0.004 | 0.05 | 0.03 | –0.01 |
| (–0.06, 0.07) | (–0.02, 0.11) | (–0.02, 0.07) | (–0.08, 0.05) | |
| LS mean difference from placebo (95% CI) | 0.02 | 0.06 | 0.04 | N/A |
| (–0.08, 0.11) | (–0.03, 0.15) | (–0.04, 0.12) | ||
| Hepatocellular injury | ||||
| ALT, U/L | ||||
| LS mean change from baseline (95% CI) | 0.7 | 1.7 | 1.2 | 1.1 |
| (–12.7, 14.1) | (–11.8, 15.3) | (–8.3, 10.8) | (–11.5, 13.7) | |
| LS mean difference from placebo (95% CI) | –0.4 | 0.6 | 0.1 | N/A |
| (–18.7, 18.0) | (–18.0, 19.3) | (–15.7, 16.0) | ||
| AST, U/L | ||||
| LS mean change from baseline (95% CI) | –5.4 | 0.9 | –2.3 | 5.7 |
| (–18.9, 8.0) | (–12.7, 14.4) | (–11.9, 7.3) | (–7.0, 18.4) | |
| LS mean difference from placebo (95% CI) | –11.2 | –4.9 | –8.0 | N/A |
| (–29.8, 7.5) | (–23.7, 13.9) | (–24.1, 8.1) | ||
| Lipid metabolism | ||||
| Total cholesterol, mg/mL | ||||
| LS mean change from baseline (95% CI) | –15.4 | –6.3 | –10.9 | 2.3 |
| (–28.2, –2.7)* | (–18.8, 6.2) | (–19.7, –2.0)* | (–9.4, 14.0) | |
| LS mean difference from placebo (95% CI) | –17.7 | –8.6 | –13.2 | N/A |
| (–35.1, –0.4)* | (–25.7, 8.5) | (–27.8, 1.5) | ||
| LDL‐C, mg/mL | ||||
| LS mean change from baseline (95% CI) | –13.7 | –11.6 | –12.6 | –4.0 |
| (–24.2, –3.1)* | (–21.7, –1.5)* | (–19.8, –5.4)‡ | (–13.6, 5.6) | |
| LS mean difference from placebo (95% CI) | –9.6 | –7.5 | –8.6 | N/A |
| (–24.1, 4.8) | (–21.4, 6.3) | (–20.7, 3.5) | ||
P < 0.05, † P < 0.01, ‡ P < 0.001 (analysis of covariance model with treatment group, ALP level [strata] and treatment group by ALP level interaction as factors, and baseline value as a covariate). All P values are nominal (not adjusted for multiplicity).
Abbreviation: N/A, not applicable.
No clear trends were observed for LS mean changes in FGF‐19 and FGF‐21 (Table 3; Supporting Table S3).
Markers of Cholestasis and Hepatocellular Injury
Changes in levels of ALP, GGT, total or conjugated bilirubin, ALT, and AST from baseline to week 13/ET did not differ between the maralixibat and placebo groups (Table 3; Supporting Table S3).
Levels of autotaxin, a marker shown to correlate with severity of cholestatic itch in PBC,27 decreased from baseline to week 13/ET in the maralixibat groups and increased in the placebo group (Table 3; Supporting Table S3). The LS mean reductions in autotaxin levels from baseline to week 13/ET were –167.7 ng/mL (95% CI, –275.6, –59.8) in the maralixibat 10 mg group and –82.5 ng/mL (95% CI, –188.9, 23.9) in the maralixibat 20 mg group compared with an LS mean increase of 62.8 ng/mL (95% CI, –42.5, 168.1) in the placebo group (Table 3; Supporting Table S3).
Lipid Metabolism
Total cholesterol decreased from baseline to week 13/ET in the maralixibat groups, with a greater LS mean reduction in the 10 mg group (–15.4 mg/mL; 95% CI, –28.2, –2.7) than in the 20 mg group (–6.3 mg/mL; 95% CI, –18.8, 6.2) (Table 3; Supporting Table S3). In contrast, total cholesterol increased in the placebo group (LS mean change, 2.3 mg/mL; 95% CI, –9.4, 14.0). Reductions from baseline in LDL‐C were observed in the maralixibat and placebo groups at week 13/ET; LS mean reductions were numerically greater with maralixibat 10 mg (–13.7 mg/mL; 95% CI, –24.2, –3.1) and 20 mg (–11.6 mg/mL; 95% CI, –21.7, –1.5) than with placebo (–4.0 mg/mL; 95% CI, –13.6, 5.6).
Quality of Life, Sleep, and Overall Treatment Benefit
Comparisons of changes in PBC‐40 domain scores in the maralixibat and placebo groups did not indicate any clinically meaningful improvements in favor of maralixibat for the itch, emotional, cognitive, symptoms, social, or fatigue domains (Table 2; Supporting Table S2).
Changes in MOS‐Sleep Scale domain scores and the proportion of patients achieving optimal sleep did not differ between the maralixibat and placebo groups (Table 2; Supporting Table S2).
Despite no difference in the primary outcome of the study, more patients in the maralixibat groups perceived a treatment benefit at week 13 than in the placebo group as assessed by responses on the PIC scale (maralixibat overall, 89.7%; placebo, 68.2%; P = 0.034). The response rate as assessed on the PGTB scale was numerically greater in the maralixibat overall group (79.5%) than the placebo group (63.6%) (Supporting Table S4).
Post Hoc Analyses
There were no strong correlations between percentage changes from baseline in Adult ItchRO™ weekly sum score and corresponding changes in levels of sBA, C4, or FGF‐19 at any assessed time point in the maralixibat or placebo groups (Supporting Table S5).
A response to treatment across the overall study population was observed in proportionately more patients who had abnormally high sBA levels at baseline (34.5%, 10/29 patients) than in those with baseline sBA levels 3 times ULN or less (5.4%, 2/37 patients).
SAFETY OUTCOMES
Duration of exposure (mean ± SD) to all doses of maralixibat was 88.5 ± 14.29 days and to placebo was 87.9 ± 15.50 days (Supporting Fig. S1).
Treatment‐Emergent AEs
In the maralixibat overall and placebo groups, AEs were reported in 97.6% and 70.8% of patients, respectively (Table 4); the incidence of AEs in the maralixibat groups did not appear to be dose related. Of the 41 patients with AEs receiving maralixibat, the maximum severity of reported AEs was mild in 31.7%, moderate in 53.7%, and severe in 14.6%. AEs were mild or moderate in severity in all patients with AEs receiving placebo (58.8% and 41.2%, respectively). The AEs reported in 10% or more of patients in either the maralixibat overall or placebo groups, respectively, were diarrhea (61.9%; 25.0%), abdominal pain (23.8%; 4.2%), abdominal pain upper (23.8%; 8.3%), nausea (23.8%; 16.7%), cough (11.9%; 0.0%), headache (11.9%; 33.3%), abdominal distension (7.1%; 12.5%), and pruritus (2.4%; 12.5%). No deaths were reported during the study.
Table 4.
Summary of Treatment‐Emergent AEs
| Patients With AEs, n (%) | Maralixibat 10 mg (n = 20) | Maralixibat 20 mg (n = 21) | Maralixibat Overall (n = 42) | Placebo (n = 24) |
|---|---|---|---|---|
| Any AE | 19 (95.0) | 21 (100.0) | 41 (97.6) | 17 (70.8) |
| Any AE potentially related to the drug | 15 (75.0) | 15 (71.4) | 31 (73.8) | 11 (45.8) |
| Maximum severity of AE | ||||
| Mild | 8 (40.0) | 5 (23.8) | 13 (31.0) | 10 (41.7) |
| Moderate | 8 (40.0) | 13 (61.9) | 22 (52.4) | 7 (29.2) |
| Severe | 3 (15.0) | 3 (14.3) | 6 (14.3) | 0 (0.0) |
| AE leading to study drug discontinuation | 1 (5.0) | 0 (0.0) | 2 (4.8) | 0 (0.0) |
| Serious AE | 2 (10.0) | 1 (4.8) | 3 (7.1) | 0 (0.0) |
| Serious AE potentially related to the drug | 1 (5.0) | 0 (0.0) | 1 (2.4) | 0 (0.0) |
| AE leading to death | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Most common AEs (occurring in ≥10% of patients), number of patients with >1 AE, n (%) | ||||
| Gastrointestinal disorders | 15 (75.0) | 18 (85.7) | 34 (81.0) | 13 (54.2) |
| Diarrhea | 14 (70.0) | 11 (52.4) | 26 (61.9) | 6 (25.0) |
| Abdominal pain | 4 (20.0) | 5 (23.8) | 10 (23.8) | 1 (4.2) |
| Abdominal pain upper | 4 (20.0) | 6 (28.6) | 10 (23.8) | 2 (8.3) |
| Nausea | 5 (25.0) | 4 (19.0) | 10 (23.8) | 4 (16.7) |
| Abdominal distension | 3 (15.0) | 0 (0.0) | 3 (7.1) | 3 (12.5) |
| General disorders and administration site conditions | 4 (20.0) | 4 (19.0) | 8 (19.0) | 2 (8.3) |
| Chills | 2 (10.0) | 0 (0.0) | 2 (4.8) | 0 (0.0) |
| Musculoskeletal and connective tissue disorders | 5 (25.0) | 3 (14.3) | 8 (19.0) | 5 (20.8) |
| Pain in extremity | 0 (0.0) | 3 (14.3) | 3 (7.1) | 0 (0.0) |
| Back pain | 2 (10.0) | 0 (0.0) | 2 (4.8) | 0 (0.0) |
| Muscle spasms | 2 (10.0) | 0 (0.0) | 2 (4.8) | 0 (0.0) |
| Nervous system disorders | 4 (20.0) | 3 (14.3) | 8 (19.0) | 9 (37.5) |
| Headache | 3 (15.0) | 2 (9.5) | 5 (11.9) | 8 (33.3) |
| Respiratory, thoracic, and mediastinal disorders | 7 (35.0) | 1 (4.8) | 8 (19.0) | 1 (4.2) |
| Cough | 4 (20.0) | 1 (4.8) | 5 (11.9) | 0 (0.0) |
| Skin and subcutaneous tissue disorders | 1 (5.0) | 2 (9.5) | 3 (7.1) | 5 (20.8) |
| Pruritus | 0 (0.0) | 1 (4.8) | 1 (2.4) | 3 (12.5) |
One patient randomized to receive maralixibat 10 mg was down‐titrated to 5 mg owing to tolerability issues. This patient had moderate AEs of interest, including diarrhea, abdominal pain, nausea, and vomiting; all AEs were related to the study drug, and one AE of abdominal pain led to study discontinuation. AEs were coded using Medical Dictionary for Regulatory Activities, version 16.0, and graded as mild, moderate, or severe in the opinion of the site investigator reporting the AE, using the Common Terminology Criteria for AEs, version 4.0.
Two patients in the maralixibat overall group discontinued the study drug owing to an AE. One patient discontinued owing to moderate abdominal pain while receiving the 5‐mg dose of maralixibat during dose escalation. The second patient discontinued owing to severe diarrhea during stable dosing with maralixibat 10 mg. These events were considered related to and possibly related to the study drug, respectively. In the placebo group, no patients discontinued the study drug owing to an AE.
Serious AEs were reported in 3 patients receiving maralixibat. In the maralixibat 10‐mg group, severe myocardial infarction, not considered to be treatment related, was reported in 1 patient, and severe abdominal pain, considered possibly treatment related, was reported in another patient. Three serious AEs (a severe pleural effusion event and two severe gastrointestinal hemorrhage events), none of which were considered treatment related, were reported by a third patient in the maralixibat 20‐mg group. No serious AEs were reported in patients receiving placebo.
Gastrointestinal AEs
The overall incidence of gastrointestinal AEs was higher in the maralixibat groups (20 mg, 81.0%; 10 mg, 75.0%) than in the placebo group (50.0%) (Supporting Table S6). Gastrointestinal AEs were reported in proportionally fewer patients during stable dosing (maralixibat overall, 31.7%; placebo, 8.7%) than during dose escalation (maralixibat overall, 61.9%; placebo, 45.8%). The majority of gastrointestinal AEs had resolved by the end of the study without dose interruption or drug discontinuation.
Vital Signs
There were no clinically meaningful changes in vital signs in any treatment group.
Discussion
Inhibition of ileal bile acid reuptake in this 13‐week, randomized, double‐blind, placebo‐controlled trial of maralixibat in adults with PBC did not significantly improve pruritus compared with placebo. However, the lessons learned from this rigorously designed and executed trial are indispensable for understanding both the importance of the design of trials with a primary endpoint of pruritus and the therapeutic window of bile acid reuptake inhibition as a therapeutic strategy for cholestatic pruritus.
In this study, pruritus improved significantly from baseline to week 13/ET in both the maralixibat and placebo groups, with LS mean changes in ItchRO™ weekly sum scores of –26.5 and –23.4, respectively. The magnitude of this improvement from baseline (maralixibat overall, 52%; placebo, 47%), based on the means at baseline and week13/ET in ItchRO™ weekly sum scores, was similar to that reported in trials of medications recommended in practice guidelines for cholestatic pruritus, such as naltrexone and rifampin.7, 28 This finding unquestionably highlights both the importance of having a placebo group and that the placebo effect can be large when using subjective patient‐reported outcomes as the primary endpoint. Without this pronounced placebo effect, the observed reduction in pruritus in the maralixibat group may have been viewed as beneficial. Perhaps one way of minimizing the placebo effect could be the use of a more objective surrogate measure of itch, such as the biomarker autotaxin, which has been shown to correlate with itch severity and may have some prognostic value.27 Indeed, in the present study, the observed reductions in autotaxin levels with maralixibat and the increases with placebo support the improvement of pruritus with active treatment. Accounting for the placebo effect in clinical trials of cholestatic pruritus is difficult. The effect of placebo reported in previous cholestatic pruritus trials is inconsistent, worsening in one trial and improving in another.29, 30 The placebo effect is hypothesized to be mediated through dopamine release that occurs in the psychosocial setting of expectation of reward. Using pharmaceuticals to inhibit the placebo effect is not yet a validated study technique. However, studies involving patient‐reported outcomes should specifically educate investigators/coordinators regarding their interactions with patients in order to minimize placebo effects.
Hegade et al. 31 recently published results from a small placebo‐controlled trial of a different IBAT inhibitor for the treatment of pruritus in patients with PBC. In that crossover design trial, using a numeric rating scale from 0 to 10, the authors reported that pruritus improved by 57% in the study drug group and by 23% in the placebo group. The major differences between Hegade et al.’s results and the findings of the present study are the percentage improvement in pruritus associated with placebo (23% versus 47%, respectively) and the duration of the treatment period (2 weeks versus 13 weeks, respectively). As previously mentioned, the difference in placebo effect between Hegade et al.’s study and the present study could be due to the unwitting influence of the two different study teams, but another likely factor is the different study designs employed as the present study was a parallel group and the Hegade et al. study had a crossover with placebo run‐in design. The placebo run‐in may have served to absorb some of the placebo effect, assuming the initial optimistic expectations of patients are self‐limited. Maintaining blinding to the study intervention is difficult in a crossover design, and unblinding is the antidote to the placebo effect. The main side effect of IBAT inhibition in both studies was gastrointestinal disturbance, and in Hegade et al.’s study there were 19 gastrointestinal AEs on treatment versus five on placebo. While gastrointestinal disturbance is congruent with the mechanism of action and the known effects of excess bile salts in the lumen of the colon, such a prominent side effect may unblind many patients. With a crossover study design, patients receiving a medication with a characteristic side effect may readily distinguish between active treatment and placebo, and therefore only the treatment period before the crossover may be blinded fairly. Analysis of only the first treatment arm (before crossover) in the Hegade et al. study would also eliminate the potential confounder of a therapeutic carryover effect.
Another explanation for the different results between the two trials may be the potencies and doses of the drugs. In the present study, reductions in sBA levels and increases in C4 levels (a measure of de novo bile acid synthesis) were observed in the maralixibat groups but not in the placebo group. This may indicate that there is some pharmacologic engagement of the intended target at the doses used in this study. An evaluation of whether such target engagement reached a maximum was not possible in this study setting. Of note, studies with maralixibat in other patient populations found more robust treatment effects with no apparent increases in AEs at doses up to 100 mg daily. Indeed, separation in findings between active treatment and placebo was observed starting at 30 mg daily dose equivalents. Therefore, higher doses of IBAT inhibitors could be investigated in the future to increase the effect size, especially given the benign safety profile shown in this PBC population.
It was also determined in a post hoc analysis that proportionately more patients with an sBA level >3 times ULN at baseline had a treatment response than those with an sBA level ≤3 times ULN at baseline. Thus, assessing pretreatment sBA levels may help predict which patients would respond best to maralixibat. The baseline sBA level was not reported in ng/mL by Hegade et al.31 However, Hegade et al. reported a greater percentage drop in sBA levels compared with the present study, which could be due to differences in either drug potency or baseline sBA levels in the two populations studied.
In the present study, gastrointestinal AEs in the maralixibat groups were reported in proportionately fewer patients during the stable dosing period than during the dose‐escalation period (weeks 2‐4 after treatment initiation), suggesting that gastrointestinal AEs may attenuate with increased duration of IBAT inhibition. Slower dose escalation could potentially be used as a strategy to administer higher doses with better gastrointestinal tolerance and potentially higher efficacy.
In conclusion, maralixibat was not associated with statistically significant improvements in pruritus in patients with PBC compared with placebo. However, the placebo effect observed in this study was large enough (47%) to mask the expected effect of an IBAT inhibitor. This fortuitously named CLARITY trial has led to a clearer vision of how future trials of drugs that inhibit bile acid reabsorption to treat cholestatic pruritus should be carefully designed to minimize the placebo effect as well as gastrointestinal side effects. Study design, dose range, dosing scheme, and patient selection are all important factors that should be carefully considered in future trials.
Author names in bold designate shared co‐first authorship.
Supporting information
Acknowledgment
Dr. David Gothard and Dr. Elizabeth Gandhi, employees of Oxford PharmaGenesis, Oxford, United Kingdom, provided writing assistance for this publication. Editorial assistance in formatting, proofreading, copy editing, and fact checking was also provided by Oxford PharmaGenesis, with funding provided by Shire International GmBH. Jürgen Wiehn and Thomas Jaecklin from Shire International GmbH also reviewed and edited the manuscript for scientific accuracy. Jana Steinmetz from Premier Research was the study statistician. Trial recruitment in Birmingham, United Kingdom, used the National Institute for Health Research clinical research facilities.
Research supported by Lumena Pharmaceuticals, one of the Shire group of companies. Shire Crossref Funder Registry identifier: http://dx.doi.org/10.13039/100007343.
ClinicalTrials.gov identifier NCT01904058
The data sets, including redacted study protocol, redacted statistical analysis plan, and individual participant’s data behind the results reported in this article, are available to researchers who provide a methodologically sound proposal after deidentification in compliance with applicable privacy laws, data protection, and requirements for consent and anonymization. Data requests should follow the process outlined in the Data Sharing section on Shire’s website and should be directed to clinicaltrialdata@shire.com.
Potential conflict of interest: Dr. Pockros advises, received grants from, and is on the speakers’ bureau for Intercept, Gilead, and AbbVie; he received grants from and consults for Shire; he advises, received grants from, and consults for Prometheus; he consults for Tobira Therapeutics. Dr. Bowlus received grants from and consults for GlaxoSmithKline, CymaBay, Intercept, and Gilead; he received grants from Shire and consults for Conatus, Eli Lilly, Parvus, AbbVie, Bristol‐Myers Squibb, Merck, NGM Biopharmaceuticals, and Takeda. Dr. Levy received grants from Gilead, Intercept, GlaxoSmithKline, Genkytoek, Genfit, CymaBay, Enanta, Novartis, AbbVie, Merck, NGM Biopharmaceuticals, Shire, and Tobira Therapeutics. Dr. Patanwala is on the speaker’s bureau and received grants from Intercept and Falk and consults for Bristol‐Myers Squibb, Gilead, Janssen‐Cilag, and Norgine Pharmaceuticals. Dr. Luketic received grants from and consults for Genfit; he received grants from Intercept and consults for GlaxoSmithKline, AbbVie, Bristol‐Myers Squibb, Gilead, Idenix Pharmaceuticals, Merck, and NGM Biopharmaceuticals. Dr. Apostol and Ms. Gu are employed by and own stock in Shire. Dr. Hirschfield consults for GlaxoSmithKline, Norvartis, CymaBay, Intercept, Biotie Therapies, Falk, NGM Biopharmaceuticals, and Shire. Dr. Bacon received grants from, advises, and is on the speaker’s bureau for Merck; he advises, is on the speaker’s bureau for, and received grants from AbbVie; he advises, is on the speaker’s bureau, on the Data and Safety Monitoring Board for, and received grants from Gilead; he is on the speaker’s bureau and consults for Intercept and is on the speakers’ bureau for Valeant. Dr. Kennedy previously owned stock in, was employed by, and consulted for Shire. Dr. Medendorp consults for Lumena. Dr. Dorenbaum was employed by Lumena Pharmaceuticals at the time of this study. Dr. Novak was an employee of Lumena, one of the Shire group of companies, at the time of this study. Dr. Jones consults for GlaxoSmithKline, Intercept, Lumena, Novartis, Pfizer, and Shire. Dr. Mayo received grants from Gilead Sciences, Intercept, Lumena, NGM Biopharmaceuticals, and Salix Pharmaceuticals, and consulted for Intercept Pharmaceuticals. Dr. Vuppalanchi has nothing to report.
References
- 1. Beuers U, Gershwin ME, Gish RG, Invernizzi P, Jones DE, Lindor K, et al. Changing nomenclature for PBC: from ‘cirrhosis’ to ‘cholangitis’. J Hepatol 2015;63:1285‐1287. [DOI] [PubMed] [Google Scholar]
- 2. Purohit T, Cappell MS. Primary biliary cirrhosis: pathophysiology, clinical presentation and therapy. World J Hepatol 2015;7:926‐941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Poupon RE, Chretien Y, Chazouilleres O, Poupon R, Chwalow J. Quality of life in patients with primary biliary cirrhosis. Hepatology 2004;40:489‐494. [DOI] [PubMed] [Google Scholar]
- 4. Montagnese S, Nsemi LM, Cazzagon N, Facchini S, Costa L, Bergasa NV, et al. Sleep‐wake profiles in patients with primary biliary cirrhosis. Liver Int 2013;33:203‐209. [DOI] [PubMed] [Google Scholar]
- 5. Bergasa NV. The pruritus of cholestasis. J Hepatol 2005;43:1078‐1088. [DOI] [PubMed] [Google Scholar]
- 6. Bergasa NV. The itch of liver disease. Semin Cutan Med Surg 2011;30:93‐98. [DOI] [PubMed] [Google Scholar]
- 7. European Association for the Study of the Liver . EASL Clinical Practice Guidelines: management of cholestatic liver diseases. J Hepatol 2009;51:237‐267. [DOI] [PubMed] [Google Scholar]
- 8. Combes B, Carithers RL Jr, Maddrey WC, Lin D, McDonald MF, Wheeler DE, et al. A randomized, double‐blind, placebo‐controlled trial of ursodeoxycholic acid in primary biliary cirrhosis. Hepatology 1995;22:759‐766. [PubMed] [Google Scholar]
- 9. Poupon RE, Lindor KD, Cauch‐Dudek K, Dickson ER, Poupon R, Heathcote EJ. Combined analysis of randomized controlled trials of ursodeoxycholic acid in primary biliary cirrhosis. Gastroenterology 1997;113:884‐890. [DOI] [PubMed] [Google Scholar]
- 10. Rudic JS, Poropat G, Krstic MN. Bjelakovic G, Gluud C. Ursodeoxycholic acid for primary biliary cirrhosis. Cochrane Database Syst Rev 2012;12:CD000551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lindor KD, Gershwin ME, Poupon R, Kaplan M, Bergasa NV, Heathcote EJ. American Association for Study of Liver Diseases. Primary biliary cirrhosis. Hepatology 2009;50:291‐308. [DOI] [PubMed] [Google Scholar]
- 12. Kremer AE, Namer B, Bolier R, Fischer MJ, Oude Elferink RP, Beuers U. Pathogenesis and management of pruritus in PBC and PSC. Dig Dis 2015;33(Suppl. 2):164‐175. [DOI] [PubMed] [Google Scholar]
- 13. Keller B, Dorenbaum A, Wynne D, Gedulin B, Setchell K, Olek E, et al.Effect of apical sodium‐dependent bile acid transporter (ASBT) inhibition on serum and fecal bile acids in healthy volunteers. In: VII Falk Gastro‐Conference: Falk Symposia 194: XXIII International Bile Acid Meeting; October 8–9, 2014; Freiburg, Germany.
- 14. Huang HC, Tremont SJ, Lee LF, Keller BT, Carpenter AJ, Wang CC, et al. Discovery of potent, nonsystemic apical sodium‐codependent bile acid transporter inhibitors (Part 2). J Med Chem 2005;48:5853‐5868. [DOI] [PubMed] [Google Scholar]
- 15. Tremont SJ, Lee LF, Huang HC, Keller BT, Banerjee SC, Both SR, et al. Discovery of potent, nonsystemic apical sodium‐codependent bile acid transporter inhibitors (Part 1). J Med Chem 2005;48:5837‐5852. [DOI] [PubMed] [Google Scholar]
- 16. Bhat BG, Rapp SR, Beaudry JA, Napawan N, Butteiger DN, Hall KA, et al. Inhibition of ileal bile acid transport and reduced atherosclerosis in apoE‐/‐ mice by SC‐435. J Lipid Res 2003;44:1614‐1621. [DOI] [PubMed] [Google Scholar]
- 17. Bradley T, Keller BT, Nikoulina S, Nazarenkov N, Gedulin B. LUM001, an inhibitor of ASBT, improves liver function and tissue pathology in a rat cholestasis model. Hepatology 2014;60:275A‐276A. [Google Scholar]
- 18. Schukfeh N, Metzelder ML, Petersen C, Reismann M, Pfister ED, Ure BM, et al. Normalization of serum bile acids after partial external biliary diversion indicates an excellent long‐term outcome in children with progressive familial intrahepatic cholestasis. J Pediatr Surg 2012;47:501‐505. [DOI] [PubMed] [Google Scholar]
- 19. Datta DV, Sherlock S. Cholestyramine for long term relief of the pruritus complicating intrahepatic cholestasis. Gastroenterology 1966;50:323‐332. [PubMed] [Google Scholar]
- 20. Di Padova C, Tritapepe R, Rovagnati P, Rossetti S. Double‐blind placebo‐controlled clinical trial of microporous cholestyramine in the treatment of intra‐ and extra‐hepatic cholestasis: relationship between itching and serum bile acids. Methods Find Exp Clin Pharmacol 1984;6:773‐776. [PubMed] [Google Scholar]
- 21. Reshetnyak VI. Primary biliary cirrhosis: clinical and laboratory criteria for its diagnosis. World J Gastroenterol 2015;21:7683‐7708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Elman S, Hynan LS, Gabriel V, Mayo MJ. The 5‐D itch scale: a new measure of pruritus. Br J Dermatol 2010;162:587‐593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cicione C, Degirolamo C, Moschetta A. Emerging role of fibroblast growth factors 15/19 and 21 as metabolic integrators in the liver. Hepatology 2012;56:2404‐2411. [DOI] [PubMed] [Google Scholar]
- 24. Kremer AE, Martens JJ, Kulik W, Rueff F, Kuiper EM, van Buuren HR, et al. Lysophosphatidic acid is a potential mediator of cholestatic pruritus. Gastroenterology 2010;139:1008‐1018, 1018.e1001. [DOI] [PubMed] [Google Scholar]
- 25. Jacoby A, Rannard A, Buck D, Bhala N, Newton JL, James OF, et al. Development, validation, and evaluation of the PBC‐40, a disease specific health related quality of life measure for primary biliary cirrhosis. Gut 2005;54:1622‐1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Spritzer K, Hays R. MOS‐Sleep Scale: A Manual for Use and Scoring, Version 1.0. Los Angeles, CA:RAND; 2003. [Google Scholar]
- 27. Wunsch E, Krawczyk M, Milkiewicz M, Trottier J, Barbier O, Neurath MF, et al. Serum autotaxin is a marker of the severity of liver injury and overall survival in patients with cholestatic liver diseases. Sci Rep 2016;6:30847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tandon P, Rowe BH, Vandermeer B, Bain VG. The efficacy and safety of bile acid binding agents, opioid antagonists, or rifampin in the treatment of cholestasis‐associated pruritus. Am J Gastroenterol 2007;102:1528‐1536. [DOI] [PubMed] [Google Scholar]
- 29. Mayo MJ, Handem I, Saldana S, Jacobe H, Getachew Y, Rush AJ. Sertraline as a first‐line treatment for cholestatic pruritus. Hepatology 2007;45:666‐674. [DOI] [PubMed] [Google Scholar]
- 30. Bergasa NV, McGee M, Ginsburg IH, Engler D. Gabapentin in patients with the pruritus of cholestasis: a double‐blind, randomized, placebo‐controlled trial. Hepatology 2006;44:1317‐1323. [DOI] [PubMed] [Google Scholar]
- 31. Hegade VS, Kendrick SF, Dobbins RL, Miller SR, Thompson D, Richards D, et al. Effect of ileal bile acid transporter inhibitor GSK2330672 on pruritus in primary biliary cholangitis: a double‐blind, randomised, placebo‐controlled, crossover, phase 2a study. Lancet 2017;389:1114‐1123. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials