

Abstract
Kindler syndrome is a rare autosomal recessive inherited disease characterized by infantile acral bullae, progressive poikiloderma, cutaneous atrophy, photosensitivity and various forms of mucosal involvement. In this paper, we report a case of a 49-year-old Greek Caucasian male aiming to emphasize the importance of genetic analysis as a gold standard of diagnosis.
INTRODUCTION
Kindler syndrome (KS) is a rare autosomal recessive genodermatosis, first described in 1954 by Kindler Theresa [1]. Since then, about 250 cases have been reported worldwide [2]. The disease is associated with mutations in the FERMT1 (KIND1) gene, located on the short arm of chromosome 20 (20p12.3) [3]. KS is characterized by a combination of features of skin blistering and poikiloderma.
Since 2007, KS is classified in the Epidermolysis bullosa (EB) group, with variable levels of skin separation and trauma-induced skin fragility [3]. We present here a case of KS in a Greek patient, which was diagnosed only at late age.
CASE REPORT
A 49-year-old Greek Caucasian male, born to non-consanguineous, skin-healthy parents, presented to our Department with a history of photosensitivity with sunburns at the slightest exposure to sunlight and repeated blistering on the hands and feet since birth.
In addition, he reported severe solid-food dysphagia from early adulthood due to oesophageal strictures. He also suffered from urethral and anal stenosis with resulting constipation.
Physical examination revealed poikiloderma involving the V area of the upper chest, the face and the neck; onychodystrophy (Fig. 1); palmoplantar punctate hyperkeratosis (Fig. 2). The dorsum of the hands and feet had marked cigarette paper-like wrinkling (Fig. 3).
Figure 1:
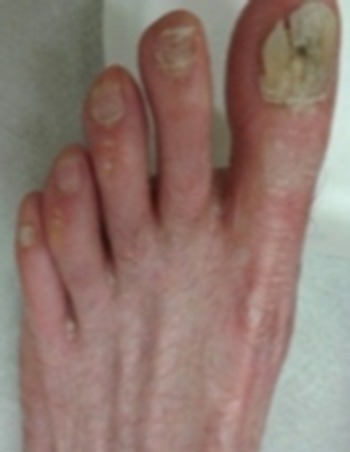
Onychodystrophy.
Figure 2:
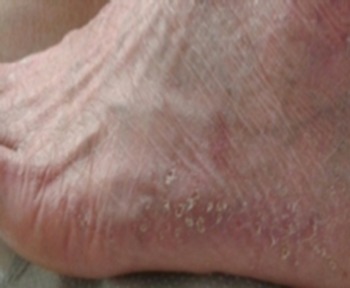
Plantar punctate hyperkeratosis.
Figure 3:
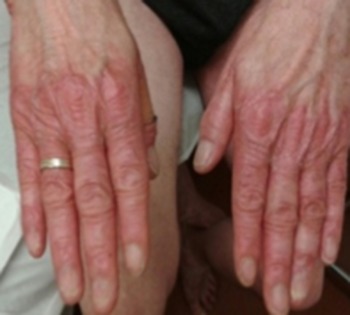
Skin atrophy over the dorsa of the hand.
Examination of the oral cavity showed white hyperkeratotic papules in the buccal mucosa, severe periodontitis, telangiectasias in gums and desquamative gingivitis (Fig. 4). Xerostomia and microstomia due to fibrosis were also evident.
Figure 4:
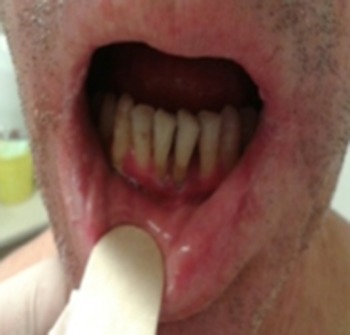
Telangiectasias in gums, desquamative gingivitis.
Ophthalmologic investigation revealed keratoconjunctivitis, conjunctival scarring, ectropion of the lower lid (Fig. 5), corneal thinning and neovascularization.
Figure 5:
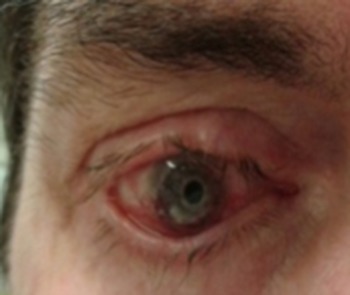
Keratoconjunctivitis, ectropion of the lower lid.
All biochemical, haematological and immunological tests were unremarkable. Any previous lab investigation of urine and faecal porphyrins was bland. In addition, molecular genetic testing for Rothmund–Thompson syndrome detected no pathogenic mutation in the RECQL4 gene.
Histopathological examination of a biopsy taken from an atrophic skin lesion revealed hyperkeratosis, epidermal atrophy, dilated blood vessels with mild lymphohistiocytic infiltration and pigmentary incontinence in the dermis. Direct immunofluorence did not reveal any deposition of immunoglobulins or complement. Immunofluorescence mapping revealed a duplication of the dermo-epidermal junction zone and variable split levels, highly suggestive of KS.
Based on these findings, mutation analysis of the FERMT1 gene with DNA from the patient was initiated after his informed consent. The mutation c.676dupC, p.Q226PfsX16 in exon 5 was identified in a homozygous state. Based on clinical findings and molecular analysis, the diagnosis of KS was confirmed.
DISCUSSION
Kindler syndrome is clinically characterized by the presence of minor trauma-induced blisters at birth or within the first days of life, which regress with age. Photosensitivity, poikiloderma, skin fragility and diffuse cutaneous atrophy especially in sun-exposed areas tend to appear in infancy or early childhood. The photosensitivity usually decreases over time, whereas poikiloderma and atrophy are progressive [4].
Mucous membrane involvement is common and may lead to severe disability, due to stenoses of the mucosal cavities. Other features include pseudosyndactyly Ainhum, nail dystrophy, palmoplantar keratoderma, ectropion, keratoconjuctivitis and conjuctival scarring [5].
The histopathological analysis of atrophic skin lesions demonstrates non-specific features, whereas transmission electron microscopy of KS skin reveals major disorganization of the basement membrane, single or multiple cleavage planes at the level of the cutaneous basement membrane and reduplicaton of the lamina densa [3, 5].
KS diagnosis can be established by immunofluorescence mapping and / or detecting mutations in the FERMT1 gene [6]. The FERMT1 gene encodes the kindlin-1 protein by keratinocytes and its loss leads to decreased dermal-epidermal adhesion. To date, more than 70 unique pathogenic variants have been identified in the FERMT1 gene [2].
In 2005, Angelova–Fischer et al. proposed five major and two minor clinical diagnostic criteria, as well as associated findings for the diagnosis of KS [8]. The major proposed criteria include acral blistering in infancy and childhood, progressive poikiloderma, skin atrophy, abnormal photosensitivity, gingival fragility and/or swelling. The minor proposed criteria are syndactyly and mucosal involvement (anal, oesophageal, urethral, laryngeal stenosis). The associated findings are nail dystrophy, ectropion of the lower lid, palmopantar keratoderma, pseudoainhum, leukokeratosis of the lips, squamous cell carcinoma, anhidrosis/hypodrosis, skeletal abnormalities, and dental caries/periodontitis. In the presence of four major criteria the diagnosis is ‘certain’. The presence of three major and two minor criteria makes the clinical diagnosis ‘probable’ and the diagnosis is considered to be ‘likely’ if two major criteria and two minor criteria or associated findings are present [7]. The above proposed algorithm is not yet validated against laboratory findings (Mutation analysis in FERMT1 gene). Therefore, it can only be useful in the clinical praxis when genetic control is not available.
KS must be differentiated from epidermolysis bullosa, Rothmund–Thompson syndrome and other congenital poikilodermatous and photosensitive conditions [3, 5]. Rothmund–Thompson syndrome is characterized by poikiloderma and photosensitivity which are also associated with hypogonadism, short stature, cataract and sparse hair [4–6].
KS has been described in individuals or families mainly of Arab origin, as well as from Iran, Pakistan, India and Turkey. The disease has been also identified in European individuals, especially of British Caucasian, Italian, Albanian and Serbian origin [2, 6, 8].
Here, we report a case in a Greek patient. The patient presented five major and one minor of the proposed clinical diagnostic criterion and four associated findings. The final Diagnosis based on FERMT1 Gene Mutation Analysis. The KS-characteristic immunofluorescence staining pattern combined with the homozygous FERMT1 mutation c.676dupC, p.Q226PfsX16 verified final the clinical diagnosis. To the best of our knowledge, this mutation was found to be recurrent in the Balkans [9].
Patients with KS have a normal life span, but they have an increased susceptibility for the development of malignancies including squamous cell carcinoma of the lip, hard palate and acral skin, as well as transitional cell carcinoma of the bladder, that have a severe course[2, 10]. Deficiency of kindlin-1 protein and consequent β1 integrin deactivation results in aberrant cell adhesion, proliferation and chemotaxis and thus, in carcinogenesis. Its correlation, though, with epithelial malignancies has to be clarified with further research [2].
Treatment of KS is mainly symptomatic with emphasis on management of mucosal involvement and associated complications. Avoiding skin trauma and sun exposure and using moisturizers and sunscreens are recommended [2] as well skin cancer screening.
ACKNOWLEDGEMENTS
None.
CONFLICT OF INTEREST STATEMENT
None declared.
FUNDING
None.
ETHICAL APPROVAL
No ethical approval required.
INFORMED CONSENT
The patient has given written consent for publication of this case report.
GUARANTOR
Corresponding author: Dr Evangelia Kalloniati.
REFERENCES
- 1. Kindler T. Congenital poikiloderma with traumatic bulla formation and progressive cutaneous atrophy. Br J Dermatol 1954;66:104–11. [DOI] [PubMed] [Google Scholar]
- 2. Youssefian L, Vahidnezhad H, Uitto J. Kindler syndrome. GeneReviews In: Adam MP, Ardinger HH, Pagon RA, et al., eds.. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle, 2016,1993–2018. Mar 3 [Updated 2016 Dec 1]. [PubMed] [Google Scholar]
- 3. Mendes L, Nogueira L, Vilasboas V, Talhari C, Talhari S, Santos M. Kindler syndrome—report of two cases. An Bras Dermatol 2012;87:779–81. 10.1590/S0365-05962012000500020. [DOI] [PubMed] [Google Scholar]
- 4. Ghosh SK, Bandyopathy D, Das J, Chatterjee G, Sarkar S. Kindler’s syndrome: a case series of three Indian children. Indian J Dermatol 2010;55:393–7. 10.4103/0019-5154.74568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Handa N, Kachhawa D, Jain VK, Rao P, Das A. Kindler’s syndrome: a tale of Two Siblings. Indian J Dermatol 2016;61:468 10.4103/0019-5154.185767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Amirchaghmaghi M, Moeintaghavi A, Rasekhi J, Mozafari PM, Dalirsani Z, Jafarian AH. Kindler syndrome: a case report from Iran. J Dent Mater Tech 2014;3:134–8. [Google Scholar]
- 7. Angelova-Fischer I, Kazandjieva J, Vassileva S, Dourmishev A. Kindler syndrome: a case report and proposal for clinical diagnostic criteria. Acta Dermatovenerol Alp Panonica Adriat 2005;14:61–7. [PubMed] [Google Scholar]
- 8. Techanukul T, Sethuraman G, Zlotogorski A, Horev L, Macarov M, Trainer A, et al. Novel and recurrent FERMT gene mutations in kindler syndrome. Acta Derm Venereol 2011;91:267–70. 10.2340/00015555-1063. [DOI] [PubMed] [Google Scholar]
- 9. Kiritsi D, He Y, Pasmooij AM, Onder M, Happle R, Jonkman MF, et al. Revertant mosaicism in a human skin fragility disorder results from slipped mispairing and mitotic recombination. J Clinical Investigation 2012;122:1742–6. 10.1172/JCI61976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. HemavathyBhaskar Y, MuraliGopikaManoharan GV. Kindler’s syndrome—a rare case report . J Dental Med Sciences 2016;15:64–7. [Google Scholar]