

Abstract
Circular RNAs (circRNAs) are a class of RNAs that is under increasing scrutiny, although their functional roles are debated. We analyzed RNA-seq data of 348 primary breast cancers and developed a method to identify circRNAs that does not rely on unmapped reads or known splice junctions. We identified 95,843 circRNAs, of which 20,441 were found recurrently. Of the circRNAs that match exon boundaries of the same gene, 668 showed a poor or even negative (R < 0.2) correlation with the expression level of the linear gene. In silico analysis showed only a minority (8.5%) of circRNAs could be explained by known splicing events. Both these observations suggest that specific regulatory processes for circRNAs exist. We confirmed the presence of circRNAs of CNOT2, CREBBP, and RERE in an independent pool of primary breast cancers. We identified circRNA profiles associated with subgroups of breast cancers and with biological and clinical features, such as amount of tumor lymphocytic infiltrate and proliferation index. siRNA-mediated knockdown of circCNOT2 was shown to significantly reduce viability of the breast cancer cell lines MCF-7 and BT-474, further underlining the biological relevance of circRNAs. Furthermore, we found that circular, and not linear, CNOT2 levels are predictive for progression-free survival time to aromatase inhibitor (AI) therapy in advanced breast cancer patients, and found that circCNOT2 is detectable in cell-free RNA from plasma. We showed that circRNAs are abundantly present, show characteristics of being specifically regulated, are associated with clinical and biological properties, and thus are relevant in breast cancer.
It is a sign of the times that the ubiquitous use of massively parallel sequencing data has delivered a parade of new insights in the cancer field and has enriched our genomic vocabulary with events like chromothripsis, kataegis, and mutational and rearrangement signatures (Stephens et al. 2011; Maher and Wilson 2012; Nik-Zainal et al. 2012, 2016; Alexandrov et al. 2013). Sequencing RNA has had less of an impact on this vocabulary, with many reports concerning traditional gene expression analysis. However, depending on the methodology of generating the sequencing library, RNA-seq has the potential to study the large variety of RNA species, including noncoding RNAs, fusion transcripts, known and novel isoforms, and, recently gaining attention, circular RNAs (circRNAs). This class of RNA was discovered many decades ago (Hsu and Coca-Prados 1979), and circRNAs were long considered idiosyncrasies of the splicing machinery processing precursor mRNA into mature mRNA. More recent studies showed an unanticipated abundance of circRNAs (Salzman et al. 2012; Memczak et al. 2013) in (normal and malignant) human cells and became particularly interesting for the cancer research field with the description (Hansen et al. 2013; Memczak et al. 2013) of a circRNA that functions as a highly potent miR-7 sponge. miR-7 has a well-described role in several malignancies, including breast cancer, and functions as a tumor suppressor in most cancers (for review, see Zhao et al. 2015) but has also been reported (Foekens et al. 2008) as a potential tumor promoter in breast cancer. Other circRNAs and additional regulatory transcriptional roles have subsequently been described in cancer (Salzman et al. 2013; Guo et al. 2014; Li et al. 2015b; Kristensen et al. 2018). Because circRNAs lack a free 5′ or 3′ end, such molecules escape exonucleic acid degrading enzymes, making them more stable (Memczak et al. 2013) than their linear counterparts. Therefore, circRNAs represent potentially useful biomarker candidates for diagnosis and therapy-monitoring; indeed, cell-free circRNAs are present in exosomes (Li et al. 2015a) and saliva (Bahn et al. 2015). In breast cancer, little has been described except for one study (Nair et al. 2016) using the The Cancer Genome Atlas (TCGA) data bank. However, this cohort has a huge limitation because the RNA-seq data were prepared using a poly(A) selection step, thereby omitting the majority of circRNAs (as these lack a poly(A) tail).
Here we describe the identification of an extensive catalog of circRNAs in a large cohort of 348 primary breast tumors, using RNA-seq data obtained via random-primed cDNA synthesis (Smid et al. 2016), likely preserving all the circRNAs. We developed a circRNA mapping algorithm that, in contrast to previous identification methods (Salzman et al. 2012; Memczak et al. 2013; Guo et al. 2014; Nair et al. 2016; Szabo and Salzman 2016), does not rely on unmapped reads or on known splice junctions and that was applied directly on transcriptome sequence BAM files, thereby allowing the identification of circRNAs in a genome-wide and annotation-independent (Szabo and Salzman 2016) fashion.
Results
Identification of a plethora of circRNAs in primary breast cancer
In total, 95,843 circRNAs were identified (Fig. 1), of which 27% (n = 25,783) had a start and end position exactly matching to an exon belonging to the same gene (Fig. 2A). The vast majority (79%) of all circRNAs were not recurrent (i.e., only found in one sample). The number of circRNAs per sample (Fig. 2B) ranged from 37 to 7105 (median, 966). For recurrent circRNAs, found in at least two samples, the range per sample was 33 to 5269 (median, 834.5). Figure 2B also shows that the number of (recurrent) circRNAs is significantly higher in estrogen receptor (ER)-negative compared with ER-positive breast cancers (Mann–Whitney U test, P < 1 × 10−5 for both all and recurrent circRNAs). Because of the extraordinary abundance of candidate circRNAs, we focused on the—still sizeable—number of recurrently found circRNAs (n = 20,441) (total number in second bar of Fig. 2A). The most frequent recurring region in our cohort was the well-characterized (Hansen et al. 2013; Memczak et al. 2013; Kristensen et al. 2018) circRNA of CDR1, which was found in 339 out of 348 cases. Other previously reported and validated (Salzman et al. 2012) circRNAs such as CAMSAP1, FBXW4, MAN1A2, RNF220, ZBTB44, and XIST were also identified in our cohort. A full list of identified circRNAs is provided in Supplemental Table S1.
Figure 1.
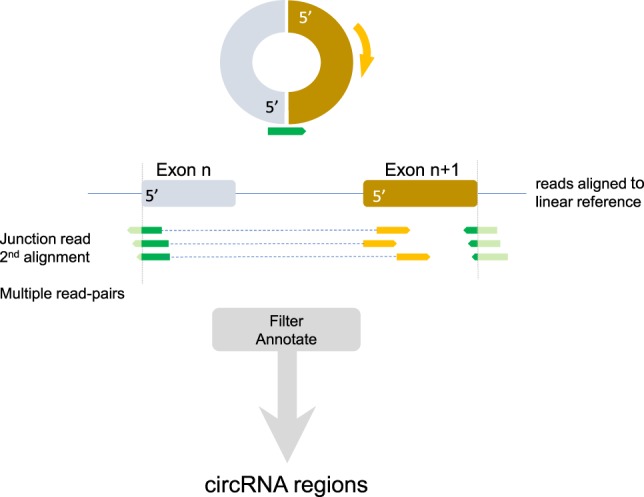
Schematic overview of identifying circular RNA (circRNA) regions. Assuming a circRNA molecule is present, a sequence read crossing the junction (green arrow) and its read-mate (gold arrow) would map to a linear reference in the manner depicted. The junction read would get multiple alignments, and the read-mate would be located in between the position of the junction read. Multiple read-pairs at the same junction strengthen the support for the circRNA. Subsequent additional filtering (details are in the Methods section) and annotation produced the list of circRNA regions.
Figure 2.

General characteristics of circRNAs in primary breast cancer. (A) Numbers of unique and recurrent circRNAs. Purple and gold indicate the number of circRNAs that, respectively, did or did not have a start and end position of a circRNA region exactly matching the start and end position of an exon of the same gene. (B) The number of circRNAs per sample, grouped by ER status. In black, the total number of circRNAs; in peach, the number of recurrent (identified in at least two samples) circRNAs. (C) Violin plots of the intron size (in log base pair) of noncircular regions and those located directly upstream of or downstream from a circRNA region.
General characteristics
Recurrent circRNAs were distributed across the genome, with one region on Chromosome 11 showing many closely spaced circRNAs (Supplemental Fig. S1). This region contains MALAT1, a highly abundant long noncoding RNA that is also frequently mutated in breast cancer (Nik-Zainal et al. 2016). Next, we evaluated the intron sizes upstream of and downstream from the circRNAs that match exon boundaries. Confirming previously reported results (Jeck et al. 2013; Zhang et al. 2014; Ivanov et al. 2015), Figure 2C shows that introns next to circRNA regions are significantly larger than introns not adjacent to circRNAs: on average 2.33 and 2.27 times larger, respectively, for introns upstream of and downstream from the circRNA (Mann–Whitney U test, both P < 1 × 10−5).
Next, we correlated the number of circRNA reads per circRNA with the expression of the respective full, linear gene, because a previous report in a limited cell line panel reported no genome-wide correlation between the circular and linear counterpart (Salzman et al. 2013). To avoid spurious correlations, only those circRNAs found in at least 50 samples were considered (n = 1625), and data were first normalized using the trimmed mean of M-values (TMM) (Robinson and Oshlack 2010). Correlations are listed in Supplemental Table S1 and ranged between −0.34 and 0.97, with 210 circRNAs showing a negative correlation to the linear gene in which the circRNA is located. When considering the average and standard deviation of the distribution of all correlation coefficients, 30 circRNAs are at the low end of the distribution (P < 0.05), showing a correlation below R = −0.182. Finally, by using GENCODE information, the position of annotated start codons was matched to the circRNA positions. circRNAs recurring in at least 50 samples showed an almost threefold higher than expected presence of a start codon compared with circRNAs that were not recurring (χ2, P < 0.0001; 557 circRNAs; expected 207.3 circRNAs).
Are circRNAs distinct molecules, specifically regulated, or splicing residues?
Many of the circRNAs that are positively correlated with the expression level of the full-length linear transcript are thought to be a residue of splicing. Figure 3A shows an example; the circRNA of exons 3, 4, and 5 of the WDR1 gene (Chr 4: 10,097,711–10,103,986, with a correlation coefficient of R = 0.66 to overall gene expression) exactly matches the difference between the known linear isoforms of this gene (Ensembl transcript ENST00000499869 includes exons 3, 4, and 5; ENST00000502702 lacks these exons). As current quantification methods do not take circRNAs into account, reads originating from the circular molecule are erroneously included in the read count of a linear isoform, resulting in an overestimation of the overall expression level. Another comprehensive example of a circRNA as splicing residue is shown in Figure 3B for ESR1. Full-length ESR1 (ENST00000206249) has eight exons, whereas ENST00000406599 is a splice variant of ESR1 that skips exons 2 to 5. circRNAs are found for several of these exons, indicating that they are likely splicing residues. We speculate that a single splice event from exons 1 to 6 generates an RNA molecule containing exons 2–5, from which a multitude of distinct circRNAs can be derived. In total, 23 patients show both a circESR1 exon 2–3 and a circESR1 of exon 4–5. If these circRNAs are derived from the same RNA molecule, the linear transcript would be ENST00000406599. A sequential model, in which first exons 2 and 3 are spliced out would prohibit the formation of a circRNA molecule of exons 3 to 4. However, we observed circESR1 exon 2–3 in 110 patients and circESR1 exon 3–4 in 64 patients, with 29 patients showing both these circRNAs. These must be derived from separate RNA molecules.
Figure 3.

circRNAs are not just residues of splicing. (A) Sashimi plot of the number of reads that are aligned to WDR1, showing only the reads that span exons. In red are the normal exon–exon reads; in purple, the reads that span the circular junction. The line and boxes indicate the exons of the gene (the whole gene is not shown). (B) Isoform of ESR1. The arcs indicate the number of samples that have a particular circRNA. (C) Two isoforms of CREBBP that are known in the first five exons (other isoforms are described, but these start downstream from exon 5). Exon 2 (purple box) is an identified circRNA that is not a remainder of a splicing event.
To investigate whether or not in general circRNAs should be considered splicing residues, we systematically evaluated how many of the identified circRNAs exactly match those exons that make up the difference between known linear isoforms of a gene. By using the GENCODE annotation for each gene, every possible known combination of spliced exons was matched to our circRNA catalog. Of the 25,783 circRNAs that matched to exons of the same gene, only 2193 (8.5%) exactly matched exons known to differ between described isoforms of a gene. This was 16.9% for the circRNAs with a correlation coefficient of R > 0.5 to linear gene expression. This suggests that the vast majority of circRNAs that matched to exons of the same gene are generated by yet-unknown splicing events of the gene.
Because the majority of circRNAs did not match to known spliced exons, we manually inspected several highly recurrent circRNAs in the UCSC Genome Browser. For example, two isoforms of CREBBP are described (ENST00000262367 and ENST00000382070) that differ in the presence of exon 5 (of note, there are 10 additional known transcripts, but all of these transcripts start downstream from exon 5). However, we observed exon 2 as circRNA (Chr 16: 3,850,297–3,851,009) that was present in 160 patients (Fig. 3C), indicating that this circRNA is either specifically generated or is a splicing residue of a yet-undescribed isoform of CREBBP that skips this exon. Visual inspection of 10 samples that had high levels of circCREBBP exon 2 (at least 30 circular junction reads) showed two samples that each had one read that crossed the junction from exon 1 to exon 3, whereas the other samples showed no evidence of an isoform that skipped exon 2. This favors the notion that the circRNA of exon 2 is not a byproduct of splicing at this location.
Finally, we matched publicly available circRNA lists to gather (indirect) evidence of functional roles for circRNAs. Rybak-Wolf and colleagues reported (Rybak-Wolf et al. 2015) 4522 circRNAs in the mammalian brain that were evolutionarily conserved between human and mouse, which is considered an indication of function (Barbosa-Morais et al. 2012; Merkin et al. 2012). Of these, 2259 circRNAs (49.9%) were also present in our catalog. In addition, 3271 circRNAs were reported in an MCF-7 breast cancer cell line panel (Tarrero et al. 2018). In total, 922 circRNAs showed (increased) expression in estrogen-stimulated MCF-7 cells compared with cells cultured in hormone-deprived medium, of which 733 circRNAs (79.5%) were present in our list. A poor correlation (R < 0.2) with the linear transcript was observed in our data for 78 of these circRNAs, suggesting independent regulation from their linear gene instead of ER-induced overall higher expression of all transcripts from that gene.
Validation of circRNAs
Besides the fact that we detected several already published circRNAs, thereby in part validating our method, we performed RT-PCR on a previously established independent cDNA pool of 100 primary breast tumors to confirm expression of three circRNAs, namely, RERE (circRNA Chr 1: 8,541,214–8,614,686), CNOT2 (circRNA Chr 12: 70,278,132–70,311,017), and CREBBP (circRNA Chr 16: 3,850,297–3,851,009), all of which were poorly correlated with their linear counterpart. Figure 4 shows the PCR fragment sizes; expected and observed sizes were 89 and 155 bp for the small and bigger CNOT2 fragment, 100 bp for RERE, and 91 bp for CREBBP. The primer pair for CNOT2 was able to amplify the circRNA of exon 2 to exon 3 of CNOT2 but also the circRNA of exons 2, 3, and 4 of this gene (a circRNA that was also identified in the RNA-seq cohort).
Figure 4.
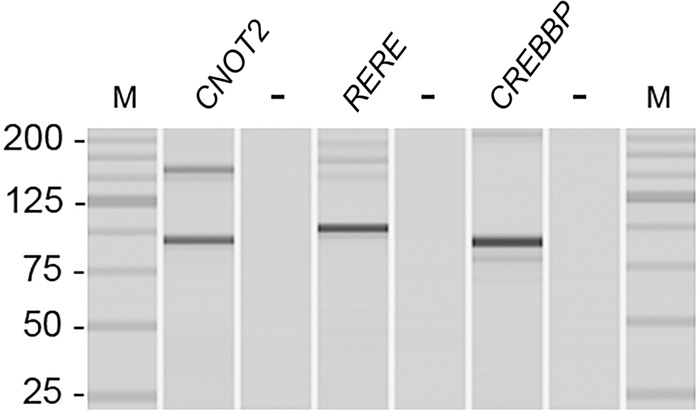
PCR products of circRNAs. PCR product sizes of circRNAs visualized using the MultiNA Microchip Electrophoresis System. (M) DNA size marker (25-bp fragment ladder); (−) the negative control (genomic DNA).
A different primer pair to PCR circCNOT2 showed additional fragments in addition to the expected fragments of 140 and 206 bp (Supplemental Fig. S2A). Sanger sequence analysis confirmed the circular junction sequence from exon 3 to exon 2 of CNOT2 (Supplemental Fig. S2B) and from exon 4 to exon 2 (Supplemental Fig. S2C). After using BLAST to identify the sequence, the largest excised PCR fragment was found to contain an additional exon of CNOT2 (Chr 12: 70,294,237–70,294,293) located between exon 2 and exon 3 that is not present in most isoforms of CNOT2 (Supplemental Fig. S2D). The sequence showed exon 3, across the circular junction to exon 2, but reading through the location where the reverse primer was located, continuing the whole of exon 2, the additional exon, and ending in exon 3 again (Supplemental Fig. S2D). A likely explanation for this observation is that during cDNA generation, the RT polymerase generated a linear cDNA molecule containing multiple copies of the circular transcript (Supplemental Fig. S2E). In summary, the investigated circRNAs were all confirmed to be truly present in primary breast cancer.
Functional relevance of circRNAs in breast cancer cells
The potential functional relevance of the validated circCNOT2 and circCREBBP transcripts, which were both poorly correlated with their corresponding linear transcript, was evaluated in breast cancer cell lines. First, expression levels of circCNOT2 and circCREBBP were established in a panel of 55 cell lines, showing variable levels (Supplemental Fig. S3). Next, an siRNA was designed to specifically target the circular junction of circCNOT2 in both MCF-7 (moderate expression level) and BT-474 (high expression level). This siRNA reduced expression of circCNOT2 by 76% in MCF-7 and 71% in BT-474 breast cancer cells, relative to cells transfected with a nontargeting control (NTC), which resulted in significantly reduced viability of both MCF-7 and BT-474 cells (Student's t-test, P < 1 × 10−5 and P = 4.94 × 10−4, respectively) (Fig. 5).
Figure 5.
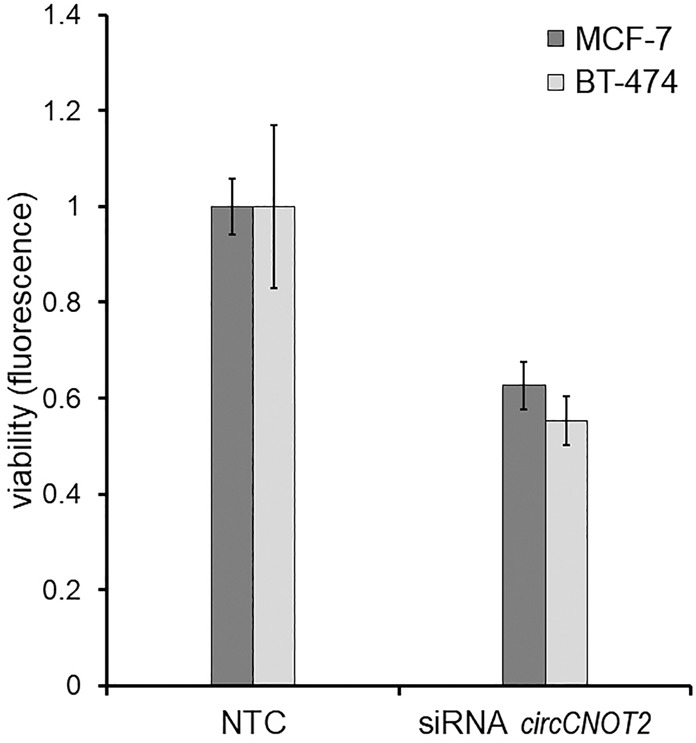
siRNA-mediated knockdown of circCNOT2 affects viability in breast cancer cells. The effect of reduced circCNOT2 expression on viability is shown in MCF-7 and BT-474 cells. Both cell lines show a significant decrease in viability (P < 0.01) following circCNOT2 knockdown relative to cells transfected with nontargeting control (NTC). Error bars, SD of five wells.
circRNAs in driver genes
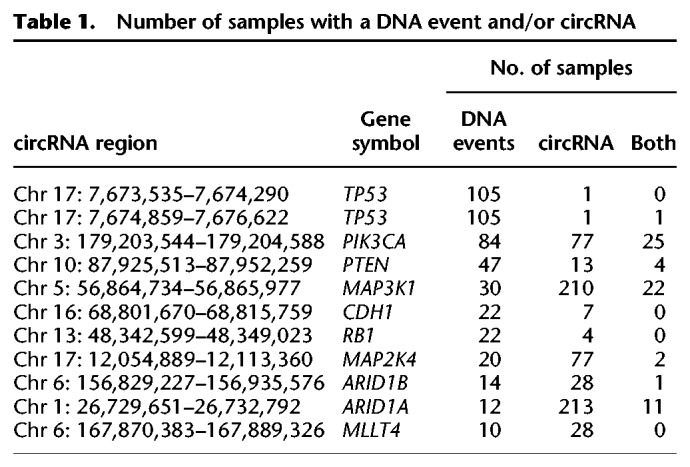
We matched our previously reported breast cancer driver gene list (Nik-Zainal et al. 2016) to our circRNA list. In total, 235 recurrent circRNAs were identified in 54 breast cancer driver genes. To integrate the data and obtain sufficient observations for analysis, we selected samples for which we had both RNA and genomic DNA sequencing results available and selected the genes with somatically acquired genetic events (mutations, copy number variants, and rearrangements) in at least 10 patients, yielding a list of 10 genes; TP53, PIK3CA, PTEN, MAP3K1, CDH1, RB1, MAP2K4, ARID1B, ARID1A, and MLLT4. For genes with multiple circRNAs, the circular region with the highest recurrence was chosen for analysis, with the exception of TP53, for which we only found two circRNAs in just one sample each (see Table 1). Only for MAP2K4 was mutual exclusivity observed between the presence of a somatic mutation or a circRNA in this cohort, in which 20 samples had a somatic mutation, 77 samples had a circRNA (Chr 17: 12,054,889–12,113,360), and only two samples had both a mutation and a circRNA (P = 0.025, CoMEt exact test) (Leiserson et al. 2015). For PIK3CA, 25 patients showed a DNA event and a circRNA, three patients with a copy number aberration (amplification), and 22 patients with a base substitution in PIK3CA. These substitutions were located in four hotspots, p.H1047 (13 cases), p.E545 (n = 4), p.E542 (n = 3), and p.E726 (n = 2). None of these hotspots was located in the circRNA region that was found in these samples.
Table 1.
Number of samples with a DNA event and/or circRNA
Breast cancer relevance
To investigate common biology in the samples, we used multiple correspondence analysis (MCA) to find naturally occurring subgroups. MCA is a generalized principle component analysis, suitable for categorical data. We used recurrent circRNAs (at least 50 cases) with the junction annotated to exons of the same gene (n = 1625) and labeled these per sample as circular or not circular, based on the presence or absence of junction reads in a sample. The main patient groups were, not unexpectedly, divided by ER status (Fig. 6A, left), whereas within circRNAs, the main division was whether or not the gene had a circRNA (Fig. 6A, right). Additional variation within the circRNAs was explained by the level of recurrence of the circRNAs (see Supplemental Fig. S4). Next, the presence/absence of circRNAs in a sample was used to cluster all samples into groups with distinct circRNA profiles. We used the gap statistic (Tibshirani et al. 2001) to determine the optimal number of sample groups, yielding six clusters (Fig. 6B; Supplemental Fig. S5). We evaluated these six sample groups on ER and tumor infiltrating lymphocyte (TIL) status (Fig. 6C,D), number of circRNAs (Fig. 6E), and outcome for the patients in the clusters (Fig. 6F). Samples in cluster 1 and 3 were predominantly ER-negative, whereas ER positivity was predominantly present in groups 2, 4, 5, and 6. TIL status was established using a previously reported gene expression signature (Massink et al. 2015; Smid et al. 2016), labeling samples as high-TIL if the average expression of the TIL signature genes fell into the top quartile (n = 87, 45 ER-negative and 42 ER-positive) (Fig. 6D, respectively, labeled as red and orange). High-TIL cases were significantly (χ2 P < 1 × 10−5) more often present in clusters 1 (71% of cases) and 3 (45% of cases). Furthermore, the number of circRNAs per sample clearly distinguished the six clusters (Fig. 6E), showing a decreasing number of circRNAs from clusters 1 to 6. Finally, for a subset of 186 patients, relapse-free survival data were available; a survival plot for the six clusters (Fig. 6F) showed that the major difference was between clusters 1 and 3, which are both predominantly ER-negative. Although the number of events was low, direct comparison of cluster 1 with cluster 3 showed a significant difference in survival curves (log rank, P = 0.04).
Figure 6.

Analysis of sample groups according to circRNA presence. Multiple correspondence analysis (MCA) was used to find naturally occurring groups in the circRNA data. In an MCA plot, samples and circRNAs are projected onto the same plane, in which the relative distance to either the samples or the circRNAs is meaningful. The 0,0 point corresponds to a sample or circRNA with an average profile. (A, left) Samples are colored according to ER status: red, ER-positive; black, ER-negative. (Right) Purple and green indicate genes with or without circRNA expression, respectively. (B) Clustering identified samples with similar circRNA profiles; samples in the MCA plot are colored according to the cluster to which the sample belongs. (C) ER status (purple, ER-positive; peach, ER-negative) and (D) TIL status of the six sample groups: Red and orange are high-TIL cases and blue and green are low-TIL cases for ER-negative and ER-positive, respectively. (E) Number of circRNAs per sample group. (F) Relapse-free survival plot by sample group. (N) number of patients; (F) number of patients who relapse; (x-axis) months; (y-axis) the cumulative probability of relapse-free survival.
Differentially expressed circRNAs
We investigated if circRNA expression levels were associated with clinically relevant features of primary breast cancer, such as presence of TILs, the tumor's stromal content, proliferation, and hypoxia status. These features were inferred from generated (stroma; see Methods) or reported (Winter et al. 2007; Massink et al. 2015; Smid et al. 2016) gene expression signatures. By using these, we grouped our samples in a similar manner as explained earlier for the TIL status (Fig. 6D), labeling samples as high if the average expression of the signature genes fell into the top quartile. To identify significantly differentially expressed circRNAs, we compared the top quartile of samples to the remaining samples separately for the ER-positive and ER-negative cases. circRNAs with FDR-corrected P-values <0.05 and a fold-change greater than two were selected. Of these, the circRNAs that had a negative correlation with the linear gene expression were considered of particular interest and are listed in Table 2. Several of these circRNAs may thus potentially play a role in, or are at least connected to, the tumors that show hypoxic characteristics (e.g., circKMT2C) or accumulate in highly proliferative cells (e.g., circRERE, circATXN2), whereas, for example, circASH1L and circPCH3 may be generated by surrounding stromal cells or infiltrating cells.
Table 2.
Fold-change of circRNAs in top quartile of samples versus remaining samples in ER-subgroups of tumors

Clinical relevance
One of the reasons CNOT2 was selected for validation was because of the poor correlation with expression of the linear gene (R = −0.09, P = 0.34). This was more prominent in ER-positive (R = −0.14) compared with ER-negative cases (R = 0.097). We validated this finding by making use of in-house array data (Smid et al. 2008) for linear CNOT2 expression and a quantitative RT-PCR assay to measure circCNOT2 (exon 2–3 Chr 12: 70,278, 132–70,311,017). The Spearman's correlation in ER-positive cases of circCNOT2 with linear CNOT2 was 0.079 (n = 187, P = 0.28) and in ER-negative cases Rs = 0.42 (n = 111, P = 2.9 × 10−6), again showing absence of correlation in ER-positive cases. Thus, the role of circCNOT2 apparently differs between ER-positive and ER-negative cases, and in the ER-negative cases, the significance of circCNOT2 cannot be easily segregated from the linear counterpart. Therefore, we evaluated two different ER-positive breast cancer cohorts (Supplemental Table S3 shows clinical characteristics) for the potential clinical value of circCNOT2. We studied progression-free survival of aromatase inhibitor (AI) therapy in a multicenter cohort of 84 ER-positive patients who received this treatment for advanced disease. Quantitative reverse transcriptase–PCR (RT-qPCR) levels of circCNOT2 showed a significant hazard ratio (HR) of 1.75 (95% confidence interval 1.32–2.33, P = 1.06 × 10−4), whereas RT-qPCR levels of linear CNOT2 were not significant: HR 1.28, P = 0.187. Figure 7A shows a survival curve after grouping patients’ circCNOT2-levels into three equally sized groups. A similar analysis in another cohort (Sieuwerts et al. 2005), which included patients receiving first-line tamoxifen treatment (n = 295 patients), did not show a significant HR (0.97, P = 0.57) for circCNOT2 or for linear CNOT2 (HR 1.16, P = 0.21).
Figure 7.

Clinical evaluation and presence in plasma samples. (A) Kaplan–Meier survival curve of progression-free survival for AI therapy in which patients were grouped in three equally sized groups based on their circCNOT2 expression: red, blue, and green indicate the samples with high, intermediate, and low expression. The x-axis is in months; y-axis depicts the cumulative probability of progression-free survival on AI therapy. The P-value is the log-rank test for trend. (B) Expression levels of circCNOT2 in plasma samples. Four metastatic breast cancer patients were evaluated. The y-axis depicts delta-Ct values of circCNOT2 relative to GUSB. Error bars, SD of two measurements.
As circular molecules are expected to be more stable than their linear counterparts, we explored whether circCNOT2 is a potential candidate as minimally invasive biomarker. To this end, we used cell-free RNA (cfRNA) from plasma samples of four breast cancer patients and amplified circCNOT2 by RT-qPCR. All samples showed detectable and variable levels of circCNOT2 (Fig. 7B), indicating that detection of circRNAs in plasma seems attainable in this exploratory setting.
Discussion
To our knowledge, we are the first to analyze RNA sequencing data using random-primed cDNA libraries from a large primary breast cancer cohort for the presence of circRNAs, using a method that does not rely on unmapped reads. Previously, Nair and colleagues (Nair et al. 2016) analyzed TCGA RNA-seq data that were obtained using a poly(A)-based method. Although we identified 25,783 circRNAs that matched with an exon boundary of the same gene, Nair et al. (2016) reported 2146 circRNAs when we applied the same selection criteria as for our data set, and after transferring the hg37 coordinates (Nair et al. 2016) to hg38 (our data set), only 45 circRNAs were found that had the exact same start and end coordinates in both data sets. Thus, a random-primed method identifies many more circRNAs than when using poly(A)-selected material. On the other hand, there seem to be many uniquely identified circRNAs in these data sets. This could stem from differences in the methodology to detect or report the circRNAs but also could reflect the fact that many circRNAs are nonrecurrent.
We showed that circRNAs are found throughout the genome and have significantly larger-sized introns located directly adjacent to the region on the genome that borders the circRNA, as also reported previously (Jeck et al. 2013; Zhang et al. 2014; Ivanov et al. 2015). Based on the presence/absence of circRNAs, six groups of samples were observed that differed in their ER status, TIL content, number of circRNAs, and prognosis. This indicates that there appears to be a functional biology associated with the biogenesis of circRNA molecules or at least a biology that cancerous cells can use to their advantage. Whether or not the circRNAs themselves serve that function or whether the process that generates differences in circRNA levels is the cause for the results presented here remains unknown at this time. The fact that several circRNAs were found differentially expressed in breast cancer subgroups, although these circRNAs are negatively correlated with the expression of the linear gene from which the circRNA is derived, corroborates the notion of functional circRNAs. Further experimentation is required to investigate the functional relationship of the differentially expressed circRNAs in the hypoxia, proliferation, stroma, and TIL phenotypes.
Although a synthetic circRNA construct including an internal ribosome entry site (IRES) can be translated (Wang and Wang 2015), current literature (Jeck et al. 2013; Guo et al. 2014; Szabo and Salzman 2016; Liang et al. 2017; Liu et al. 2018; Wang et al. 2018; Zeng et al. 2018) describes mostly noncoding functions for circRNAs, for example, as miRNA sponge circCDR1 (Hansen et al. 2013; Memczak et al. 2013; Zhao et al. 2015; Kristensen et al. 2018) and circZNF91 (Guo et al. 2014), whereas associative evidence was reported (Rybak-Wolf et al. 2015) of evolutionarily conserved circRNAs between human and mouse. Further support that circRNAs may be specifically generated and regulated was derived from a study using (estrogen-stimulated) MCF-7 cells (Tarrero et al. 2018), showing higher levels of H3K36me3 (posttranscriptional histone modification) and a higher number of Ago binding sites in circularizing exons.
Here, we contribute to the search for relevant circRNAs in three ways; first, expression levels of circRNAs that are not, or even negatively, correlated with the linear transcript of the gene may point to an intentional process. Although differences in degradation rates between the circular and linear isoforms may influence the correlation, we did not find systemic evidence for this (Supplemental Fig. S6). Furthermore, for genes that generate several distinct circRNAs, correlations can vary, indicating that degradation of the linear transcript cannot be the only explanation for the observed correlations. Second, circRNAs that include the start codon could potentially influence the expression of the linear gene, because the linear transcript from which the circRNA was spliced is now forced to use another start codon for its translation; if none is available, the transcript may be degraded. For example, circCNOT2 (exon 2–3 Chr 12: 70,278,132–70,311,017) contains the start codon of the consensus (Pruitt et al. 2009) transcript (CCDS31857.1). Annotation shows that exon 1 is untranslated and both exon 2 and exon 4 start with the methionine codon (ATG). Thus, the linear transcript wherein exon 1 is ligated to exon 4 lacks the start codon from exon 2 and may use the ATG in exon 4 for its translation. Third, we observed that circRNAs matching exon boundaries of the same gene rarely (8.5%) overlap with known spliced exons. It could be that our analysis overlooked possible splice variants from the GENCODE annotation (both HAVANA and Ensembl exon annotations were included), but if the concordance is indeed this low, two scenarios may be applicable: Either circRNAs are still mostly a remnant of splicing, implying that many more transcript isoforms of genes exist, or otherwise circRNAs are specifically generated, implying that they do have a biological role. The observations of variable expression levels of circCNOT2 and circCREBBP in cell lines and especially the effect of circCNOT2 knockdown on cell viability corroborate a biological role for circRNAs. Future studies are needed to systematically evaluate if the correlation between a circRNA and its linear transcript, the presence of a start codon, and/or known splice junctions are reliable criteria to prioritize circRNAs of interest.
Regardless, we were able to show clinical potential for circCNOT2 by showing its association with the response to AI therapy. Knowing that circular molecules are not targeted by exonucleases, these molecules may be suitable candidates to be detected in cell-free environments (Li et al. 2015a), and in a pilot experiment, we showed that circCNOT2 can indeed be detected in cfRNA from plasma samples of breast cancer patients. As such, circCNOT2 could prove to be a useful biomarker to choose the right type of therapy or to monitor disease in a minimally invasive manner. Furthermore, we observed that very likely because of the strand displacement activity of the reverse transcriptase during cDNA generation, multiple concatemeric copies of a single circular molecule are made, contributing to the sensitive detection of circRNAs. In conclusion, we have demonstrated the abundance and potential roles of circRNAs in primary breast cancer. The methodology and selection criteria we used may help in making more sense of the seeming chaos and disorder existing in the flow from DNA to RNA to protein. circRNAs show the potential to function as relevant actors in the transcriptional regulation of RNA in addition to their promise as stable biomarkers that can be used for disease progression.
Methods
Sequencing
Internal review boards of each participating institution approved collection and use of samples of all patients in this study. RNA-seq data were generated by our consortium (Nik-Zainal et al. 2016; Smid et al. 2016) for 348 primary breast cancer tumors that are available through the European Genome-phenome Archive under accession number EGAS00001001178. Sequence protocols of the samples were previously described in detail (Nik-Zainal et al. 2016); in short, total RNA after gDNA removal, clean-up, and depletion of ribosomal RNA using duplex-specific nuclease (DSN) treatment was used as input for random-primed cDNA synthesis. Paired-end (75 bases) sequencing was performed on an Illumina HiSeq 2000. The resulting FASTQ files were mapped to GRCh38 using STAR (version 2.4.2a) (Dobin et al. 2013), and the resulting BAM files were sorted and indexed using Sambamba (version 0.6.6) (Tarasov et al. 2015; https://github.com/lomereiter/sambamba/). Gene annotation was derived from GENCODE Release 23 (https://www.gencodegenes.org/).
Identification of circRNAs
A detailed explanation of the methodology to identify circRNA reads, including the Perl script, is stated in the Supplemental Methods. The script is also available at https://bitbucket.org/snippets/MSmid/Le949d/identify-circularrna-reads. In short, the method developed here uses sequence reads that have a “secondary alignment” (SA) tag. When using paired-end sequence data and assuming a circRNA molecule is present (Fig. 1, top), the sequence read that aligns over the crossing of the junction (green arrows) would “point toward” its read-mate (orange arrow) somewhere in the circle. Aligning these reads to the linear reference (Fig. 1, middle), the junction read will get an SA tag and will be assigned to two locations if and only if this is the one and unique alignment configuration the STAR software can find. The read-mate aligns somewhere in between these two locations. Finding additional read-pairs showing this configuration, with a breakpoint at the exact same location, strengthens the evidence for circular transcripts. Only regions with at least five reads crossing the circular junction were included. After filtering (for details, see Supplemental Methods), GENCODE annotation was used to obtain the exon locations of genes that exactly matched to the circular region. For each sample, STAR also gives the raw read counts for all genes. These were normalized (TMM implemented in edgeR) (Robinson and Oshlack 2010) and used to correlate with the number of junction reads of the circular transcripts.
Multiple correspondence analysis
Because many genes only show a linear transcript in many samples, standard cluster analysis to identify groups of samples with similar circRNA-related biology is problematic because of the many missing values. Thus, the circRNA data were considered categorical using “circular” or “not circular” if a circRNA was present or absent in a sample. These categorical data are suitable for an MCA, a generalization of a principle component analysis. The MCA generates a combined plot that shows both patients and circRNAs such that patients/circRNAs that have similar patterns are closer together. R-packages “ade4,” “canceracm,” and “cluster” were used to perform the MCA and determine the optimum number of clusters. The latter was determined using the clusGap option (k-means to partition the samples) in the cluster package. R version 3.4.1 was used (R Core Team 2017).
Reverse transcription, PCR, and Sanger sequencing
Candidate circRNAs were selected and primers were designed such that a PCR would only yield a product when the RNA was circular, whereas in the linear situation, the primers would be divergent. Primer sequences are listed in Supplemental Table S2.
First, total RNA, isolated with RNA-Bee according to the manufacturer's instructions (CS105B, TEL TEST) was reverse transcribed into cDNA with the RevertAid H Minus first-strand cDNA synthesis kit (K1632; Thermo Fisher Scientific), followed by a RNase H step (AM2293; Ambion). Next, circRNAs were real-time PCR amplified at 10 ng input in a final volume of 25 µL using 40 PCR cycles and an annealing temperature of 67°C with 330 nM of each primer and SensiFAST SYBR Lo-ROX mastermix (BIO-94050, Bioline), followed by a final 5-min extension at 72°C, in a MX3000P (Agilent Technologies). PCR products were visualized using a MultiNA microchip electrophoresis system (Shimadzu).
For sequencing, PCR fragments were separated on a standard agarose gel and were excised from gel using the QIAquick gel extraction kit from Qiagen according to manufacturer's protocol. The sequencing reaction contained 2 µL of gel-extracted PCR product, 1 µL BigDye terminator v3.1 reaction mix (Thermo Fisher Scientific), 1× BigDye terminator sequencing buffer (Thermo Fisher Scientific), and 0.16 µM of sequencing primer in a final volume of 10 µL and was performed using an ABI2720 thermal cycler according to the following protocol: one step for 2 min at 96°C and 25 cycles of 30 sec at 96°C , 30 sec at 58°C, and 2 min at 72°C. Subsequently, the sequencing product was precipitated with absolute ethanol and 3 M of NaAc, resuspended in 20 µL of Hi-Di formamide (Thermo Fisher Scientific), and ran on an ABI3130XL genetic analyzer (Thermo Fisher Scientific).
RT-qPCR
After RNA isolation and cDNA synthesis performed as described above, circCNOT2 (Chr 12: 70,278,132–70,311,018) and circCREBBP (Chr 16: 3,850,297–3,851,009) transcripts were real-time PCR amplified at 10 ng input in a final volume of 25 µL in 40 PCR cycles and an annealing temperature of 60°C with 200 nM of each primer and 100 nM Fam-labeled TaqMan MGB probe that covers the circular junction (Thermo Fisher Scientific) (Supplemental Table S2) in SensiFAST Probe Lo-ROX mastermix (BIO-84020, Bioline) using a MX3000P (Agilent Technologies). Levels were quantified relative to the average expression of three reference genes (HPRT1, HMBS, and TBP) (Supplemental Table S2) using the delta Cq method (dCq = 2(average Cq reference genes–Cq target gene)) (Schmittgen and Livak 2008). A serially diluted cDNA pool (Sieuwerts et al. 2014) of 100 independent breast tumor samples (containing both ER-positive/-negative and ERBB2-positive cases) was included in each experiment to evaluate the linear amplification and efficiencies for all genes and absence of amplification in the absence of reverse transcriptase. Samples in the cDNA pool were independent from the cases that were used for the RNA-seq cohort.
Detection of circRNAs in plasma
cfRNA was isolated with the Maxwell RSC miRNA tissue kit (Promega) adapted for plasma according the manufacturer's instructions. One milliliter of EDTA plasma of different metastatic breast cancer patients was used. These patients provided written informed consent. Six microliters of the resulting 50 µL cfRNA (3.8–7 ng RNA/µL) was used to generate 20 µL cDNA with the SuperScript IV VILO cDNA synthesis kit (Thermo Fisher Scientific). Next, 2 µL of the cDNA was preamplified in the presence of 0.50 nM of the reverse primers of the hydrolysis probe assays for circCNOT2 and GUSB as a reference marker during 15 cycles with TaqMan preamp mastermix (Thermo Fisher Scientific). Finally, 0.5 µL of the preamplified product was measured real-time with the hydrolysis probe assays (200 nM forward primer, 200 nM reverse primer, and 100 nM FAM-labeled hydrolysis MGB probe) during 40 cycles with SensiFAST Probe Lo-ROX mastermix (BioLine) in a final qPCR volume of 25 µL in a MX3000P qPCR machine (Agilent Technologies).
siRNA-mediated knockdown of circRNAs and cell viability assay
All cell lines in this study were established to be genetically unique and monoclonal and of correct identity by performing STR profiling using the PowerPlex 16 system (Promega). MCF-7 and BT-474 were plated at 60%–70% confluency in six-well plates and transfected with 50 nM ON-TARGETplus siRNA targeting circCNOT2 (Horizon Discovery) using 4 µL (MCF-7) or 8 µL (BT-474) DharmaFECT 1 (Horizon Discovery) following the manufacturer's instructions. Used sequences (5′-3′) were as follows: sense, AAAGAUAGGGAGACGUGGUUU; antisense, 5′-PACCACGUCUCCCUAUCUUUUU. The ON-TARGETplus nontargeting pool and On-TARGETplus human UBB smart pool were included in each experiment as negative and positive controls, respectively (Horizon Discovery). After 24 h of transfection, cells were trypsinized, counted, and seeded in quintuplicate at 20,000 cells per well in 96-well plates. Cell viability was determined using the CellTiter-Blue cell viability assay (Promega) at day 0 and day 3. Viability measurements at day 3 were corrected for baseline viability values by subtracting the average measurement of day 0.
Gene expression signatures
We used several signatures: a TIL and proliferation signature (Smid et al. 2016), a hypoxia signature (Winter et al. 2007), and a stroma-specific signature using public data GSE5847 (Gene Expression Omnibus) (Boersma et al. 2008). We performed a paired t-test to obtain genes significantly higher expressed in microdissected stroma (FDR < 0.05 and fold-change >1.7). For all signatures, genes that were up-regulated in the category of interest were matched to our data set, and the average expression of the signature genes was calculated per sample. Samples were labeled as high-TIL (or stroma, proliferation, hypoxia) if the average expression of the signature genes fell into the top quartile. To identify significantly differentially expressed circRNAs, we compared the top quartile of samples versus the rest, per ER-group. circRNAs were only included when detected in >50% of the samples and matched known exon locations of the same gene. Analyses were performed using BRB-ArrayTools developed by Dr. Richard Simon and the BRB-ArrayTools development team. circRNAs were considered significant when the FDR corrected P-value was below 0.05 and the fold-change greater than 2.
Breast cancer cohort treated with endocrine therapy
RT-qPCR was performed on a linear and circular isoform of CNOT2 (Chr 12: 70,278,132–70,311,017) in a first-line TAM (Sieuwerts et al. 2005) and a first-line AI cohort to study the predictive value of circCNOT2 on therapy response. The AI cohort was a multicenter cohort consisting of 30 patients from Erasmus MC, Rotterdam; 35 patients from The Netherlands Cancer Institute, Amsterdam; and 19 patients from the Translational Cancer Research Unit, Antwerp (Belgium). All 295 patients in the TAM cohort are patients of the Erasmus MC, Rotterdam. Patient characteristics are listed in Supplemental Table S3.
Statistical analyses
STATA version 14 was used to perform the statistical tests that are indicated in the text. P-values are two-sided, corrected for multiple testing when necessary, and considered significant below 0.05.
Supplementary Material
Acknowledgments
We thank the Erasmus MC Cancer Computational Biology Center for giving access to their IT infrastructure and the software that was used for the computations and data analysis in this study. We thank Sandra Albassam for her help with the first versions of the script to identify circular regions. We thank Maurice P.H.M. Jansen, Jean C. Helmijr, Inge de Kruijff, and Manouk K. Bos for their help in evaluating plasma samples that were gathered in the EU-FP7 CareMore (nr 601760) project. We thank for technical support Miriam Ragle Aure and Anita Langerød of the Oslo University Hospital, Norway; Ewan Birney of the European Bioinformatics Institute, UK; and Stefania Tommasi of the IRCCS Istituto Tumori “Giovanni Paolo II,” Bari, Italy. We thank the Oslo Breast Cancer Research Consortium (OSBREAC), Norway (https://www.ous-research.no/home/kgjebsen/home/14105) for contributing patient samples and Sabine Linn and Marleen Kok of The Netherlands Cancer Institute for contributing samples for the AI cohort. Finally, we thank all members of the ICGC Breast Cancer Working Group. This work has been funded through the ICGC Breast Cancer Working group by the Breast Cancer Somatic Genetics Study (a European research project funded by the European Community's Seventh Framework Programme (FP7/2010-2014) under the grant agreement number 242006) and the Triple Negative project funded by the Wellcome Trust (grant reference 077012/Z/05/Z). F.G.R.-G. and S.M. were funded by BASIS. J.A.F. was funded through an ERC Advanced Grant (ERC-2012-AdG-322737) and ERC Proof-of-Concept Grant (ERC-2017-PoC-767854). K.U. was funded by the Daniel den Hoed Foundation. S.N.-Z. is a Wellcome Beit Fellow and personally funded by a Wellcome Trust Intermediate Fellowship (WT100183MA). A.L.R. is partially supported by the Dana-Farber/Harvard Cancer Center SPORE in Breast Cancer (NIH/NCI 5 P50 CA168504-02). A.M.S. was supported by Cancer Genomics Netherlands (CGC.nl) through a grant from the Netherlands Organization of Scientific research (NWO). M. Smid was supported by the EU-FP7-DDR response project. C.D. was supported by a grant from the Breast Cancer Research Foundation. J.E. was funded by The Icelandic Centre for Research (RANNIS).
Author contributions: M. Smid, S.M.W., and J.W.M.M. wrote the main paper. M. Stratton, S.M., S.N.-Z., H.G.S., J.A.F., and J.W.M.M. were involved in the strategy and supervision of the project. Experiments were performed by S.M.W., F.G.R.-G., V.d.W., A.M.S., W.J.C.P.-v.d.S., M.v.d.V.-D, A.v.G., and J.S. M. Smid, A.M.S., K.U., S.M.W., S.N.-Z., J.S., M.J.v.d.V., A.L.R., A.B., H.R.D., F.C.G.J.S., A.V., A.B.-D., and J.W.M.M. analyzed data. Samples and/or clinical data were contributed by J.A.F., J.W.M.M., A.L.R., C.A.P., A.M.T., C.C., P.N.S., F.C.G.J.S., P.T.S., S.R.L., S.v.L., C.D., A.P., J.E., A.B., A.V.-S., A.P.F., S.K., T.K., A.V., A.B.-D., G.M., R.S., and G.G.G.M.v.d.E.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.238121.118.
References
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. 2013. Signatures of mutational processes in human cancer. Nature 500: 415–421. 10.1038/nature12477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahn JH, Zhang Q, Li F, Chan TM, Lin X, Kim Y, Wong DT, Xiao X. 2015. The landscape of microRNA, piwi-interacting RNA, and circular RNA in human saliva. Clin Chem 61: 221–230. 10.1373/clinchem.2014.230433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa-Morais NL, Irimia M, Pan Q, Xiong HY, Gueroussov S, Lee LJ, Slobodeniuc V, Kutter C, Watt S, Colak R, et al. 2012. The evolutionary landscape of alternative splicing in vertebrate species. Science 338: 1587–1593. 10.1126/science.1230612 [DOI] [PubMed] [Google Scholar]
- Boersma BJ, Reimers M, Yi M, Ludwig JA, Luke BT, Stephens RM, Yfantis HG, Lee DH, Weinstein JN, Ambs S. 2008. A stromal gene signature associated with inflammatory breast cancer. Int J Cancer 122: 1324–1332. 10.1002/ijc.23237 [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foekens JA, Sieuwerts AM, Smid M, Look MP, de Weerd V, Boersma AW, Klijn JG, Wiemer EA, Martens JW. 2008. Four miRNAs associated with aggressiveness of lymph node-negative, estrogen receptor-positive human breast cancer. Proc Natl Acad Sci 105: 13021–13026. 10.1073/pnas.0803304105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Agarwal V, Guo H, Bartel DP. 2014. Expanded identification and characterization of mammalian circular RNAs. Genome Biol 15: 409 10.1186/s13059-014-0409-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J. 2013. Natural RNA circles function as efficient microRNA sponges. Nature 495: 384–388. 10.1038/nature11993 [DOI] [PubMed] [Google Scholar]
- Hsu MT, Coca-Prados M. 1979. Electron microscopic evidence for the circular form of RNA in the cytoplasm of eukaryotic cells. Nature 280: 339–340. 10.1038/280339a0 [DOI] [PubMed] [Google Scholar]
- Ivanov A, Memczak S, Wyler E, Torti F, Porath HT, Orejuela MR, Piechotta M, Levanon EY, Landthaler M, Dieterich C, et al. 2015. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep 10: 170–177. 10.1016/j.celrep.2014.12.019 [DOI] [PubMed] [Google Scholar]
- Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE. 2013. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19: 141–157. 10.1261/rna.035667.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen LS, Hansen TB, Venø MT, Kjems J. 2018. Circular RNAs in cancer: opportunities and challenges in the field. Oncogene 37: 555–565. 10.1038/onc.2017.361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiserson MD, Wu HT, Vandin F, Raphael BJ. 2015. CoMEt: a statistical approach to identify combinations of mutually exclusive alterations in cancer. Genome Biol 16: 160 10.1186/s13059-015-0700-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zheng Q, Bao C, Li S, Guo W, Zhao J, Chen D, Gu J, He X, Huang S. 2015a. Circular RNA is enriched and stable in exosomes: a promising biomarker for cancer diagnosis. Cell Res 25: 981–984. 10.1038/cr.2015.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Huang C, Bao C, Chen L, Lin M, Wang X, Zhong G, Yu B, Hu W, Dai L, et al. 2015b. Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol 22: 256–264. 10.1038/nsmb.2959 [DOI] [PubMed] [Google Scholar]
- Liang HF, Zhang XZ, Liu BG, Jia GT, Li WL. 2017. Circular RNA circ-ABCB10 promotes breast cancer proliferation and progression through sponging miR-1271. Am J Cancer Res 7: 1566–1576. [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Lu C, Zhou Y, Zhang Z, Sun L. 2018. Circular RNA hsa_circ_0008039 promotes breast cancer cell proliferation and migration by regulating miR-432-5p/E2F3 axis. Biochem Biophys Res Commun 502: 358–363. 10.1016/j.bbrc.2018.05.166 [DOI] [PubMed] [Google Scholar]
- Maher CA, Wilson RK. 2012. Chromothripsis and human disease: piecing together the shattering process. Cell 148: 29–32. 10.1016/j.cell.2012.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massink MP, Kooi IE, Martens JW, Waisfisz Q, Meijers-Heijboer H. 2015. Genomic profiling of CHEK2*1100delC-mutated breast carcinomas. BMC Cancer 15: 877 10.1186/s12885-015-1880-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, et al. 2013. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495: 333–338. 10.1038/nature11928 [DOI] [PubMed] [Google Scholar]
- Merkin J, Russell C, Chen P, Burge CB. 2012. Evolutionary dynamics of gene and isoform regulation in mammalian tissues. Science 338: 1593–1599. 10.1126/science.1228186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair AA, Niu N, Tang X, Thompson KJ, Wang L, Kocher JP, Subramanian S, Kalari KR. 2016. Circular RNAs and their associations with breast cancer subtypes. Oncotarget 7: 80967–80979. 10.18632/oncotarget.13134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, et al. 2012. Mutational processes molding the genomes of 21 breast cancers. Cell 149: 979–993. 10.1016/j.cell.2012.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC, et al. 2016. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534: 47–54. 10.1038/nature17676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Harrow J, Harte RA, Wallin C, Diekhans M, Maglott DR, Searle S, Farrell CM, Loveland JE, Ruef BJ, et al. 2009. The consensus coding sequence (CCDS) project: identifying a common protein-coding gene set for the human and mouse genomes. Genome Res 19: 1316–1323. 10.1101/gr.080531.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2017. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna: https://www.R-project.org/. [Google Scholar]
- Robinson MD, Oshlack A. 2010. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11: R25 10.1186/gb-2010-11-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybak-Wolf A, Stottmeister C, Glažar P, Jens M, Pino N, Giusti S, Hanan M, Behm M, Bartok O, Ashwal-Fluss R, et al. 2015. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol Cell 58: 870–885. 10.1016/j.molcel.2015.03.027 [DOI] [PubMed] [Google Scholar]
- Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO. 2012. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One 7: e30733 10.1371/journal.pone.0030733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO. 2013. Cell-type specific features of circular RNA expression. PLoS Genet 9: e1003777 10.1371/journal.pgen.1003777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3: 1101–1108. 10.1038/nprot.2008.73 [DOI] [PubMed] [Google Scholar]
- Sieuwerts AM, Meijer-van Gelder ME, Timmermans M, Trapman AM, Garcia RR, Arnold M, Goedheer AJ, Portengen H, Klijn JG, Foekens JA. 2005. How ADAM-9 and ADAM-11 differentially from estrogen receptor predict response to tamoxifen treatment in patients with recurrent breast cancer: a retrospective study. Clin Cancer Res 11: 7311–7321. 10.1158/1078-0432.CCR-05-0560 [DOI] [PubMed] [Google Scholar]
- Sieuwerts AM, Lyng MB, Meijer-van Gelder ME, de Weerd V, Sweep FC, Foekens JA, Span PN, Martens JW, Ditzel HJ. 2014. Evaluation of the ability of adjuvant tamoxifen-benefit gene signatures to predict outcome of hormone-naive estrogen receptor-positive breast cancer patients treated with tamoxifen in the advanced setting. Mol Oncol 8: 1679–1689. 10.1016/j.molonc.2014.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smid M, Wang Y, Zhang Y, Sieuwerts AM, Yu J, Klijn JG, Foekens JA, Martens JW. 2008. Subtypes of breast cancer show preferential site of relapse. Cancer Res 68: 3108–3114. 10.1158/0008-5472.CAN-07-5644 [DOI] [PubMed] [Google Scholar]
- Smid M, Rodríguez-Gonzalez FG, Sieuwerts AM, Salgado R, Prager-Van der Smissen WJ, Vlugt-Daane MV, van Galen A, Nik-Zainal S, Staaf J, Brinkman AB, et al. 2016. Breast cancer genome and transcriptome integration implicates specific mutational signatures with immune cell infiltration. Nat Commun 7: 12910 10.1038/ncomms12910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. 2011. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144: 27–40. 10.1016/j.cell.2010.11.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo L, Salzman J. 2016. Detecting circular RNAs: bioinformatic and experimental challenges. Nat Rev Genet 17: 679–692. 10.1038/nrg.2016.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. 2015. Sambamba: fast processing of NGS alignment formats. Bioinformatics 31: 2032–2034. 10.1093/bioinformatics/btv098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarrero LC, Ferrero G, Miano V, De Intinis C, Ricci L, Arigoni M, Riccardo F, Annaratone L, Castellano I, Calogero RA, et al. 2018. Luminal breast cancer-specific circular RNAs uncovered by a novel tool for data analysis. Oncotarget 9: 14580–14596. 10.18632/oncotarget.24522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibshirani R, Walther G, Hastie T. 2001. Estimating the number of clusters in a data set via the gap statistic. J R Stat Soc B 63: 411–423. 10.1111/1467-9868.00293 [DOI] [Google Scholar]
- Wang Y, Wang Z. 2015. Efficient backsplicing produces translatable circular mRNAs. RNA 21: 172–179. 10.1261/rna.048272.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Xiao Y, Wu L, Ma D. 2018. Comprehensive circular RNA profiling reveals the regulatory role of the circRNA-000911/miR-449a pathway in breast carcinogenesis. Int J Oncol 52: 743–754. 10.3892/ijo.2018.4265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter SC, Buffa FM, Silva P, Miller C, Valentine HR, Turley H, Shah KA, Cox GJ, Corbridge RJ, Homer JJ, et al. 2007. Relation of a hypoxia metagene derived from head and neck cancer to prognosis of multiple cancers. Cancer Res 67: 3441–3449. 10.1158/0008-5472.CAN-06-3322 [DOI] [PubMed] [Google Scholar]
- Zeng K, He B, Yang BB, Xu T, Chen X, Xu M, Liu X, Sun H, Pan Y, Wang S. 2018. The pro-metastasis effect of circANKS1B in breast cancer. Mol Cancer 17: 160 10.1186/s12943-018-0914-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XO, Wang HB, Zhang Y, Lu X, Chen LL, Yang L. 2014. Complementary sequence-mediated exon circularization. Cell 159: 134–147. 10.1016/j.cell.2014.09.001 [DOI] [PubMed] [Google Scholar]
- Zhao J, Tao Y, Zhou Y, Qin N, Chen C, Tian D, Xu L. 2015. MicroRNA-7: a promising new target in cancer therapy. Cancer Cell Int 15: 103 10.1186/s12935-015-0259-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.