

Abstract
Background
Refractory diseases, including bacterial infections, are causing huge economic losses in dairy farming. Despite efforts to prevent and treat those diseases in cattle, including the use of antimicrobials, it is not well controlled in the field. Several inflammatory cytokines, including tumor necrosis factor alpha (TNF-α), play important roles in disease progression; thus, blocking these cytokines can attenuate the acute and sever inflammation and may be a novel strategy for treatment. However, biological drugs targeting inflammatory cytokines have not been used in cattle. Therefore, in this study, bovine sTNFR1 and sTNFR2 IgG1 Fc-fusion proteins (TNFR1-Ig and TNFR2-Ig) were produced, and their anti-inflammatory functions were analyzed in vitro, to develop decoy receptors for bovine TNF-α.
Results
Both TNFR1-Ig and TNFR2-Ig were shown to bind with TNF-α, and TNFR2-Ig showed higher affinity toward TNF-α than TNFR1-Ig. We next stimulated murine fibroblast-derived cells (L929 cells) with TNF-α to induce cell death and analyzed cell viability in the presence of TNFR-Ig proteins. Both TNFR1-Ig and TNFR2-Ig suppressed TNF-α-induced cell death, significantly improving cell viability. In addition, cell death induced by TNF-α was suppressed, even at low TNFR2-Ig concentrations, suggesting TNFR2-Ig has higher activity to suppress TNF-α functions than TNFR1-Ig. Finally, to examine TNFR2-Ig’s anti-inflammatory, we cultured peripheral blood mononuclear cells from cattle with TNF-α in the presence of TNFR2-Ig and analyzed the gene expression and protein production of the inflammatory cytokines IL-1β and TNF-α. TNFR2-Ig significantly reduced the gene expression and protein production of these cytokines. Our results suggest that TNFR2-Ig inhibits inflammatory cytokine kinetics by blocking TNF-α to transmembrane TNFR, thereby attenuating excessive inflammation induced by TNF-α.
Conclusions
Collectively, the findings of this study demonstrated the potential of TNFR2-Ig as a novel therapeutic for inflammatory diseases, such as bovine clinical mastitis. Further investigation is required for future clinical application.
Keywords: Cattle, TNF-α, TNF receptor, Decoy receptor, Cell death, Inflammatory cytokines
Background
In dairy farming and the livestock industry worldwide, numerous infectious diseases threaten animal health and productivity. There are effective controlling strategies, such as vaccination, for some diseases. However, various refractory diseases, including bacterial infections such as paratuberculosis, mycoplasmosis, and mastitis remain serious issues [1, 2]. In these diseases, inflammation is a common feature, and many types of inflammatory cytokines, such as interleukin (IL)-1, IL-6, and tumor necrosis factor alpha (TNF-α) have been strongly associated with disease progression [3, 4]. Although the current treatment for these inflammatory bacterial diseases mainly depends on antibiotics, many issues remain to be resolved. While antibiotics are typically effective for certain bacteria, if applied adequately, sometimes they are not effective because the bacteria have developed a mechanism to escape from the lethal effect of antimicrobials. Moreover, the improper use of antibiotics causes environmental problems, such as antibiotic-resistant bacteria and milk pollution [5]. In European countries, predonisolone, one of the steroids, are used to reduce inflammation [6]. However, it has been indicated that steroid administration reduce milk production and decrease prolactin release [7]. Therefore, developing novel therapeutic methods for these refractory bacterial infections is required.
As mentioned, inflammatory cytokines are critical factors in bacterial infection progression. Thus, inhibiting these cytokines may be promising as part of an overall control strategy. TNF-α is produced by fibroblasts and epithelial cells as well as activated immune cells. Immediately upon microbial invasion, it serves various functions, such as activation of immune cells and cytotoxicity to tumor cells [8, 9]. TNF-α constitutes a trimer and is expressed on cell membranes [9, 10]. Membrane-expressed TNF-α (mTNF-α) is cleaved by a TNF-α converting enzyme and released into the bloodstream as soluble TNF-α (sTNF-α) [11]. The receptors of TNF-α are classified as type 1 (TNFR1) and type 2 (TNFR2). While TNFR1 is expressed on various cell types, the existence of TNFR2 is limited to immune and epithelial cells [8, 9]. Extracellular domains of both receptors contain similar motifs, including cysteine-rich regions where TNF-α binds. While TNFR1 can be activated by both mTNF-α and sTNF-α, activation of TNFR2 is mediated by only mTNF-α, despite the fact that sTNF-α can also bind TNFR2 [9, 12]. The intracellular domain of TNFR1 contains a death domain (DD); in response to TNF-α binding, the TNFR1-associated death domain protein (TRADD) interacts with DD immediately. There are two different cascades known to be downstream of TRADD. In the canonical pathway, TRADD interacts with various adaptor molecules, such as Receptor-Interacting serine/threonine-Protein Kinase 1 (RIPK1) and TNF receptor associated factor 2 (TRAF2), resulting in NF-κB activation [13]. NF-κB promotes the transcription not only of cytokine genes, including IL-1 and TNF-α, but also of Cellular Inhibitor of Apoptosis Protein 1 (c-IAP1) and c-IAP2 to prevent apoptosis-inducible pathways [14–16]. In contrast, in the non-canonical pathway, the Fas-associated DD protein (FADD) interacts with TRADD and induces apoptosis via Caspase 8 and 10 activation [13]. On the other hand, regarding TNFR2, TRAF2 interacts directly with TNFR2’s intracellular domain upon mTNF-α-stimuli. This process then leads to NF-κB activation, regulatory T cells proliferation, and cytotoxic T cell function suppression [17, 18]. Accordingly, TNF-α greatly contributes to the pathogenesis of inflammatory diseases.
On the contrary, soluble TNFRs (sTNFRs) might reduce inflammation as “decoy receptors” by competitively inhibiting TNF-α/membrane TNFR (mTNFR) interactions [19]. sTNFRs consist of only the extracellular domains of TNFRs, which are cleaved from the membrane by proteases and released into the bloodstream. In inflammatory diseases, the concentrations of sTNFR in the bloodstream appear to increase in correlation with disease progression [20, 21]. Therefore, sTNFRs should suppress excess immune responses and inflammation.
In human inflammatory diseases, such as rheumatoid arthritis, inhibiting TNF-α with anti-TNF-α antibodies or sTNFR-fusion proteins has been gaining increasing attention as a promising treatment strategy. For example, sTNFR2-fusion proteins are widely used for rheumatoid arthritis, with high therapeutic performance [22–24]. However, in the veterinary field, few reports exist of therapeutic strategies targeting inflammatory cytokines and the application of biologicals. In this study, Fc-fusion proteins of bovine sTNFR were developed, and their anti-apoptotic and anti-inflammatory properties were evaluated in vitro using recombinant bovine TNF-α based assays.
Results
Generation of recombinant bovine TNFR-Ig and Cont-Ig
To evaluate the TNF-α decoy receptor’s efficacy for bovine inflammatory diseases, we first prepared recombinant bovine TNFR-Ig, consist of the extracellular domains of bovine TNFR1 or 2 and the Fc domain of bovine IgG1. In addition, as a negative control, Cont-Ig, consists of the signal peptide domain of bovine TNFR2, and the Fc domain of bovine IgG1, was generated using the Expi293 Expression System. Figure 1 shows the expressions and purities of each recombinant protein, as determined by SDS-PAGE and Western blotting with the anti-bovine IgG Fc antibody (Fig. 1a and b). TNFR1-Ig, TNFR2-Ig, and Cont-Ig were detected at approximately 50, 70, and 30 kDa, respectively. Each recombinant protein was detected at double the molecular weight in the absence of 2ME, confirming that these proteins formed dimers (Fig. 1b).
Fig. 1.

Generation of recombinant bovine TNFR1-Ig and TNFR2-Ig (a) SDS-PAGE, (b) Western blot analysis of purified proteins using anti-bovine IgG antibody
Binding of TNFR-Igs to bovine TNF-α
The binding of TNFR1-Ig and TNFR2-Ig to bovine TNF-α was analyzed by enzyme-linked immunosorbent assay (ELISA). TNF-α-dose dependent increases in absorbance were observed with TNFR1-Ig and TNFR2-Ig application, whereas changes in absorbance were not detected when using Cont-Ig, indicating that both TNFR1-Ig and TNFR2-Ig can capture bovine TNF-α (Fig. 2). In addition, TNFR2-Ig showed 11-fold at 6.25 ng/mL of TNF-α and 21-fold absorbance at 25 ng/mL of TNF-α compared to TNFR1-Ig, suggesting that TNF-α’s affinity toward TNFR2-Ig was higher than that of TNFR1-Ig.
Fig. 2.
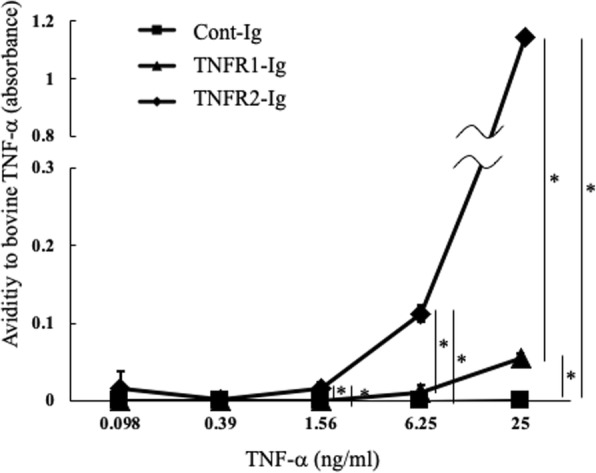
Avidities of TNFR1 and R2-Ig to bovine TNF-α The avidities of TNFR-Ig to bovine TNF-α were analyzed using ELISA using biotinylated anti-bovine TNF-α antibody. Briefly, TNFR-Igs or Cont-Ig was fixed on ELISA plate and recombinant bovine TNF-α (0.098–25 ng/ml) was added. After the incubation (room temperature) and washing, biotinylated anti-bovine TNF-α antibody was added. TNFR2-Ig showed significantly higher avidity to bovine TNF-α than that showed by TNFR1-Ig (n = 3), * p < 0.05
Effect of TNFR-Igs on cell death by TNF-α
TNF-α induces cell death by activating Caspase 8 and 10, via the DD, when captured by mTNFR1, which is expressed on cell membranes [9]. Therefore, we investigated whether TNFR-Ig could suppress cell death in L929 cells and bovine PBMCs driven by TNF-α using the Realtime-Glo MT Cell Viability Assay. In L929 cells, treatment with TNFR1-Ig and TNFR2-Ig significantly enhanced cell viability compared to Cont-Ig; cell viability of TNFR2-Ig-treated L929 cells tended to be higher than those of TNFR1-Ig-treated cells, indicating that TNFR2-Ig could suppress cell death by capturing TNF-α more efficiently than TNFR1-Ig (Fig. 3). In bovine PBMCs, however, stimulation with TNF-α did not induce cell death at all, and TNFR-Ig treatment did not affect cell viability (data not shown). Considering these results, TNFR2-Ig appears to have higher affinity toward TNF-α and can inhibit TNF-α function compared to TNFR1-Ig in L929 cells. Therefore, we proceeded with experiments only using TNFR2-Ig.
Fig. 3.
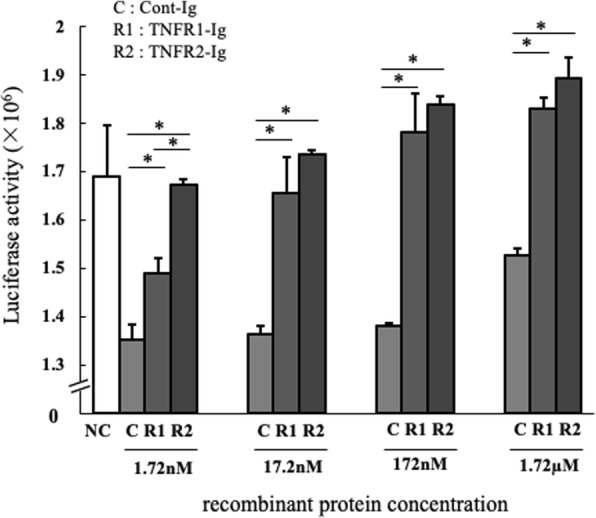
Effect of TNFR-Igs on cell death by TNF-α L929 cells were cultured with recombinant bovine TNF-α, actinomycin-D, and TNFR1-Ig, TNFR2-Ig, or Cont-Ig under several concentrations. Cell viabilities were assessed using the RealTime-Glo™ MT Cell Viability Assay according to the manufacturer’s instructions. Both TNFR-Ig, but particularly TNFR2-Ig, significantly reduced cell death induced by TNF-α. The assays were conducted in triplicate. * p < 0.05
Effect of TNFR2-Ig on inflammatory cytokine kinetics by TNF-α
It has been reported that TNF-α can induce not only cell death but also NF-κB activation when captured by mTNFR1 and mTNFR2 [14], which can lead to inflammatory cytokine production. Therefore, we investigated whether TNFR2-Ig could reduce the gene expression and production of inflammatory cytokines from bovine PBMCs, driven by TNF-α. Treatment with TNFR2-Ig significantly reduced both gene expression and protein production of IL-1β and TNF-α, induced by stimulation with TNF-α (Figs. 4 and 5). These results indicated that TNFR2-Ig trapped soluble TNF-α and competitively inhibited the interaction between TNF-α/mTNFR, and could modulate the inflammatory response triggered by TNF-α.
Fig. 4.
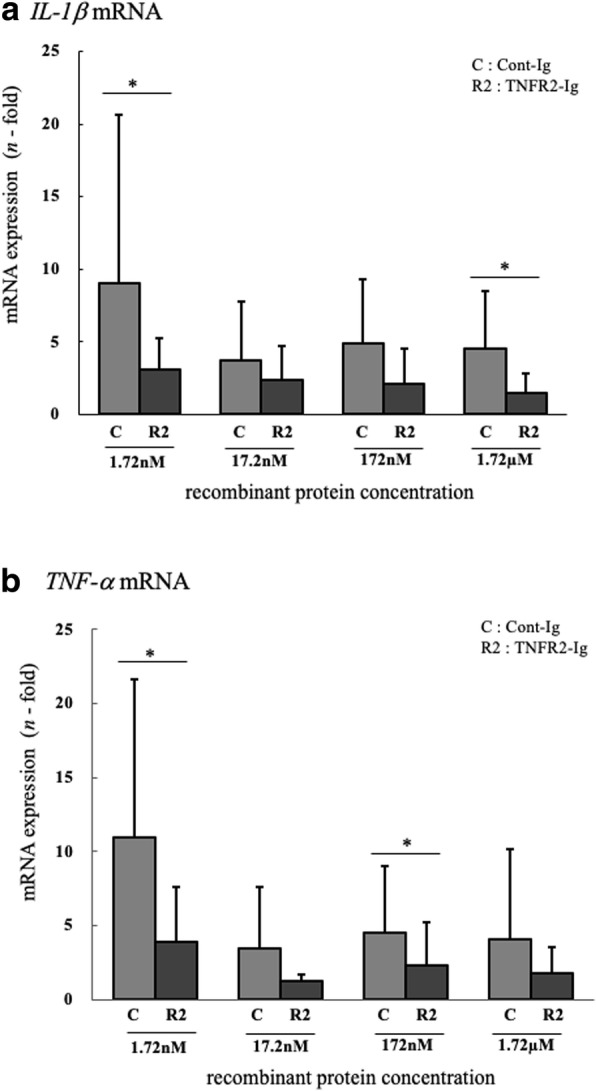
Effect of TNFR2-Ig on TNF-α-induced inflammatory cytokine gene expressions PBMCs were cultured with recombinant bovine TNF-α, actinomycin-D, and TNFR2-Ig or Cont-Ig. The gene expression of (a) IL-1β mRNA and (b) TNF-α mRNA in bovine PBMCs were assessed using real-time PCR. The results of IL-1β and TNF-α mRNA expression are presented as a ratio to their expressions in untreated PBMCs (n = 6). * p < 0.05
Fig. 5.
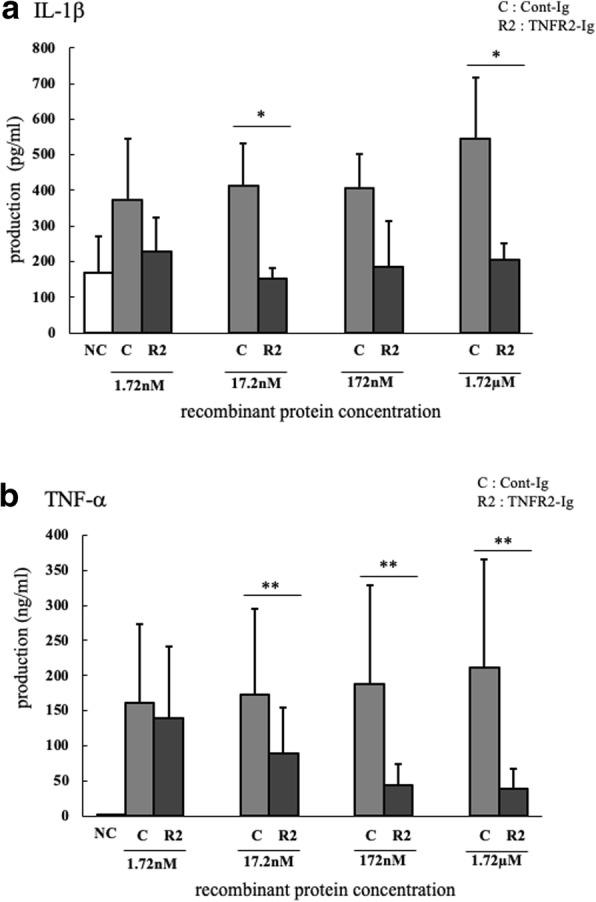
Effect of TNFR2-Ig on TNF-α-induced inflammatory cytokine productions. PBMCs (n = 6) were cultured with recombinant bovine TNF-α, actinomycin-D, and TNFR2-Ig, or Cont-Ig under several concentrations. IL-1β and TNF-α in the cultural supernatant were assessed using ELISA. TNFR2-Ig significantly suppressed the production of both cytokines induced by bovine TNF-α. ** p < 0.01, * p < 0.05
Discussion
TNF-α is produced immediately upon pathogen-invasion, including bacterial infections, and induces various immune responses such as cell proliferation and inflammation [8, 9]. It has been previously reported that, in the veterinary field and particularly in bovine infectious diseases, numerous bacteria such as Escherichia coli and Mycoplasma bovis can induce the prompt release of TNF-α [25]. In human clinical medicine, soluble TNFR (sTNFR) seems capable of suppressing TNF-α bioactivities by competitively inhibiting TNF-α/membrane TNFR (mTNFR) interactions. In this study, we established soluble bovine TNFRs Fc-fusion proteins (TNFR-Ig) and demonstrated that these proteins possess these inhibitive features as well as the potential to be novel therapeutic treatments for the inflammatory diseases mentioned above.
In our experiments, we showed that both TNFR1-Ig and TNFR2-Ig can capture bovine TNF-α, and that TNFR2-Ig has much higher affinity toward TNF-α than TNFR1-Ig. According to previous reports, the affinities of human TNF-α and TNFR are still controversial. In some reports, TNFR1 seemed have greater affinity toward TNF-α than TNFR2 [26], while there have also been opposite suggestions [27]. These contradictions may depend on whether TNF-α and TNFR are membrane-expressed or in their soluble form. Regarding human mTNFR, it has been reported that mTNFR1 was higher in affinity toward sTNF-α than mTNFR2 [28]. However, there is little information of the affinities between sTNFR and sTNF-α. In this study, regarding bovine sTNFR, the affinity toward sTNF-α seemed much higher for sTNFR2 than for sTNFR1. Nevertheless, we only measured the bindings of sTNFRs and sTNF-α by ELISA, so further analyses, such as evaluation of bonding and dissociation constants, are required. Moreover, additional experiments using mTNF-α are needed to evaluate whether TNFR-Ig can inhibit mTNF-α as well as sTNF-α.
When TNF-α binds mTNFR1, Caspase 8 and 10 are activated via the DD, resulting in apoptosis [13]. While both TNFR1-Ig and TNFR2-Ig, and particularly TNFR2-Ig, significantly reduced cell death in L929 cells triggered by TNF-α, regarding bovine PBMCs, neither TNF-α or TNFR-Ig affected cell viabilities at all. To explain these different responses between L929 cells and PBMCs, we present two hypotheses. The first is that this is because of the difference of mTNFR1 functions on each cell. L929 cells have been reported to be very susceptible to the cytotoxicity of TNF-α, and generally used for functional analysis of TNF-α [29, 30]. When TNF-α binds to mTNFR1, it promotes the formation of the death domain/TRADD complex. Typically, this complex would activate NF-κB via recruitment of other adaptor molecules such as RIPK1 and TRAF2, which induces inflammatory responses or cell proliferations [13]. However, in some cases, although the mechanisms are still unclear, the death domain/TRADD complex induces apoptosis via activation of caspases caused by RIP1K ubiquitination deficiency [31, 32]. Although TNFR1’s cell type-dependent functions are poorly understood, we might uncover the mechanisms underlying the different responses between L929 cells and PBMCs by analyzing the activation of downstream pathways of the death domain/TRADD complex. The second hypothesis focuses on the receptor types expressed on each cell. While only mTNFR1 is expressed on L929 cells, PBMCs express both mTNFR1 and mTNFR2 [8, 9]. When mTNF-α captured TNF-α, it activates NF-κB and promotes the transcriptions of c-IAP1 and c-IAP2, which result in the suppression of apoptosis-inducible pathway [15, 16], hence cell death triggered by mTNFR1 was mediated. Further experiments, such as expression analysis of mTNFR, or the evaluation of the activities of related molecules like Caspase 8, NF-κB, or c-IAP, are warranted to verify these hypotheses.
As previously described, TNF-α can trigger cytokine gene transcriptions, including IL-1, IL-6, and TNF-α via activation of NF-κB. We demonstrated that both gene expressions and productions of IL-1β and TNF-α in PBMCs triggered by the stimulation of TNF-α were significantly reduced by treatment of TNFR2-Ig, indicating that TNFR2-Ig can mediate inflammatory responses driven by TNF-α by competitively inhibit TNF-α/mTNFR interactions.
In this study, we proposed the potential of TNFR2-Ig to be a novel method to control inflammatory diseases in cattle. However, our experiments were based on recombinant bovine TNF-α; thus, our results could not be necessarily applied to infection situations because disease progressions are mediated by more complex interactions between pathogens and host immunity. Hence, further experiments using bacterial antigens or live bacteria, and bovine mammary epithelial cells are required to support TNFR2-Ig’s efficacy. Moreover, TNFR2-Ig’s function is no more than suppressing inflammation and diseases are not eradicable by TNFR2-Ig treatment alone because the target of this approach is not pathogen elimination. Hence, the administration of TNFR2-Ig would be for attenuation of acute and severe inflammation, such as E. coli-mediated peracute mastitis. On the other hand, the immunosuppressive effect of TNFR2-Ig is limited to the inhibition of TNF-α, and should be much more moderate than that driven by other immunosuppressive agents, such as steroids. Considering clinical application, therefore, careful and further experiments are required to ascertain the therapeutic potential. In addition, combining TNFR2-Ig with existing approaches such as antibiotics should be investigated.
Conclusions
Collectively, this study indicates that both TNFR1-Ig and TNFR2-Ig were shown to bind with TNF-α, and TNFR2-Ig showed higher affinity toward TNF-α than TNFR1-Ig. Both TNFR1-Ig and TNFR2-Ig suppressed TNF-α-induced cell death and significantly improved cell viabilities. Interestingly, cell death induced by TNF-α was suppressed even in low concentrations of TNFR2-Ig, suggesting that TNFR2-Ig has higher activity to suppress the TNF-α functions than TNFR1-Ig. Furthermore, TNFR2-Ig significantly reduced inflammatory cytokine genes’ expressions and their protein production. These results suggest that TNFR2-Ig inhibits inflammatory cytokine kinetics via blocking of TNF-α to transmembrane TNFR, and thereby, attenuate excessive inflammation induced by TNF-α. Overexpression of TNF-α is a major death-causing factor in several bovine inflammatory diseases. This study demonstrated TNFR2-Ig’s potential as a novel therapeutic for the bovine inflammatory diseases such as mastitis, arthritis, and pneumonia. Further investigations are required for clinical application in the future.
Methods
Cell cultures
Expi293F™ cells1 (A14635) were cultured in Expi293™ Expression Medium1 at 37 °C, 125 rpm, and 8% CO2. Murine fibroblast-derived L929 cells1 (ATCC CCL-1) were cultured in low glucose Dulbecco’s Modified Eagle Medium (D-MEM)1 containing 10% heat-inactivated fetal bovine serum (FBS)2 at 37 °C and 5% CO2. Bovine peripheral blood mononuclear cells (PBMCs) were isolated from the venous blood of healthy cattle (maintained at the Field Science Center for Northern Biosphere, Hokkaido University) by density gradient centrifugation in Percoll solution.3 Purified PBMCs were cultured in RPMI medium4 containing 10% heat-inactivated FBS, 0.01% L-glutamine, 200 U/mL penicillin, and 200 μg/mL streptomycin5 at 37 °C and 5% CO2.
Cloning of cDNA encoding the extracellular domain fragment of bovine TNFR1 and TNFR2
Total RNA was extracted from the isolated PBMCs of healthy cattle using TRI reagent®6 according to the manufacturer’s instructions. Remnant DNA was removed from the RNA samples with DNase I (amplification grade)5 treatment for 10 min at 65 °C. The cDNA was synthesized using 1 μg of RNA with PrimeScript Transcriptase7 according to the manufacturer’s instructions. The signal peptide and the extracellular domains of the bovine TNFR1 and TNFR2 genes were predicted using the software systems SignalP (http://www.cbs.dtu.dk/services/SignalP/) and SOSUI (http://harrier.nagahama-i-bio.ac.jp/sosui/sosui_submit.html) respectively, based on the sequences registered in the GeneBank database (BC113278 and NM001040490) (Fig. 6). The extracellular domain fragments of bovine TNFR1 and TNFR2 and the signal peptide domain of TNFR2 cDNA (as a negative control) were amplified by PCR using the specific primers shown in Table 1. The cycling conditions consisted of initial denaturation at 94 °C for 5 min, followed by 40 cycles of 94 °C for 15 s, 55–45 °C at the rate of 1.0 °C/cycle for 30 s, and 68 °C for 1 min, and final extension at 68 °C for 7 min. Each PCR amplicon was inserted into the cloning site of a modified pCXN2.1 (+) - bovine IgG1 Fc plasmid (kindly provided by Dr. Takehiko Yokomizo, Juntendo University, Tokyo, Japan) that contained the Fc fragment of bovine IgG1 at the C terminal, and transformed into a competent HST08 E.coli strain. Recombinant plasmids were purified with the FastGene Xpress Plasmid PLUS Kit,8 according to the manufacturer’s instructions. The recombinant plasmids were named pCXN2.1 (+) - TNFR1-Ig, pCXN2.1 (+) - TNFR2-Ig, and pCXN2.1 (+) - Cont-Ig.
Fig. 6.

Amino acid sequence of bovine TNFR1 and TNFR2 genes. Amino acid sequence of (a) Bovine TNFR1 and (b) bovine TNFR2 genes are shown. The single underline in TNFR1 indicates the DD. The dotted line in TNFR2 indicates a signal peptide. Transmembrane regions are indicated using double line. Predicted extracellular domains are shadowed
Table 1.
Primers used for amplification of each gene in this study

Expression of recombinant soluble bovine TNFR1-Ig and TNFR2-Ig fusion proteins
Recombinant plasmids were transfected into maintained Expi293F™ cells using ExpiFectamine™ 293 Reagent1, ExpiFectamine™ 293 Transfection Enhancer I1, and II1 according to the manufacturer’s instructions. Culture supernatants containing TNFR-Ig or Cont-Ig were harvested for 7 days after transfection, and the recombinant proteins were purified with Ab-Capture ExTra™.9 The purified proteins were then concentrated by ultrafiltration using Amicon Ultra.10 To confirm TNFR-Ig and Cont-Ig expression, Western blot analysis was performed using the peroxidase-labeled anti-bovine IgG Fc rabbit antibody,11 and purities were analyzed by SDS-PAGE. The quantities of the recombinant proteins were determined by sandwich ELISA with the anti-bovine IgG Fc rabbit antibody11 and the peroxidase-labeled anti-bovine IgG Fc rabbit antibody11.
Bovine TNF-α binding assay
The binding of TNFR-Ig to bovine TNF-α was analyzed using the anti-bovine TNF-α antibody. Briefly, TNFR-Ig or Cont-Ig was fixed on 96-well plates for ELISA12 for 24 h with phosphate buffered saline (PBS). After washing each well three times with PBS, incubation buffer [IB; PBS containing 0.05% Tween 20 and 1% Bovine Serum Albumin (BSA)4] was added and incubated for 1 h. The solution was then washed five times with PBS containing 0.05% Tween 20 (PBS-T), and the recombinant bovine TNF-α was added (0.098 ng/mL-25 ng/mL).13 After 2 h incubation, samples were washed five times with PBS-T and reacted with the biotinylated anti-bovine TNF-α rabbit antibody13 for 1 h. After washing for five times with PBS-T, peroxidase-labeled Pierce™ NeutrAvidin™ Protein1 was added and incubated for 1 h. Finally, the mixture was washed five times with PBS-T and reacted with the TMB one component substrate14 for 10 min in the dark. The reaction was quenched with 0.18 M H2SO4 and the absorbance was measured at 450 nm. Each incubation was performed at room temperature. The assay was performed in triplicate.
Cell viability assay
L929 cells (3 × 104/0.2 mL) were pre-cultured for 24 h on LIMITRAC® 96-well plates.15 After cultivation, cells were cultured 24 h with recombinant bovine TNF-α [300 μg/mL (17.2 nM)], actinomycin-D (1 μg/mL)4 and TNFR1-Ig or TNFR2-Ig or Cont-Ig at varying concentrations (1.72 nM, 17.2 nM, 172 nM, 1.72 μM). Cell viabilities were measured using the RealTime-Glo™ MT Cell Viability Assay16 according to the manufacturer’s instructions. The assay was performed in triplicate.
Expression analysis of bovine interleukin-1β and TNF-α mRNA
PBMCs (1.5 × 106/0.2 mL) from 6 healthy cattle were cultured for 4 h with recombinant bovine TNF-α [300 μg/mL (17.2 nM)], actinomycin-D (1 μg/mL)4 and TNFR1-Ig or TNFR2-Ig or Cont-Ig at varying concentrations (1.72 nM, 17.2 nM, 172 nM, 1.72 μM). As a calibrator sample, PBMCs from each cattle were cultured in the absence of TNF-α, actinomycin-D, and TNFR2-Ig or Cont-Ig. Total RNA from cultured PBMCs was extracted and used to synthesize cDNA as described above. Real-time quantitative RT-PCR was performed using the LightCycler480® SystemII17 and SYBR® Premix DimerEraser™ 7 according to the manufacturer’s instructions. The primers and target genes are shown in Table 1. The cycling conditions consisted of initial denaturation at 95 °C for 30 s, followed by 45 cycles of 95 °C for 5 s, 60 °C for 30 s and 72 °C for 30 s. A final melting curve analysis was performed from 65 °C to 95 °C at a rate of 0.1 °C/sec (continuous acquisition) to evaluate each primer pair for specificity to confirm that only one product was amplified. Each sample was tested in duplicate, and the relative expression level of IL-1β and TNF-α mRNA were analyzed using the 2-ΔΔCT method [33].
IL-1β and TNF-α ELISA
PBMCs (5 × 105/0.2 mL) from 6 healthy cattle were cultured for 24 h with recombinant bovine TNF-α [300 μg/mL (17.2 nM)], actinomycin-D (1 μg/mL)4 and TNFR1-Ig or TNFR2-Ig or Cont-Ig at varying concentrations (1.72 nM, 17.2 nM, 172 nM, 1.72 μM). IL-1β and TNF-α in the cultural supernatant were measured by ELISA using IL-1beta ELISA Reagent Kit, bovine1 and TNF-α Bovine TNF alpha Do-It-Yourself ELISA13, respectively, according to the manufacturer’s instructions.
Statistics
The differences between the groups were examined using the Wilcoxon signed rank test. For multiple group comparisons, the Steel-Dwass’ test or Tukey’s test was performed depending on the sample numbers. A p-value less than 0.05 was considered statistically significant.
Acknowledgments
We are grateful to Dr. Hideyuki Takahashi, Dr. Yasuyuki Mori, and Dr. Tomio Ibayashi for their valuable advice and discussions. We would like to thank Enago for the English language review.
Funding
This work was supported by grants-in-aid for Scientific Research from Japan Society for the Promotion of Science (JSPS), by the Research Project for Improving Animal Disease Prevention Technologies to Combat Antimicrobial Resistance 2017–2021 FY, and by grants from the Project of the NARO, Bio-oriented Technology Research Advancement Institution (Research program on development of innovative technology; number 26058 BC to SK and the special scheme project on regional developing strategy; grant 16817557 to SK). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
All datasets are available in the main manuscript.
Abbreviations
- BSA
Bovine Serum Albumin
- c-IAP1
Cellular Inhibitor of Apoptosis Protein 1
- DD
Death domain
- D-MEM
Dulbecco’s Modified Eagle Medium
- ELISA
Enzyme-linked immunosorbent assay
- FADD
Fas-associated death domain protein
- FBS
Fetal bovine serum
- IL
Interleukin
- mTNFR
Transmembrane TNFR
- mTNF-α
Membrane-expressed TNF-α
- PBMC
Peripheral blood mononuclear cells
- PBS
Phosphate buffered saline
- PBS-T
PBS containing 0.05% Tween 20
- RIPK1
Receptor-Interacting serine/threonine-Protein Kinase 1
- sTNFR2
Soluble TNF receptor 2
- sTNF-α
Soluble TNF-α
- TNFR1
Associated death domain protein
- TNFR1
TNF receptor 1
- TNFR2
TNF receptor 2
- TNF-α
Tumor necrosis factor alpha
- TRADD
TNFR1-associated death domain protein
- TRAF2
TNF receptor associated factor 2
Authors’ contributions
SK and KO were responsible for the conception and design of the study. SF, TO, NM, and AT performed the experiments. SF, SK, TO, YS, SM, and KO analyzed the data. SF, SK, TO, NM, YS and SM provided intellectual input, laboratory materials, reagents and/or analytic tools. SF wrote the manuscript. SK, TO, NM, YS, SM, and KO contributed to the revision of the manuscript. All authors reviewed and approved the final manuscript.
Ethics approval and consent to participate
All experimental procedures were conducted following approval from the local committee for animal studies according to the Hokkaido University (17–0024). Collection of blood samples from healthy (non-treated) cattle is announced as simple general permission procedure for the use of blood samples for assays.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Thermo Fisher Scientific, Waltham, MA, USA
Cell Culture Technologies LLC, Gravesano, Ticino, Switzerland
GE Healthcare UK Ltd., Amersham Place, Little Chalfont, Buckinghamshire, England
Sigma-Aldrich, St. Louis, MO, USA
Invitrogen, Carlsbad, CA, USA
Molecular Research Center, Inc., Montgomery Road, Cincinnati, OH, USA
TaKaRa, Shiga, Japan
NIPPON Genetics Co., Ltd., Tokyo, Japan
ProteNova, Tokushima, Japan
Merck, Summit Drive, Burlington, MA, USA
Rockland Immunochemicals, Pottstown, PA, USA
Sumitomo Bakelite Co., Ltd., Tokyo, Japan
Kingfisher Biotech, Inc., Westgate Drive Suite, Saint Paul, MN, USA
Bethyl Laboratories, Inc., P.P. Box, Montgomery, TX, USA
GreinerBio-One GmbH, Bad Haller, Kremsmünster, Austria
Promega, Fitchburg, WI, USA
Roche Diagnostics, Mannheim, Germany
Contributor Information
Sotaro Fujisawa, Email: s.fujisawa@vetmed.hokudai.ac.jp.
Satoru Konnai, Phone: (81) 11 706 5216, Email: konnai@vetmed.hokudai.ac.jp.
Tomohiro Okagawa, Email: okagawa@vetmed.hokudai.ac.jp.
Naoya Maekawa, Email: maekawa@vetmed.hokudai.ac.jp.
Akina Tanaka, Email: moa.1108@outlook.jp.
Yasuhiko Suzuki, Email: suzuki@czc.hokudai.ac.jp.
Shiro Murata, Email: murata@vetmed.hokudai.ac.jp.
Kazuhiko Ohashi, Email: okazu@vetmed.hokudai.ac.jp.
References
- 1.Barkema HW, Orsel K, Nielsen SS, Koets AP, Rutten VPMG, Bannantine JP, Keefe GP, Kelton DF, Wells SJ, Whittington RJ, Mackintosh CG, Manning EJ, Weber MF, Heuer C, Forde TL, Ritter C, Roche S, Corbett CS, Wolf R, Griebel PJ, Kastelic JP, De Buck J. Knowledge gaps that hamper prevention and control of Mycobacterium avium subspecies paratuberculosis infection. Transbound Emerg Dis. 2018;65:125–148. doi: 10.1111/tbed.12723. [DOI] [PubMed] [Google Scholar]
- 2.Nicholas RAJ, Fox LK, Lysnyansky I. Mycoplasma mastitis in cattle: to cull or not to cull. Vet J. 2016;216:142–147. doi: 10.1016/j.tvjl.2016.08.001. [DOI] [PubMed] [Google Scholar]
- 3.Benoit M, Desnues B, Mege JL. Macrophage polarization in bacterial infections. J Immunol. 2008;181:3733–3739. doi: 10.4049/jimmunol.181.6.3733. [DOI] [PubMed] [Google Scholar]
- 4.Imanishi J. Expression of cytokines in bacterial and viral infections and their biochemical aspects. J Bio Chem. 2000;127:525–530. doi: 10.1093/oxfordjournals.jbchem.a022636. [DOI] [PubMed] [Google Scholar]
- 5.Sharma C, Rokana N, Chandra M, Singh BP, Gulhane RD, Gill JPS, Ray P, Puniya AK, Panwar H. Antimicrobial resistance: its surveillance, impact, and alternative management strategies in dairy animals. Front Vet Sci. 2018;4:237. doi: 10.3389/fvets.2017.00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sipka A, Gurjar A, Klaessig S, Duhamel GE, Skidmore A, Swinkels J, Cox P, Schukken Y. Prednisolone and cefapirin act synergistically in resolving experimental Escherichia coli mastitis. J Dairy Sci. 2013;96(7):4406–4418. doi: 10.3168/jds.2012-6455. [DOI] [PubMed] [Google Scholar]
- 7.Ponchon B, Zhao X, Ollier S, Lacasse P. Relationship between glucocorticoids and prolactin during mammary gland stimulation in dairy cows. J Dairy Sci. 2017;100(2):1521–1534. doi: 10.3168/jds.2016-11490. [DOI] [PubMed] [Google Scholar]
- 8.Aggarwal B. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3:745–756. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- 9.Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 10.Akdis M, Aab A, Altunbulakli C, Azkur K, Costa RA, Crameri R, Duan S, Eiwegger T, Eljaszewicz A, Ferstl R, Frei R, Garbani M, Globinska A, Hess L, Huitema C, Kubo T, Komlosi Z, Konieczna P, Kovacs N, Kucuksezer UC, Meyer N, Morita H, Olzhausen J, O'Mahony L, Pezer M, Prati M, Rebane A, Rhyner C, Rinaldi A, Sokolowska M, Stanic B, Sugita K, Treis A, van de Veen W, Wanke K, Wawrzyniak M, Wawrzyniak P, Wirz OF, Zakzuk JS, Akdis CA. Interleukins (from IL-1 to IL-38), interferons, transforming growth factor β, and TNF-α: receptors, functions, and roles in diseases. J Allergy Clin Immunol. 2016;138:984–1010. doi: 10.1016/j.jaci.2016.06.033. [DOI] [PubMed] [Google Scholar]
- 11.Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP. A metalloproteinase disintegrin that releases tumor-necrosis factor a from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 12.Grell M, Douni E, Wajant H, Löhden M, Clauss M, Maxeiner B, Georgopoulos S, Lesslauer W, Kollias G, Pfizenmaier K, Scheurich P. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell. 1995;83:793–802. doi: 10.1016/0092-8674(95)90192-2. [DOI] [PubMed] [Google Scholar]
- 13.Varfolomeev E, Vucic D. Intracellular regulation of TNF activity in health and disease. Cytokine. 2018;101:26–32. doi: 10.1016/J.CYTO.2016.08.035. [DOI] [PubMed] [Google Scholar]
- 14.Praga M, Morales E. Renal damage associated with proteinuria. Kidney Int. 2002;62:42–46. doi: 10.1046/j.1523-1755.62.s82.9.x. [DOI] [PubMed] [Google Scholar]
- 15.Van Antwerp DJ, Martin SJ, Verma IM, Green DR. Inhibition of TNF-induced apoptosis by NF-κB. Trends Cell Biol. 1998;8:107–111. doi: 10.1016/S0962-8924(97)01215-4. [DOI] [PubMed] [Google Scholar]
- 16.Wang CY, Mayo MW, Korneluk RG, Goeddel DV, NF-κB Antiapoptosis BAS. Induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress Caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 17.Ji W, Li Y, Wan T, Wang J, Zhang H, Chen H, Min W. Both internalization and AIP1 association are required for tumor necrosis factor receptor 2-mediated JNK signaling. Arterioscler Thromb Vasc Biol. 2012;32:2271–2279. doi: 10.1161/ATVBAHA.112.253666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ward-Kavanagh LK, Lin WW, Šedý JR, Ware CF. The TNF receptor superfamily in co-stimulating and co-inhibitory responses. Immunity. 2016;44:1005–1019. doi: 10.1016/j.immuni.2016.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Waetzig GH, Rosenstiel P, Arlt A, Till A, Brautigam K, Schafer H, Rose-John S, Seegert D, Schreiber S, Bräutigam K, Schäfer H. Soluble tumor necrosis factor (TNF) receptor-1 induces apoptosis via reverse TNF signaling and autocrine transforming growth factor-beta1. FASEB J. 2005;19:91–93. doi: 10.1096/fj.04-2073fje. [DOI] [PubMed] [Google Scholar]
- 20.Spoettl T, Hausmann M, Klebl F, Dirmeier A, Klump B, Hoffmann J, Herfarth H, Timmer A, Rogler G. Serum soluble TNF receptor I and II levels correlate with disease activity in IBD patients. Inflamm Bowel Dis. 2007;13:727–732. doi: 10.1002/ibd.20107. [DOI] [PubMed] [Google Scholar]
- 21.van der Poll T, Jansen J, van Leenen D, von der Möhlen M, Levi M, ten Cate H, Gallati H, ten Cate JW, van Deventer SJ. Release of soluble receptors for tumor necrosis factor in clinical Sepsis and experimental Endotoxemia. J Infect Dis. 1993;168:955–960. doi: 10.1093/infdis/168.4.955. [DOI] [PubMed] [Google Scholar]
- 22.Feldmann M, Maini RN. ANTI-TNFα therapy of rheumatoid arthritis : what have we learned? Annu Rev Immunol. 2001;19:163–196. doi: 10.1146/annurev.immunol.19.1.163. [DOI] [PubMed] [Google Scholar]
- 23.Feldmann M, Maini RN. TNF defined as a therapeutic target for rheumatoid arthritis and other autoimmune diseases. Nat Med. 2003;9:1245–1250. doi: 10.1038/nm939. [DOI] [PubMed] [Google Scholar]
- 24.Moreland LW, Schiff MH, Baumgartner SW, Tindall EA, Fleischmann RM, Bulpitt KJ, Weaver AL, Keystone EC, Furst DE, Mease PJ, Ruderman EM, Horwitz DA, Arkfeld DG, Garrison L, Burge DJ, Blosch CM, Lange MLM, McDonnell ND, Weinblatt ME. Etanercept therapy in rheumatoid arthritis. A randomized, controlled trial Ann Intern Med. 1999;130:478–486. doi: 10.7326/0003-4819-130-6-199903160-00004. [DOI] [PubMed] [Google Scholar]
- 25.Gondaira S, Higuchi H, Iwano H, Nakajima K, Kawai K, Hashiguchi S, Konnai S, Nagahata H. Cytokine mRNA profiling and the proliferative response of bovine peripheral blood mononuclear cells to mycoplasma bovis. Vet Immunol Immunopathol. 2015;165:45–53. doi: 10.1016/j.vetimm.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 26.Grell M, Wajant H, Zimmermann G, Scheurich P. The type 1 receptor (CD120a) is the high-affinity receptor for soluble tumor necrosis factor. Proc Natl Acad Sci U S A. 1998;95:570–575. doi: 10.1073/pnas.95.2.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tartaglia LA, Pennica D, Goeddel DV. Ligand passing: the 75-kDa tumor necrosis factor (TNF) receptor recruits TNF for signaling by the 55-kDa TNF receptor. J Biol Chem. 1993;268:18542–18548. [PubMed] [Google Scholar]
- 28.Lang I, Füllsack S, Wyzgol A, Fick A, Trebing J, Arana JAC, Schäfer V, Weisenberger D, Wajant H. Binding studies of TNF receptor superfamily (TNFRSF) receptors on intact cells. J Biol Chem. 2016;291:5022–5037. doi: 10.1074/jbc.M115.683946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flick DA, Gifford GE. Comparison of in vitro cell cytotoxic assays for tumor necrosis factor. J Immunol Methods. 1984;68:167–175. doi: 10.1016/0022-1759(84)90147-9. [DOI] [PubMed] [Google Scholar]
- 30.Humphreys DT, Wilson MR. Modes of L929 cell death induced by TNF-α and other cytotoxic agents. Cytokine. 1999;11:773–782. doi: 10.1006/cyto.1998.0492. [DOI] [PubMed] [Google Scholar]
- 31.Cottin V, Doan JES, Riches DWH. Restricted localization of the TNF receptor CD120a to lipid rafts: a novel role for the death domain. J Immunol. 2002;168:4095–4102. doi: 10.4049/jimmunol.168.8.4095. [DOI] [PubMed] [Google Scholar]
- 32.Ting AT, Bertrand MJM. More to life than NF-κB in TNFR1 signaling. Trends Immunol. 2016;37:535–545. doi: 10.1016/j.it.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All datasets are available in the main manuscript.