

Abstract
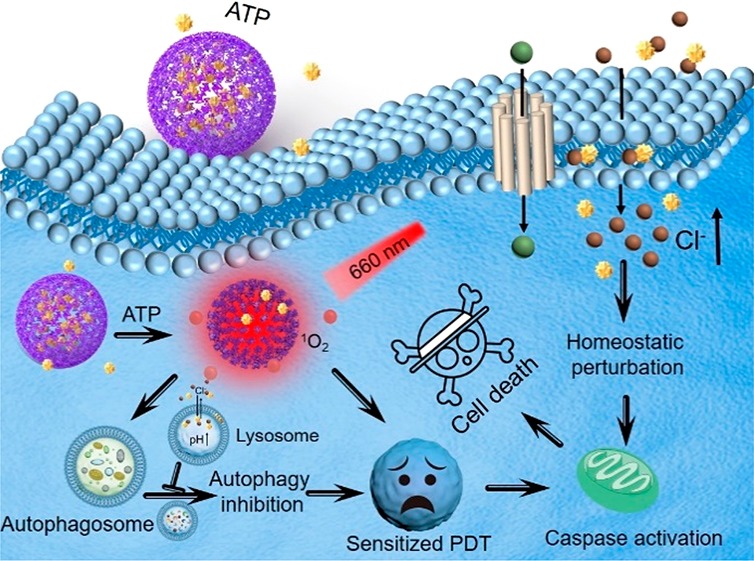
In this article, an adenosine-triphosphate-regulated (ATP-regulated) ion transport nanosystem [SQU@PCN, porphyrinic porous coordination network (PCN) incorporated with squaramide (SQU)] was designed and synthesized for homeostatic perturbation therapy (HPT) and sensitizing photodynamic therapy (PDT) of tumors. It was found that this nanotransporter SQU@PCN easily accumulated in tumor sites while avoiding metabolic clearance and side effects. In response to a high expression of ATP in the tumor, SQU@PCN was decomposed because of the strong coordination of ATP with metal ligand of PCN. Subsequently, incorporated SQU was released and then simultaneously transported chloride ions across membrane of the cell and lysosome along with the chloride ion concentration gradient. On one hand, influx of chloride ions by SQU increased intracellular ion concentration, which disrupted ion homeostasis and further induced tumor cell apoptosis. On the other hand, SQU-medicated coupling transport of H+/Cl– across the lysosomal membrane alkalized the lysosome, resulting in inhibition of autophagy. This SQU-mediated autophagy inhibition would sensitize PCN-based PDT since activated autophagy by traditional PDT would resist and weaken the therapeutic efficacy. In vivo animal test results revealed that combined HPT and sensitized PDT could realize tumor eradication while blocking metastasis, which provided a paradigm for complementary multimodal tumor treatment.
Short abstract
The synthetic SQU@PCN nanotransporter achieved homeostatic perturbation therapy by transporting Cl− into the cell and sensitized photodynamic therapy by inhibiting autophagy in tumor cells.
1. Introduction
Very recently, as a safe and noninvasive treatment of cancer, photodynamic therapy (PDT) has attracted more and more attention, which is capable of producing highly toxic reactive oxygen species (ROS) under light stimulation.1−3 The strong oxidative property of ROS results in photodamage of proteins and genes, eventually contributing to cancer cell death.4 Unfortunately, a lot of evidence supports that resistance mechanisms of cancer cells will be simultaneously activated during PDT,5−8 which reduces therapeutic efficiency of PDT.9,10 Generally, autophagy as a homeostatic lysosome-dependent catabolic process is activated for relieving cancer cell press and is regarded as a very important defense mechanism.11,12 Also, it has been documented that enhanced tolerance by autophagy plays a crucial role for resistance of cancer cells in response to multiple treatments, including chemotherapy, radiotherapy, and phototherapy.13−15 A great quantity of research work has confirmed that enhanced antitumor effects of phototherapy can be achieved by pharmacological autophagy inhibition or knockout of autophagy-related genes.16−19 However, taking into account hysteresis of small molecule inhibitors and the instability of gene delivery, further applications of these two strategies are limited. Thus, there is an urgent need to develop an alternative strategy to improve the effectiveness of phototherapy by suppressing autophagy.
Maintaining ion homeostasis via transmembrane anion transport is essential for diverse physiological processes, such as cell proliferation, cellular signaling, cell membrane potentials, and osmotic pressure, etc.20−24 As one of the most abundant cellular anions in the physiological system, the chloride ion has a stable extracellular concentration (∼120 mM), cytosolic concentration (5–40 mM), and lysosomal concentration (∼80 mM), which is maintained by transmembrane ion transporters and channels.25,26 Perturbation of cellular chloride ion concentration via transporters will lead to perturbation of ion homeostasis that further induces cell apoptosis.27−31 In addition, disrupting pH gradients of the lysosome mediated by the chloride ion transporter is able to inhibit autophagy because of reduced lysosomal enzyme activity in the alkalized environment.32,33 In other words, the introduction of a chloride ion transport system to disorder the intracellular ion concentration of tumor cells can achieve both cell apoptosis and autophagy suppression, which is attractive for cancer treatment. However, the majority of chloride ion transporters are nonspecific and lipophilic small molecules,34−36 which hinder their biomedical applications. It is necessary for ion transporters to achieve intelligent ion transport with efficient delivery as well as stimulus response transport activity in a specific target.
In view of strong coordination of phosphate and metal ions37,38 and the high expression of ATP in the tumor environment,39,40 here, an ATP-regulated ion transport nanosystem (SQU@PCN) was designed and synthesized by incorporating squaramide (SQU) into a porphyrinic porous coordination network (PCN) with a simple one-pot method for homeostatic perturbation therapy (HPT) and sensitizing photodynamic therapy (PDT) of the tumor. As illustrated in Scheme 1, after SQU@PCN accumulated in the tumor site by enhanced permeability and retention effect (EPR), high expression of ATP in the tumor might disintegrate PCN to release encapsulated SQU. As a chloride ion transporter, SQU could both cause cell apoptosis for HPT and inhibit autophagy by disrupting cellular ion homeostasis. In addition, PCN can produce plenty of ROS to damage tumor cells for PDT under near-infrared light (NIR) irradiation. Importantly, SQU-medicated autophagy inhibition would sensitize PDT, which would greatly improve the therapeutic efficacy of PDT. Ultimately, synergistic effects of PCN-mediated PDT and SQU-mediated HPT effectively achieved comprehensive tumor suppression.
Scheme 1. Schematic Illustration of SQU@PCN Preparation and the Tumor Cell Death Process by Homeostatic Perturbation Therapy and Sensitized Photodynamic Therapy.

2. Results and Discussion
The synthesis of SQU@PCN was according to the procedures of the reported literature with some modifications.41 The size and morphology of as-synthesized SQU@PCN were measured by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). As shown in Figure 1A,B, we could find that SQU@PCN was a monodisperse uniform spherical nanoparticle with a size of about 155 nm, which was similar to that of PCN exhibited in Figure S1. In addition, dynamic light scattering (DLS) (Figure S2) indicated that obtained SQU@PCN nanoparticles had a positive potential of about 22 mV and a narrow diameter distribution (polydispersity index, PDI 0.03) at approximately 160 nm, roughly consistent with the value measured by electron microscopy. In contrast with results of PCN, the slight increase in the values of both potential and diameter should be attributed to the encapsulation of SQU by coordination and π–π stacking. The loading was about 7.4% calculated on the basis of the standard curve of SQU (Figure S3), which was well-matched with the data determined by thermogravimetric analysis (TGA) (Figure S4). In addition, the patterns of SQU@PCN were identical to that of PCN from a characteristic peak comparison of powder X-ray diffraction (PXRD) (Figure S5), illustrating that the modified one-pot synthesis method did not have much impact on the crystal structure of PCN while successfully achieving high loading of SQU. Moreover, as presented in Figure 1C, the hydrodynamic diameter and PDI of SQU@PCN fluctuated within the error range during the 7 day soaking in water. This indicated the good stability of nanoparticles, which would be beneficial for subsequent applications.
Figure 1.

(A) SEM and (B) TEM image of SQU@PCN. (C) Hydrodynamic size and PDI of SQU@PCN during 7 days of soaking in water. (D) PXRD patterns of SQU@PCN after treatment with or without ATP at different times. (E) Release of SQU from SQU@PCN in different HEPES buffer solutions. (F) Detailed process of SQU@PCN-mediated chloride transport. (a) ATP-responsive SQU release from SQU@PCN. (b) Chloride transport of released SQU in POPC vesicle. (c) Chloride transport mechanism of released SQU by mobile carrier and ion exchange. (G) Chloride efflux after treatment with different samples at different times. (H) Chloride efflux mediated by SQU@PCN after treatment with or without ATP at different times. (I) ROS production of different samples in the solution containing ATP.
Because of the strong coordination between phosphate and metal ions,37,38 the degradation behavior of SQU@PCN was explored in the presence of substances containing phosphate like ATP. From the observation of TEM images (Figure S6), the surface and morphology of SQU@PCN had been partially swelled and melted after treatment in ATP (1 mg/mL) solution for 48 h, implying ATP-induced collapse of the PCN framework. This conclusion was further confirmed by patterns of PXRD in Figure 1D. In a solution containing ATP, the crystal structure of SQU@PCN deteriorated with the prolongation of processing time until the crystal peak was completely lost after 4 h of treatment. In sharp contrast, SQU@PCN always kept its crystallinity in ATP-free solution. Together, all the above phenomena implied the degradation characteristics of SQU@PCN in response to ATP. Furthermore, we quantitatively detected degradation of SQU@PCN over time considering that tetrakis (4-carboxyphenyl) porphyrin (TCPP), a decomposition product of SQU@PCN, is a fluorescent molecule. The absorption peak of TCPP at 420 nm in dialysate with ATP significantly rose with time (Figure S7A), while absorption of TCPP was hardly detected in the absence of ATP (Figure S7B). The corresponding released percentage of TCPP was shown in Figure S7C. Compared to 2% TCPP release in the control group, the released percentage of TCPP in ATP solution approached 32% within 6 h. These results not only confirmed the good stability of SQU@PCN nanoparticles but also indicated an ATP-triggered rapid and sensitive degradation property, which provided a prerequisite as an excellent drug delivery for achieving site-specific drug “burst release”. Subsequently, the release of incorporated SQU in the presence of ATP was detected by a UV/vis absorption spectrometer. As expected, the release of SQU presented a positive correlation of time and reached the maximum of approximately 80% within 1 h in the HEPES buffer containing ATP (Figure 1E). Little release of SQU indicated a negligible leak and stability of loading. Such sensitive release of SQU in response to a simulated tumor microenvironment with high expression of ATP was helpful for improving drug utilization while avoiding side effects during therapy.
After successfully certifying ATP-responsive SQU release, the transport ability of SQU@PCN with ATP was assessed. As depicted in Figure 1F, chloride efflux in 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) vesicles containing 489 mM NaCl and 1 mg/mL ATP were prepared and then dispersed in an external solution with 489 mM NaNO3 and 1 mg/mL ATP. After the addition of samples, a chloride-selective electrode was used to monitor ion efflux from the vesicle interior. Incorporated SQU was released from nanotransporter SQU@PCN in response to ATP, which could bind chloride ions by hydrogen bonding because of the strong anion-binding ability and transport chloride ions across lipid bilayers along with ion concentration gradients via mobile-carrier and anion-exchange mechanisms (Figure 1F).42 As expected, the ranking of transport activity was SQU > SQU@PCN ≫ PCN in a short time (30 min) (Figure 1G). Chloride efflux mediated by SQU@PCN was less than that of SQU, which was ascribed to time-related ATP-triggered SQU release. This explanation was further verified by comparing chloride transport of SQU@PCN and SQU for a long time. At 1 and 2 h, nearly 100% chloride efflux by SQU@PCN implied that a majority of the encapsulated SQU release accelerated ion transport. For the group treated with PCN, a small amount of chloride detected by the ion-selective electrode (ISE) might be due to the slow leakage of chloride from POPC vesicles. In addition, we also studied the effect of ATP and SQU@PCN concentration on ion transport efficiency (Figure 1H). In the absence of ATP, the transport effect of SQU@PCN was poor, and less than 25% of the chloride ions were transported across POPC even at 2 h. This was far worse than the reported result of about 100% transport in 120 s mediated by pure SQU.33 The limited transport ability suggested that incorporated SQU into nanoparticles had severely suppressed the ion transport activity of SQU. However, once ATP was added, the efficiency of transport greatly improved and exhibited a trend of increase with time and NP concentration, demonstrating time- and concentration-related transport activity of SQU@PCN. Collectively, all results indicated that SQU@PCN could only implement time- and concentration-positive ion transport in the presence of ATP. Perfectly, such a property endowed SQU@PCN with the ability to achieve site-specific and controllable ion transport with little side effects.
It was known that ROS played an important role in PDT of cancer. Hence, ROS generation of SQU@PCN should be detected by using 2′,7′-dichlorofluorescin diacetate (DCFH-DA), a ROS probe which could emit green fluorescence after being oxidized by ROS. As expected, SQU@PCN had significant fluorescence enhancement over time under light irradiation as observed in Figure 1I. After irradiation for 60 s, the relative fluorescence intensity of SQU@PCN was nearly 55 times stronger than that of the control group without light, indicating that light triggered plenty of ROS production. The slight difference between SQU@PCN + hv and PCN + hv might be attributed to the effect of the incorporated SQU in the crystal structure of PCN. Inspired by the outstanding ROS production ability of SQU@PCN, its performance in vitro was further explored. As shown in Figure 2A, similar to the group treated with PCN + hv, the SQU@PCN + hv group could observe a large area of bright green fluorescence, which implied a large amount of ROS generation in vitro. However, there was almost no green fluorescence in all other control groups. These phenomena were in agreement with the results tested in solution, illustrating that SQU@PCN could produce sufficient ROS for PDT under the stimulation of light.
Figure 2.

(A) In vitro ROS production after cells were incubated with different samples. “hv” refers to light irradiation with 660 nm laser at 30 mW/cm2 for 3 min. Scale bar: 20 μm. Change of intracellular chloride ion concentration with MQAE as a probe after treatment with different (B) time, (C) concentration, and (D) samples. **P < 0.01 and ***P < 0.001.
With confirmation of the excellent ATP-regulated ion transport capacity of SQU@PCN, its ion transport behavior was studied in tumor cells with overexpressed ATP. In general, HeLa cells were precultured with a chloride fluorescent probe, N-(ethoxycarbonylmethyl)-6-methoxyquinolinium bromide (MQAE), whose fluorescence intensity proportionally decreases with the increase of intracellular chloride ions. Then, fluorescence of cellular MQAE was tested by a microplate reader after different treatments. From the result displayed in Figure 2B, fluorescence of MQAE gradually decreased with the prolongation of culture time of SQU@PCN at a given concentration of nanoparticles, indicating an increase of cellular Cl– concentration over time. This also showed that the nanotransporter could realize efficient ion transport in vitro. More importantly, because of the best effect of MQAE fluorescent quenching at 4 h of culture, it was chosen as an optimal culture time for subsequent experiments. In addition, as proven in Figure 2C, Cl– concentration transported by SQU@PCN exhibited a correlation with concentration of nanoparticles at the determined culture time (4 h) mentioned above. Namely, when the concentration of nanoparticles was higher, more chloride ions were transported. Of special note, nearly 30% of MQAE fluorescence was quenched after treatment with 60 mg/L SQU@PCN, and the quenching effect of MQAE fluorescence was not obvious by further increasing the concentration of nanoparticles. Hence, 60 mg/L SQU@PCN nanoparticles were considered as the most suitable concentration for cellular ion transport. Then, an experiment was conducted to compare the transport activities of different materials at defined incubation time (4 h) and nanoparticle concentration (60 mg/L). In contrast with all control groups, cells treated with SQU@PCN were the group with lowest percentage of MQAE fluorescence (Figure 2D), illustrating the best ion transport activity. All the data above revealed the fact that released SQU from ATP-induced SQU@PCN degradation could maximize cellular chloride ion transport at optimized time and concentration conditions, which laid the foundation for regulating intracellular physiological changes triggered by ion transport.
As documented in the literature,33 the transfer of chloride ions from the lysosome was accompanied by hydrogen ions, which meant a change of lysosomal pH. With motivation from the ability of SQU@PCN to efficiently transport chloride ions in vitro, the change of lysosomal pH during the ion transport process was explored in depth. For the monitoring of the lysosomal acidity, LysoSensor Green DND-189 was adopted as its fluorescence is enhanced with acidity. As observed in Figure 3A, cells treated with PCN and PBS exhibited bright green fluorescence while fluorescence in SQU@PCN and SQU groups severely attenuated. This indicated that SQU@PCN nanoparticles like a simple small molecule SQU could lead to an increase of lysosomal pH in the process of transporting chloride ions via penetrating the lysosomal membrane. From this, we verified that lysosomal pH was associated with chloride ion transport and could be regulated by chloride ion transporters. In addition, we found that green fluorescence gradually diminished with time as shown in Figure 3B, illustrating that transport of chloride ions mediated by SQU@PCN did cause lysosomal alkalization. More importantly, the degree of lysosomal alkalization could be regulated by controlling incubation time of SQU@PCN. In particular, it was of great significance for the manipulating autophagic process because of the close relationship between lysosomal pH and autophagy.43 This meant that SQU@PCN serving as chloride ion transporter had the potential to manipulate the autophagic process by coupling H+/Cl– transport in the lysosome.
Figure 3.

Detection of lysosome pH after cells were (A) incubated with different samples and (B) treated with SQU@PCN at different time. Scale bar: 20 μm. (C) Western blot analysis of cells treated with PCN in the presence or absence of NIR irradiation for LC3 I/II and P62 proteins and (D) corresponding gray-scale analysis result of strips. (E) In vitro cytotoxicity of PCN against HeLa cells under NIR irradiation with or without CQ. “hv” refers to light irradiation with 660 nm laser at 30 mW/cm2 for 5 min.
Subsequently, the influence of SQU@PCN on autophagy was investigated in vitro. In general, the microtubule-associated protein 1 light chain 3-II (LC3-II) and p62 serving as autophagic markers are often used to monitor autophagy. The levels of LC3-II increase, and the levels of p62 decrease during autophagy induction. However, when autophagy is inhibited, both levels of proteins increase. According to a report in the literature,5 photodamage like PDT was able to induce autophagy. Indeed, as tested by Western blot in Figure 3C, cells treated with PCN + hv presented obviously increased expression of LC3-II and decreased expression of p62 compared with the group without irradiation. The corresponding gray-scale analysis results (Figure 3D) further illustrated nearly twice the LC3-II increment and p62 reduction in the PCN + hv group. Such light irradiation (660 nm, 30 mW/cm2, 5 min) conditions had a negligible effect on autophagy considering nontoxicity against cells even at twice the dose of light (Figure S8). Therefore, we could draw a conclusion that PCN-based PDT induced autophagy. Furthermore, tumor cells were pretreated with chloroquine (CQ), a well-known autophagy inhibitor, before PCN-induced PDT to clarify the effect of induced autophagy on PDT efficiency. The result of the MTT assay in Figure 3E showed that cell viability treated with PDT +hv was higher than that of PDT + hv+ CQ with a nontoxic concentration of CQ (Figure S9), which suggested that autophagy activated by PDT inhibited therapeutic efficiency because of the resistance of autophagy-related tolerance.15 In other words, PDT effects could be improved by reagents which were able to inhibit autophagy.
Interestingly, SQU as an ion transporter had been documented to disrupt the autophagic process by increasing lysosomal pH and inhibiting lysosomal cathepsin activity.33 We proved above that SQU@PCN serving as a nanotransporter led to an increase of lysosomal pH by coupling H+/Cl– transport in the lysosome. Thus, this forced us to explore if SQU@PCN could disrupt autophagy. In the experiment, we selected a specific HeLa cell line, GFP-LC3/HeLa cells, which stably express GFP-LC3 fusion protein (GFP, green fluorescent protein; and LC3, microtubule-associated light chain 3 protein). Typically, GFP-LC3 exhibits homogeneous green fluorescence in the cytoplasm, but it appears as green fluorescent spots due to accumulation on the autophagosome membrane upon autophagy. As shown in Figure 4A, obvious yellow fluorescence spots in the PCN + hv group reflected the formation of the autolysosome due to smooth fusion of GFP-LC3-reprsented autophagosomes and LysoTracker Red-represented lysosomes. This phenomenon further indicated that PCN-based photodynamic therapy could activate autophagy. In contrast, GFP-LC3/HeLa cells incubated with SQU@PCN displayed more green spots and little red fluorescence as compared with that of the control group, revealing disruption of the autophagic process by damaging lysosomes. Moreover, from the observation of the SQU@PCN +hv group, there was almost no autolysosome formation, indicating that SQU@PCN could still suppress PDT-induced autophagy under light irradiation. The phenomenon similar to SQU demonstrated that SQU@PCN acting as a nanotransporter was able to successfully exert autophagy inhibition by blocking the formation of autolysosomes in vitro whether under light or nonlight conditions. For a further illustration of lysosomal-disrupted autophagy inhibition by SQU@PCN, bio-TEM of HeLa cells was carried out after different treatments with 660 nm irradiation. In Figure 4B, cells treated with PCN displayed much more autolysosomes than the control group, indicating autophagy induction by PCN-based PDT. In contrast, SQU@PCN-treated cells were observed with the obvious decrease of autolysosomes in comparison with all other groups, which should attribute to autophagy inhibition of SQU@PCN by blocking the formation of autolysosomes. In addition, the autophagy suppression effect of SQU@PCN was also confirmed by Western blot (Figure 4C). As expected, cells only treated with SQU were observed the improved levels of LC3-II and p62, fully indicating its autophagy inhibition. Compared with the control group, PCN with light presented increased levels of LC3-II and decreased levels of p62, which further implied autophagy induction during PDT as proven above. However, when SQU was incorporated into PCN, autophagy was suppressed because of increased levels of LC3-II and P62. The same conclusion was also reflected in values by gray-scale analysis of strips in Figure 4D. These suggested that SQU@PCN could act as an inhibitor to impair the autophagic process triggered by PCN with light; as a consequence, subsequent efficiency of PDT was significantly improved.
Figure 4.

(A) Study of autolysosome formation by CLSM with GFP-LC3 HeLa cells after treatment with different samples. The lysosome was stained with LysoTracker Red. Scale bar: 20 μm. (B) Bio-TEM image of HeLa cells with different treatments under NIR irradiation. Yellow arrows refer to autolysosome. Scale bar (top panel): 10 μm. Scale bar (bottom panel): 4 μm. (C) Western blot analysis of cells with different treatments under NIR irradiation for LC3 I/II and P62 proteins and (D) corresponding gray-scale analysis result of strips. (E) In vitro cytotoxicity of different samples against HeLa cells with or without NIR irradiation. ***P < 0.001. (F) Live/dead cell staining assay with different treatments against HeLa cells. Dead cells were stained with PI (red), and live cells were stained with Calcein-AM (green). “hv” refers to light irradiation with a 660 nm laser at 30 mW/cm2 for 5 min.
Under these premises above, the toxicity of SQU@PCN against tumor cells was further assessed. Then, we designed and performed a series of experiments, including MTT assay and live/dead cell staining assay. As shown in Figure 4E, the nontoxicity of PCN indicated that PCN was suitable as a carrier to achieve SQU delivery in vivo. Additionally, except for the inhibition autophagy proven above, SQU as an ion transporter was found with a high cytotoxicity (IC50 = 1.36 mg/L) at low concentration due to dysregulation of cellular ion homeostasis. Once incorporated into PCN nanoparticles, SQU@PCN exhibited similar toxic effects as SQU, which were ascribed to released SQU in response to highly expressed ATP of tumor cells. Moreover, SQU@PCN exhibited a specific tumor killing effect when compared to the little toxicity in normal cells (Figure S10) with low expression of ATP. In sharp contrast, cells treated with SQU@PCN were found the highest toxicity and the lowest IC50 of all after being irradiated with a 660 nm laser. Compared with only HPT with SQU or PDT with PCN, the ability of SQU@PCN to induce cell death was better than their sum, implying an excellent cancer cell killing effect of synergistic treatment. Similarly, matched with the results of the MTT assay, cells stained with Calcein-AM and PI showed a similar trend of toxicity. The ratio of red and green fluorescence (green and red fluorescence refers to dead and living cells) was highest in the SQU@PCN + hv group of all (Figure 4F), further suggesting that it had the best effect for damaging tumor cells.
To further study the mechanism of material-induced tumor cell death, we conducted experiments with annexin V-FITC/PI and JC-1. As displayed in Figure 5A, cells had migrated to Q2 and Q3 quadrants after treatment with SQU@PCN + hv, PCN + hv, and SQU + hv as well as SQU@PCN. Q2 and Q3 quadrants represent bound annexin V and uptake of PI, which is an important feature of apoptosis. Hence, we could draw a conclusion that PCN-based PDT and SQU-based HPT were apoptosis-induced cells death. Of special note, SQU@PCN with light displayed the highest proportion of apoptotic cells, nearly 67.8% as the data of statistical analysis in Figure S11. This phenomenon indicated that the synergistic effect of PDT and HPT with SQU@PCN showed greater superiority than the single one, which was also matched with the result of toxicity in vitro. In addition, since the decreased mitochondrial membrane potential is a marker of apoptosis, we also detected the change of mitochondrial membrane potential to rule out the mechanism of cell death via necrosis. JC-1 was used as a membrane-potential-sensitive probe, which emits red fluorescence as a polymer (J-aggregates) when the mitochondrial membrane potential is high and green fluorescence as a monomer when the mitochondrial membrane potential is low. When the ratio of green and red fluorescence is higher, the mitochondrial membrane potential rises more, which means there is more apoptosis. As expected, the ranking of green and red fluorescence ratio was SQU@PCN + hv > PCN + hv > SQU + hv ≈ SQU@PCN > PCN ≈ control + hv from the observation in Figure 5B. These groups with higher ratio than the control group revealed apoptosis as the main mechanism of cell death. Also, consistent with the results via cell apoptosis assay, the SQU@PCN + hv group with green fluorescence was the group that displayed the best performance to induce apoptosis of tumor cells.
Figure 5.

Cell apoptosis assay by (A) flow cytometry with annexin V-FITC/PI staining and (B) CLSM with JC-1. Scale bar: 20 μm. (C) Western blot analysis of cells treated with different samples under NIR irradiation for LC3 I/II, P62, and caspase 3 proteins and (D) corresponding gray-scale analysis result of strips. (E) In vitro cytotoxicity of SQU@PCN against HeLa cells in HEPES buffer solution or Cl–-free HEPES solution or Na+-free HEPES solution. (F) In vitro cytotoxicity of SQU@PCN and PCN against HeLa cells under NIR irradiation in Cl–-free HEPES solution. **P < 0.01. “hv” refers to light irradiation with a 660 nm laser at 30 mW/cm2 for 5 min.
In view of the dual role (autophagy inhibition and apoptotic induction) of SQU in our nanosystem, it was necessary to clearly illustrate the contribution of autophagy inhibition by SQU for the excellent antitumor effect of SQU@PCN in vitro as proven above. Hence, we designed an experiment to rule out the toxic interference of SQU itself. In the experiment, a cell-permeable pan-caspase inhibitor (ZVAD-FMK) was used to only shield apoptosis induction of SQU at the selected concentration (1.36 mg/L). In detail, SQU and ZVAD-FMK were cocultured with HeLa cells for 4 h. Then, PCN was added and cultured for another 4 h. Finally, the cells were subjected to light irradiation (660 nm, 30 mW/cm2) for 5 min and analyzed by Western blot. On the basis of the results in Figure 5C, SQU still possessed the ability to inhibit autophagy because of the high expression of LC3-II and p62 proteins. However, its apoptosis induction was completely inhibited by the ZVAD-FMK inhibitor because of the scarce expression of caspase 3. This implied that apoptosis inhibitors had no effect on autophagy induction of SQU because of independent pathways between autophagy inhibition and apoptotic induction according to the report in the literature.33 In contrast with PCN + hv group, the SQU + PCN + hv group was observed a higher overexpression of caspase 3 and LC3-II as well as a lower overexpression of p62 at the same concentration of PCN, indicating that SQU could inhibit PDT-induced autophagy for increasing apoptosis of tumor cells. In other words, autophagy inhibition of SQU made a contribution to outstanding toxic effects of SQU@PCN. The corresponding gray-scale analysis (Figure 5D) of the strip illustrated the same conclusion. Furthermore, when cells were incubated with SQU@PCN at various concentrations in HEPES buffer or analogous buffers without Cl– or Na+ (Cl–-free HEPES or Na+-free HEPES), cells in HEPES buffer were tested a higher toxicity while there was a negligible toxicity in Cl–-free HEPES and Na+-free HEPES (Figure 5E). These results suggested that the toxicity of SQU@PCN was closely related to extracellular concentration of ions, confirming that cell death was caused by both SQU-medicated chloride transport and concomitant sodium channel-medicated Na+ ions as reported in the literature.33 More importantly, on the basis of nontoxicity SQU@PCN against tumor cells in Cl–-free solution, we could shield the toxic effect of SQU in Cl–-free solution so that it only exerts autophagy inhibition in vitro. Therefore, the effect of sensitizing PDT mediated by SQU@PCN could be performed in Cl–-free solution where apoptosis induction of SQU was shielded. In Cl–-free HEPES buffer, the cell viability treated with SQU@PCN under light was only 22% at the concentration of 50 mg/L (Figure 5F), which was obviously lower than that of PCN. This further demonstrated that inhibited autophagy did significantly enhance the effect of PDT.
Upon confirmation of the excellent cancer cell damage effect of SQU@PCN in vitro, we were encouraged to further evaluate its therapeutic effect in vivo. First, for a demonstration of the superiority of SQU@PCN as a nanotransporter relative to a small-molecule transporter in vivo, its acute systemic toxicity was studied by analyzing indicators in the blood of mice after 24 h postinjection. As expected, there was no significant differences in the main parameters of blood biochemistry and hematology between the SQU@PCN group and PBS group (Figure 6A), indicating a negligible systemic toxicity and biocompatibility of SQU@PCN. However, different from the two groups, the mice treated with SQU displayed an obviously high level of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) (Figure 6B,C). This demonstrated not only the toxicity of the small-molecule transporter to normal liver tissue due to its nonspecificity but also the advantage of SQU@PCN as a nanotransporter in blood circulation. In addition, the hemolysis assay (Figure S12) also implied the biocompatibility of SQU@PCN nanoparticles, which laid the foundation for further clinical application.
Figure 6.

(A) Main hematological and biochemical parameters of mice after treatment with PBS, SQU@PCN, and SQU. Concentration of (B) ALT and (C) AST in blood of mice after treatment with PBS, SQU@PCN, and SQU. **P < 0.01. (D) Fluorescence imaging in vivo after intravenous injection with SQU@PCN and ex vivo tissues after 36 h postinjection. Ki, kidney; Li, liver; Tu, tumor; Lu, lung; He, heart; and Sp, spleen. (E) Detailed operation process of antitumor treatment against HeLa tumor-bearing mice with SQU@PCN in vivo. (F) Relative body weight and (G) relative tumor volume of mice during 14 days of treatment. (H) Photographs and (I) weight of tumor tissues ex vivo obtained on the 14th day. Scale bar: 1 cm. ***P < 0.001. “hv” refers to light irradiation with a 660 nm laser at 220 mW/cm2 for 8 min.
By aid of self-fluorescence of SQU@PCN, we investigated its targeting and enrichment in vivo. After intravenous administration with SQU@PCN, the mice were imaged at predetermined times using an imaging system. Because of the proper particle size distribution of SQU@PCN which was beneficial for accumulation in the tumor site by the EPR effect, we could obviously observe that the fluorescence signal gradually enhanced in the tumor region with time (Figure 6D). At 12 h after administration, fluorescence intensity was the strongest; that was, accumulation of SQU@PCN nanoparticles reached a maximum at 12 h. Greatly, it was especially important to provide an accurate time for PDT in vivo. Subsequently, fluorescence was slowly weakened because of metabolism, but the tumor region still retained strong fluorescence. This phenomenon implied that SQU@PCN nanoparticles had a strong retention capacity in the tumor, which contributed to the acquisition of long-term treatment. Such a trend of fluorescence change was also reflected in the results of statistical analysis (Figure S13). After 36 h, the major organs and tumor tissue were obtained by dissecting the mouse. As shown in Figure 6D, tumor tissue ex vivo was found with strong fluorescence, confirming the accumulation and retention of SQU@PCN in the tumor. The corresponding fluorescence intensity value also verified the conclusion (Figure S14). In addition, as a metabolic organ, it was normal to observe fluorescence in the liver and spleen.44 We had certificated the nontoxicity of nanoparticles against liver cells, NCTC cells (Figure S15).
Inspired by the strong targeting and accumulation ability of the nanoparticle, the antitumor effect of SQU@PCN was further studied in vivo. The detailed operation process of antitumor treatment against HeLa tumor-bearing mice with SQU@PCN in vivo was described in Figure 6E. First, the living animal model was built by subcutaneously injecting HeLa cells at the back of BALB/c nude mice. When the tumor volume reached about 100 mm3, different samples with the same dose of TCPP (7.5 mg/kg) and SQU (0.55 mg/kg) were intravenously administrated. After 12 h postinjection, the mice in some groups were subjected to light irradiation (660 nm, 220 mW/cm2) for 8 min. As displayed in Figure 6F, the relative body weight of mice treated with SQU@PCN fluctuated within the normal range, indicating that administrated SQU@PCN did not cause significant systemic toxicity to mice during treatment. Additionally, from the observation in Figure 6G, the relative tumor volume of mice was not much different in the PBS group with light or not, demonstrating that only light irradiation did not have much effect on the tumor growth. Compared with the change trend in the SQU + hv group, tumor volume growth of SQU@PCN was slow, indicating that SQU@PCN as a nanotransporter was more conductive to enrichment and performing HPT at the tumor site while avoiding metabolic clearance and damage to normal tissues. Incredibly, when mice treated with SQU@PCN were irradiated by a 660 nm laser, the tumor growth was comprehensively suppressed because of the almost unchanged relative tumor volume. Such a superior antitumor effect resulted from synergistic treatment between PCN-based PDT and SQU-based HPT. After 14 days of treatment, the obtained tumor tissues were photographed and weighed. In Figure 6H, the size of the tumor exhibited a similar trend as the relative tumor volume, which also coincided with the weight of the tumor as shown in Figure 6I. They conveyed the same conclusion that the performance of the SQU@PCN + hv group was the best of all regardless of size and weight, further illustrating the superiority of the material design and treatment strategy in treatment in vivo. In addition, upon analysis of the slice data of tumors (Figure 7A), the SQU@PCN + hv group had the fewest tumor cells (blue), which showed the best induced tumor death effect of all. The same conclusion could also be drawn from the immunofluorescence assay. Mice treated with SQU@PCN + hv displayed the strongest apoptotic cells signal (Tunnel) and the weakest cell proliferation signal (Ki67). In particular, to better understand the mechanism of synergistic therapy between PDT and HPT in vivo, we also detected the expression of LC3-II and p62 in tumor tissues by immunofluorescence assay. As proven by Western blot in Figure 4C, PCN-based PDT was able to induce autophagy due to the enhanced fluorescence of LC3-II and weakened fluorescence of p62. However, once SQU was loaded into PCN, fluorescence of LC3-II and p62 were enhanced. This indicated significant autophagy suppression, which eventually resulted in improvement of the PDT effect. Thence, we verified the process in vivo that light-triggered ROS produced by SQU@PCN induced autophagy, and then, released SQU completely inhibited autophagy by coupling transport H+/Cl–, which led to the great sensitization of PDT compared with traditional PDT.
Figure 7.

(A) H&E staining and the immunofluorescence signal (green) of Tunnel, Ki67, LC3 II, and P62 of tumor tissues. Blue fluorescence: cell nuclear stained with DAPI. (B) Liver metastasis after 30 days postinjection. (B1, B2) Photographs of liver ex vivo and metastatic nodules (yellow circles). (B3, B4) H&E staining of liver tissues and partially enlarged images. “hv” refers to light irradiation with a 660 nm laser at 220 mW/cm2 for 8 min.
Considering the death caused by cancer recurrence due to the high invasion and metastasis feature of HeLa tumors,45 metastasis of all groups in the liver was analyzed to evaluate late recurrence on the 16th day after termination of treatment. As observed in Figure 7B, all groups could find different numbers of metastatic nodules except for the SQU@PCN + hv group (Figure 7B1,B2). The same conclusion was also drawn from images of the hematoxylin and eosin (H&E) staining (Figure 7B3,B4). These results indicated that an effective synergistic treatment strategy with SQU@PCN could completely eliminate the tumor in situ while blocking metastasis.
3. Conclusion
In summary, we had successfully constructed an ATP-regulated ion transporter nanosystem for homeostatic perturbation therapy and sensitized photodynamic therapy. The high-loading nanoparticles (SQU@PCN) were simply and efficiently synthesized by a one-pot method. Moreover, such a method of delivering hydrophobic ion transporter SQU achieved the enrichment of SQU in tumor sites while avoiding metabolic clearance and toxic side effects on normal tissues during blood circulation. In response to the tumor microenvironment with highly expressed ATP, the SQU@PCN nanotransporter could quickly disintegrate and release incorporated small molecule SQU. Then, SQU as an effective ion transporter mediated the influx of extracellular chloride ions. Dysregulation of intracellular chloride ion concentration induced apoptosis-related tumor cell death. On the other hand, coupled transport of H+/Cl– by SQU across the lysosomal membrane resulted in autophagy inhibition via increasing lysosomal pH. Excellently, inhibition of autophagy made up for the loss of PDT efficiency by phototherapy-activated autophagy, significantly sensitizing traditional PDT. Hence, such a strategy of synergistic treatment between PCN-based PDT and SQU-based HPT showed outstanding in situ tumor eradication while blocking the metastasis from the source, which provided novel ideas for clinical cancer treatment research.
4. Experimental Section
Materials
Cholesterol and chloroquine (CQ) were supplied by Sigma-Aldrich Co. Ltd. Octaethylene glycol monododecyl ether and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) were supplied by TCI. Zirconyl chloride octahydrate (ZrOCl2·8H2O) and benzoic acid (BA) were purchased from Sinopharm Chemical Reagent Co., Ltd. Tetrakis (4-carboxyphenyl) porphyrin (TCPP) was synthesized according to the previous report.41 The synthesis of squaramide (3,4-bis(4-(trifluoromethyl)phenylamino)cyclobut-3-ene-1,2-dione, SQU) was based on the steps in the previous literature.42 LysoTracker Red, LysoSensor Green DND-189, and annexin V-FITC/PI cell apoptosis kit were supplied by Yeasen, Shanghai, China. The 2′,7′-dichlorofluorescin diacetate (DCFH-DA), LysoTracker Red, N-(ethoxycarbonylmethyl)-6-methoxyquinolinium bromide (MQAE), and ZVAD-FMK were obtained from Beyotime Institute of Biotechnology. JC-1 and Calcein-AM were purchased from 4A Biotech Co., Ltd.
Instrumentation
TEM images were taken by a Tecnai G20 S-TWIN instrument. SEM photos were imaged by a field-emission scanning electron microscope (Sigma). The PXRD pattern was detected by a Rigaku MiniFlex 600 X-ray diffractometer with Cu (Kα = 1.5418 Å). The hydrodynamic size and ζ potential were measured by dynamic light scattering (DLS) on a PSS Z3000 instrument. Confocal microscopy images were carried out on a confocal laser scanning microscope (CLSM) (Nikon C1-si TE2000) and processed by EZ-C1 software. The flow cytometric assay was tested in flow cytometry (BD FACSAria III). Thermogravimetric analysis (TGA) was determined by a Pyris1 thermogravimetric analyzer (PerkinElmer). Intracellular pH and live/dead cell staining assay were carried out by fluorescence inverted microscope (Olympus U-HGLGPS). The in vivo imaging experiment was carried out on IVIS imaging systems (PerkinElmer). The light source (660 nm) used for PDT was obtained from Beijing Laserwave Optoelectronics Technology. Co., Ltd.
Synthesis of SQU@PCN Nanoparticles
SQU@PCN nanoparticles were synthesized by a one-pot method. In detail, the mixture of ZrOCl2·8H2O (300 mg, 0.93 mmol), TCPP (100 mg, 0.13 mmol), BA (2.8 g, 23 mmol) and SQU (50 mg, 0.13 mmol) was dissolved in 100 mL of DMF. Then, the solution was sealed and stirred in an oil bath at 90 °C in the dark. After 5 h, the mixture was purified by centrifugation with 12 000 rpm for 20 min and washed with DMF 3 times. Finally, the obtained SQU@PCN nanoparticles were suspended in DMF in the dark for further use. As the control, PCN was synthesized using the same method without SQU.
Disintegration of SQU@PCN in Response to ATP
A 1 mg/mL SQU@PCN NP solution was dispersed in HEPES buffer (pH 7.4) with or without ATP (1 mg/mL). After being incubated for the scheduled time (0.5, 1, 2, 4, and 6 h), the solution was centrifuged to obtain the supernatant. The absorption change of TCPP at 517 nm in the supernatant was detected via UV/vis absorption spectrometer. The amount of TCPP released was calculated according to the standard curve of TCPP absorption. In addition, the obtained sediments after centrifugation were dried in vacuum overnight and tested to obtain PXRD patterns.
Release of SQU in Response to ATP
A 1 mL of SQU@PCN NPs (1 mg/mL) was placed in a dialysis bag (MWKO 3500 Da). Then, the bag was soaked in HEPES buffer (pH 7.4) containing 1 mg/mL ATP or not and shaken at 37 °C. At the preset time, 1 mL of dialysate was removed and replaced with the same amount of fresh solution. The absorbance of SQU at 343 nm in dialysate was measured by a UV/vis absorption spectrometer. The cumulative release of SQU was calculated according to the standard curve of SQU.
Chloride Transport Assay
The procedures were conducted with a typical membrane transport method reported in the literature.33 First, a mixture of POPC and cholesterol in a mass ratio of 7:3 was dissolved in CHCl3 and then dried under vacuum overnight. The obtained mixture was added into phosphate buffer (pH 7.2) containing 489 mM NaCl and 1 mg/mL ATP, followed by hydrating in a vortex. Subsequently, the solution was subjected to freeze and thaw nine times in liquid nitrogen and water. After aging for 30 min, the solution was squeezed out using an Avanti mini extruder with a 200 nm polycarbonate membrane. Finally, the unilamellar vesicles were purified by dialysis with MWCO 14 KD.
The POPC vesicles obtained above were suspended in phosphate buffer (pH 7.2) containing 489 mM NaNO3 and 1 mg/mL ATP. Then, different concentrations of SQU@PCN NPs with a concentration of 1 mM lipid per SQU were added. The potential change caused by chloride ion transport was recorded by a chloride-selective electrode. At 120 min, the vesicles were lysed with 100 μL of octaethylene glycol monododecyl ether (0.232 mM) in solution (7:1 = water: DMSO). The final potential of the chloride ion was read. The potential value was converted to chloride ion concentration on the basis of a standard curve and further calculated into percentages where the initial value was defined as 0% and the final chloride reading (at 120 min) defined as 100%. As for chloride transport experiments in different materials, the procedures were similar to the method above.
ROS Production
The ROS generation ability of samples was evaluated in the presence of ATP. Because of DCFH being able to react with ROS to emit green fluorescence, it served as the probe for ROS detection. In general, 10 μL of DCFH (20 μM) was added into samples with equivalent TCPP concentration (20 mg/L). Then, the mixture was subjected to irradiation (660 nm, 30 mW/cm2) or not for a given time. Finally, the fluorescence of samples at 525 nm was tested by fluorescence spectrometer.
Cell Culture
Human cervical cancer (HeLa) cells, 3T3 cells, and NCTC clone 1469 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% antibiotics (penicilline–streptomycin, 10 000 U/mL) at 37 °C in a humidified atmosphere containing 5% CO2. In addition to adding 5 μg/mL puromycin to DMEM medium, stably transfected HeLa cells containing the EGFP-LC3-fusion gene were of the same culture conditions as the normal HeLa cells.
In Vitro ROS Detection
HeLa cells were seeded and incubated in small dishes for 24 h. Then, different samples with an equal amount of TCPP or SQU concentration (TCPP, 30 mg/L; SQU, 2.2 mg/L) were added. After coculture with cells for another 4 h, cells were washed with PBS 3 times, followed by staining with DCFH-DA for 30 min. Finally, cells were irradiated with 660 nm panel light (30 mW/cm2) for 3 min and then observed by CLSM.
Intracellular Cl– Detection
HeLa cells were seeded and incubated in a 96-well plate for 24 h. Then, medium containing 10 mM MQAE was added and coincubated with cells for 1 h. After washing with PBS, cells were cultured with different samples with an equal amount of TCPP or SQU concentration (TCPP, 30 mg/L; SQU, 2.2 mg/L) for 4 h. The fluorescence value of MQAE was read by a microplate reader (λex = 350 nm, λem = 460 nm). The same method was used to detect Cl– concentration mediated by SQU@PCN after treatment with different culture times or different culture concentrations.
Detection of Lysosomal pH
HeLa cells were seeded and incubated in a 6-well plate for 24 h. Then, different samples with an equal amount of TCPP or SQU concentration (TCPP, 30 mg/L; SQU, 2.2 mg/L) were added and cocultured with cells for 12 h. Hereafter, cells were washed with PBS to remove excess samples and stained with 15 μM LysoSensor Green DND-189 for 2 h. After washing, cells were observed by fluorescence inverted microscope. Similarly, this method was also used to detect the lysosome pH after treatment with SQU@PCN at different culture times.
Study of Autolysosome Formation
HeLa cells stably expressing GFP-LC3 protein were seeded and incubated in a small dish for 24 h. Then, the medium was replaced with the fresh ones containing different samples with an equal amount of TCPP or SQU concentration (TCPP, 30 mg/L; SQU, 2.2 mg/L). After 4 h, cells were washed with PBS three times and irradiated with a 660 nm laser for 3 min. After that, cells were stained with 100 nM LysoTracker Red for 1 h. The formation of autolysosome was eventually observed by CLSM.
Cytotoxicity Measurements
The cytotoxicity in vitro against HeLa cells was tested by multiple tests, including MTT assay, JC-1 assay, live/dead cell staining assay, and cell apoptosis assay.
For the MTT assay, Hela cells were seeded and cultured in a 96-well plate for 24 h. Then, various samples at an equivalent concentration (TCPP, 30 mg/L; SQU, 2.2 mg/L) were added for another 4 h of culture. Subsequently, the cells were subjected to irradiate (660 nm, 30 mW/cm2) or not for 5 min. After 24 h, 20 μL of MTT (5 mg/mL) was added for 4 h of incubation. The supernatant was removed, and 150 μL of DMSO was added for dissolving the formazan. Finally, the absorbance wavelength of cells at 570 nm was read using the microplate reader (BIO-RAD 550). The cell viability was calculated as follows: OD (samples)/OD (control) × 100%, where OD (control) and OD (samples) refer to the optical density at 570 nm with samples or not, respectively.
Then, a live/dead cell staining assay was performed to evaluate cytotoxicity via fluorescence inverted microscope. In detail, after seeding HeLa cells in a 6-well plate, 100 μL of various samples containing equivalent TCPP or SQU concentration (TCPP, 30 mg/L; SQU, 2.2 mg/L) was added and further cultured with cells for 4 h. Subsequently, cells in some groups were subjected to the light irradiation (660 nm, 30 mW/cm2) for 5 min. After 2 h, cells were washed and added with 1 μL of Calcein-AM (4 × 10–6 M) and 10 μL of PI (4 × 10–6 M) in each well for 15 min of dyeing. Finally, cells were observed and analyzed by fluorescence inverted microscope.
For a study of the mechanism of cell lethality in depth, the cell apoptosis assay was performed with the annexin V-FITC/PI cell apoptosis kit via flow cytometry. HeLa cells were seeded in a 6-well plate, followed by incubation with various samples with equivalent TCPP or SQU concentration (TCPP, 30 mg/L; SQU, 2.2 mg/L) for 4 h. Then, the cells were washed and irradiated for 5 min (660 nm, 30 mW/cm2). After 2 h of incubation, cells were digested with trypsin and collected. Finally, cells were stained with 2 μL of annexin V-FITC and 4 μL of PI for 20 min, followed by analysis via flow cytometry.
In addition to the cell apoptosis assay, the JC-1 staining assay was also conducted to illustrate apoptosis-induced death. After HeLa cells were seeded in a small dish, various samples with equivalent TCPP or SQU concentration (TCPP, 30 mg/L; SQU, 2.2 mg/L) were added for 4 h of coculture. Then, cells were washed and stained with 3 μg/mL JC-1 for 15 min. The fluorescence signal of treated cells was observed by CLSM.
Fluorescence Imaging in Vivo
All live animal experiments were conducted in accordance with the criterion of the Institutional Animal Care and Use Committee of the Animal Experiment Center of Wuhan University (Wuhan, China). The living tumor model was built by subcutaneous injection with HeLa cells in the back of female nude mice. When tumor volume reached 200 mm3, SQU@PCN NPs at a dose of 6 mg/kg were intravenously injected into mice. At the given time (1, 2, 3, 4, 6, 9, 12, 24, and 36 h), the administered mice were imaged by an IVIS spectrum imaging system. The mice were sacrificed 36 h later. The main organs and tumors were collected for tissue imaging.
In Vivo Antitumor Studies
When tumor volume reached nearly 100 mm3, HeLa tumor-bearing mice were randomly divided into 7 groups with 8 mice per group. Different groups were treated with different materials at the equivalent dose of 6 mg/kg TCPP and 0.44 mg/kg SQU. Mice in some groups were subjected with irradiation for 8 min (660 nm, 220 mW/cm2) after 12 h postinjection. Then, the length (L) and width (W) of the tumor, as well as weight of mice, were measured every day in the whole treatment. The tumor volume was defined as the following formula: L × W2/2. After treatment for 14 days, four mice in each group were sacrificed and dissected to obtain tumors for hematoxylin and eosin (H&E) staining analysis and immunofluorescence (Tunnel, Ki67, LC3, and p62) evaluation. Meanwhile, the obtained tumors were photographed and weighted. On the 16th day after the end of treatment, the remaining four mice in each group were dissected to obtain the liver. Then, HeLa tumor metastasis in the liver was assessed by observing metastatic nodules and analyzing H&E-stained sections.
Hemolysis Assay
The whole blood from mice was dispersed in PBS buffer. Then, the solution was centrifuged to obtain red blood cells (RBCs). Soon afterward, 0.1 mL of RBC solution was added into 0.4 mL of SQU@PCN solution at different concentrations. After incubation at 37 °C for 30 min, all samples were centrifuged to evaluate the hemolysis, in which the RBC in PBS served as the negative control, and the RBC in distilled water served as the positive control. The corresponding concentrations of NP solution without RBCs were used as another control.
Statistical Analysis
The data was analyzed by a three-sample Student’s test to assess the statistical difference between two groups. A value of P < 0.05 was regarded as significant statistical difference.
Safety Statement
All operations described here were according to Standard laboratory security protocols. No unexpected or unusually high safety hazards were encountered.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (51833007, 51690152, and 21721005).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.8b00822.
Additional experimental figures including electron microscopy, hydrodynamic size, standard curve, thermogravimetric analysis, PXRD patterns, fluorescence test, cell viability, statistical data, and hemolysis test (PDF)
Author Contributions
† S.-S.W. and L.Z. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Li S. Y.; Cheng H.; Xie B. R.; Qiu W. X.; Zeng J. Y.; Li C. X.; Wan S. S.; Zhang L.; Liu W. L.; Zhang X. Z. Cancer Cell Membrane Camouflaged Cascade Bioreactor for Cancer Targeted Starvation and Photodynamic Therapy. ACS Nano 2017, 11, 7006–7018. 10.1021/acsnano.7b02533. [DOI] [PubMed] [Google Scholar]
- Li S. Y.; Cheng H.; Xie B. R.; Qiu W. X.; Li C. X.; Li B.; Cheng H.; Zhang X. Z. Mitochondria Targeted Cancer Therapy Using Ethidium Derivatives. Mater. Today Chem. 2017, 6, 34–44. 10.1016/j.mtchem.2017.09.002. [DOI] [Google Scholar]
- Chen Z. X.; Liu M. D.; Zhang M. K.; Wang S. B.; Xu L.; Li C. X.; Gao F.; Xie B. R.; Zhong Z. L.; Zhang X. Z. Interfering with Lactate-Fueled Respiration for Enhanced Photodynamic Tumor Therapy by a Porphyrinic MOF Nanoplatform. Adv. Funct. Mater. 2018, 28, 1803498. 10.1002/adfm.201803498. [DOI] [Google Scholar]
- Felsher D. W. Cancer Revoked: Oncogenes as Therapeutic Targets. Nat. Rev. Cancer 2003, 3, 375–380. 10.1038/nrc1070. [DOI] [PubMed] [Google Scholar]
- Reiners J. J.; Agostinis P.; Berg K.; Oleinick N. L.; Kessel D. H. Assessing Autophagy in the Context of Photodynamic Therapy. Autophagy 2010, 6, 7–18. 10.4161/auto.6.1.10220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessel D.; Reiners J. J. Apoptosis and Autophagy After Mitochondrial or Endoplasmic Reticulum Photodamage†. Photochem. Photobiol. 2007, 83, 1024–1028. 10.1111/j.1751-1097.2007.00088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buytaert E.; Callewaert G.; Vandenheede J. R.; Agostinis P. Deficiency in Apoptotic Effectors BAX and BAK Reveals an Autophagic Cell Death Pathway Initiated by Photodamage to the Endoplasmic Reticulum. Autophagy 2006, 2, 238–240. 10.4161/auto.2730. [DOI] [PubMed] [Google Scholar]
- Wei M. F.; Chen M. W.; Chen K. C.; Lou P. J.; Lin S. Y.; Hung S. C.; Hsiao M.; Yao C. J.; Shieh M. J. Autophagy Promotes Resistance to Photodynamic Therapy-Induced Apoptosis Selectively in Colorectal Cancer Stem-Like Cells. Autophagy 2014, 10, 1179–1192. 10.4161/auto.28679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coupienne I.; Bontems S.; Dewaele M.; Rubio N.; Habraken Y.; Fulda S.; Agostinis P.; Piette J. NF-kappaB Inhibition Improves the Sensitivity of Human Glioblastoma Cells to 5-Aminolevulinic Acid-Based Photodynamic Therapy. Biochem. Pharmacol. 2011, 81, 606–616. 10.1016/j.bcp.2010.12.015. [DOI] [PubMed] [Google Scholar]
- Domagala A.; Stachura J.; Gabrysiak M.; Muchowicz A.; Zagozdzon R.; Golab J.; Firczuk M. Inhibition of Autophagy Sensitizes Cancer Cells to Photofrin-Based Photodynamic Therapy. BMC Cancer 2018, 18, 210. 10.1186/s12885-018-4126-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E. Deconvoluting the Context-Dependent Role for Autophagy in Cancer. Nat. Rev. Cancer 2012, 12, 401–410. 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G.; Mariño G.; Levine B. Autophagy and the Integrated Stress Response. Mol. Cell 2010, 40, 280–293. 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X.; De Vera M. E.; Buchser W. J.; de Vivar Chavez A. R.; Loughran P.; Stolz D. B.; Basse P.; Wang T.; Van Houten B.; Zeh H. J.; Lotze M. T. Inhibiting Systemic Autophagy during Interleukin 2 Immunotherapy Promotes Long-term Tumor Regression. Cancer Res. 2012, 72, 2791–2801. 10.1158/0008-5472.CAN-12-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Hou N.; Faried A.; Tsutsumi S.; Kuwano H. Inhibition of Autophagy Augments 5-Fluorouracil Chemotherapy in Human Colon Cancer in Vitro and in Vivo Model. Eur. J. Cancer 2010, 46, 1900–1909. 10.1016/j.ejca.2010.02.021. [DOI] [PubMed] [Google Scholar]
- Wei M. F.; Chen M. W.; Chen K. C.; Lou P.-J.; Lin S. Y. F.; Hung S. C.; Hsiao M.; Yao C. J.; Shieh M. J. Autophagy Promotes Resistance to Photodynamic Therapy-Induced Apoptosis Selectively in Colorectal Cancer Stem-Like Cells. Autophagy 2014, 10, 1179–1192. 10.4161/auto.28679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z.; Yan Y.; Hu K.; Zou Y.; Li Y.; Ma R.; Zhang Q.; Cheng Y. Autophagy Inhibition Enabled Efficient Photothermal Therapy at a Mild Temperature. Biomaterials 2017, 141, 116–124. 10.1016/j.biomaterials.2017.06.030. [DOI] [PubMed] [Google Scholar]
- Wu X.; Wu Y.; Wang Z.; Liu L.; Sun C.; Chen Y.; Wang C. A Cascade-Targeting Nanocapsule for Enhanced Photothermal Tumor Therapy with Aid of Autophagy Inhibition. Adv. Healthcare Mater. 2018, 7, 1800121. 10.1002/adhm.201800121. [DOI] [PubMed] [Google Scholar]
- Ren X.; Chen Y.; Peng H.; Fang X.; Zhang X.; Chen Q.; Wang X.; Yang W.; Sha X. Blocking Autophagic Flux Enhances Iron Oxide Nanoparticle Photothermal Therapeutic Efficiency in Cancer Treatment. ACS Appl. Mater. Interfaces 2018, 10, 27701–27711. 10.1021/acsami.8b10167. [DOI] [PubMed] [Google Scholar]
- Lin J.; Huang Z.; Wu H.; Zhou W.; Jin P.; Wei P.; Zhang Y.; Zheng F.; Zhang J.; Xu J.; Hu Y.; Wang Y.; Li Y.; Gu N.; Wen L. Inhibition of Autophagy Enhances the Anticancer Activity of Silver Nanoparticles. Autophagy 2014, 10, 2006–2020. 10.4161/auto.36293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen T. W.; Andersen O. S.; Roux B. Energetics of ion conduction through the gramicidin channel. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 117–122. 10.1073/pnas.2635314100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKinnon R. Potassium Channels and the Atomic Basis of Selective Ion Conduction (Nobel Lecture). Angew. Chem., Int. Ed. 2004, 43, 4265–4277. 10.1002/anie.200400662. [DOI] [PubMed] [Google Scholar]
- Doyle D. A.; Cabral J. M.; Pfuetzner R. A.; Kuo A.; Gulbis J. M.; Cohen S. L.; Chait B. T.; MacKinnon R. The Structure of the Potassium Channel: Molecular Basis of K+ Conduction and Selectivity. Science 1998, 280, 69–77. 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Bayrhuber M.; Meins T.; Habeck M.; Becker S.; Giller K.; Villinger S.; Vonrhein C.; Griesinger C.; Zweckstetter M.; Zeth K. Structure of the Human Voltage-Dependent Anion Channel. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 15370–15375. 10.1073/pnas.0808115105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCoursey T. E.; Chandy K. G.; Gupta S.; Cahalan M. D. Voltage-Gated K+ Channels in Human T Lymphocytes: a Role in Mitogenesis?. Nature 1984, 307, 465. 10.1038/307465a0. [DOI] [PubMed] [Google Scholar]
- Stauber T.; Jentsch T. J. Chloride in Vesicular Trafficking and Function. Annu. Rev. Physiol. 2013, 75, 453–477. 10.1146/annurev-physiol-030212-183702. [DOI] [PubMed] [Google Scholar]
- Gould D. J. Adv. Nurs. 1998, 28 (3), 680–682. [Google Scholar]
- Ko S. K.; Kim S. K.; Share A.; Lynch V. M.; Park J.; Namkung W.; Van Rossom W.; Busschaert N.; Gale P. A.; Sessler J. L.; Shin I. Synthetic Ion Transporters Can Induce Apoptosis by Facilitating Chloride Anion Transport into Cells. Nat. Chem. 2014, 6, 885–892. 10.1038/nchem.2021. [DOI] [PubMed] [Google Scholar]
- Saha T.; Gautam A.; Mukherjee A.; Lahiri M.; Talukdar P. Chloride Transport through Supramolecular Barrel-Rosette Ion Channels: Lipophilic Control and Apoptosis-Inducing Activity. J. Am. Chem. Soc. 2016, 138, 16443–16451. 10.1021/jacs.6b10379. [DOI] [PubMed] [Google Scholar]
- Saha T.; Hossain M. S.; Saha D.; Lahiri M.; Talukdar P. Chloride-Mediated Apoptosis-Inducing Activity of Bis(sulfonamide) Anionophores. J. Am. Chem. Soc. 2016, 138, 7558–7567. 10.1021/jacs.6b01723. [DOI] [PubMed] [Google Scholar]
- Ren C.; Ding X.; Roy A.; Shen J.; Zhou S.; Chen F.; Yau Li S. F.; Ren H.; Yang Y. Y.; Zeng H. A Halogen Bond-Mediated Highly Active Artificial Chloride Channel with High Anticancer Activity. Chem. Sci. 2018, 9, 4044–4051. 10.1039/C8SC00602D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X. H.; Peng C. C.; Sun X. X.; Chen W. H. Synthesis, Anionophoric Activity and Apoptosis-Inducing Bioactivity of Benzimidazolyl-Based Transmembrane Anion Transporters. Eur. J. Med. Chem. 2018, 152, 115–125. 10.1016/j.ejmech.2018.04.036. [DOI] [PubMed] [Google Scholar]
- Hosogi S.; Kusuzaki K.; Inui T.; Wang X.; Marunaka Y. Cytosolic Chloride Ion Is a Key factor in Lysosomal Acidification and Function of Autophagy in Human Gastric Cancer Cell. J. Cell. Mol. Med. 2014, 18, 1124–1133. 10.1111/jcmm.12257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busschaert N.; Park S. H.; Baek K. H.; Choi Y. P.; Park J.; Howe E. N. W.; Hiscock J. R.; Karagiannidis L. E.; Marques I.; Felix V.; Namkung W.; Sessler J. L.; Gale P. A.; Shin I. A Synthetic Ion Transporter That Disrupts Autophagy and Induces Apoptosis by Perturbing Cellular Chloride Concentrations. Nat. Chem. 2017, 9, 667–675. 10.1038/nchem.2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson N. R.; Fineran P. C.; Leeper F. J.; Salmond G. P. C. The Biosynthesis and Regulation of Bacterial Prodiginines. Nat. Rev. Microbiol. 2006, 4, 887–899. 10.1038/nrmicro1531. [DOI] [PubMed] [Google Scholar]
- Li H.; Valkenier H.; Judd L. W.; Brotherhood P. R.; Hussain S.; Cooper J. A.; Jurček O.; Sparkes H. A.; Sheppard D. N.; Davis A. P. Efficient, Non-Toxic Anion Transport by Synthetic Carriers in Cells and Epithelia. Nat. Chem. 2016, 8, 24–32. 10.1038/nchem.2384. [DOI] [PubMed] [Google Scholar]
- Santacroce P. V.; Davis J. T.; Light M. E.; Gale P. A.; Iglesias-Sánchez J. C.; Prados P.; Quesada R. Conformational Control of Transmembrane Cl– Transport. J. Am. Chem. Soc. 2007, 129, 1886–1887. 10.1021/ja068067v. [DOI] [PubMed] [Google Scholar]
- Deng J.; Wang K.; Wang M.; Yu P.; Mao L. Mitochondria Targeted Nanoscale Zeolitic Imidazole Framework-90 for ATP Imaging in Live Cells. J. Am. Chem. Soc. 2017, 139, 5877–5882. 10.1021/jacs.7b01229. [DOI] [PubMed] [Google Scholar]
- Yu C. J.; Wu S. M.; Tseng W. L. Magnetite Nanoparticle-Induced Fluorescence Quenching of Adenosine Triphosphate–BODIPY Conjugates: Application to Adenosine Triphosphate and Pyrophosphate Sensing. Anal. Chem. 2013, 85, 8559–8565. 10.1021/ac400919j. [DOI] [PubMed] [Google Scholar]
- Song X. R.; Li S. H.; Dai J.; Song L.; Huang G.; Lin R.; Li J.; Liu G.; Yang H. H. Polyphenol-Inspired Facile Construction of Smart Assemblies for ATP- and pH-Responsive Tumor MR/Optical Imaging and Photothermal Therapy. Small 2017, 13, 1603997. 10.1002/smll.201603997. [DOI] [PubMed] [Google Scholar]
- Chen W. H.; Liao W. C.; Sohn Y. S.; Fadeev M.; Cecconello A.; Nechushtai R.; Willner I. Stimuli-Responsive Nucleic Acid-Based Polyacrylamide Hydrogel-Coated Metal-Organic Framework Nanoparticles for Controlled Drug Release. Adv. Funct. Mater. 2018, 28, 1705137. 10.1002/adfm.201705137. [DOI] [Google Scholar]
- Park J.; Jiang Q.; Feng D.; Mao L.; Zhou H. C. Size-Controlled Synthesis of Porphyrinic Metal-Organic Framework and Functionalization for Targeted Photodynamic Therapy. J. Am. Chem. Soc. 2016, 138, 3518–3525. 10.1021/jacs.6b00007. [DOI] [PubMed] [Google Scholar]
- Busschaert N.; Kirby I. L.; Young S.; Coles S. J.; Horton P. N.; Light M. E.; Gale P. A. Squaramides as Potent Transmembrane Anion Transporters. Angew. Chem. 2012, 124, 4502–4506. 10.1002/ange.201200729. [DOI] [PubMed] [Google Scholar]
- Zheng W.; Wei M.; Li S.; Le W. Nanomaterial-Modulated Autophagy: Underlying Mechanisms and Functional Consequences. Nanomedicine 2016, 11, 1417–1430. 10.2217/nnm-2016-0040. [DOI] [PubMed] [Google Scholar]
- Tsoi K. M.; MacParland S. A.; Ma X. Z.; Spetzler V. N.; Echeverri J.; Ouyang B.; Fadel S. M.; Sykes E. A.; Goldaracena N.; Kaths J. M.; Conneely J. B.; Alman B. A.; Selzner M.; Ostrowski M. A.; Adeyi O. A.; Zilman A.; McGilvray I. D.; Chan W. C. W. Mechanism of Hard-Nanomaterial Clearance by the Liver. Nat. Mater. 2016, 15, 1212–1221. 10.1038/nmat4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J.; Wu Q.; Zheng W.; Li L.; Mei W. Microwave-Assisted Synthesis of Polypyridyl Ruthenium(ii) Complexes as Potential Tumor-Targeting Inhibitors against the Migration and Invasion of HeLa Cells Through G2/M Phase Arrest. RSC Adv. 2017, 7, 26625–26632. 10.1039/C7RA00658F. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.