

Abstract
Background
Sarcoidosis is a systemic disease of unknown etiology. The disease mechanisms are largely speculative and may include the role microbial patterns that initiate and drive an underlying immune process. The aim of this study was to characterize the microbiota of the lung of patients with sarcoidosis and compare its composition and diversity with the results from patients with other interstitial lung disease (ILD) and historic healthy controls.
Methods
Patients (sarcoidosis, n = 31; interstitial lung disease, n = 19) were recruited within the PULMOHOM study, a prospective cohort study to characterize inflammatory processes in pulmonary diseases. Bronchoscopy of the middle lobe or the lingula was performed and the recovered fluid was immediately sent for analysis of the pulmonary microbiota by 16sRNA gene sequencing. Subsequent bioinformatic analysis was performed to compare the groups.
Results
There were no significant differences between patients with sarcoidosis or other ILDs with regard to microbiome composition and diversity. In addition, the abundance of the genera Atopobium, Fusobacterium, Mycobacterium or Propionibacterium were not different between the two groups. There were no gross differences to historical healthy controls.
Conclusion
The analysis of the pulmonary microbiota based on 16sRNA gene sequencing did not show a significant dysbiosis in patients with sarcoidosis as compared to other ILD patients. These data do not exclude a microbiological component in the pathogenesis of sarcoidosis.
Electronic supplementary material
The online version of this article (10.1186/s12931-019-1013-2) contains supplementary material, which is available to authorized users.
Keywords: Microbiome, Sarcoidosis, Interstitial lung disease, Inflammation, Infection
Background
Sarcoidosis is a systemic disease that is characterized by typical histopathology with noncaseating granuloma formation in affected organs. While most organs can be affected, the pulmonary manifestation is the most frequent manifestation. The clinical presentation is heterogenous, what makes diagnostic procedures often difficult. This heterogeneity also points toward complex and possible multiple disease mechanisms. The pathomechanisms of sarcoidosis are still not entirely clear but involve antigen-driven CD4+ T-cell activation with macrophage attraction and granuloma formation. There are several hypotheses about the causative antigen that initiates and drives the underlying immune process. Some studies indicated that autoimmunity may play a role in these processes [1, 2]. A potential microbial compound has been discussed in the last decades. While classical culture-dependent microbiology did not unravel a clear candidate, remnant microbial molecules were identified in sarcoidosis patients, many data sets point towards the involvement of mycobacterial species [2–4]. In addition, also other microbial classes such as Haemophilus, Atopobium and Fusobacterium may show alterations in their abundance in sarcoidosis [5, 6].
The development of technologies to characterize the microbiota of a given habitat offers opportunities to further understand the role of microorganisms in disease development. Microbiome analysis is based on the analysis of microbial genes or transcripts. Current techniques use 16sRNA gene sequencing or whole genome shot-gun sequencing with subsequent bioinformatic analysis. Only a few studies have been performed in sarcoidosis and suggested that there are no or little differences in the microbial composition between sarcoidosis and control groups [6–8]. In addition, a few studies have also characterized the microbiota in other interstitial lung disease (ILD) [9, 10].
The aim of this study was to characterize the microbiota in the lungs of patients with sarcoidosis and to compare these data with results from other interstitial lung diseases and historical healthy controls. The study aimed to show whether there is unique phenotype of the sarcoidosis microbiome as compared to a heterogenous group of other ILDs. We performed analysis of the microbiome by 16sRNA-gene sequencing and biostatistical analysis.
Methods
Study population
Patients were recruited within the PULMOHOM study, a prospective cohort study to characterize inflammatory processes in pulmonary diseases. Patients with suspected sarcoidosis or interstitial lung diseases (ILD) undergoing bronchoscopy participated in the study. Patient were selected based on clinical and radiological finding indicating high probability for these disease entities.
Patients were further diagnosed by standard diagnostic procedures including bronchoalveolar lavage, biopsy, CT scan and serological markers. The final diagnosis was used to segregate the patients in the diagnostic groups (sarcoidosis, ILD).
The PULMOHOM study was approved by the ethics board of the Landesärztekammer des Saarlandes and informed written consent was obtained from all patients.
Bronchoalveolar lavage (BAL) and processing
Bronchoscopy was performed as described earlier [11] and BAL of a segment of the middle lobe or the lingula was performed by instillation of 5 times 20 mL of sterile saline and recovered fluid was immediately sent for further analysis.
DNA isolation and 16sRNA gene sequencing
PCRs of V1/V2 and V3/V4 variable regions of 16S rRNA genes were performed (5–25 ng template, 80 mM Tris-HCL, 20 mM (NH4)2SO4, 0.2% Tween-20, 2.5 mM MgCl2, 0.2 mM of each dNTP, 2.5 U HotFirePol (Solis BioDyne)) using 200 pmol of each primer (V1/V2: forw, 5′-agagtttgatcctggctcag-3′ and rev, 5′-tgctgcctcccgtaggagt-3′; V3/V4: forw, 5′-cctacgggnggcwgcag-3′ and rev, 5′-gactachvgggtatctaatcc-3′) with Illumina universal TruSeq adaptor sequences attached at the 5′-end. PCRs were performed in a thermocycler starting with 15 min 95 °C followed by 35 cycles 95 °C 1 min, 54 °C 1 min, 72 °C 1 min and a 5 min final extension at 72 °C. Amplicons were purified with MagSi-NGS PREP Plus beads (Steinbrenner, Wiesenbach, Germany), diluted, pooled and sequenced on the Illumina MiSeq (v3 chemistry: 2 × 300 bp paired-end) following the manufacturer’s instructions aiming at 50,000 reads per sample.
Biostatistical analysis of microbiome data
Read quality reports were generated from the FASTQ files using FastQC (version 0.11.7) and a summary was created using MultiQC (version 1.6). Operational taxonomic units (OTUs) were defined for both amplicons separately using LotuS (version 1.565). OTUs are clusters of sequences grouped together based on sequence similarity; one sequence is chosen to represent the complete cluster [12]. All obtained OTU sequences were additionally searched in the BLAST nr/nt database (created on 04.08.2014) using BLASTn (version 2.6.0+) with minimal query coverage per HSP and identity cutoff of 80% and by saving maximal 10 hits per query. For each OTU, the best hit was kept by sorting the associated hits by query coverage per HSP and percentage identity. For each of the remaining hits, the subject accession ID was extracted and searched in the NCBI database to retrieve the assigned taxon ID using EDirect (version 9.50). The taxon ID was searched in the NCBI taxonomy database to get the associated taxonomic lineage. For each amplicon, the OTUs were filtered to remove likely contaminants. The OTUs were flagged if they had “Eukaryota” in the superkingdom taxon name derived from the BLAST hits, or if they were present in the NTC sample, or if they had a count below 1 in all non-control samples (i.e. all samples except the NTC sample). All flagged OTUs were then removed from the OTU count table of the non-control samples. An overview of the taxonomic composition of all kept OTUs was created per amplicon over all samples and for each sample indication group using Krona (version 2.5). In the following, only OTUs kept after the filtering step were considered and the analysis were performed for both amplicons (V1/V2 and V3/V4) separately.
The R-package “ampvis2” (version 2.3.2) was used to plot rarefaction curves (number of reads vs. the number of observed OTUs, step size 500), and to compute and plot the alpha-diversity values for all provided measures (i.e. “observed”, “shannon”, “simpson”, and “invsimpson”). The alpha-diversity values between the indication groups were compared using the Wilcox test (two-sided, confidence level 0.95) from the R-package coin (version 1.2–2).
For CLR-transformation, zero imputation was applied to the OTU count matrix using the method “cmultRepl” from the R-package “zCompositions” (version 1.1.1, parameters: method = “CZM”, label = 0, output = “prop”). Then, the sample values were CLR-transformed using the method “clr” from the R-package “compositions” (version 1.40–1). Principal component analysis (PCA) was applied to the resulting matrix (method “prcomp”, parameters: center = TRUE, scale. = FALSE). Also, a heatmap was created using hierarchical clustering (Euclidean distance, complete linkage) to re-order the samples and OTUs.
For the genera Atopobium, Fusobacterium, Mycobacterium and Propionibacterium, the corresponding OTUs were identified (based on the taxonomy assigned by LotuS). A heatmap showing only the CLR-transformed counts of these OTU was plotted and the samples were re-ordered using hierarchical clustering (Euclidean distance, complete linkage).
The R-package “ALDEx2” (version 1.8.0) was used for differential analysis of the OTU counts by comparing the indication groups. The counts were first CLR-transformed using the “aldex.clr” method (parameter: denom = “iqlr”). Then, the effect sizes were calculated using the method “aldex.effect”: Finally, the Wilcoxon test was applied.
The code used for the analysis can be found at https://github.com/VGalata/16S_Sarcoidosis. The original sequencing data are available at https://www.ncbi.nlm.nih.gov/sra/PRJNA510549.
Results
Patient characterization
The characteristics of the study population are presented in Table 1. Based on the radiologic evaluation, patients were classified into the Scadding group: 0 (1), I (10), II (13), III (4) and IV (3). As compared to the ILD control groups, patients with sarcoidosis were significantly younger and had a lower FEV1/VC ratio. The number of lymphocytes in the BAL was significantly elevated and the CD4/CD8 ratio was increased. The group of ILD patients comprised individuals with rheumatoid-related ILD (n = 2), sclerodermia-associated ILD (n = 1), drug-related ILD (n = 3), hypersensitivity pneumonitis (n = 2), idiopathic pulmonary fibrosis (n = 3), nonspecific interstitial pneumonia (NSIP, n = 7), and cryptogenic organizing pneumonia (COP, n = 1).
Table 1.
Patient characteristics
| Clinical characteristics | Sarcoidosis | Interstitial lung disease | p-value |
|---|---|---|---|
| Number of subjects, n (%) | 31 (62%) | 19 (38%) | – |
| Female, n (%) | 14 (45%) | 8 (42%) | – |
| Male, n (%) | 17 (55%) | 11 (58%) | – |
| Age (years), median (range) | 51 (22–79) | 66 (57–82) | 0.0003 |
| Smoking | |||
| Non-smokers, n (%) | 18 (58%) | 6 (31.5%) | 0.06 |
| Current-smokers, n (%) | 7 (22.5%) | 2 (10.5%) | 0.25 |
| Former-smokers, n (%) | 6 (19.5%) | 9 (47%) | 0.05 |
| BMI, median (range) | 27 (17–37.1) | 24.7 (16.8–34.9) | 0.5 |
| Pulmonary function test | |||
| FEV1%, median (range) | 81.5 (61.5–126.8) | 66.8 (49–109.6) | 0.66 |
| FEV1/VC, median (range) | 79.8 (66.2–86.5) | 85.7 (61.4–108.8) | 0.02 |
| TLC (l), median (range) | 5.54 (4.17–8.38) | 4.2 (2.56–7.99) | 0.26 |
| ERV (l), median (range) | 0.78 (0.21–1.39) | 0.55 (0.12–1.97) | 0.89 |
| RV (l), median (range) | 2.12 (1.12–4.54) | 1.76 (1.41–2.87) | 0.60 |
| VC (l), median (range) | 3.34 (1.9–5.61) | 2.34 (0.88–5.6) | 0.26 |
| Bronchoscopy, BAL | |||
| BAL recovery %, median | 50 | 55 | |
| Neutrophils %, median | 9 | 14 | 0.05 |
| Macrophages %, median | 49 | 62 | 0.01 |
| Lymphocytes %, median | 38 | 25.5 | 0.02 |
| Eosinophils %, median | 1 | 2 | 0.19 |
| Basophils %, median | 0 | 0 | |
| CD4/CD8 ratio, median | 3.2 | 0.8 | 0.03 |
| Laboratory | |||
| CRP mg/dl, median (range) | 4.2 (0.4–83.9) | 5.4 (0.6–326.3) | 0.44 |
| Leukocytes (1/μl), median (range) | 5.65 (3–13.8) | 7.55 (3.4–13.9) 0.12 | |
Microbiome features of sarcoidosis and ILD patients
16sRNA gene sequencing was applied to characterize the lung microbiota of 31 patients with sarcoidosis and 19 with other interstitial lung diseases. The median number of reads was 10,951 (control: 10,023; sarcoidosis: 11,118) for V1/V2 and 51,916 (control: 47,312; sarcoidosis: 53,879) for V3/V4, respectively (Fig. 1). In total, 1212 (V1/V2) and 1743 (V3/V4) OTUs were defined. The original sequencing data are available at https://www.ncbi.nlm.nih.gov/sra/PRJNA510549.
Fig. 1.

Number of raw reads per sample for V1/V2 and V3/V4. The samples were grouped by their indication (“Control” and “Sarcoidosis”). The distribution of the read counts is shown by boxplots
The percentage of counts belonging to OTUs removed during the filtering step was variable. While these percentage values were below 40% for most of the V1/V2 samples, in the V3/V4 samples, the fraction of counts was above 75% for more than half of the samples (Additional file 1: Figure S1 A and B). The largest fraction of OTU counts in V1/V2 samples belonged to OTUs present in the control (NTC) sample. In the V3/V4 samples, however, the largest fraction belonged to OTUs present in the NTC sample and was classified as contamination (based on the BLAST hits). In the phylogenetic tree of all OTUs, most removed OTUs were classified as contaminants (Additional file 1: Figure S2 A and B). Also, most of these OTUs were placed in a separate subtree distant from the remaining OTUs. The V3/V4 samples demonstrated a higher frequency of OTUs flagged as contaminants than the V1/V2 samples.
Composition of the microbiota
The taxonomic composition of the remaining OTUs is illustrated in Fig. 2 (the interactive version of the plot can be found in the online supplement). In total, 902 and 685 OTUs remained after the filtering step in the V1/V2 and V3/V4 datasets, respectively. For both amplicons, a similar distribution of the most frequent genera was found. In V1/V2, 231 unique genera were found where four taxa had a frequency of above 2%: Prevotella (37 OTUs, 4.1%), Corynebacterium (28 OTUs, 3.1%), Actinomyces (25 OTUs, 2.77%), and Streptococcus (24 OTUs, 2.66%). In V3/V4, 194 unique genera were observed with 5 taxa having a frequency above 2%: Prevotella (46 OTUs, 6.72%), Actinomyces (33 OTUs, 4.82%), Streptococcus (31 OTUs, 4.53%), Treponema (19 OTUs, 2.77%), and Corynebacterium (15 OTUs, 2.19%). In 232 (25.72%) and 146 (21.31%) cases the genus was unknown in the V1/V2 and V3/V4 datasets, respectively. There was no significant difference between patients with sarcoidosis and ILD.
Fig. 2.
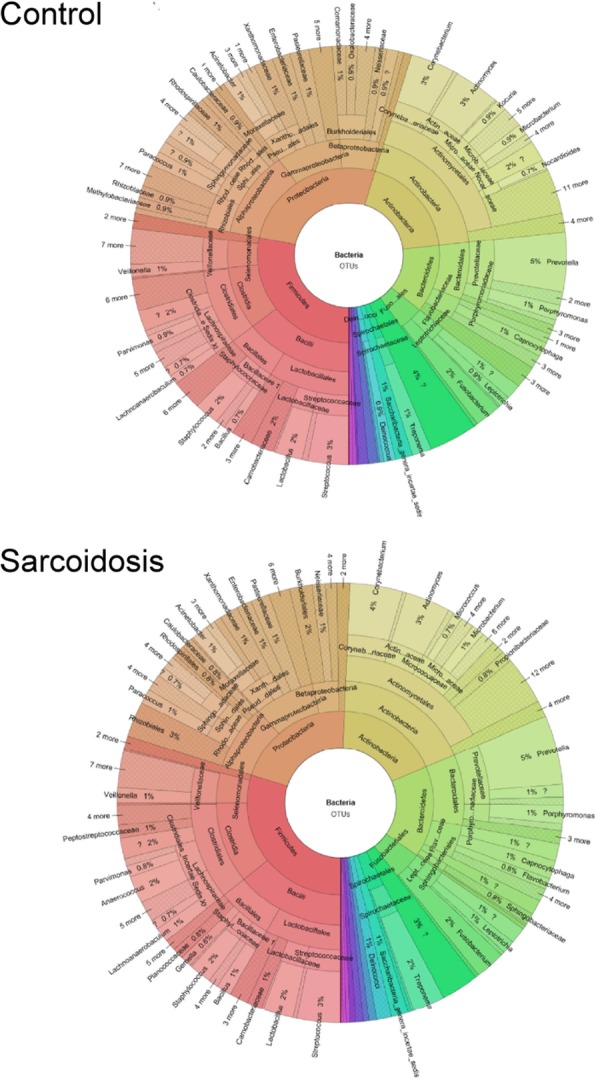
Taxonomic composition of the dataset based on taxa assigned by LotuS. Only OTUs not removed during the filtering process were considered
Richness and diversity
Rarefaction analysis was performed and demonstrated no trend of lower richness (number of OTUs) in one of the groups (Fig. 3 a and b). For only some samples, the curve showed a saturation shape, i.e. the richness most of these samples cannot be considered as completely sampled. For both amplicons and all tested alpha-diversity measures, the alpha-diversity distribution was not significantly different between the two groups (two-sided Wilcoxon test, alpha = 0.05, Fig. 3 c and d).
Fig. 3.
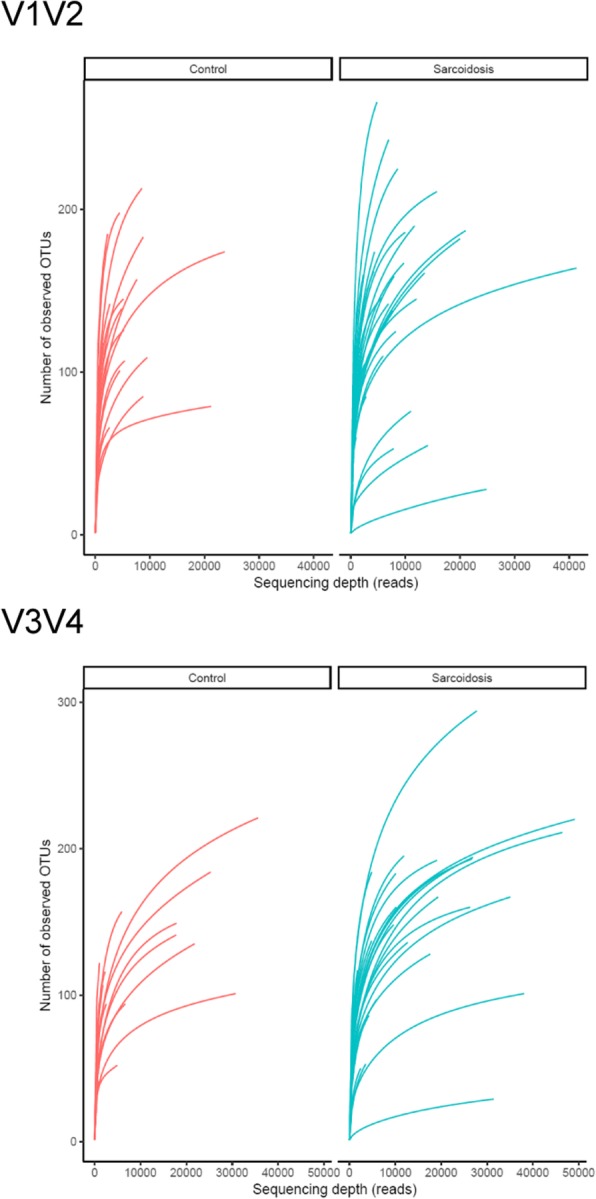
Rarefaction curves (a) for V1/V2 and (b) for V3/V4, and alpha-diversity (c) for V1/V2 and (d) for V3/V4. The rarefaction curves were plotted for both sample groups separately. The alpha-diversity plot shows the distribution (boxplot) and individual sample values (circles), and whether the distributions were found significantly different between the sample groups (“NS” means not significant)
Unsupervised analysis of sample clusters
The PCA analysis on the CLR-transformed OTU counts did not show any notable clusters of samples related to the disease groups or smoker status (Additional file 1: Figure S3 a and b). The variance explained by the two first principal components was approximately 37.48% for V1/V2 and 41.66% for V3/V4). The same holds for the heatmaps, where the samples were grouped by hierarchical clustering, using all OTUs (Fig. 4) and only OTUs assigned to genus Atopobium, Fusobacterium, Mycobacterium or Propionibacterium (Additional file 1: Figure S4).
Fig. 4.

Heatmap of CLR-transformed OTU counts for V1/V2. The samples and OTUs were re-ordered by hierarchical clustering and additional annotation was added to the resulting sample tree – the sample group label (“Control” or “Sarcoidosis”) and smoker status (“never”, “former”, “current”)
Differential analysis of OTUs
To identify OTUs that significantly differ between the groups, a differential analysis was performed and showed that none of the OTUs had an effect size with an absolute value larger than 0.5, and only 2 and 4 OTUs had a p-value below 0.05 for the V1/V2 and V3/V4 amplicons, respectively: OTU_90 (Staphylococcus) and OTU_15 (Actinomyces) in V1/V2, and OTU_75 (Mogibacterium), OTU_20 (Actinomyces), OTU_220 (Saccharibacteria_genera_incertae_sedis) and OTU_22 (Atopobium) in V3/V4 (Fig. 5). These data indicate that there are no meaningful differences with respect to specific OTUs between sarcoidosis and other ILD in the present study.
Fig. 5.
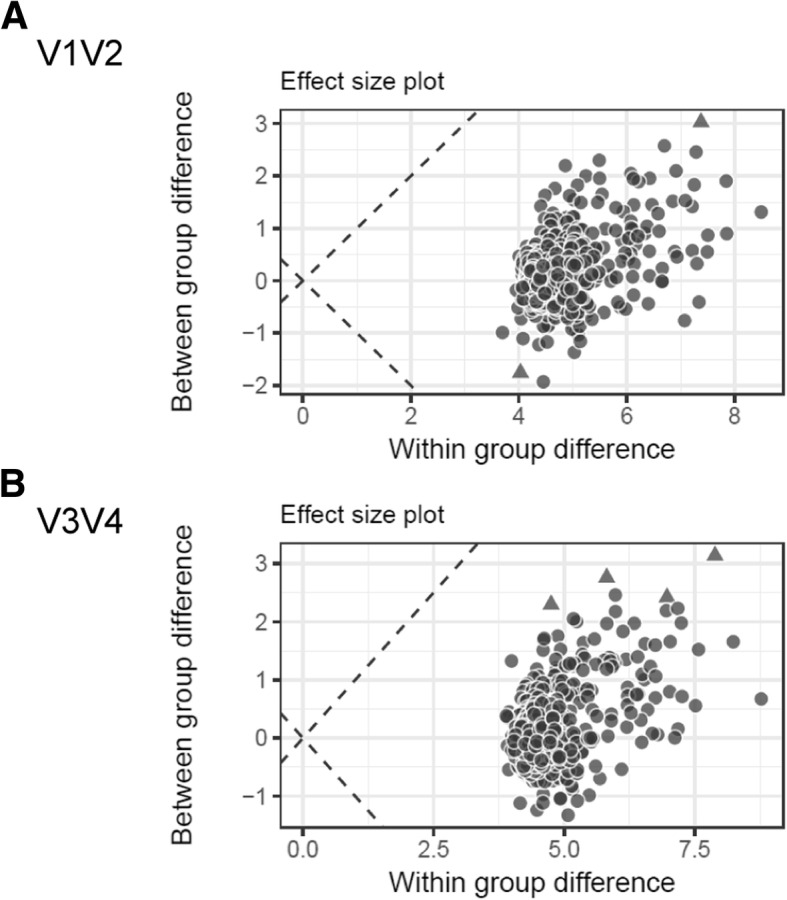
Results of differential analysis of OTUs for (a) V1/V2 and (b) V3/V4. The effect size plot shows the within and between group differences. The dashed lines correspond to points where the within and between group differences would be the same
Discussion
The present study characterized the pulmonary microbiome of patients with sarcoidosis in comparison to patients with ILD and historical healthy controls. The main findings were that there are no significant differences between the sarcoidosis and the ILD groups with regard to the overall structure, to the diversity, and to differential analysis.
It has been hypothesized for a long time that microorganisms have a role in the development of sarcoidosis [13, 14]. While there is no evidence that sarcoidosis is an infectious disease, DNA or other components of microorganisms have been detected in sarcoidosis biosamples. There is evidence that DNA from Propionibacterium acnes [15], peptides or DNA [16] from Mycobacterium are correlated with the active sarcoidosis [3]. In addition, significantly more patients with sarcoidosis show the presence of IgG directed to recombinant mycobacterial katG in serum [17]. Which exact roles these microbial patterns could play in the pathogenesis is not clear, one view is that they could initiate the formation of persistent, granulomatous inflammation. The present study applies microbiome analysis based on 16sRNA gene sequencing to characterize the microbiota in sarcoidosis BAL samples. We found no significant difference between the numbers of OTUs, the composition or the diversity between patients with sarcoidosis and ILD. Of note, about 20% of the OTUs could not be assigned to a genus. Whether these unknown OTUs could include disease-specific species is unknown. Similar results were obtained in most other microbiome studies addressing this disease [7, 8, 18]. A study investigated different approaches to samples lung specimens, performed whole-genome sequencing and found enrichment of microbes in tissue (fungi and Cladosporiaceae), BAL (Aspergillus), however, “no microbial lineage was enriched in more than one sample type after correction for multiple comparisons” [7]. A 16sRNA gene sequencing approach was used to compare the microbiota in BAL from patients with rheumatoid arthritis, sarcoidosis and healthy controls. The microbiota of patient groups were less diverse than that from healthy controls but similar to each other [18]. One study compared the microbiota of sarcoidosis patients with those from interstitial lung disease, Pneumocystis pneumonia and healthy controls and found no differences in the overall composition and diversity between these groups [8].
To compare the microbiota from sarcoidosis patients with those from healthy controls we used historical data from our studies [11] and from other researchers [19, 20]. This historical approach is limited to the comparison of the gross structure of the microbiota. In the present study, the lung microbiota in sarcoidosis patients is composed of the major groups Firmicutes, Proteobacteria, Acinetobacter, Bacteroidetes, Fusobacteriales, and Spirochaetales. Healthy controls from an earlier study using T-FRLP, cloning and sequencing revealed the presence of these groups. Based on the methodological differences, a direct comparison is difficult [11]. One study in healthy individuals investigated the difference of the microbiome depending on the topology of the sampling and found a qualitatively equivalent composition of the microbiota [19]. Quantitative differences may have been caused by methodological differences. Another study with material from healthy individuals shows similar results (dominance of Bacteroides, Firmicutes, Proteobacteria, and Fusobacteria). In contrast, a recent study in 71 patients with sarcoidosis, 15 patients with idiopathic pulmonary fibrosis and 10 healthy controls found a significant difference in the redundancy analysis between the sarcoidosis and the healthy control group [6]. The genera Atopobium and Fusobacterium were found more frequently in the sarcoidosis group and also an association with the Scadding type was reported.
This and other studies on th microbiome did not unravel a disease-characteristic change of the pulmonary microbiota in the lungs of patients with sarcoidosis, while some few other studies did find differences. It is currently unclear whether this indicates that microbial patterns have no role in the development of the disease or whether the methodologies applied have limitations that preclude the repeated validation of such alterations of the microbiota.
The present study has strengths and weaknesses: One issue is the selection of the optimal source specimen for the microbiome analysis. As pathomechanisms of sarcoidosis likely takes place within the distal lung parenchyma, BAL might be a favorable material. The study of Clark and colleagues showed small but highly variable differences between sample types [7]. A larger number of patients could have allowed to associate specific changes of the microbiota with clinical phenotypes. Due to the limitations in sample size, it was not possible to discriminate patients by phenotype (Scadding groups) or disease severity. We chose to compare sarcoidosis patients with patients with interstitial lung disease because both groups share at least some pathomechanisms and we sought to identify sarcoidosis-specific changes of the microbiota. The ILD group is heterogenous and comprises several specific disease entities, which likely increases the variability of the results from this group. We could not include a healthy-control group due to restrictions imposed by the local ethics committee. The comparison between actual and historical data is difficult and likely not conclusive. The analysis of 16sRNA genes has its inherent limitations such as limited resolution and lower sensitivity as compared to shotgun metagenomic sequencing [7]. In contrast, analysis of the pulmonary microbiome is more difficult with metagenomic approaches due to the low abundance of microbial DNA.
Conclusion
We could not detect a significant difference between the lung microbiota of patients with sarcoidosis as compared to patients with other interstitial lung diseases.
Additional file
Figure S1. Percentage of OTU counts per sample covered by OTUs not removed (“kept”) or discarded during the filtering step for (A) V1/V2 and (B) V3/V4. OTUs were removed if they were considered as contaminants (flag “contamination”), were present in the NTC sample (flag “controls”), had a count below 1 in non-control samples (i.e. all samples except the NTC sample; flag “count”), or any combination thereof. Figure S2. Phylogenetic OTU tree constructed by LotuS for (A) V1/V2 and (B) V3/V4. The OTUs were colored based on whether they were kept (flag “kept”) or discarded (flags “contamination”, “controls”, “counts”, or any combination thereof) during the filtering step. OTUs were removed if they were considered as contaminants (flag “contamination”), were present in the NTC sample (flag “controls”), or had a count below 1 in non-control samples (i.e. all samples except the NTC sample; flag “count”). Figure S3. PCA plots for (A) V1/V2 and (B) V3/V4. The plots show all samples using the two first principal components. The percentage of the variance explained by each principal component can be found in the axis title. The samples were colored with respect to their indication group. Figure S4. Heatmap of CLR-transformed OTU counts for (A) V1/V2 and (B) V3/V4 only for OTUs assigned by LotuS to the genus Atopobium, Fusobacterium, Mycobacterium or Propionibacterium. The samples were grouped by hierarchical clustering and additional annotation was added to the resulting tree – the sample group label (“Control” or “Sarcoidosis”) and smoker status (“never”, “former”, “current”). (ZIP 1170 kb)
Acknowledgements
The authors thank Martina Seibert for the management of the clinical investigation and Christina Lo Porto and Beate Schmitt for excellent technical assistance conducting the wet lab work.
Funding
This work has been supported by grants from the Mukoviszidose e.V. and the Saarland University to Robert Bals.
Availability of data and materials
The original sequencing data are available at https://www.ncbi.nlm.nih.gov/sra/PRJNA510549. The code used for the analysis can be found at https://github.com/VGalata/16S_Sarcoidosis.
Abbreviations
- BAL
Bronchoalveolar lavage
- CLR
Centered log ratio
- COPD
Cryptogenic organizing pneumonia
- CT
Computer tomography
- ILD
Interstitial lung disease
- NSIP
Nonspecific interstitial pneumonia
- OTU
Operational taxonomic unit
- PCR
Polymerase chain reaction
- RNA
Ribonucleic acid
Authors’ contributions
The author contribution is as follow: AB collected clinical data and biosamples, GV analyzed data and processes biosamples, VG performed bioinformatic analysis, KR collected clinical data and biosamples, CB planned the study and analyzed the data, CH analyzed data and organized the biobank, JW performed sequence analyses, ST performed sequence analyses, HS planned the study and analyzed data, AK performed bioinformatic analysis, RB designed and planned the study, analyzed data. All authors contributed in the writing, read and approved the final manuscript.
Ethics approval and consent to participate
The PULMOHOM study was approved by the ethics board of the Landesärztekammer des Saarlandes and informed written consent was obtained from all patients.
Consent for publication
not applicable.
Competing interests
There are no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Eberhardt C, Thillai M, Parker R, Siddiqui N, Potiphar L, Goldin R, Timms JF, Wells AU, Kon OM, Wickremasinghe M, et al. Proteomic analysis of Kveim reagent identifies targets of cellular immunity in sarcoidosis. PLoS One. 2017;12(1):e0170285. doi: 10.1371/journal.pone.0170285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahmadzai H, Cameron B, Chui JJ, Lloyd A, Wakefield D, Thomas PS. Peripheral blood responses to specific antigens and CD28 in sarcoidosis. Respir Med. 2012;106(5):701–709. doi: 10.1016/j.rmed.2012.01.012. [DOI] [PubMed] [Google Scholar]
- 3.Song Z, Marzilli L, Greenlee BM, Chen ES, Silver RF, Askin FB, Teirstein AS, Zhang Y, Cotter RJ, Moller DR. Mycobacterial catalase-peroxidase is a tissue antigen and target of the adaptive immune response in systemic sarcoidosis. J Exp Med. 2005;201(5):755–767. doi: 10.1084/jem.20040429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen ES, Wahlstrom J, Song Z, Willett MH, Wiken M, Yung RC, West EE, McDyer JF, Zhang Y, Eklund A, et al. T cell responses to mycobacterial catalase-peroxidase profile a pathogenic antigen in systemic sarcoidosis. J Immunol. 2008;181(12):8784–8796. doi: 10.4049/jimmunol.181.12.8784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kosikowska U, Rybojad P, Stepien-Pysniak D, Zbikowska A, Malm A. Changes in the prevalence and biofilm formation of Haemophilus influenzae and Haemophilus parainfluenzae from the respiratory microbiota of patients with sarcoidosis. BMC Infect Dis. 2016;16(1):449. doi: 10.1186/s12879-016-1793-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zimmermann A, Knecht H, Hasler R, Zissel G, Gaede KI, Hofmann S, Nebel A, Muller-Quernheim J, Schreiber S, Fischer A. Atopobium and fusobacterium as novel candidates for sarcoidosis-associated microbiota. Eur Respir J. 2017;50(6). [DOI] [PubMed]
- 7.Clarke EL, Lauder AP, Hofstaedter CE, Hwang Y, Fitzgerald AS, Imai I, Biernat W, Rekawiecki B, Majewska H, Dubaniewicz A, et al. Microbial lineages in sarcoidosis. A metagenomic analysis tailored for low-microbial content samples. Am J Respir Crit Care Med. 2018;197(2):225–234. doi: 10.1164/rccm.201705-0891OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garzoni C, Brugger SD, Qi W, Wasmer S, Cusini A, Dumont P, Gorgievski-Hrisoho M, Muhlemann K, von Garnier C, Hilty M. Microbial communities in the respiratory tract of patients with interstitial lung disease. Thorax. 2013;68(12):1150–1156. doi: 10.1136/thoraxjnl-2012-202917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takahashi Y, Saito A, Chiba H, Kuronuma K, Ikeda K, Kobayashi T, Ariki S, Takahashi M, Sasaki Y, Takahashi H. Impaired diversity of the lung microbiome predicts progression of idiopathic pulmonary fibrosis. Respir Res. 2018;19(1):34. doi: 10.1186/s12931-018-0736-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang Y, Ma SF, Espindola MS, Vij R, Oldham JM, Huffnagle GB, Erb-Downward JR, Flaherty KR, Moore BB, White ES, et al. Microbes are associated with host innate immune response in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2017;196(2):208–219. doi: 10.1164/rccm.201607-1525OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zakharkina T, Heinzel E, Koczulla RA, Greulich T, Rentz K, Pauling JK, Baumbach J, Herrmann M, Grunewald C, Dienemann H, et al. Analysis of the airway microbiota of healthy individuals and patients with chronic obstructive pulmonary disease by T-RFLP and clone sequencing. PLoS One. 2013;8(7):e68302. doi: 10.1371/journal.pone.0068302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen NP, Warnow T, Pop M, White B. A perspective on 16S rRNA operational taxonomic unit clustering using sequence similarity. NPJ Biofilms Microbiomes. 2016;2:16004. doi: 10.1038/npjbiofilms.2016.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valeyre D, Prasse A, Nunes H, Uzunhan Y, Brillet P-Y, Müller-Quernheim J. Sarcoidosis. Lancet. 2014;383(9923):1155–1167. doi: 10.1016/S0140-6736(13)60680-7. [DOI] [PubMed] [Google Scholar]
- 14.Celada LJ, Hawkins C, Drake WP. The etiologic role of infectious antigens in sarcoidosis pathogenesis. Clin Chest Med. 2015;36(4):561–568. doi: 10.1016/j.ccm.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishige I, Eishi Y, Takemura T, Kobayashi I, Nakata K, Tanaka I, Nagaoka S, Iwai K, Watanabe K, Takizawa T, et al. Propionibacterium acnes is the most common bacterium commensal in peripheral lung tissue and mediastinal lymph nodes from subjects without sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2005;22(1):33–42. [PubMed] [Google Scholar]
- 16.Ishige I, Usui Y, Takemura T, Eishi Y. Quantitative PCR of mycobacterial and propionibacterial DNA in lymph nodes of Japanese patients with sarcoidosis. Lancet. 1999;354(9173):120–123. doi: 10.1016/S0140-6736(98)12310-3. [DOI] [PubMed] [Google Scholar]
- 17.Gabrilovich MI, Walrath J, van Lunteren J, Nethery D, Seifu M, Kern JA, Harding CV, Tuscano L, Lee H, Williams SD, et al. Disordered toll-like receptor 2 responses in the pathogenesis of pulmonary sarcoidosis. Clin Exp Immunol. 2013;173(3):512–522. doi: 10.1111/cei.12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scher JU, Joshua V, Artacho A, Abdollahi-Roodsaz S, Ockinger J, Kullberg S, Skold M, Eklund A, Grunewald J, Clemente JC, et al. The lung microbiota in early rheumatoid arthritis and autoimmunity. Microbiome. 2016;4(1):60. doi: 10.1186/s40168-016-0206-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dickson RP, Erb-Downward JR, Freeman CM, McCloskey L, Falkowski NR, Huffnagle GB, Curtis JL. Bacterial topography of the healthy human lower respiratory tract. mBio. 2017;8(1). [DOI] [PMC free article] [PubMed]
- 20.Dickson RP, Erb-Downward JR, Martinez FJ, Huffnagle GB. The microbiome and the respiratory tract. Annu Rev Physiol. 2016;78:481–504. doi: 10.1146/annurev-physiol-021115-105238. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Percentage of OTU counts per sample covered by OTUs not removed (“kept”) or discarded during the filtering step for (A) V1/V2 and (B) V3/V4. OTUs were removed if they were considered as contaminants (flag “contamination”), were present in the NTC sample (flag “controls”), had a count below 1 in non-control samples (i.e. all samples except the NTC sample; flag “count”), or any combination thereof. Figure S2. Phylogenetic OTU tree constructed by LotuS for (A) V1/V2 and (B) V3/V4. The OTUs were colored based on whether they were kept (flag “kept”) or discarded (flags “contamination”, “controls”, “counts”, or any combination thereof) during the filtering step. OTUs were removed if they were considered as contaminants (flag “contamination”), were present in the NTC sample (flag “controls”), or had a count below 1 in non-control samples (i.e. all samples except the NTC sample; flag “count”). Figure S3. PCA plots for (A) V1/V2 and (B) V3/V4. The plots show all samples using the two first principal components. The percentage of the variance explained by each principal component can be found in the axis title. The samples were colored with respect to their indication group. Figure S4. Heatmap of CLR-transformed OTU counts for (A) V1/V2 and (B) V3/V4 only for OTUs assigned by LotuS to the genus Atopobium, Fusobacterium, Mycobacterium or Propionibacterium. The samples were grouped by hierarchical clustering and additional annotation was added to the resulting tree – the sample group label (“Control” or “Sarcoidosis”) and smoker status (“never”, “former”, “current”). (ZIP 1170 kb)
Data Availability Statement
The original sequencing data are available at https://www.ncbi.nlm.nih.gov/sra/PRJNA510549. The code used for the analysis can be found at https://github.com/VGalata/16S_Sarcoidosis.