

Abstract
To understand how complex genetic networks perform and regulate diverse cellular processes, the function of each individual component must be defined. Comprehensive phenotypic studies of mutant alleles have been successful in model organisms in determining what processes depend on the normal function of a gene. These results are often ported to newly sequenced genomes by using sequence homology. However, sequence similarity does not always mean identical function or phenotype, suggesting that new methods are required to functionally annotate newly sequenced species. We have implemented comparative analysis by high-throughput experimental testing of gene dispensability in Saccharomyces uvarum, a sister species of Saccharomyces cerevisiae. We created haploid and heterozygous diploid Tn7 insertional mutagenesis libraries in S. uvarum to identify species-dependent essential genes, with the goal of detecting genes with divergent functions and/or different genetic interactions. Comprehensive gene dispensability comparisons with S. cerevisiae predicted diverged dispensability at 12% of conserved orthologs, and validation experiments confirmed 22 differentially essential genes. Despite their differences in essentiality, these genes were capable of cross-species complementation, demonstrating that trans-acting factors that are background-dependent contribute to differential gene essentiality. This study shows that direct experimental testing of gene disruption phenotypes across species can inform comparative genomic analyses and improve gene annotations. Our method can be widely applied in microorganisms to further our understanding of genome evolution.
The ability to accurately predict gene function based on DNA sequence similarity is a valuable tool, especially in the current stage of genomic research in which an increasing number of genomes are being sequenced. It has become crucially important to predict gene function based on sequence similarity because of the lack of experimentally determined functional information associated with each newly sequenced genome. Most functional predictive methods rely on similarities of DNA sequence homology, coexpression patterns, or protein structure to help assign function to uncharacterized genes, using genes in which known functions have been previously characterized (Eisen 1998; Usadel et al. 2009). However, these methods come with their own set of limitations and often produce a substantial number of predictive errors, highlighting the importance of implementing experimental methods to directly test gene function in previously uncharacterized genomes to improve current methods of annotation.
The gold standard of gene function characterization relies on observing phenotypes of targeted deletions of predicted coding sequences to probe the contributions of each gene to specific biological processes. To get a global view of gene function within an organism, several genome-wide deletion collections have been created in model species, particularly in bacteria and yeast (Winzeler et al. 1999; Baba et al. 2006; de Berardinis et al. 2008; Porwollik et al. 2014), including highly diverged species (Kim et al. 2010; Schwarzmüller et al. 2014) as well as different strains within a species (Dowell et al. 2010). These systematic deletion collections are powerful tools for investigating gene function, biological pathways, and genetic interactions, especially in the genetic workhorse Saccharomyces cerevisiae, in which gene function characterization and gene dispensability comparisons have been extensively performed among various deletion collections of yeast (Tong et al. 2001; Dowell et al. 2010; Kim et al. 2010; Costanzo 2016). These studies have identified ∼17% of essential genes to be differentially essential between highly diverged species (S. cerevisiae and Schizosaccharomyces pombe) and have discovered 6% of essential genes (57) that are differentially essential even between two strains of S. cerevisiae.
However, considerable effort and resources are required to create these targeted, systematic libraries, and they are not a practical approach for interrogating a wide range of nonstandard genetic backgrounds in a high-throughput manner. Alternative approaches to targeted gene deletion libraries are transposon-based mutagenesis methods used to create random insertional mutant collections, eliminating requirements for a priori knowledge about defined coding regions and providing information about partial loss-of-function or gain-of-function mutations. Random insertional profiling has been widely applied across various species and has been instrumental in understanding virulence genes, stress tolerance mechanisms, and even tumor suppressor genes in mice (van Opijnen and Camilli 2013; DeNicola et al. 2015; Weerdenburg et al. 2015; Yung et al. 2015; de la Rosa et al. 2017; Coradetti et al. 2018). In yeasts in particular, transposon libraries have provided useful information about gene function, growth inhibiting compounds, and essential functional protein domains (Ross-Macdonald et al. 1999; Gangadharan et al. 2010; Oh et al. 2010; Guo et al. 2013b; Michel et al. 2017; Zhao et al. 2017; Price et al. 2018; Segal et al. 2018; Zhu et al. 2018).
Despite this growing body of literature, the genetic mechanisms explaining differences across species are still poorly understood. Here we use a random insertional method to assay gene dispensability using approximately 50,000 mutants in Saccharomyces uvarum, a species that diverged from S. cerevisiae 20 million years (Myr) ago and whose coding sequences are ∼20% divergent from those of S. cerevisiae (Kellis et al. 2003; Scannell et al. 2007; Dujon 2010). These species can mate with one another to create hybrids, allowing us to explore the genetic basis for possible differential gene dispensability between them using genetic toolsets established in S. cerevisiae. Genes with different dispensability patterns between these two species could be explained by divergent gene function and/or genetic interactions, providing a model for investigating genome evolution between two diverged species of yeast. We successfully validate a subset of predicted differentially essential genes required for growth in rich media, establishing the utility of our mutagenesis approach in prioritizing genes for testing viability (Guo et al. 2013a; Michel et al. 2017). In what follows, for genes that emerge from our analyses as differentially essential between the reference strains of S. cerevisiae and S. uvarum, we refer to them as species-specific; rigorously speaking, a comprehensive study across populations will be necessary to establish whether a given essentiality pattern is truly common to the entirety of the respective species. Together, our data make clear that our Tn7 transposon mutagenesis library serves as a valuable resource for studying the S. uvarum genome and that our approach is a powerful framework for comparative functional genomics studies across newly sequenced, previously uncharacterized species.
Results
Generating Tn7 insertional libraries in S. uvarum to predict essential and nonessential genes
One of the most straightforward mutant phenotypes to characterize is cell viability, which reveals if a given gene is involved in an essential cellular process. Therefore, we first sought to characterize gene essentiality in S. uvarum, with the aim of identifying genes that are differentially essential between S. cerevisiae and S. uvarum. Instead of creating a library of individual knockout strains, we applied a high-throughput approach of creating random insertional mutants and leveraged the power of sequencing to identify the insertion sites in a pooled collection. The Tn7 mutagenesis library approach described by Kumar et al. (2004) was used to create a collection of S. uvarum mutant strains and has been previously described by Caudy et al. (2013). Briefly, in vitro transposition of the Tn7 transposon was performed in a plasmid library containing random S. uvarum genomic fragments (Fig. 1A). The Tn7 transposon was designed to carry a clonNat resistance marker that carries stop codons in all reading frames near both termini. The interrupted genomic fragments were excised out of the plasmid and integrated at their corresponding genomic positions in the reference strain background of S. uvarum, each of which is expected to produce a truncation when inserted within a coding region (Fig. 1B). The plasmid library contains approximately 50,000 unique genomic insertion sites; we integrated the library into a diploid strain and, separately, a haploid MATa strain at 10× coverage (additional details can be found in the Supplemental Material). Pools of mutants from each Tn7 library were grown in liquid nutrient-rich media as described in the Methods. Insertion sites were determined using sequencing methods as described in detail in the Supplemental Material along with DNA sequencing library preparation protocols (Fig. 1C).
Figure 1.

Schematic of Tn7 transposon mutagenesis library and insertion identification in S. uvarum. (A) Simplified representation of in vitro transposition of the Tn7 transposon into a plasmid library containing random S. uvarum genomic DNA fragments. Approximately 50,000 plasmids containing the Tn7 transposon were pooled together to form the final library. (B) Illustration of the Tn7-containing excised portion of the plasmid integrated into haploid and diploid yeast through homologous recombination. Approximately 500,000 NatR clones of each ploidy were pooled into two separate pools (haploid pool and diploid pool). (C) Design of Tn7-seq libraries used to identify insertion sites through sequencing. Reads containing Tn7 sequence are enriched (PCR off common flanking region of the Tn7 and Illumina adapter sequences) and mapped to the genome to identify insertion sites.
We cataloged transposon insertion mutants on the basis of sequenced insertion sites that could be detected after mutagenesis and outgrowth of haploid and diploid S. uvarum, and we also tabulated those mutants present in both pools. We found these insertion sites to be evenly distributed throughout the S. uvarum genome, as illustrated in Supplemental Figure S1 (detailed information about overall sequencing coverage is listed in Supplemental Table S4). Once the insertion sites were determined in both libraries, we counted the number of insertion sites in each annotated open reading frame (Methods). Supplemental Table S11 summarizes the number of insertion sites and the number of genes that contain insertion sites within each library, including the initial plasmid library. The number of insertion sites from each library that fell into each annotated S. uvarum gene is listed in Supplemental Table S5. Of the 5908 annotated genes, a total of 5315 (90%) genes harbored insertion sites that were identified in at least one library. Comparisons between shared genes and unique genes harboring insertion sites are illustrated in Supplemental Figure S2.
Because by definition, loss-of-function mutants in genes essential in S. uvarum would not be viable in the haploid of this species, we used our observations of detected transposon mutants in this strain as a jumping-off point for inferences of gene essentiality. We first tested whether orthologs of the known essential set in S. cerevisiae would be depleted for insertion sites in the S. uvarum haploid, as would be predicted if the essentiality of most genes were conserved between the species. Consistent with this notion, we identified a significant reduction in the number of inserts present in known S. cerevisiae essential genes in the haploid S. uvarum library (Wilcoxon test P < 2.2 × 10−16, essential average inserts/kb = 0.88, SD = 1.28 vs. nonessential average inserts/kb = 4, SD = 4.38) (Supplemental Fig. S3), indicating that essential genes are effectively targeted by this approach. However, because of the nature of the library, insertional events at different positions across a gene may result in a partial loss of function (Sadhu et al. 2018). Because essential genes may still tolerate some insertions, we instead relied on comparisons between the diploid and haploid libraries to make inferences about gene essentiality. Specifically, we calculated an insertion ratio using the number of inserts per gene in the haploid library divided by the number of inserts in the diploid library, which inherently normalizes for the length of the gene (Methods). By using the insertion ratio as a metric, we tested for significant differences between S. uvarum genes whose orthologs were essential and nonessential in S. cerevisiae. In our analyses, we used S. uvarum intergenic regions as a control: Intergenic regions between convergently oriented genes are expected to largely not be essential, and thus, we expected the distribution of intergenic regions to be similar to that of nonessential genes.
Figure 2A reports the distribution of detected insertion mutants in S. uvarum haploids and diploids (quantified by the insertion ratio) for each feature type. As predicted, we found that S. uvarum orthologs of known S. cerevisiae nonessential genes generally behaved similarly in our mutant pools to S. uvarum intergenic regions. However, the distribution of insertion ratios from these orthologs of nonessential genes had a left shoulder resembling the distribution among orthologs of S. cerevisiae essential genes. We hypothesized that this population of genes depleted for insertions in S. uvarum haploids was likely to reflect S. uvarum–specific essential genes. Likewise, we also noted a righthand tail (corresponding to highly abundant transposon mutants in S. uvarum haploids) in the distribution of insertion ratios among S. uvarum orthologs of S. cerevisiae essential genes, suggesting that some were in fact not essential in S. uvarum. The differences between orthologs of S. cerevisiae essential genes and nonessential gene insertion ratios were significant, as well the differences between orthologs of essential genes and intergenic regions (Wilcoxon test P < 2.2 × 10−16) (Fig. 2B). By using the insertion ratio, we formulated a prediction of each gene as either essential or nonessential in S. uvarum using a null distribution to rank genes above or below a cut-off metric of 0.25 (for details, see Supplemental Material). By using this cut-off value, 1170 genes were categorized as predicted essential genes. We applied an additional cut-off metric (more details in the Methods) to remove a class of low-coverage genes, resulting in a total number of 718 (13%) predicted essential genes and 3838 (65%) genes that are predicted nonessential, with 1299 genes (22%) undetermined (genes without inserts in the diploid library). We proceeded to characterize each gene set and validate the dispensability of each of the predicted gene categories.
Figure 2.

Insertion ratio distributions of S. uvarum intergenic regions and known S. cerevisiae essential and nonessential genes. (A) Density plots displaying the distribution of insertion ratios across three feature types: S. uvarum intergenic regions between Watson-and-Crick–oriented genes ranging from 7 kb–500 bp (gray) and S. uvarum genes whose orthologs are known S. cerevisiae essential (Sc_E in blue) and nonessential genes (Sc_NE in red). The dashed line represents an insertion ratio of 0.25 and defines the cut-off value to classify essential and nonessential genes. (B) Box plots of insertion ratios by feature type described in plot A. Significant insertion ratio differences exist between known S. cerevisiae essential and nonessential genes and between S. uvarum intergenic regions. (Wilcoxon tests Sc_E:Su_Intergenic P < 2.2 × 10−16, Sc_E:Sc_NE P < 2.2 × 10−16, Sc_NE:Su_Intergenic P = 7.08 × 10−6).
Analysis of predicted gene dispensability
The predicted gene list of S. uvarum essential genes was compared with known essential genes lists from both S. cerevisiae and S. pombe to determine the amount of conservation that exists between orthologs across diverged species. Of the predicted 718 S. uvarum essential genes, 297 genes (42%) were shared among all three sets, with a total of 487 genes (68%) shared with at least one other set. Furthermore, nine genes whose essentiality was specific to particular S. cerevisiae strains, including four genes specific to the S288C strain and five genes that are specific to the ∑1278b strain, were inferred by our analysis to be essential in S. uvarum (Supplemental Fig. S4). Similar to what has been previously shown in S. cerevisiae, predicted essential genes in S. uvarum were more likely to be unique, with 91% of essential genes (656/718) being present in single copy compared with 76% of nonessential genes (2736/3604). Additionally, comparisons between Gene Ontology (GO) molecular function terms of essential gene sets from both species showed significant enrichment (P-value <0.01) for fundamental biological functions. Processes such as DNA replication/binding and RNA and protein biosynthesis, as well as structural constituents of the ribosome and cytoskeleton, were enriched in both predicted S. uvarum essential genes and those known to be essential in S. cerevisiae (Supplemental Table S6). In contrast, nonessential genes were significantly (P-value <0.01) enriched for regulatory functions (transcription factor activity) and conditional responsive processes, such as transmembrane transporter activity and cell signaling (kinase activity) (Supplemental Table S7). We conclude that many of the features of the predicted essential genes in S. uvarum are similar to confirmed essential genes in other species.
We next sought to validate experimentally the predictions of essentiality we had made from our transposon mutagenesis. We note that interactions with different auxotrophic markers may potentially influence deletion phenotypes. To limit these potential effects, we used as few markers as practical and aimed to use prototrophs when possible. Also, all phenotypes in the validation experiments were tested by sporulating a heterozygous diploid; so if any markers were required for the phenotype, deviations in the segregation pattern would have been apparent.
We first focused on genes whose orthologs were known to be essential in S. cerevisiae and that we had likewise predicted to be essential in S. uvarum. For each of 13 such cases, we sporulated the respective heterozygous deletion strain and performed tetrad analysis for cell viability; the results confirmed essentiality for 12 (92%) of the 13 strains (Supplemental Table S12). One example of a confirmed essential gene can be found in Figure 3A, which illustrates the genomic positions of all insertion sites across a genomic locus of Chromosome V that contains essential and nonessential genes. The color of the gene outline reports the predicted dispensability, which is determined by their insertion ratio. For example, the gene BRR2, a RNA-dependent RTPase RNA helicase, had an insertion ratio of 0.130 and was predicted to be a conserved essential gene (Fig. 3C). The tetrad analysis of a BRR2 heterozygous deletion strain displayed a two viable:two inviable segregation pattern in both S. cerevisiae and S. uvarum, validating this gene as a conserved essential gene (Fig. 3B). Images of all other confirmed essential genes are in Supplemental Figure S5. We also tested three genes known to be nonessential in S. cerevisiae and predicted by our analysis to be nonessential in S. uvarum, confirming all three (100%) as nonessential in both species (Supplemental Fig. S6; Supplemental Table S12).
Figure 3.

Validation of conserved essential and nonessential genes. (A) Mapped chromosomal insertion positions are plotted across Chromosome V. Haploid inserts are indicated in red; diploid inserts, in blue; and overlapping inserts, in purple. Genes indicated across the top are outlined according to predicted dispensability and filled in if confirmed. (B) Tetrad analysis of a confirmed conserved essential gene brr2Δ in S. cerevisiae and S. uvarum. Segregants containing brr2Δ alleles are inviable in both species. (C) Table indicating the insertion ratio (number of haploid inserts by the number of diploid inserts) per gene. The final column lists the predicted classification: (NE) nonessential; (E) essential; (N/A) no data.
Additionally, we obtained an independent set of S. uvarum haploid deletion strains (see Methods), which was used as a validated nonessential gene set. Out of the total 356 genes that were viable upon deletion in this collection, our analysis of transposon mutants inferred 346 to be nonessential in S. uvarum (97%), providing further support to the predictions from our method.
Gene dispensability comparisons of orthologous pairs between S. cerevisiae and S. uvarum
Our main goal of this project was to identify genes that were differentially essential in a species-dependent manner. To make direct comparisons of dispensability between S. cerevisiae and S. uvarum, we narrowed our analysis to 4543 orthologous genes for which we had data in the S. uvarum data set (Supplemental Table S8). Overall, our predicted patterns of essentiality in S. uvarum were consonant with the known behavior in S. cerevisiae for 88% (4016/4543) of these genes. The remaining 12% of orthologs were predicted to differ in essentiality between the two species, with 304 (7%) of these genes only essential in S. uvarum and 221 (5%) genes only essential in S. cerevisiae (Supplemental Fig. S7A). We note that the former could represent an overestimate of the count of predicted essential genes specific to S. uvarum in that it is inferred from the lack of detected insertion sites in our haploid S. uvarum libraries in genes not previously characterized as essential in S. cerevisiae. All predicted genes that differ in dispensability are listed in Supplemental Table S9.
To analyze further our predictions of differential essentiality between the species, we compiled a list of 222 genes whose respective orthologs were known to be essential in S. cerevisiae and predicted to be nonessential in S. uvarum; we also formulated more stringent cutoffs for our transposon mutant library analysis (see Methods) to yield a similarly sized set of genes (220 total) inferred to be essential in S. uvarum and known to be nonessential in S. cerevisiae. By using this list, we determined the proportion of inferred S. cerevisiae–specific and S. uvarum–specific essential genes annotated for each function by performing GO term finder using the molecular function ontology. Among the results, reported in Supplemental Figure S7B, were enrichments for structural constituents of the ribosome among genes predicted to be essential only in S. uvarum, as well as for RNA polymerase activity among genes inferred to be essential only in S. cerevisiae. Full lists of significant (P-value <0.01) GO enrichment molecular function terms for each species individually are listed in Supplemental Table S10. We also hypothesized that genetic interaction patterns could distinguish genes that were predicted to be differentially essential between S. cerevisiae and S. uvarum. Toward this end, we tabulated combined interaction degree scores for all orthologous genes between yeast, worm, flies, mice, and humans. We first compared interaction degrees between known essential and nonessential genes and found the former to be increased, as previously reported (Costanzo 2016; Supplemental Fig. S8). Next, we formulated a comparison between two gene sets: those predicted to be essential in S. uvarum and known to be nonessential in S. cerevisiae, on the one hand, and those categorized as nonessential in both species, on the other hand. We found a significant increase in the number of combined interactions in the former (P-value = 1.99 × 10−7), suggesting that having more interactions may be predictive of essential function. Analyses of expression could not account for the differences in essentiality (Supplemental Fig. S13).
We next set out to confirm experimentally a subset of predicted essential genes within each species by sporulating heterozygous deletion strains to determine the viability pattern of the segregants. As an example of a confirmed S. uvarum–specific essential gene from these experiments, SSQ1, which is required for assembly of iron/sulfur clusters into proteins, is illustrated in Figure 4. Figure 5 displays an example of a gene confirmed to be essential in S. cerevisiae but not S. uvarum: VTC4, a gene involved in the regulation of membrane trafficking. Overall, we confirmed a total of 22 S. uvarum–specific and S. cerevisiae–specific essential genes (tetrad analysis can be found in Supplemental Figs. S9, S10, respectively). We found a variety of growth phenotypes associated with deletions in the permissive species background of confirmed species-specific essential genes. Some deletions resulted in poor growth, whereas others did not show a growth defect when compared to wild type. All combined tetrad analysis results are reported in Supplemental Figure S11; these results include genes for which sporulation experiments did not validate the inferences of species-specific essentiality from our transposon mutagenesis, which we refer to as false positives. Supplemental Table S12 summarizes the total number of genes confirmed in each category. We note the higher false-positive rate in the species-specific essential gene categories; as above, in part this likely reflects incorrect calls of essentiality in S. uvarum in which we detected no transposon mutants in our haploid libraries as a product of low coverage, rather than the inviability of the respective mutants.
Figure 4.

Validation of the S. uvarum–specific essential gene SSQ1. (A) Mapped chromosomal insertion positions are plotted across Chromosome X. Haploid inserts are indicated in red; diploid inserts, in blue; and overlapping inserts, in purple. Genes indicated across the top are outlined according to predicted dispensability and filled in if confirmed. Light blue filling indicates a gene that is essential in S. uvarum and nonessential in S. cerevisiae (confirmed E_NE). (B) Tetrad analysis of a heterozygous ssq1Δ::KanMX strain displaying inviable segregants containing the ssq1Δ allele in S. uvarum. (C) Tetrad analysis of a heterozygous ssq1Δ::KanMX strain in S. cerevisiae containing viable segregants plated on YPD and G418. (D) Table indicating the insertion ratio (number of haploid inserts by the number of diploid inserts) per gene. The final column lists the predicted classification: (NE) nonessential; (E) essential; (N/A) no data.
Figure 5.

Validation of the S. cerevisiae–specific essential gene VTC4. (A) Mapped chromosomal insertion positions are plotted across Chromosome XII. Haploid inserts are indicated in red; diploid inserts, in blue; and overlapping inserts, in purple. Genes indicated across the top are outlined according to predicted dispensability and filled in if confirmed. Light pink filling indicates a gene that is essential in S. cerevisiae and nonessential in S. uvarum (confirmed NE_E). (B) Tetrad analysis of a heterozygous vtc4Δ strain displaying viable segregants with the vtc4Δ allele in S. uvarum plated on YPD and G418. (C) Tetrad analysis of a heterozygous vtc4Δ allele in S. cerevisiae resulting in inviable segregants. (D) Table indicating the insertion ratio (number of haploid inserts by the number of diploid inserts) per gene. The final column lists the predicted classification: (NE) nonessential; (E) essential; (N/A) no data.
That said, in some cases, validation failures could be explained by errors in genome annotation not errors in our insertional library results. For example, the gene DRE2, which functions in cytosolic iron–sulfur protein biogenesis, was predicted to be a nonessential gene but was confirmed as an essential gene through tetrad analysis. Manual inspection revealed that all the haploid insertions were clustered at the 5′ end of the gene (Supplemental Fig. S12). In protein alignments of DRE2 between S. cerevisiae and S. uvarum, we noted an annotated start codon in S. uvarum upstream of the annotated start codon in S. cerevisiae. These data suggest that the gene was misannotated in S. uvarum and instead shares the methionine start position further downstream. By using the reannotated gene coordinates, we would correctly classify DRE2 as essential in S. uvarum because the haploid insertions were no longer included in the open reading frame. This example highlights the utility of our method to improve gene annotation in addition to characterizing gene essentiality.
Paralog divergence and duplicate gene loss explain some background effects on differential gene essentiality
We next set out to investigate genetic background effects that could be contributing to differences in gene dispensability between S. cerevisiae and S. uvarum. One explanation could be genetic redundancy caused by gene duplications, such that a gene is nonessential in one species because of the presence of a paralog, whereas the other species contains only a single copy. To investigate this possibility, we began investigating genes that both differed in dispensability and harbored a paralog. Of the 222 S. cerevisiae–specific essential genes, 11 were known to have paralogs. Our initial analysis identified the Ras activator CDC25 as a S. cerevisiae–specific essential gene (nonessential in S. uvarum). CDC25 is a paralog of SDC25, which contains a premature stop codon in the reference strain of S. cerevisiae and other laboratory strains (Folch-Mallol et al. 2004).
We performed complementation assays by cloning S. uvarum alleles of both paralogs into a CEN/ARS plasmid and testing whether the S. uvarum alleles could rescue the inviable phenotype of segregants from a heterozygous cdc25Δ deletion in S. cerevisiae. We found that SDC25 from S. uvarum was functional and that both SDC25 and CDC25 alleles from S. uvarum could complement a cdc25Δ deletion in S. cerevisiae (Table 1). Although we did not test for complementation in the S. uvarum background, the results from the complementation assays show that CDC25 is required for growth in this strain of S. cerevisiae because of the lack of redundancy as a consequence of the nonfunctional copy of SDC25. We expect that this relationship may be unique to those S. cerevisiae strains in which SDC25 is a pseudogene, but we nonetheless consider it a satisfying validated mechanism for the background dependence of CDC25 essentiality.
Table 1.
Viability summary of gene deletions and complementation assays

Following this same logic, we hypothesized that CDC25 nonessentiality in S. uvarum could be attributed to the redundancy provided by the functional copy of SDC25 in this species. To test this idea, we created an S. uvarum mutant heterozygous for both cdc25Δ and sdc25Δ and performed segregation analysis on the dissected tetrads (Table 1). Unexpectedly, the segregation pattern of a double mutant displayed a lethal phenotype for not only the double mutant but also the single sdc25Δ mutant. We confirmed this result by constructing an sdc25Δ heterozygous mutant in S. uvarum and found a 2:2 segregation pattern showing that SDC25 is an essential gene in S. uvarum. We conclude that of the two paralogs, one is essential in our S. cerevisiae strain and the other in S. uvarum, representing a novel case of a swap in essentiality.
Divergent gene dispensability is largely caused by trans-effects
Although genetic redundancy or gene loss is a possible explanation for a fraction of differentially essential genes, the remaining much larger portion of this class of genes remained unexplained. Another hypothesis to explain differences in essentiality is gene function divergence between these two species. We therefore proceeded to further investigate the remaining differentially essential genes for functional differences. For a subset of these genes, we performed complementation assays in both species to test for divergent function. We cloned five S. cerevisiae alleles and their promoters from the list of S. uvarum–specific essential genes (SAC3, TUP1, CCM1, SSQ1, and AFT1) and seven S. uvarum alleles from the list of S. cerevisiae–specific essential genes (ALR1, SHR3, CDC25, INN1, LCD1, SEC24, VTC4) and tested each allele's ability to rescue the inviability caused by deletion of the corresponding ortholog in the alternate species (Fig. 6). The results from these complementation tests revealed that all genes are able to complement the inviable phenotype in the other species, suggesting that the differences in essentiality are more likely to be caused by trans-acting changes rather than functional differences of protein-coding regions.
Figure 6.
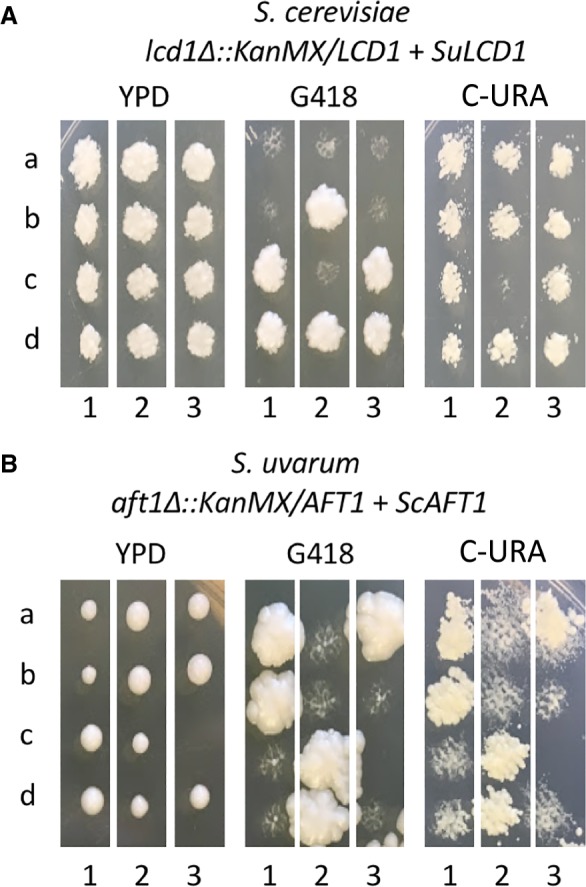
Complementation assay confirming two examples of genes that differ in essentiality but complement the viability phenotype in both genetic backgrounds. Letters a through d represent four spores, whereas numbers 1 through 3 indicate three tetrads. Tetrads were dissected on YPD and were replica plated to G418 to indicate which spore contains the deletion and to C-URA to indicate the presence of the plasmid expressing the indicated gene. (A) S. cerevisiae lcd1Δ::KanMX strain containing a plasmid with the S. uvarum allele of LCD1. (B) S. uvarum aft1Δ::KanMX strain containing a plasmid with the S. cerevisiae allele of AFT1.
Discussion
In this study, we applied a comparative functional genomics approach to investigate how genetic background influences gene dispensability between the reference strains of two diverged species of yeast. By using insertional integration comparisons between haploid and diploid pools of mutants, we prioritized genes to validate as predicted essential, nonessential, and differentially essential gene categories in S. uvarum. We predicted ∼12% of orthologs to differ in dispensability between S. uvarum and S. cerevisiae and validated 22 genes in this category. However, most genes that differ in dispensability have retained their function between these two species, suggesting that differences in gene dispensability are likely because of trans-acting changes rather than the direct result of divergent coding sequence.
Specifically, our comparison of orthologous genes between S. cerevisiae and S. uvarum revealed that a majority of genes maintain conserved dispensability requirements (88%), whereas 12% of orthologs are predicted to be essential in one species but not the other. We confirmed 93% (15/16) of predicted conserved categories of essentiality and 49% (27/55) of genes predicted to be differentially essential. Although applying a less restrictive insertion ratio cut-off value includes more genes to be characterized in this category, it also increases the likelihood of false positives and creates a challenge for the correct validation of this category type. Although our rate of confirmed genes in this category was lower than the conserved category, we correctly identified a subset of genes that are differentially dispensable, despite the moderately dense insertional profile of the library and a less restrictive cut-off value applied to include more genes to be classified as this type. Further analysis of predicted species-specific essential genes revealed enriched GO terms of molecular functions involved in structural constituent of the ribosome and DNA binding, although more precise analysis of functional enrichments may require more thorough validation to remove the influence of false positives. Finally, we used yeast genetic tools to test hypotheses about genetic background effects that contribute to differences in essentiality. We find that differences can be explained by paralog divergence and trans-acting factors.
Applying a random insertional approach has proved to be useful in functionally profiling S. uvarum and will be useful for studying other understudied species, with the goal of adding information to gene annotation methods. Although this study was performed in standard rich media laboratory conditions, it is easily amenable for testing stress conditions, other nutrient sources, and naturally relevant conditions. This library can be applied to probe previously unannotated genes or even proto-genes for functional acquisition, because it is not restricted to a priori assumptions of genic boundaries. The identification of synthetic lethal interactions can also be determined by performing insertional profiling in the background of a particular mutation of interest relatively quickly and economically. Similarly, the library could be generated in different strain backgrounds to confirm which phenotypes are truly species-specific versus which might vary within the species. Additionally, pooled competition experiments en masse can be used to determine the frequency of particular insertional mutants, providing quantitative measurements of cellular fitness across conditions. Such a strategy could be efficiently used using computational approaches to prioritize experimental conditions that are most likely to probe the most valuable phenotypic information for further functional characterization (Guan et al. 2010).
Gene regulation also plays a large role in evolution and is crucial for responding to environmental change (Carroll 2005). In previous studies, we aimed to experimentally characterize differences in gene expression patterns between S. cerevisiae and S. uvarum and discovered species-specific responses to osmotic stress, peroxisome biogenesis, and autophagy, suggesting that each species may have been exposed to different selective pressures within their respective evolutionary histories (Caudy et al. 2013; Guan et al. 2013). We did not find that genes with different gene expression patterns between species were more likely to be differentially essential. Instead, trans-genetic interactions dominate. Identifying the molecular basis of these trans-effects can now be undertaken, potentially revealing principles of genetic interactions across species.
Methods
Strains, plasmids, and primers
The strains, plasmids, and primers used in this study are listed in Supplemental Tables S1 through S3, respectively. All S. uvarum strains are derivatives of the sequenced strain CBS 7001 (previously sometimes called Saccharomyces bayanus or S. bayanus var uvarum), and all S. cerevisiae strains are of S288C background. Unless specified below, yeast strains were grown at 25°C for S. uvarum strains and 30°C for S. cerevisiae strains in media prepared according to standard recipes.
Construction of the Tn7 mutagenesis library
The construction of the Tn7 plasmid library has been previously described in detail and was obtained from the Caudy laboratory (Caudy et al. 2013). Briefly, this mutagenesis approach uses a plasmid library of S. uvarum genomic DNA containing random Tn7 transposon insertions. The construct has a selectable marker for transformation into yeast, allowing the selection of disruption alleles.
To make the plasmid library, genomic DNA was isolated and fragmented by sonication to an average length of 3 kb from a ρ0 S. uvarum strain. The ends of the DNA were blunted and cloned into the pZero Blunt vector (Invitrogen). Approximately 50,000 colonies were recovered from the transformation into E. coli DH5α strain. The transformants were scraped from kanamycin plates and pooled for plasmid purification. A version of the Tn7 transposon was constructed by amplifying the promoter from the Tet-on pCM224 (Bellí et al. 1998). The cassette of the Tet-on promoter and the ClonNAT resistance gene was amplified using PCR primers containing lox and BamHI sites and cloned into the BamHI site of the NEB vector pGPS3. This transposon construct was inserted into the S. uvarum genomic DNA library in vitro using a transposon kit from NEB. Initial selection (50,000 colonies) was on ClonNAT/Zeo. HindIII and XbaI were used to digest the pZero backbone to release the linearized genomic DNA for efficient recombination. The library was then used to transform a haploid S. uvarum strain (ACY12) and a diploid strain (YMD1228) using a modified transformation protocol optimized for S. uvarum (Caudy et al. 2013). Transformant colonies were plated to YPD-ClonNat plates and allowed to grow for 5 d at 25°C. A total of approximately 500,000 colonies were scraped for each pool. Each final pool was well mixed at a 1:1 ratio with 50% glycerol, and 2-mL aliquots were stored at −80°C.
Pooled growth of Tn7 S. uvarum libraries
To determine the initial complexity of the integrated pools, genomic DNA was extracted directly from the glycerol stocks of both haploid and diploid pools using the Hoffman and Winston method (Hoffman and Winston 1987). Additionally, we inoculated 500 µL of both libraries in separate YPD flasks for 24 h to recover mutants after 24 h of growth. Furthermore, to collect samples over time, we competed both pools under sulfate-limiting conditions in chemostats for approximately 30 generations at 25°C. A large-volume, ∼300 mL, sulfate-limited chemostat (Gresham et al. 2008) was inoculated with a single 2 mL glycerol stock sample of each pool. After allowing the chemostat to grow at 25°C without dilution for ∼24 h, fresh media was added to the chemostat at a rate of 0.17 h−1. This pooled growth assay was repeated twice, each including five time points with O.D. and dilution rate measurements, as well as collected cell pellets for DNA extractions using the modified Hoffman–Winston prep referenced above. Early time points from this pooled growth assay were included in our analysis here because we found it to be largely overlapping with the rich media collection, and we found that the additional sequencing coverage improved our overall results.
Tn7 sequencing library preparation
Sequencing libraries were prepared by first extracting genomic DNA from pools of each library grown in YPD and sulfate limited conditions. Genomic DNA libraries were prepared for Illumina sequencing using a Tn7-seq protocol described previously (Wetmore et al. 2015). Briefly, the Covaris was used to randomly fragment DNA to ∼200–800 bp in length. The fragments were blunt ended, and A-tails were added to the fragments to ligate the Illumina adapter sequences. Custom index primers (listed in Supplemental Table S3) targeting Tn7-specific sequence and Illumina adapter sequence were used to enrich for genomic DNA with Tn7 insertion sites. The barcoded libraries were quantified on an Invitrogen Qubit fluorometer and submitted for 150-bp paired-end sequencing on an Illumina HiSeq 2000 by JGI. This method was also applied to make the plasmid library from linearized plasmid DNA.
Sequencing analysis
Sequencing reads from the FASTQ files were trimmed to remove Tn7-specific sequences and adapter sequences, restricting the minimal length of reads to 36 bp using Trimmomatic (Bolger et al. 2014) and a FASTX toolkit. Trimmed FASTQ files were aligned against the reference strain of S. uvarum (CBS 7001) using the Burrows–Wheeler aligner (BWA) with standard filters applied (Li and Durbin 2009). Specifically, nonuniquely mapping reads, reads in which the pair did not map, reads with a mapping quality less than 30, and PCR/optical duplicate reads were filtered out; the SAMtools (Li et al. 2009) C-50 filter was applied as recommended for reads mapped with BWA. To limit the insertional analysis to actively growing cells, SAM files were merged from the later time points in the growth assays of each pool using SAMtools. The sequence coverage of the nuclear genome ranged from 70× to 300× (Supplemental Table S4). Insertion sites were determined from SAM files using a custom Ruby script (Supplemental Code). Insertion sites that had 10 reads or more were processed through a custom Python script that counted the number of insertion events in each coding region across the genome (Supplemental Code). This pipeline was applied to both libraries, and further comparisons were made between the pools to determine essential genes.
Predicting gene dispensability between species
To determine a list of predicted essential genes, comparisons were made between the haploid and diploid libraries. We calculated an insertion ratio by dividing the number of insertions in the haploid pool by the number in the diploid pool. This direct comparison inherently accounts for the length of the gene, because the length is constant in both libraries. Therefore, a decrease in insertion sites in the haploid library indicates a reduction in the presence of mutants containing insertional sites that impact cellular viability. Ratios closer to zero represent insertional mutants that reduce the frequency of haploids harboring insertional sites in a coding region that is required for cellular growth.
To make an insertion ratio cut-off value to categorize essential and nonessential genes, we analyzed the distribution of insertion ratios within intergenic regions between 500 bp and 7 kb in length and positioned between Watson-and-Crick–oriented coding regions (so chosen because these are less likely to contain promoter sequences). The distribution of the insertion ratio calculated for these regions was similar to that of known nonessential genes in S. cerevisiae. Therefore, we used this distribution to rank the insertion ratios of all coding regions and set a cut-off value to 0.25, in which 20% of the insertion ratio of coding regions fell below the intergenic distribution, which was similar to the kernel density estimates of known S. cerevisiae essential genes. The kernel density estimates were computed in R and visualized using ggplot2 (Wickham 2009; R Core Team 2017). To remove a class of low-coverage genes in the essential gene category, we applied an additional cut-off value. Because the difference between zero and one with a gene that is longer has a lower weighted difference than a shorter gene, we calculated the difference between the diploid pool and haploid pool and normalized this value to the length of the gene (normalized difference). Genes with less than a normalized difference of two were removed from the essential category.
Validating predicted essential and nonessential genes
We validated predicted essential genes by creating S. uvarum heterozygous diploid deletion mutants using primers listed in the Supplemental Table S3. Primers containing 50 bp of homology upstream of and downstream from each candidate open reading frame were used to amplify the KanMX cassette from the pRS400 plasmid. The PCR product was used to integrate into the S. uvarum genome using an S. uvarum–specific transformation protocol. The proper integration of the construct was validated through clone-purifying positive colonies and extracting genomic DNA to perform PCR using diagnostic primers listed in Supplemental Table S3. The diagnostic primers were designed to target ∼150 bp upstream of and ∼150 bp downstream from the open reading frame to identify wild-type and drug-marker alleles. Positive clones were sporulated for 3–5 d at 25°C, and eight tetrads were screened for 2:2 viable segregation. Images were taken after 4 d of growth on YPD plates. Mutants conferring nonessential phenotypes were replicated on G418 plates, and images were taken after 4 d of growth at 25°C (Supplemental Figs. S9, S10). This method was also applied to making double mutants. A collection of 440 MATα S. uvarum strains was generated by standard methods in the Rine laboratory and used as confirmed nonessential genes.
Cross-species complementation assays
To determine if genes are diverging in gene function or in other trans-acting factors, we performed cross-species complementation assays with species-specific essential genes. Essential genes that were S. cerevisiae–specific were tested in a heterozygous diploid deletion strain from the SGA marker collection. Alleles of each S. cerevisiae essential gene and their promoters were amplified from S. cerevisiae and S. uvarum genomes and cloned into a CEN ARS plasmid. PCR using Phusion DNA polymerase was used to amplify 500 bp upstream of and 5 bp downstream from the stop codon of each gene from S. cerevisiae and S. uvarum. Each gene was cloned into pIL37 by Gibson assembly using primers listed in Supplemental Table S3 using standard methods (Thomas et al. 2015). All plasmids used in this study are listed in Supplemental Table S2. The S. cerevisiae heterozygous diploid deletion strains were transformed with a plasmid containing a corresponding allele from each species and selected on C-URA plates. Similarly, S. uvarum–specific essential genes were also tested by making each heterozygous diploid deletion strain ura3Δ/ura3Δ and transformed with a plasmid containing a corresponding S. cerevisiae allele from the MoBY-ORF collection (Ho et al. 2009).
Transformed strains were sporulated for 5 d at 30°C and 25°C for the S. cerevisiae and S. uvarum species, respectively, and tetrad analysis was performed on YPD plates. After 3 d of growth, plates were replica plated on C-URA and YPD + G418 plates and imaged after 2 d of growth (Supplemental Fig. S15).
Data access
The transposon insertion data in this study have been submitted to the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra/) under accession number SRP115313. All custom scripts are available as Supplemental Code.
Supplementary Material
Acknowledgments
We thank Jasper Rine for generously sharing S. uvarum deletion strains, Noah Hanson and Kolena Dang for technical assistance with tetrad dissections, Daniel Chee for his help with optimizing the Python code, and Jeremy Stone for his help with the Ruby script that was used for the insertional analysis pipeline. We thank M.K. Raghuraman and Harmit Malik for their helpful comments on the manuscript and we thank Sergey Kryazhimskiy for his helpful insight about data analysis. This work was supported by National Science Foundation (NSF) grant 1516330. M.R.S. was funded by NSF GSRF DGE-1256082 and the Robert D. Watkins Graduate Research Fellowship from the American Society for Microbiology. M.J.D. is supported in part by a faculty scholars grant from the Howard Hughes Medical Institute. M.J.D. is also a senior fellow in the Genetic Networks program at the Canadian Institute for Advanced Research. Sequencing resources were provided by the U.S. Department of Energy, JGI CSP project 1460. A.A.C. was supported by an Open Operating Grant from the Canadian Institutes for Health Research and by a discovery grant from the Natural Sciences and Engineering Research Council of Canada. A.A.C. is the Canada Research Chair of Metabolomics for Enzyme Discovery.
Author contributions: M.R.S., C.P., A.A.C., and M.J.D. conceptualized the project. M.R.S., C.P., F.C., and B.T.H. performed the experiments. M.R.S. and M.J.D. performed analysis. J.M.S., R.B.B., A.P.A., and A.A.C. provided resources. M.J.D. supervised the project. M.R.S. and M.J.D. drafted the manuscript with editing assistance from all authors. M.R.S. performed data visualization. A.A.C., M.J.D., A.P.A., and R.B.B. acquired funding.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.232330.117.
References
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2: 2006.0008 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellí G, Garí E, Piedrafita L, Aldea M, Herrero E. 1998. An activator/repressor dual system allows tight tetracycline-regulated gene expression in budding yeast. Nucleic Acids Res 26: 942–947. 10.1093/nar/26.4.942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll SB. 2005. Evolution at two levels: on genes and form. PLoS Biol 3: e245 10.1371/journal.pbio.0030245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caudy AA, Guan Y, Jia Y, Hansen C, DeSevo C, Hayes AP, Agee J, Alvarez-Dominguez JR, Arellano H, Barrett D, et al. 2013. A new system for comparative functional genomics of Saccharomyces yeasts. Genetics 195: 275–287. 10.1534/genetics.113.152918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coradetti ST, Pinel D, Geiselman GM, Ito M, Mondo SJ, Reilly MC, Cheng Y-F, Bauer S, Grigoriev IV, Gladden JM, et al. 2018. Functional genomics of lipid metabolism in the oleaginous yeast Rhodosporidium toruloides. eLife 7: e32110 10.7554/eLife.32110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M. 2016. Systems biology: a yeast global genetic interaction map. Nat Methods 13: 904 10.1126/science.aaf1420 [DOI] [Google Scholar]
- de Berardinis V, Vallenet D, Castelli V, Besnard M, Pinet A, Cruaud C, Samair S, Lechaplais C, Gyapay G, Richez C, et al. 2008. A complete collection of single-gene deletion mutants of Acinetobacter baylyi ADP1. Mol Syst Biol 4: 174 10.1038/msb.2008.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Rosa J, Weber J, Friedrich MJ, Li Y, Rad L, Ponstingl H, Liang Q, de Quirós SB, Noorani I, Metzakopian E, et al. 2017. A single-copy Sleeping Beauty transposon mutagenesis screen identifies new PTEN-cooperating tumor suppressor genes. Nat Genet 49: 730–741. 10.1038/ng.3817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNicola GM, Karreth FA, Adams DJ, Wong CC. 2015. The utility of transposon mutagenesis for cancer studies in the era of genome editing. Genome Biol 16: 229 10.1186/s13059-015-0794-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowell RD, Ryan O, Jansen A, Cheung D, Agarwala S, Danford T, Bernstein DA, Rolfe PA, Heisler LE, Chin B, et al. 2010. Genotype to phenotype: a complex problem. Science 328: 469 10.1126/science.1189015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujon B. 2010. Yeast evolutionary genomics. Nat Rev Genet 11: 512–524. 10.1038/nrg2811 [DOI] [PubMed] [Google Scholar]
- Eisen JA. 1998. Phylogenomics: improving functional predictions for uncharacterized genes by evolutionary analysis. Genome Res 8: 163–167. 10.1101/gr.8.3.163 [DOI] [PubMed] [Google Scholar]
- Folch-Mallol JL, Martínez LM, Casas SJ, Yang R, Martínez-Anaya C, López L, Hernández A, Nieto-Sotelo J. 2004. New roles for CDC25 in growth control, galactose regulation and cellular differentiation in Saccharomyces cerevisiae. Microbiology 150: 2865–2879. 10.1099/mic.0.27144-0 [DOI] [PubMed] [Google Scholar]
- Gangadharan S, Mularoni L, Fain-Thornton J, Wheelan SJ, Craig NL. 2010. DNA transposon Hermes inserts into DNA in nucleosome-free regions in vivo. Proc Natl Acad Sci 107: 21966–21972. 10.1073/pnas.1016382107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gresham D, Desai MM, Tucker CM, Jenq HT, Pai DA, Ward A, DeSevo CG, Botstein D, Dunham MJ. 2008. The repertoire and dynamics of evolutionary adaptations to controlled nutrient-limited environments in yeast. PLoS Genet 4: e1000303 10.1371/journal.pgen.1000303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Dunham M, Caudy A, Troyanskaya O. 2010. Systematic planning of genome-scale experiments in poorly studied species. PLoS Comput Biol 6: e1000698 10.1371/journal.pcbi.1000698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Dunham MJ, Troyanskaya OG, Caudy AA. 2013. Comparative gene expression between two yeast species. BMC Genomics 14: 33 10.1186/1471-2164-14-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Park JM, Cui B, Humes E, Gangadharan S, Hung S, FitzGerald PC, Hoe K-L, Grewal SIS, Craig NL, et al. 2013a. Integration profiling of gene function with dense maps of transposon integration. Genetics 195: 599–609. 10.1534/genetics.113.152744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Sheng Q, Li J, Ye F, Samuels DC, Shyr Y. 2013b. Large scale comparison of gene expression levels by microarrays and RNAseq using TCGA data. PLoS One 8: e71462 10.1371/journal.pone.0071462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho CH, Magtanong L, Barker SL, Gresham D, Nishimura S, Natarajan P, Koh JLY, Porter J, Gray CA, Andersen RJ, et al. 2009. A molecular barcoded yeast ORF library enables mode-of-action analysis of bioactive compounds. Nat Biotechnol 4: 369–377. 10.1038/nbt.1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman CS, Winston F. 1987. A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of Escherichia coli. Gene 57: 267–272. 10.1016/0378-1119(87)90131-4 [DOI] [PubMed] [Google Scholar]
- Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES. 2003. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature 423: 241–254. 10.1038/nature01644 [DOI] [PubMed] [Google Scholar]
- Kim D-U, Hayles J, Kim D, Wood V, Park H-O, Won M, Yoo H-S, Duhig T, Nam M, Palmer G, et al. 2010. Analysis of a genome-wide set of gene deletions in the fission yeast Schizosaccharomyces pombe. Nat Biotechnol 28: 617–623. 10.1038/nbt.1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Seringhaus M, MC Biery, RJ Sarnovsky, Umansky L, Piccirillo S, Heidtman M, K-H Cheung, CJ Dobry, MB Gerstein, et al. 2004. Large-scale mutagenesis of the yeast genome using a Tn7-derived multipurpose transposon. Genome Res 14: 1975–1986. 10.1101/gr.2875304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel AH, Hatakeyama R, Kimmig P, Arter M, Peter M, Matos J, Virgilio CD, Kornmann B. 2017. Functional mapping of yeast genomes by saturated transposition. eLife 6: e23570 10.7554/eLife.23570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Fung E, Schlecht U, Davis RW, Giaever G, St. Onge RP, Deutschbauer A, Nislow C. 2010. Gene annotation and drug target discovery in Candida albicans with a tagged transposon mutant collection. PLoS Pathog 6: e1001140 10.1371/journal.ppat.1001140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porwollik S, Santiviago CA, Cheng P, Long F, Desai P, Fredlund J, Srikumar S, Silva CA, Chu W, Chen X, et al. 2014. Defined single-gene and multi-gene deletion mutant collections in Salmonella enterica sv Typhimurium. PLoS One 9: e99820 10.1371/journal.pone.0099820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Wetmore KM, Waters RJ, Callaghan M, Ray J, Liu H, Kuehl JV, Melnyk RA, Lamson JS, Suh Y, et al. 2018. Mutant phenotypes for thousands of bacterial genes of unknown function. Nature 557: 503–509. 10.1038/s41586-018-0124-0 [DOI] [PubMed] [Google Scholar]
- R Core Team. 2017. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: https://www.R-project.org/. [Google Scholar]
- Ross-Macdonald P, Coelho PSR, Roemer T, Agarwal S, Kumar A, Jansen R, Cheung K-H, Sheehan A, Symoniatis D, Umansky L, et al. 1999. Large-scale analysis of the yeast genome by transposon tagging and gene disruption. Nature 402: 413–418. 10.1038/46558 [DOI] [PubMed] [Google Scholar]
- Sadhu MJ, Bloom JS, Day L, Siegel JJ, Kosuri S, Kruglyak L. 2018. Highly parallel genome variant engineering with CRISPR–Cas9. Nat Genet 50: 510–514. 10.1038/s41588-018-0087-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scannell DR, Butler G, Wolfe KH. 2007. Yeast genome evolution: the origin of the species. Yeast 24: 929–942. 10.1002/yea.1515 [DOI] [PubMed] [Google Scholar]
- Schwarzmüller T, Ma B, Hiller E, Istel F, Tscherner M, Brunke S, Ames L, Firon A, Green B, Cabral V, et al. 2014. Systematic phenotyping of a large-scale Candida glabrata deletion collection reveals novel antifungal tolerance genes. PLoS Pathog 10: e1004211 10.1371/journal.ppat.1004211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal ES, Gritsenko V, Levitan A, Yadav B, Dror N, Steenwyk JL, Silberberg Y, Mielich K, Rokas A, Gow NAR, et al. 2018. Gene essentiality analyzed by in vivo transposon mutagenesis and machine learning in a stable haploid isolate of Candida albicans. mBio 9: e02048-18 10.1128/mBio.02048-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S, Maynard ND, Gill J. 2015. DNA library construction using Gibson Assembly. Nat Methods 12: 1098 10.1038/nmeth.f.384 [DOI] [Google Scholar]
- Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, Pagé N, Robinson M, Raghibizadeh S, Hogue CW, Bussey H, et al. 2001. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294: 2364–2368. 10.1126/science.1065810 [DOI] [PubMed] [Google Scholar]
- Usadel B, Obayashi T, Mutwil M, Giorgi FM, Bassel GW, Tanimoto M, Chow A, Steinhauser D, Persson S, Provart NJ. 2009. Co-expression tools for plant biology: opportunities for hypothesis generation and caveats. Plant Cell Environ 32: 1633–1651. 10.1111/j.1365-3040.2009.02040.x [DOI] [PubMed] [Google Scholar]
- Van Opijnen T, Camilli A. 2013. Transposon insertion sequencing: a new tool for systems-level analysis of microorganisms. Nat Rev Microbiol 11: 435–442. 10.1038/nrmicro3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerdenburg EM, Abdallah AM, Rangkuti F, Abd El Ghany M, Otto TD, Adroub SA, Molenaar D, Ummels R, ter Veen K, van Stempvoort G, et al. 2015. Genome-wide transposon mutagenesis indicates that Mycobacterium marinum customizes its virulence mechanisms for survival and replication in different hosts. Infect Immun 83: 1778–1788. 10.1128/IAI.03050-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetmore KM, Price MN, Waters RJ, Lamson JS, He J, Hoover CA, Blow MJ, Bristow J, Butland G, Arkin AP, et al. 2015. Rapid quantification of mutant fitness in diverse bacteria by sequencing randomly bar-coded transposons. mBio 6: e00306-15 10.1128/mBio.00306-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. 2009. ggplot2: elegant graphics for data analysis. Springer-Verlag, New York. [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, et al. 1999. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285: 901–906. 10.1126/science.285.5429.901 [DOI] [PubMed] [Google Scholar]
- Yung MC, Park DM, Overton KW, Blow MJ, Hoover CA, Smit J, Murray SR, Ricci DP, Christen B, Bowman GR, et al. 2015. Transposon mutagenesis paired with deep sequencing of Caulobacter crescentus under uranium stress reveals genes essential for detoxification and stress tolerance. J Bacteriol 197: 3160–3172. 10.1128/jb.00382-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Anderson MT, Wu W, Mobley HL T, Bachman MA. 2017. TnseqDiff: identification of conditionally essential genes in transposon sequencing studies. BMC Bioinformatics 18: 326 10.1186/s12859-017-1745-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Gong R, Zhu Q, He Q, Xu N, Xu Y, Cai M, Zhou X, Zhang Y, Zhou M. 2018. Genome-wide determination of gene essentiality by transposon insertion sequencing in yeast Pichia pastoris. Sci Rep 8: 10223 10.1038/s41598-018-28217-z [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.