

SUMMARY
Pharmacologic targeting of components of ERK signaling in ERK-dependent tumors is often limited by adaptive resistance, frequently mediated by feedback-activation of RTK signaling and rebound of ERK activity. Here, we show that combinatorial pharmacologic targeting of ERK signaling and the SHP2 phosphatase prevents adaptive resistance in defined subsets of ERK-dependent tumors. In each tumor that was sensitive to combined treatment, p(Y542) SHP2 induction was observed in response to ERK signaling inhibition. The strategy was broadly effective in TNBC models and tumors with RAS mutations at G12, whereas tumors with RAS(G13D) or RAS(Q61X) mutations were resistant. In addition, we identified a subset of BRAF(V600E) tumors that were resistant to the combined treatment, in which FGFR was found to drive feedback-induced RAS activation, independently of SHP2. Thus, we identify molecular determinants of response to combined ERK signaling and SHP2 inhibition in ERK-dependent tumors.
In Brief
Ahmed et al. identify molecular determinants of tumor response to combined targeting with SHP2 and ERK signaling inhibitors. This strategy was effective in triple-negative breast cancer and molecularly defined subsets of RAS and BRAF-mutant tumor models. The results provide a roadmap for the translation of this strategy to the clinic.
Graphical Abstract
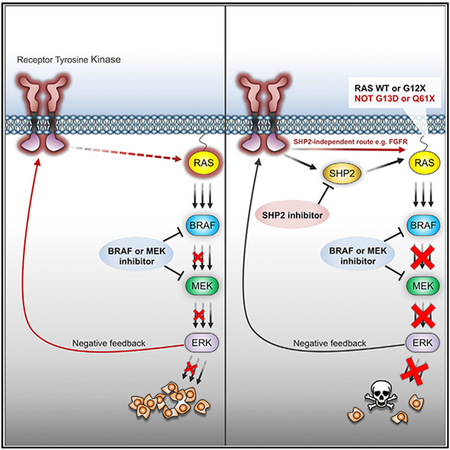
INTRODUCTION
The clinical effectiveness of therapeutic strategies targeting oncogenic signaling is often limited by mechanisms of adaptive resistance, in which initial suppression of oncogenic signaling by a drug is incomplete and temporary, followed by signaling reactivation (rebound) in the presence of the drug. Deregulated RAS/RAF/MEK/ERK signaling (extracellular signal-regulated kinase [ERK] signaling) drives growth of a large fraction of human tumors. We and others have shown that relief of negative feedback upon RAF or MEK inhibitor treatment in multiple ERK-dependent tumor contexts, promotes upregulation of various receptor tyrosine kinases (RTKs), which, in turn, activate RAS, resulting in rebound of ERK activity and development of adaptive resistance of the tumor to the inhibitor (Corcoran et al., 2012; Duncan et al., 2012; Karoulia et al., 2016; Lito et al., 2012; Montero-Conde et al., 2013; Prahallad et al., 2012; Sun et al., 2014).
The non-receptor protein tyrosine phosphatase SHP2 (PTPN11) mediates signal transduction downstream of various RTKs. It is a core component of a signaling multi-protein complex downstream of activated RTKs, which includes Grb2-associated binder (GAB) 1, GRB2, and other adaptor proteins, that promotes RAS activation by its guanine exchange factor (GEF) SOS (Dance et al., 2008; Grossmann et al., 2010). The development of small-molecule inhibitors of SHP2 provides the opportunity to potentially overcome adaptive resistance by co-targeting both oncogenic signaling and feedback-induced RTK-mediated RAS activation. Recently, SHP2 inhibition and the combination of SHP2 and ALK or MEK inhibitors were shown to have activity in tumors with deregulated ALK (Dardaei et al., 2018) or RAS (Mainardi et al., 2018; Ruess et al., 2018; Wong et al., 2018) signaling, but whether the combined SHP2 and ERK signaling inhibition would be effective in the broader context of ERK-dependent tumors is not known. Thus, we used a recently developed allosteric small-molecule inhibitor of SHP2, SHP099 (Chen et al., 2016; Garcia Fortanet et al., 2016), in an effort to identify molecular determinants of sensitivity and resistance to combined SHP2 and ERK signaling inhibition in ERK-driven tumors.
RESULTS
The Small-Molecule SHP099 Disrupts the SHP2 Signaling Complex
SHP099 was shown to bind to the closed conformation of SHP2 and inhibit its catalytic activity (Chen et al., 2016). The chemical structure and reported selectivity of this and other inhibitors used in this study are shown in Figure S1. To investigate the mechanism by which SHP099 suppresses SHP2 signaling, we treated HeLa cells with epidermal growth factor (EGF) and monitored the formation of the SHP2 signaling complex in the absence or presence of SHP099. EGF stimulation promoted the interaction of SHP2 with EGF receptor (EGFR), GAB1, GRB2, and SOS1, induced SHP2 phosphorylation at Y542 (p(Y542)SHP2), a surrogate marker of SHP2 activation downstream of RTK activation (Araki et al., 2003; Bennett et al., 1994; Dance et al., 2008), as well as pMEK and pERK. SHP099 pretreatment disrupted this interaction and diminished SHP2 phosphorylation as well as ERK signaling activation without affecting EGFR phosphorylation (Figure 1A), confirming that SHP2 functions downstream of RTK signaling and upstream of RAS. Consistent with this observation, treatment with SHP099 disrupted the SHP2/GRB2 complex and suppressed SHP2 phosphorylation as well as RAS and ERK activity in HER2- or EGFR-amplified tumor cells (Figure 1B).
Figure 1. SHP099 Disrupts the SHP2/GRB2 Complex and Suppresses RAS Activity and ERK Signaling.

(A) HeLa cells were pretreated with either DMSO or SHP099 (SHP, 10 μM) for 1 hr before stimulation with epidermal growth factor (EGF, 10 ng/mL) for the indicated times. Cell lysates were either subjected to immunoprecipitation with a SHP2 antibody, followed by immunoblotting, or immunoblotted with the indicated antibodies.
(B) The indicated RTK-overexpressing cell lines were treated with either SHP, lapatinib (LAP), or gefitinib (GEF) at 10 μM for 1 hr. Cell lysates were subjected to immunoprecipitation with a SHP2 antibody and immunoblotted for GRB2 and SHP2 or subjected to RAS-pull down assay and immunoblotted for RAS. Total cell lysates were also immunoblotted with the indicated antibodies.
Single MEK or SHP2 Inhibitor Treatments Are Accompanied by Rebound of ERK Signaling in TNBCs
Targeting ERK signaling with MEK inhibitors has shown preclinical activity in triple-negative breast cancer (TNBC) models (Hoeflich et al., 2009); however, acute inhibition of ERK activity causes relief of negative feedback, which promotes upstream RTK upregulation and RAS activation resulting in rebound of pERK in the presence of the inhibitor (Duncan et al., 2012). In a panel of TNBC cell lines treated with the MEK inhibitor trametinib, we observed suppression of pERK within 1 hr of treatment, followed by a significant pERK rebound at 24 hr (Figure 2A). Moreover, the pERK rebound was associated with upregulation of p(Y542)SHP2 in all TNBC lines analyzed (Figure 2A). These results suggested a possible role for SHP2 in mediating RTK-driven adaptive resistance to MEK inhibition in these tumor cells.
Figure 2. Combination of SHP2 and MEK Inhibitors Overcomes the Feedback-Induced pERK Rebound and Suppresses Growth of TNBC Cells.

(A) The indicated TNBC tumor lines were treated with trametinib (TRAM, 20 nM), and SHP2 and ERK activity were monitored for 24 hr by immunoblotting with the indicated antibodies.
(B) The same cell lines were treated with SHP (5 μM), and the pERK rebound was monitored for 24 hr by immunoblotting with the indicated antibodies.
(C) Crystal violet cell growth assays of the indicated TNBC cell lines treated with SHP (1 and 5 μM).
(D) The indicated TNBC tumor lines were treated with TRAM (20 nM), SHP (5 μM), or the combination, and p(Y542)SHP2 and ERK signaling were monitored for 24 hr by immunoblotting with the indicated antibodies.
(E) The indicated cell lines were treated with DMSO, TRAM (20 nM), or TRAM (20 nM) and SHP (5 μM) for 24 hr. Cell lysates were subjected to immunoprecipitation with a SHP2 antibody and immunoblotted for GAB1, GRB2, and SHP2 or were subjected to RAS-pull down assay and immunoblotted for RAS. Total lysates were subjected to immunoblotting with the indicated antibodies.
(F) Crystal violet cell growth assays assessing the effect of TRAM (20 nM), SHP (5 and 10 mM), and the combination in the indicated TNBC cell lines.
SHP2 is a critical mediator of RAS/ERK signaling in response to EGFR activation (Feng et al., 1993). EGFR expression is upregulated in the majority of TNBCs (Gumuskaya et al., 2010), and its expression has been found to correlate with that of SHP2 (Matalkah et al., 2016), consistent with the importance of SHP2 in EGFR signaling. We thus investigated the effectiveness of the SHP2 inhibitor as single agent in TNBC tumor cells. Treatment of TNBC cell lines with SHP099 resulted in only short-term inhibition and subsequent pERK rebound (Figure 2B), which was associated with minimal effect on cell growth (Figure 2C). These results suggested that treatment with SHP099 alone promoted adaptive resistance mechanisms analogous to those elicited by other ERK signaling inhibitors (i.e., reactivation of signaling because of relief from negative feedback), resulting in the inability of SHP099 to effectively suppress activated SHP2 and, consequently, the pERK rebound. Thus, in TNBC, MEK activity has been shown to mediate the pERK rebound downstream of SHP2 inhibition (Duncan et al., 2012), and conversely, we found that pERK rebound downstream of MEK inhibition is associated with SHP2 activation (Figure 2A).
Combined MEK and SHP2 Inhibition Overcomes Adaptive Resistance to Either Inhibitor Alone in TNBC Models
Although trametinib or SHP099 treatment alone resulted in transient inhibition of ERK signaling and minimal to modest inhibition of TNBC growth (Figures 2A–2C), we reasoned that combined targeting of MEK and SHP2 might prevent the development of adaptive resistance in these tumor lines. In fact, combined treatment with trametinib and SHP099 resulted in potent and durable suppression of pERK (Figure 2D), associated with disruption of the SHP2 interaction with GAB1 and GRB2 and reduction in RAS activity (Figure 2E). Moreover, the combination induced profound growth inhibition in all TNBC models tested, including EGFR-amplified (MDA-MB-468 and BT-20), RAS mutant (MDAMB-231, Hs 578T, and SUM159), neurofibromatosis type 1 (NF1) mutant (MDA-MB-157), and other TNBC cell lines (Figure 2F), suggesting that co-targeting of MEK and SHP2 could serve as a powerful therapeutic approach in TNBC, for which targeted therapeutics are currently lacking.
Various RAS Mutations Predict Different Sensitivities to Either SHP2 or Combined MEK and SHP2 Inhibition
Activating RAS mutations occur in more than 30% of human tumors (Baines et al., 2011) and have been long considered independent of upstream signaling. However, the recent development of irreversible inhibitors of RAS(G12C) revealed the dependency of this RAS mutant on RTK-driven GEF activity in cells (Janes et al., 2018; Lito et al., 2016; Ostrem et al., 2013; Patricelli et al., 2016). These findings raised the possibility that RAS(G12C) or other RAS mutations might be dependent on SHP2 activity; we thus assessed the effect of SHP099 alone or in combination with trametinib in various RAS mutant tumor cells.
We found that short-term (2 hr) treatment with SHP099 suppressed RAS activity and pERK selectively in RAS(G12X) mutant cells but not in RAS(G13D) or RAS(Q61X) mutant cells (Figures 3A, 3B, and S2A). Further, among RAS(G12C) mutant cells, H358 and MIAPaCa-2 showed greater sensitivity to SHP099 alone compared with Calu-1 or H1792 cell lines, (Figures 3B and 3C). Sensitivity to SHP099 correlated well with basal levels of RAS activity, which were greater in the SHP099-sensitive H358 and MIAPaCa-2 cells compared with the less-sensitive Calu-1 and H1792 cell lines (Figure 3D). Of note, combined trametinib and SHP099 treatment effectively suppressed ERK signaling and cell growth of all RAS(G12C) tumor lines (Figures 3B and 3C). We further found tumor lines with other RAS(G12X) mutant cell lines to be sensitive to the MEK and SHP2 combination, including RAS(G12A) or RAS(G12S) (Figures 3B and 3C), suggesting that additional RAS(G12X) mutations likely also require GEF activity and, therefore, depend on SHP2.
Figure 3. Combined MEK and SHP2 Inhibition Is Effective in KRAS (G12X) Mutant Cancer Cells.

(A) The indicated RAS(G12X), RAS(G13D), or RAS(Q61X) mutant cell lines were treated with SHP (10 μM) for 2 hr. Cell lysates were subjected to RAS-pull down assay and immunoblotted for RAS. Total lysates were subjected to immunoblotting with the indicated antibodies.
(B) RAS(G12X), RAS(G13D), or RAS(Q61X) mutant cell lines were treated with TRAM (5 nM), SHP (10 μM), or the combination, and pERK levels were detected at 2 and 24 hr by immunoblotting with the indicated antibodies. A549 and H1792 cells were treated with 1 or 10 nM TRAM, respectively.
(C) Crystal violet cell growth assays assessing the effect of TRAM (1 nM), SHP (5 and 10 μM), and the combination in the indicated RAS(G12X), RAS(G13D), and RAS(Q61X) mutant cell lines. H358 cells were treated with TRAM (5 nM) and SHP (0.1, 0.3, 1, 3, and 10 mM). A549 and H1792 cells were treated with 5 or 10 nM TRAM, respectively.
(D) RAS(G12C)-expressing tumor cells were subjected to RAS-pull down assay and immunoblotted with the indicated antibodies. See also Figure S2.
In contrast to RAS(G12X) mutant cells, tumor cells expressing RAS(G13D) or RAS(Q61X) mutations were resistant to SHP099 alone and relatively insensitive to the SHP099 and trametinib combination (Figures 3A–3C and S2A). Moreover, culturing RAS(G13D) or RAS(Q61X) mutant tumor cells under conditions of low serum did not confer sensitivity to SHP099 on either ERK signaling or cell growth (Figures S2B and S2C). Together, these data indicate that RAS(G13D) and RAS(Q61X) activate RAS in the absence of upstream RTK/SHP2 signaling, consistent with previously reported biochemical properties of these mutants (a high rate of nucleotide exchange and very low intrinsic GTPase activity, respectively [Hunter et al., 2015]).
SHP2 Inhibition Overcomes Adaptive Resistance to RAF Inhibitor in a Subset of BRAF(V600E) Colorectal and Thyroid Tumors in Which Negative Feedback Induces p(Y542)SHP2
Unlike BRAF(V600E) melanomas, BRAF(V600E) colorectal and thyroid cancers showed modest response to RAF inhibitors, due to RTK-mediated adaptive resistance resulting in pERK rebound in such tumors (Corcoran et al., 2012; Karoulia et al., 2016; Montero-Conde et al., 2013; Prahallad et al., 2012, 2015). SHP2 has been identified previously by a genetic screen as a mediator of adaptive resistance to RAF inhibitors in BRAF(V600E) colorectal cells, and pharmacologic or genetic targeting of SHP2 was shown to sensitize those cells to vemurafenib (Prahallad et al., 2015). We thus asked whether combining the RAF inhibitor vemurafenib (VEM) with SHP099 would potentiate VEM effectiveness. In BRAF(V600E) colorectal tumor lines, including RKO, WiDr, and HT-29 as well as the thyroid line 8505C, the combination of VEM and SHP099 resulted in potent suppression of the pERK rebound and marked inhibition of cell growth (Figures 4A, 4B, S3A, and S3B). Consistent with those findings, VEM treatment of BRAF(V600E) tumor cells sensitive to the combination resulted in the formation of the GRB2/SHP2 complex and in RAS activation, both of which were abrogated upon co-treatment with SHP099 (Figures 4C and 4D). In contrast, Hth104 and SW1736 thyroid and SW1417 colorectal tumor lines exhibited RAS activation and pERK rebound in response to VEM, which were completely insensitive to added SHP2 inhibition (Figures 4A, 4C, and S3A), with neither treatment antagonizing the growth of these tumor cells (Figures 4B and S3B). Of note, insensitive SW1736 cells showed less or no interaction of SHP2 with GRB2 and GAB1 compared with sensitive HT-29 cells when treated with VEM (Figure 4E).
Figure 4. A Subset of Colorectal and Thyroid BRAF(V600E) Tumor Cells Are Sensitive to Combined BRAF and SHP2 Inhibition.

(A) Colorectal BRAF(V600E) (RKO, HT-29, WiDr, and SW1417) and Thyroid BRAF(V600E) (8505C, SW1736, and Hth104) cells were treated with vemurafenib (VEM, 2 μM), SHP (5 and 10 μM), or the combination, and pERK levels were monitored for 48 hr by immunoblotting with the indicated antibodies. The 8505C and HT-29 cells were treated with 1 or 3 μM VEM, respectively.
(B) Crystal violet cell growth assays assessing the growth inhibitory effect of VEM (2 μM), SHP (5 or 10 μM), and the combination in the indicated colorectal and thyroid BRAF(V600E) cell lines.
(C) The indicated tumor cells were treated with VEM, SHP, or the combination for 24 hr then subjected to RAS-GTP pull-down assay. Total cell lysates were subjected to immunoblotting with the indicated antibodies.
(D) HT-29 and WiDr cells were treated with DMSO, VEM, SHP, or the combination for 24 hr. Cell lysates were subjected to immunoprecipitation with a SHP2 antibody and immunoblotted for GRB2 and SHP2.
(E) HT-29 and SW1736 cells were treated with VEM for 24 hr. Cell lysates were subjected to immunoprecipitation with a SHP2 antibody and immunoblotted for GAB1, GRB2, and SHP2. Total lysates were subjected to immunoblotting with the indicated antibodies.
(F) The indicated tumor cells were treated with either VEM (2 μM in BRAF(V600E)-expressing cells) or TRAM (20 nM in TNBC cells) for 24 hr, and the indicated proteins were detected by immunoblotting.
(G) Colorectal BRAF(V600E) (RKO, HT-29, and WiDr) cells were treated with dabrafenib (DAB, 100 nM) and TRAM (1 nM), SHP (10 μM), or the combination, and reactivation of ERK signaling was monitored for 24 hr by immunoblotting with the indicated antibodies.
(H) Mice bearing RKO xenografts were treated with vehicle (tumors 1–3) and DAB (30 mg/kg) and TRAM (0.25 mg/kg) for 48 hr (tumors 4–6) or DAB and TRAM followed by SHP (75 mg/kg, once daily) for 24 hr (tumors 7–9). Tumors were collected and ERK and SHP2 activity were determined by immunoblotting.
(I) RKO cells were injected subcutaneously into the flanks of nude mice (10 million cells/injection). When tumors reached 100–150 mm3 in size, the indicated treatments started. Tumors were collected after 21 days of treatment or when tumors reached approximately 1,000 mm3 and were lysed and subjected to immunoblotting with the indicated antibodies.
(J) Waterfall graph showing the fold change in tumor volume compared with baseline in mice bearing the RKO xenografts (n = 7 mice/arm) after 21 days of the indicated treatment (p values calculated using unpaired t test).
See also Figure S3.
Furthermore, we observed that in the SHP2-dependent BRAF(V600E) tumor cells, p(Y542)SHP2 was detected at basal level (“SHP2-positive”), which was further induced upon treatment with RAF inhibitor but suppressed upon treatment with SHP099 (Figure 4C). In contrast, p(Y542)SHP2 was virtually undetectable in certain BRAF(V600E) cells, in which RAS was feedback-activated independent of SHP2 (“SHP2-negative”) (Figure 4C). These findings raised the possibility that a lack of SHP2 phosphorylation may help predict independence on SHP2 for feedback-induced RAS activation and thus tumor insensitivity to combined SHP2/ERK signaling inhibition. Thus, we compared levels of p(Y542)SHP2 upon relief of negative feedback using ERK signaling inhibitors in a larger panel of cell lines. The results confirmed that, in each case (TNBC, RAS mutant, and BRAF(V600E) tumor lines), the efficacy of the combined SHP2 and ERK signaling inhibition was associated with detectable p(Y542)SHP2 (Figure 4F and not shown). These findings suggest that low levels of p(Y542)SHP2 may serve as an indicator of resistance to the combination of SHP2 and ERK signaling inhibitors.
To assess the in vivo effectiveness of combined ERK signaling and SHP2 inhibition, we treated mice carrying RKO xenografts with the triple combination of the U.S. Food and Drug Administration (FDA)–approved RAF and MEK inhibitor combination (dabrafenib and trametinib, respectively) and SHP099, after confirming it was more effective than dabrafenib and trametinib in inhibiting ERK signaling ex vivo (Figure 4G). Dabrafenib and trametinib or SHP099 alone had minimal effect on xenograft tumor growth or ERK signaling (Figures 4H–4J). However, the triple combination dabrafenib, trametinib, and SHP099 markedly suppressed p(Y542)SHP2 (Figure 4H) and ERK signaling (Figure 4I) and growth (Figure 4J) of RKO xenograft tumors, without any obvious effect on body weight (Figure S3C), providing further evidence that combined ERK signaling and SHP2 inhibition may be an effective therapeutic strategy for patients with BRAF(V600E) colorectal tumors.
ERBB Family or MET Activation Promotes Adaptive Resistance to RAF Inhibitor via SHP2-Dependent RAS Activation in BRAF(V600E) Colorectal Tumors
To dissect the molecular mechanisms underlying BRAF(V600E)-expressing thyroid and colorectal tumors with SHP2-dependent and SHP2-independent adaptive resistance to RAF inhibition (“SHP2-positive” and “SHP2-negative,” respectively), we treated cells with VEM for 48 hr, followed by different RTK inhibitors for 2 hr and examined their effect on the pERK rebound. ERBB family inhibitors (gefitinib, lapatinib, and AZD8931) potently suppressed the pERK rebound in WiDr and HT-29 cells but failed to do so in RKO cells or in any of the SHP2-negative tumor cells (Figure 5A). To identify additional RTKs beyond the ERBB family that might be drivers of feedback-induced RAS activation, we performed RTK arrays after treatment with VEM in RKO and in the SHP2-negative cells. In RKO, phosphorylation of multiple RTKs, including MET and AXL, was detected (Figures 5B, 5C, and S4). Treatment of RKO cells with the MET inhibitors crizotinib or cabozantinib, an inhibitor of both MET and AXL among other kinases, but not with the AXL inhibitor R428, potently suppressed the pERK rebound after VEM treatment (Figures 5C and 5D) as well as MET phosphorylation (Figure 5C). Together, these results argued that, in RKO negative feedback-induced RAS, activation was mediated by MET signaling through SHP2.
Figure 5. Inhibitors of Members of the ERBB Family or the MET Receptor Suppress the pERK Rebound in Colorectal BRAF(V600E)-Tumor Cells.

(A) Indicated tumor cells were treated with 2 μM VEM for 48 hr, followed by different ERBB inhibitors at different concentrations (0.2 and 2 μM) for 2 hr. Lysates were subjected to immunoblotting with the indicated antibodies. GEF, gefitinib; LAP, lapatinib; AZD, AZD8931.
(B) Cells were treated with or without 2 μM VEM for 24 hr. Levels of phosphorylated RTKs in cell lysates were detected using phospho-RTK arrays.
(C) RKO cells were treated with 2 μM VEM for 48 hr, followed by crizotinib (CRIZ, 2 μM) for 2 hr. Cell lysates were either subjected to immunoprecipitation with a MET antibody and immunoblotted for pMET or subjected to immunoblotting with the indicated antibodies.
(D) Cells were treated with 2 μM VEM for 48 hr, followed by different RTK inhibitors at different concentrations (0.2 μM or 2 μM) for 2 hr. Lysates were subjected to immunoblotting with the indicated antibodies. CRIZ, crizotinib; CABO, cabozantinib; GSK, GSK1904529A. See also Figures S4 and S5.
Feedback Activation of Fibroblast Growth Factor Receptor in Response to VEM Can Promote RAS Activation Independent of SHP2
In the SHP2-negative cells, none of the aforementioned inhibitors or the IGF-1R and insulin receptor (IR) inhibitor GSK1904529A in SW1417 cells, in which both IGF-1R and IR up-regulation were detected in the RTK array, suppressed the VEM-induced pERK rebound (Figures 5A, 5B, and 5D), suggesting that another RTK or upstream factor drives feedback-induced RAS independent of SHP2 in these cells.
Upregulation of HER3 was previously reported to mediate adaptive resistance to RAF inhibitors in BRAF(V600E) thyroid tumor cells (Montero-Conde et al., 2013). In agreement with that report, both our RTK array and western blot analysis revealed evidence of upregulated HER3 phosphorylation in all thyroid tumor cell lines analyzed, including the two SHP2-negative lines Hth104 and SW1736 (Figure 5B and S5A). The observation that HER3 upregulation coincided with feedback-induced RAS activation in SHP2-negative cells, prompted us to ask whether HER3 was able to induce the pERK rebound independently of SHP2. Thus, we treated luminal breast cancer cell lines that express HER3, including MCF7, T47D, BT474, and SK-BR-3, with the HER3 ligand neuregulin (NRG), which induced pERK and p(Y542)SHP2, similar to EGF (Figures S5B and S5C). Moreover, treatment with SHP099 blocked NRG-induced ERK activation, similarly to its inhibitory effect upon EGF stimulation (Figures S5B and S5C). These data suggested that ERK activation downstream of HER3 is mediated by SHP2 activation. However, pharmacological or shRNA-mediated targeting of HER3 had no effect on the pERK rebound after VEM treatment in Hth104 and SW1736 thyroid tumor cells (Figures 5A and S5D). These findings suggested that HER3 was unlikely to mediate the pERK rebound in response to RAF inhibitor treatment in this setting.
To search for other drivers of feedback-induced RAS activation in the SHP2-negative BRAF(V600E) cells, we carried out RNA sequencing (RNA-seq) analysis before and after treatment with VEM, with the SHP2-positive cell line WiDr used for comparison (Figure 6A). RNA-seq analysis showed no obvious differences in expression levels of known RAS activity regulators (RAS GEFs and GTPase activating proteins [GAPs]). Among multiple chemokines and cytokines upregulated in response to VEM treatment, interleukin 6 (IL-6) showed much higher expression levels in the SHP2-negative than in SHP2-positive cells, in line with a previous report implicating IL-6 in driving adaptive resistance to RAF inhibitors (Sos et al., 2014). However, treatment of SW1736 cells with IL-6 did not induce RAS or ERK activation, and pSTAT3 induction by IL-6 was not affected by SHP099 (Figures S6A and S6B). Further, small interfering RNA (siRNA)-mediated knockdown of IL-6 in SW1736 cells did not suppress the pERK rebound upon VEM treatment (Figure S6C), suggesting that IL-6 was unlikely to mediate the pERK rebound in response to RAF inhibitor treatment in this context.
Figure 6. FGFR Inhibition Overcomes Primary Resistance to RAF Inhibitor in a Subset of SHP2-Negative BRAF(V600E) Cancer Cells.

(A) Heatmaps of SHP2-negative (Hth104 and SW1736), compared with SHP2-positive (WiDr), BRAF(V600E) cells treated with VEM at 2 μM for 24 hr, showing normalized expression of genes associated with RAS activity regulation, chemokine and cytokines pathways, and RTK pathways. Heatmaps were generated with Morpheus software (Broad Institute, Cambridge, MA, USA).
(B) HT-29, WiDr, Hth104, and SW1736 cells were treated with VEM (2 μM) for 24 hr. Total cell lysates were subjected to immunoblotting with the indicated antibodies.
(C) HT-29 and SW1736 cells were treated with VEM (2 μM) for 24 hr, followed by GEF (2 μM) or BGJ398 (BGJ, 5 μM) for 2 hr, then stimulated with EGF (10 ng/mL) or FGF2 (100 ng/mL) for 10 min, respectively. Total lysates were subjected to immunoblotting with the indicated antibodies.
(D) Hth104 or SW1736 cells were treated with 2 μM VEM for 48 hr followed by either ponatinib (PON) or BGJ at increasing concentrations for 2 hr. Total lysates were subjected to immunoblotting with the indicated antibodies.
(E) Hth104 or SW1736 cells were treated with 2 μM VEM for 48 hr, followed by BGJ (5 μM) for 2 hr, then subjected to RAS-GTP pull-down assay. Total lysates were subjected to immunoblotting with the indicated antibodies.
(F) Hth104 or SW1736 cells were treated with 2 μM VEM combined with either PON (500 or 750 nM) or BGJ (1 or 5 μM) for 24 hr. Total lysates were subjected to immunoblotting with the indicated antibodies.
(G) Hth104 cells were transfected with either non-targeting control or FGFR1 siRNA (100 nM) for 24 hr then treated for either 1 or 24 hr with VEM (2 μM). FGFR1 expression and ERK phosphorylation were determined by immunoblotting.
(H) SW1736 cells were transfected with non-targeting control, FGFR1 or FGFR2 siRNA (100 nM), or the combination (75 nM each) for 24 hr, followed by treatment with VEM (2 μM/24 hr). FGFR1 and FGFR2 expression and ERK phosphorylation were determined by immunoblotting.
(I) Crystal violet cell growth assays assessing the effect of VEM (1, 2, or 4 μM), PON (100 nM), or the combination in Hth104 or SW1736 cells.
(J) Crystal violet cell growth assays assessing the effect of VEM (2 μM), BGJ (0.5 or 1 mM), or the combination in Hth104 and SW1736 cells. See also Figures S6 and S7.
We next focused on identifying candidate RTKs, whose RNA expression were increased at either basal levels or in response to VEM treatment in the SHP2-negative cell lines Hth104 and SW1736. In fact, RNA expression levels of both the fibroblast growth factor receptor 1 (FGFR1) and its ligand FGF2 were several-fold higher in these lines as compared with the SHP2-positive cell line WiDr (Figure 6A). Consistent with the RNA expression data, protein expression levels of FGFR1 and FGF2, as well as FGFR1 phosphorylation, were much higher in SW1736 and Hth104, compared with WiDr and HT-29 (Figure 6B). Moreover, SW1736 cells were also expressing high levels of FGFR2 (Figure 6B). We further found that stimulation of FGFR using FGF2 in SW1736 induced pERK without detectable p(Y542)SHP2, whereas EGFR stimulation caused a substantial induction of p(Y542)SHP2 in HT-29 cells (Figure 6C), indicating that, in SW1736 cells, FGFR signaling can drive RAS/ERK activation independent of SHP2. We next assessed the effect of combining VEM with pan-FGFR inhibitors, such as BGJ398 or ponatinib. In each case, BGJ398 or ponatinib suppressed RAS activation (Figure S7A) and the pERK rebound (Figure 6D) after 48 hr of treatment with VEM. Moreover, simultaneous treatment of VEM and ponatinib or BGJ398 suppressed RAS activity and pERK more potently compared with either compound alone (Figures 6E and 6F). These results suggested that FGFR activity mediates feedback-induced, SHP2-independent RAS activation in these cells.
Further, siRNA-mediated knockdown of FGFR1 or the double knockdown FGFR1/FGFR2, diminished the pERK rebound in Hth104 and SW1736 cells, respectively (Figures 6G and 6H). Finally, combinatorial treatments of VEM with either ponatinib or BGJ398 potently suppressed cell growth of both Hth104 and SW1736 cells (Figures 6I and 6J), arguing strongly that co-targeting of FGFR and ERK signaling may be an effective strategy for at least some SHP2-negative tumor cells.
DISCUSSION
Adaptive drug resistance is a major challenge to the clinical success of cancer therapies. Incomplete inhibition of oncogenic signaling allows survival of “drug-tolerant” tumor cells, which persist in that state for variable periods before acquiring additional genetic mutations associated with acquired drug-resistance and tumor relapse (Hata et al., 2016; Sharma et al., 2010). For example, complete suppression of ERK activity (over 85%) has been shown to be required for significant tumor response in BRAF mutant melanomas (Bollag et al., 2010). In this context, adaptive resistance is frequently associated with homeostatic mechanisms, such as negative feedback, which are mobilized upon target inhibition and lead to ERK signaling rebound in the presence of the drug. Activation of RTK signaling has been found to drive adaptive resistance in various ERK-dependent tumor contexts, including BRAF(V600E) melanoma, colorectal and thyroid cancers, and TNBC and RAS mutant tumors (Chandarlapaty, 2012; Corcoran et al., 2012; Duncan et al., 2012; Karoulia et al., 2016, 2017; Lito et al., 2012; Montero-Conde et al., 2013; Prahallad et al., 2012; Samatar and Poulikakos, 2014; Shaffer et al., 2017; Sun and Bernards, 2014). However, because adaptive resistance is mediated by various RTKs across various tumor types, or even in the same tumor, establishing effective approaches for combined targeting of ERK signaling and individual RTKs is challenging. Here, we characterized the strategy of targeting SHP2, a phosphatase that mediates RAS activation downstream of multiple RTKs to overcome adaptive resistance to ERK signaling inhibitors.
SHP2 (PTPN11) has been found to be required for full activation of RAS/ERK pathway in several contexts (Dance et al., 2008); however, the mechanistic details of how SHP2 regulates RAS activity downstream of RTK signaling remain unclear. Catalytic (phosphatase) activity of SHP2 has been shown to be critical for RAS/ERK activation, and SHP2 has been reported to dephosphorylate a number of proteins, including platelet-derived growth factor receptor (PDGFR) (Klinghoffer and Kazlauskas, 1995), EGFR(Agazie and Hayman, 2003), and GAB (Montagner et al., 2005); however, the relevant SHP2 substrate has not been conclusively identified. On the other hand, SHP2 has been shown to act as a scaffold protein recruiting GRB2/SOS complex to the membrane and promoting RAS activation (Dance et al., 2008; Grossmann et al., 2010). The allosteric SHP2 inhibitor used in our study (SHP099) both inhibits the catalytic activity and stabilizes the inactive conformation of SHP2 (Chen et al., 2016), resulting in the disruption of SHP2 interaction with other adaptor proteins, such as GRB2 and GAB1, and the concomitant decrease of RAS activity.
In this study, we used p(Y542)SHP2 as a surrogate marker for SHP2 activation. However, the regulatory role of the SHP2 C-terminal phosphorylation remains incompletely understood. It has been shown that the phosphorylation of tyrosine 542 and 580 at the C-terminal tail of SHP2 are the main recruitment events for GRB2/SOS and subsequent activation of downstream RAS/ERK signaling (Bennett et al., 1994; Vogel and Ull-rich, 1996), whereas another report showed that mutation of those sites had no functional effect on SHP2 signaling (O’Reilly and Neel, 1998). Nonetheless, in our experiments using SHP099, the interaction of SHP2 with GRB2 and GAB1 and RAS/ERK activation always correlated with phosphorylation of SHP2 at Y542, suggesting that, at least in this context, p(Y542)SHP2 can serve as a marker of SHP2 activation downstream of RTK signaling.
TNBC represents about 15% of breast tumors, and typically has a poorer outcome compared with other breast cancer sub-types because of an inherently more aggressive clinical behavior and the current lack of targeted therapeutic options (Bianchini et al., 2016). MEK inhibitors have shown preclinical activity in TNBC (Hoeflich et al., 2009), but feedback activation of upstream RTKs has been shown to limit their efficacy (Duncan et al., 2012). We show here that combined MEK and SHP2 inhibition had a profound inhibitory effect on both ERK signaling and cell growth in all TNBC lines tested, including RAS mutant and RTK-overex-pressing TNBC tumor cells, suggesting that this combination provides a powerful therapeutic strategy for patients with this aggressive tumor type (Figure 7A).
Figure 7. Schematic of the Role of SHP2 in Driving Adaptive Resistance to ERK Signaling Inhibition in Different Molecular Contexts.

(A) In tumors with deregulated ERK signaling and wild-type BRAF and RAS (as in the majority of TNBCs), negative feedback downstream of ERK suppresses RTK signaling (left). Inhibition of ERK activity by a MEK inhibitor induces upregulation of feedback-suppressed RTKs, which activate RAS resulting in incomplete inhibition of ERK activity (middle). Combined targeting of MEK and SHP2 results in potent inhibition of ERK signaling thereby overcoming adaptive resistance to the MEK inhibitor (right).
(B) In tumors with RAS mutations, relief of negative feedback of RTKs by MEK inhibitor attenuates its effect in suppressing ERK signaling (left). Sensitivity to the combination is more prominent in cells with RAS(G12) mutants (middle), whereas expression of RAS(G13D) or RAS(Q61X) is predictive of resistance (right).
(C) In tumors with BRAF(V600E) mutations, RTKs drive adaptive resistance and the pERK rebound upon treatment with a RAF inhibitor or the combination of a RAF and a MEK inhibitor (left). We identified cells sensitive (middle) and cells completely resistant (right) to the combination. In the latter, ‘‘SHP2-negative’’ cells, SHP2 activity was very low (i.e., undetectable p(Y542)SHP2), and the feedback-induced RAS activity and ERK rebound were independent of SHP2. In two such cases, we identified another RTK, FGFR, driving RAS activation independent of SHP2 (right).
Among RAS mutant tumors analyzed, we found that the efficacy of either the SHP2 inhibitor SHP099, or combined MEK and SHP2 inhibition was best in those expressing RAS mutations at G12 (Figure 7B). Recent studies revealed that cellular RAS(G12C) activity depends on RTK-mediated nucleotide exchange, and binding of an irreversible RAS(G12C) inhibitor promotes dissociation of RAS(G12C) from the GEF SOS (Janes et al., 2018; Lito et al., 2016; Ostrem et al., 2013; Patricelli et al., 2016). In our studies, RAS(G12C) tumor cells showed variable degrees of sensitivity to SHP099, which paralleled reported sensitivity of the same cell lines to the RAS(G12C) inhibitors (Lito et al., 2016; Patricelli et al., 2016), consistent with both compounds affecting a common underlying dependence of RAS(G12C) on GEF activity. The combination of MEK and SHP2 inhibition was effective in RAS(G12C) cells, consistent with previously reported data showing low, but detectable, intrinsic GTPase activity retained in such mutants (Hunter et al., 2015). We further found that SHP2 inhibition suppressed ERK activity and that combined MEK and SHP2 inhibition was effective in cells with other RAS(G12X) mutants, such as RAS(G12S) and RAS(G12A). These results suggest that, although these RAS mutants did not show substantial GTPase activity in biochemical assays as purified proteins (Hunter et al., 2015), they may still depend on nucleotide exchange in cells. Alternatively, it is possible that, in certain contexts, pERK activity is regulated by SHP2 via additional mechanisms to GEF (SOS) recruitment. Further biochemical and cell-based studies are warranted to delineate the role of RTK/SHP2 and nucleotide exchange in the regulation of the different RAS mutants in cells.
Recent studies reported on the dependency of tumors with mutant RAS on SHP2 (Fedele et al., 2018; Mainardi et al., 2018; Nichols et al., 2018; Ruess et al., 2018). Consistent with those studies, we found RAS(G12X) mutants, but not RAS(Q61X), to be dependent on SHP2. However, although Mainardi et al. (2018) reported that RAS(G13D) signals in a SHP2-dependent manner, we found that RAS(G13D)-driven ERK activity was independent of SHP2 in the cell line models analyzed, and culturing conditions at low serum levels did not confer sensitivity to SHP2 inhibition. RAS(G13D) has been found previously in biochemical assays to retain low, but detectable, intrinsic GTPase activity and, in theory, could require upstream RTK signaling to maintain its active state. However, the same biochemical studies also found that this mutant exhibited an order of magnitude higher rate of nucleotide exchange, compared with wild-type RAS (Hunter et al., 2015; Smith et al., 2013). Thus, the much greater cellular concentration of GTP compared with GDP could result in SOS-independent auto-activation (Figure 7B), consistent with our findings with RAS(G13D) tumor cells.
In BRAF(V600E) colorectal and thyroid tumors, we observed upregulation of multiple RTKs in response to ERK signaling inhibition (Figure 7C). By using a combination of pharmacological and knockdown targeting of specific RTKs to dissect the relative contribution of feedback-induced RTKs to RAS activation, we identified a role of EGFR signaling in a subset of colorectal BRAF(V600E) tumor lines, consistent with previous reports (Corcoran et al., 2012; Prahallad et al., 2012). We also identified an example of adaptive resistance to RAF inhibitors driven by another RTK, MET, rather than by the ERBB family. In each case, the tumor cells were also sensitive to combined RAF and SHP2 inhibition, indicating that SHP2 inhibition in combination with RAF and MEK inhibitors may be effective in a broader range of colorectal BRAF(V600E) tumors than combined targeting with EGFR/RAF/MEK, a drug combination recently assessed clinically in this context with modest results (Corcoran et al., 2018).
In two BRAF(V600E)-expressing tumor lines, in which both basal and RAF inhibitor-induced p(Y542)SHP2 levels were virtually undetectable (‘‘SHP2-negative’’), we identified FGFR signaling driving RAS activation in response to ERK signaling inhibition (Figure 7C). These observations are consistent with previous findings that FGFR is able to signal both dependently or independently of SHP2 in different settings (Hadari et al., 1998; Kouhara et al., 1997). In a third SHP2-negative tumor line, SW1417, selective inhibition of upregulated RTKs detected by RTK array or in our RNA-seq data (not shown) did not affect the pERK rebound after VEM treatment, indicating that another, as-yet-unknown factor mediates feedback-induced RAS activation in those cells. Together these findings raise the possibility that other RTKs, or other RAS-stimulating factors, could signal in an SHP2-independent fashion, depending on cellular context. Identifying which factors and settings drive SHP2-mediated adaptive resistance to ERK signaling inhibitors in various ERK-dependent tumors should enable the development of effective combinatorial pharmacologic strategies tailored for specific tumor contexts.
Our present findings establish that combined ERK signaling and SHP2 inhibition effectively overcome adaptive resistance to RAF and MEK inhibitors in a defined subset of ERK-dependent tumors, for which there are no presently available, targeted therapeutic options. Moreover, even though more comprehensive studies across different tumor contexts are warranted, our results suggest that expression of certain RAS mutations (G13D and Q61X) and low/undetectable p(Y542)SHP2 could serve as predictive biomarkers for resistance to the combination and thus help to select patients more likely to benefit from the addition of a SHP2 inhibitor to therapy with a RAF and/or MEK inhibitor. Thus, our findings provide a roadmap for the clinical development of this potentially powerful treatment strategy for a large portion of ERK-dependent tumors.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents should be directed to and will be fulfilled by the Lead Contact, Poulikos Poulikakos (poulikos.poulikakos@mssm.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All animals were examined prior to the initiation of studies to ensure that they were healthy and acclimated to the laboratory environment. 5–7-week-old, female athymic NCR-NU-NU (Envigo laboratories) mice were used for animal experiments. All mouse experiments were approved by the Icahn School of Medicine at Mount Sinai Animal Care and Use Committee (protocol no. IACUC-2014–0229). Mice were maintained under specific pathogen-free conditions, and food and water were provided ad libitum.
RKO cells were harvested on the day of use and injected subcutaneously in one flank per mouse (10 × 106/injection). After inoculation, mice were monitored daily, weighed every three days, and caliper measurements begun when tumors became visible. Tumor volume was calculated using the following formula: tumor volume = (D × d2)/2, in which D and d refer to the long and short tumor diameter, respectively. When tumors reached a size of 100–150 mm3, mice (n = 7) were randomized and treated with vehicle, dabrafenib (30 mg/kg, Selleckchem) and trametinib (0.25 mg/kg, Selleckchem) dissolved in 5% DMSO and 0.5% hydroxypropyl methyl cellulose and 0.2% Tween 80, SHP099 (75 mg/kg, Chemietek) dissolved in 5% DMSO and 0.5% methyl cellulose and 0.1% Tween 80, or the combination, orally once a day, based on mean group body weight. No obvious toxicities were observed in the vehicle- or drug-treated animals as assessed by difference in body weight between vehicle- and drug-treated mice. The endpoint of the experiment for survival studies was considered a tumor volume of 1,000 mm3 as per our approved protocol. Fold-change tumor growth was calculated relative to day 0 with the following formula: fold change in tumor growth = ([tumor volume at day 21 – tumor volume at day 0]/tumor volume at day 0).
Cell Lines
RKO, WiDr, MCF7, T47D, MDA-MB-468, SW1417, NCI-H358, MIAPaCa-2, NCI-H1573, NCI-H1792, SKMEL2, Calu-1, Calu-6, HCT15, SW620, HCT116, HCC1937, BT20, MDA-MB-231, MDA-MB-157, Hs 578T, HT-29, SK-BR-3, BT-474, T84, and LoVo cells were purchased from the American Type Culture Collection (ATCC). HeLa, MDA-MB-436, BT-549, and SUM159 were provided by Ramon Parsons, and SNU387 cells were provided by Amaia Lujambio (both at Icahn School of Medicine at Mount Sinai). SW1736, Hth104, and 8505C cells were provided by James Fagin (Memorial Sloan Kettering Cancer Center). Cell lines were maintained in a humidified incubator at 37° C with 5% CO2, cultured in RPMI 1640, DMEM, DMEM/F12, or F12 supplemented with 10% FBS, 2 mM glutamine and 100 IU/ml penicillin and streptomycin.
METHOD DETAILS
Western Blot, Immunoprecipitation, and RTK arrays
Cells were washed with PBS and lysed on ice for 10 min in lysis buffer (50 mM Tris [pH 7.5], 1% NP40, 150 mM NaCl, 10% glycerol, 1 mM EDTA) supplemented with protease inhibitors protease inhibitor cocktail tablets, Roche). Lysates were centrifuged at 15,000 rpm for 10 min, and the protein concentration was quantified using BCA (Pierce). Proteins were separated by NuPAGE, 4–12% Bis Tris Gel (Novex) and immunoblotted and transferred to nitrocellulose membranes (GE Healthcare) according to standard protocols. Membranes were immunoblotted overnight with antibodies against pERKT202/Y204, ERK, pSTAT3Y705, pMEKS217/221, MEK, pEGFRY1068, EGFR, pERBB3Y1289, ERBB3, FGFR1, FGFR2, GRB2, GAB1, pMETY1234/1235, MET and actin from Cell Signaling; SHP2 and IL6 from Santa Cruz Biotechnology; and pSHP2Y542, pFGFR1Y653/654, DUSP6, and SOS1 from Millipore; and FGF2 antibody was purchased from BD Biosciences. Next day, membranes were probed with anti-rabbit IgG or anti-mouse IgG secondary antibody (Cell Signaling) and chemiluminescent signals were detected on X-ray films.
For immunoprecipitations, lysates were incubated with SHP2 antibody (Santa Cruz Biotechnology) overnight at 4°C, followed by protein G agarose (Life Technologies) for 1 hr at 4°C. Samples were washed three times with lysis buffer, and sample buffer was added for subsequent immunoblot analysis.
Human phospho-RTK arrays were purchased from R&D Systems and were used according the manufacturer’s guidelines.
Active RAS Pull-Down
RAS pull down kit (Pierce) was used to determine levels of RAS-GTP, according to the manufacturer’s protocol. RAS was detected by western blot using either an antibody for KRAS (Novus Biologicals), or a pan-RAS antibody provided with the kit.
Crystal Violet Cell Growth Assays
Cells were plated in six-well plates at a density of 1–10 × 103 cells/well. The next day, cells were treated with inhibitors as indicated, in regular growth medium for 10–14 days. Growth medium with or without inhibitors was replaced every 3 days. Cells were fixed with paraformaldehyde (4%) for 5 min and then stained with 0.5% crystal violet for 30 min.
Lentiviral Production and Stable Cell Line Generation
For shRNA experiments, HER3 short hairpin RNA (shRNA) constructs were obtained from the Broad RNAi consortium (TRCN0000040109: GCC TAC CAG TTG GAA CAC TTA; and TRCN0000010344: GAA TTC TCT ACT CTA CCA TTG) and were subcloned into Tet-pLKO plasmid (Addgene #21915). Lentivirus was produced by co-transfecting HEK293T cells with Tet-pLKO plasmid containing HER3 shRNA with lentiviral packaging and envelope plasmids (psPAX2 and pMD2.G), using Lipofectamine 2000 (Invitrogen). Supernatant was collected 48 hr after transfection and filtered through a 0.45-μm filter unit (Millipore).
SW1736 and Hth104 cells stably expressing HER3 inducible shRNA were generated by transducing shHER3 lentivirus, followed by drug selection (2 μg/ml puromycin) for 1 week and treated with 1 μg/ml doxycycline to induce shRNA expression.
RNA Interference
SW1736 and Hth104 cells were seeded into six-well plates at a density of 1 × 105 cells/well. Next day, cells were transfected with ON-TARGETplus SMARTpool siRNA against IL6, FGFR1, FGFR2 or non-targeting control (NTC) siRNA (Dharmacon) using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s instructions. Cells were treated, 24 hr post-transfection, with vemurafenib (2 μM) for an additional 1 and 24 hr prior to lysis.
RNA Sequencing
Cells were seeded and incubated overnight then treated with vemurafenib (2 μM) for 24 hr. Total RNA was isolated using TRIzol (Invitrogen) from DESCRIBE SAMPLE. Poly A-tailed mRNA was selected using beads with oligo-dT, fragmented, cDNAs were created using random-hexamers and ligated with bar-coded adaptors compatible with NextSeq500. Single-end, 75 nt long reads were sequenced on the instrument in the Department of Oncological Sciences of the Icahn School of Medicine at Mount Sinai. Custom-built software was used to map the reads to the human genome (hg38) and estimate the coverage of each gene. Briefly, the reads were split into two 37-bp parts after trimming 2 nt at the 3’ end, and the parts were mapped to the genome using a suffix-array based approach. The median of coverage across the transcript was used as an estimate of gene expression. The expression values were quantile normalized and log-ratios were calculated by comparing X to the average of the controls. Unique Gene Ontology terms (GO terms) were assigned to each gene by ranking the GO terms by relevance to the biology of the response. The RNA Sequencing data was deposited in the Gene Expression Omnibus (GEO) under accession number GEO: GSE121117.
QUANTIFICATION AND STATISTICAL ANALYSIS
To assess differences in tumor growth between the different groups of mice (number of mice/group, n = 7) unpaired, two-tailed Student’s t test was used. A p value less than 0.05 was considered statistically significant. Statistical analysis was carried out using GraphPad Prism 5.
DATA AND SOFTWARE AVAILABILITY
The RNA sequencing data have been deposited in the Gene Expression Omnibus (GEO) under accession number GEO:GSE121117.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-pERKT23202/Y204 | Cell Signaling | Cat# 4370; RRID: AB_2315112 |
| anti-ERK | Cell Signaling | Cat# 4696; RRID: AB_390780 |
| anti-pSTAT3Y705 | Cell Signaling | Cat# 9145; RRID: AB_2491009 |
| anti-pMEKS217/221 | Cell Signaling | Cat# 9154; RRID: AB_2138017 |
| anti-MEK | Cell Signaling | Cat# 2352; RRID: AB_10693788 |
| anti-pEGFRY1068 | Cell Signaling | Cat# 3777; RRID: AB_2096270 |
| anti-EGFR | Cell Signaling | Cat# 4267; RRID: AB_2246311 |
| anti-pERBB3Y1289 | Cell Signaling | Cat# 4791; RRID: AB_2099709 |
| anti-ERBB3 | Cell Signaling | Cat# 12708; RRID: AB_2721919 |
| anti-FGFR1 | Cell Signaling | Cat# 9740; RRID: AB_11178519 |
| anti-FGFR2 | Cell Signaling | Cat#11835 |
| anti-GRB2 | Cell Signaling | Cat# 3972; RRID: AB_10693935 |
| anti-GAB1 | Cell Signaling | Cat# 3232; RRID: AB_2304999 |
| anti-pMETY1234/1235 | Cell Signaling | Cat# 3077; RRID: AB_2143884 |
| anti-MET | Cell Signaling | Cat# 8198; RRID: AB_10858224 |
| anti-actin | Cell Signaling | Cat# 5125; RRID: AB_1903890 |
| anti-rabbit IgG secondary | Cell Signaling | Cat# 7074; RRID: AB_2099233 |
| anti-mouse IgG secondary | Cell Signaling | Cat# 7076; RRID: AB_330924 |
| anti-SHP2 | Santa Cruz Biotechnology | Cat# sc-7384; RRID: AB_628252 |
| anti-IL6 | Santa Cruz Biotechnology | Cat# sc-57315; RRID: AB_2127596 |
| anti-pSHP2Y542 | Abeam | Cat# ab62322; RRID: AB_945452 |
| anti-DUSP6 | Abeam | Cat# ab76310; RRID: AB_1523517 |
| anti-SOS1 | Abeam | Cat#ab 140621 |
| anti-pFGFR1Y653/654 | Millipore | Cat# 06–1433; RRID: AB_10918364 |
| anti-FGF2 | BD biosciences | Cat# 610072; RRID: AB_399693 |
| anti-RAS | R&D Systems | N/A |
| anti-KRAS | Novus Biologicals | Cat# H00003845-M01; RRID: AB_1506275 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| SHP099 HCL | Selleckchem | Cat# S8278 |
| Trametinib | Selleckchem | Cat# S2673 |
| Vemurafenib | Selleckchem | Cat#S1267 |
| Dabrafenib | Selleckchem | Cat# S2807 |
| Gefitinib | Selleckchem | Cat#S1025 |
| Lapatinib | Selleckchem | Cat#S2111 |
| AZD8931 | Selleckchem | Cat#S2192 |
| Cabozantinib | Selleckchem | Cat# S4001 |
| R428 | Selleckchem | Cat# S2841 |
| GSK1904529A | Selleckchem | Cat#S1093 |
| Crizotinib | Selleckchem | Cat#S1068 |
| BGJ398 | Selleckchem | Cat#S2183 |
| Ponatinib | Selleckchem | Cat#S1490 |
| SHP099 HCL | Chemietek | Cat#CT-SHP099 |
| Doxycycline | Sigma-Aldrich | Cat# D9891 |
| Puromycin Dihydrochloride | Thermo Fisher Scientific | Cat#A1113803 |
| NP-40 | United States Biologicals | Cat# 9036–19-5 |
| Sodium Chloride | Thermo Fisher Scientific | Cat# 507517460 |
| Glycerol | Thermo Fisher Scientific | Cat# BP 2291 |
| EDTA | Sigma-Aldrich | Cat# 60004 |
| TRIS HCL PH7.5 | Thermo Fisher Scientific | Cat# 501031383 |
| Tween 80 | Sigma-Aldrich | Cat# 9490 |
| DMSO | Thermo Fisher Scientific | Cat#BP231100 |
| Hydroxypropyl methyl Cellulose | Thermo Fisher Scientific | Cat# 9004–65-3 |
| Methyl Cellulose | Sigma-Aldrich | Cat# M0512 |
| Lipofectamine 2000 | Invitrogen | Cat# 11668019 |
| Lipofectamine RNAiMAX | Invitrogen | Cat# 13778075 |
| Recombinant Human- IL-6 protein | R&D systems | Cat#206-IL-010 |
| Recombinant Human EGF protein | Invitrogen | Cat# PHG0311 |
| Recombinant Human NRG1 protein | R&D systems | Cat# 5898-NR-050 |
| Human Basic Fibroblast Growth Factor (hFGF basic/FGF2) | Cell Signaling | Cat#8910 |
| Recombinant protein G agarose | Invitrogen | Cat# 15920010 |
| RPMI 1640 Medium | Thermo Fisher Scientific | Cat# 11875119 |
| DMEM Medium | Thermo Fisher Scientific | Cat# 10313039 |
| DMEM/F12 Medium | Thermo Fisher Scientific | Cat# 11320033 |
| Ham’s F-12 Nutrient Mix | Thermo Fisher Scientific | Cat# 11765062 |
| Fetal Bovine Serum | Thermo Fisher Scientific | Cat# 16000044 |
| GlutaMAX Supplement | Thermo Fisher Scientific | Cat# 35050061 |
| Penicillin-Streptomycin | Thermo Fisher Scientific | Cat# 15140122 |
| TRIzol Reagent | Thermo Fisher Scientific | Cat# 15596026 |
| cOmplete, EDTA-free Protease Inhibitor Cocktail | Sigma-Aldrich | Cat# 5056489001 |
| Critical Commercial Assays | ||
| Human phospho-RTK arrays | R&D Systems | Cat# ARY001B |
| RAS pull down kit | Pierce | Cat# P116117 |
| Deposited Data | ||
| RNA sequencing data and analysis | NCBI GEO | GSE121117 |
| Experimental Models: Cell Lines | ||
| RKO | ATCC | Cat# CRL-2577 |
| WiDr | ATCC | Cat#CCL-218 |
| MCF7 | ATCC | Cat# HTB-22 |
| T47D | ATCC | Cat# HTB-133 |
| MDA-MB-468 | ATCC | Cat#HTB-132 |
| SW1417 | ATCC | Cat# CCL-238 |
| NCI-H358 | ATCC | Cat# CRL-5807 |
| MIAPaCa-2 | ATCC | Cat# CRL-1420 |
| NCI-H1573 | ATCC | Cat# CRL-5877 |
| NCI-H1792 | ATCC | Cat# CRL-5895 |
| SKMEL2 | ATCC | Cat# HTB-68 |
| Calu-1 | ATCC | Cat# HTB-54 |
| Calu-6 | ATCC | Cat# HTB-56 |
| HCT15 | ATCC | Cat# CCL-225 |
| SW620 | ATCC | Cat# CCL-227 |
| HCT116 | ATCC | Cat# CCL-247 |
| HCC1937 | ATCC | Cat# CRL-2336 |
| BT20 | ATCC | Cat#HTB-19 |
| MDA-MB-231 | ATCC | Cat# HTB-26 |
| MDA-MB-157 | ATCC | Cat# HTB-24 |
| Hs 578T | ATCC | Cat# CRL-7849 |
| HT-29 | ATCC | Cat# HTB-38 |
| SK-BR-3 | ATCC | Cat# HTB-30 |
| BT-474 | ATCC | Cat# HTB-20 |
| T84 | ATCC | Cat# CCL-48 |
| LoVo | ATCC | Cat# CCL-229 |
| HeLa | Dr. Ramon Parsons | N/A |
| MDA-MB-436 | Dr. Ramon Parsons | N/A |
| BT-549 | Dr. Ramon Parsons | N/A |
| SUM159 | Dr. Ramon Parsons | N/A |
| SNU387 | Dr. Amaia Lujambio | N/A |
| SW1736 | Dr. James Fagin | N/A |
| Hth1O4 | Dr. James Fagin | N/A |
| 8505C | Dr. James Fagin | N/A |
| Experimental Models: Organisms/Strains | ||
| Female athymic NCR-NU-NU | Envigo laboratories | N/A |
| Oligonucleotides | ||
| SMARTpool: ON-TARGETplus IL6 siRNA | Dharmacon | Cat# L-007993-00-0005 |
| SMARTpool: ON-TARGETplus FGFR1 siRNA | Dharmacon | Cat#L-003131-00-0005 |
| SMARTpool: ON-TARGETplus FGFR2 siRNA | Dharmacon | Cat#L-003132-00-0005 |
| Non-targeting control (NTC) siRNA | Dharmacon | Cat# D-001810-10-05 |
| Recombinant DNA | ||
| Tet-pLKO-puro plasmid | Addgene | Cat# 21915 |
| psPAX2 | Addgene | Cat# 12260 |
| pMD2.G | Addgene | Cat# 12259 |
| Software and Algorithms | ||
| GraphPad Prism 5 | GraphPad Software | N/A |
| Morpheus software | Broad Institute | N/A |
| Other | ||
| GE Healthcare Amersham Protran NC Nitrocellulose Membranes: Rolls | GE Healthcare Life Sciences | 10600006 |
| Invitrogen Novex NuPAGE 4–12% Bis-Tris Protein Gels, 1.5mm, 10 well | Thermo Fisher Scientific | NP0335BOX |
Highlights.
SHP2 drives adaptive resistance to RAF and MEK inhibitors in multiple tumors
TNBC and RAS(G12X)-expressing tumors are sensitive to combined MEK-SHP2 inhibition
Expression of RAS(G13D), RAS(Q61X), or low p(Y542)SHP2 are predictors of resistance
FGFR can drive adaptive resistance to RAF and MEK inhibitors independently of SHP2
ACKNOWLEDGMENTS
We are grateful to Amaia Lujambio, James Fagin, and Ramon Parsons for providing cell lines and Evripidis Gavathiotis for critical reading of the manuscript. We would like to thank Saboor Hekmaty for providing expertise with RNA-seq analysis. P.I.P. would like to acknowledge funding by the Dermatology Foundation, the Melanoma Research Foundation, the Melanoma Research Alliance, the Sidney Kimmel Foundation for Cancer Research, a TCI developmental award, and an NIH/NCI grant (R01CA204314). S.A.A. acknowledges a grant from the Breast Cancer Research Foundation. T.A.A. is supported by grant T32CA078207, and Z.K. would like to acknowledge the 2017 Robin Chemers Neustein Postdoctoral Fellowship.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.12.013.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Agazie YM, and Hayman MJ (2003). Molecular mechanism for a role of SHP2 in epidermal growth factor receptor signaling. Mol. Cell. Biol 23, 7875–7886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki T, Nawa H, and Neel BG (2003). Tyrosyl phosphorylation of Shp2 is required for normal ERK activation in response to some, but not all, growth factors. J. Biol. Chem 278, 41677–41684. [DOI] [PubMed] [Google Scholar]
- Baines AT, Xu D, and Der CJ (2011). Inhibition of Ras for cancer treatment: the search continues. Future Med. Chem 3, 1787–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett AM, Tang TL, Sugimoto S, Walsh CT, and Neel BG (1994). Protein-tyrosine-phosphatase SHPTP2 couples platelet-derived growth factor receptor beta to Ras. Proc. Natl. Acad. Sci. USA 91, 7335–7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchini G, Balko JM, Mayer IA, Sanders ME, and Gianni L (2016). Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol 13, 674–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, Spevak W, Zhang C, Zhang Y, Habets G, et al. (2010). Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 467, 596–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandarlapaty S (2012). Negative feedback and adaptive resistance to the targeted therapy of cancer. Cancer Discov 2, 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YN, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, Antonakos B, Chen CH, Chen Z, Cooke VG, et al. (2016). Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 535, 148–152. [DOI] [PubMed] [Google Scholar]
- Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, Brown RD, Della Pelle P, Dias-Santagata D, Hung KE, et al. (2012). EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov 2, 227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran RB, André T, Atreya CE, Schellens JHM, Yoshino T, Bend-ell JC, Hollebecque A, McRee AJ, Siena S, Middleton G, et al. (2018). Combined BRAF, EGFR, and MEK Inhibition in Patients with BRAFV600E-Mutant Colorectal Cancer. Cancer Discov 8, 428–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dance M, Montagner A, Salles JP, Yart A, and Raynal P (2008). The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cell. Signal 20, 453–459. [DOI] [PubMed] [Google Scholar]
- Dardaei L, Wang HQ, Singh M, Fordjour P, Shaw KX, Yoda S, Kerr G, Yu K, Liang J, Cao Y, et al. (2018). SHP2 inhibition restores sensitivity in ALK-rearranged non-small-cell lung cancer resistant to ALK inhibitors. Nat. Med 24, 512–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, Johnson NL, Granger DA, Jordan NV, Darr DB, et al. (2012). Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 149, 307–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedele C, Ran H, Diskin B, Wei W, Jen J, Geer MJ, Araki K, Ozerdem U, Simeone DM, Miller G, et al. (2018). SHP2 inhibition prevents adaptive resistance to MEK inhibitors in multiple cancer models. Cancer Discov 8, 1237–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng GS, Hui CC, and Pawson T (1993). SH2-containing phosphotyro-sine phosphatase as a target of protein-tyrosine kinases. Science 259, 1607–1611. [DOI] [PubMed] [Google Scholar]
- Garcia Fortanet J, Chen CH, Chen YN, Chen Z, Deng Z, Firestone B, Fekkes P, Fodor M, Fortin PD, Fridrich C, et al. (2016). Allosteric Inhibition of SHP2: Identification of a potent, selective, and orally efficacious phosphatase inhibitor. J. Med. Chem 59, 7773–7782. [DOI] [PubMed] [Google Scholar]
- Grossmann KS, Rosário M, Birchmeier C, and Birchmeier W (2010). The tyrosine phosphatase Shp2 in development and cancer. Adv. Cancer Res 106, 53–89. [DOI] [PubMed] [Google Scholar]
- Gumuskaya B, Alper M, Hucumenoglu S, Altundag K, Uner A, and Guler G (2010). EGFR expression and gene copy number in triple-negative breast carcinoma. Cancer Genet. Cytogenet 203, 222–229. [DOI] [PubMed] [Google Scholar]
- Hadari YR, Kouhara H, Lax I, and Schlessinger J (1998). Binding of Shp2 tyrosine phosphatase to FRS2 is essential for fibroblast growth factor-induced PC12 cell differentiation. Mol. Cell. Biol 18, 3966–3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata AN, Niederst MJ, Archibald HL, Gomez-Caraballo M, Siddiqui FM, Mulvey HE, Maruvka YE, Ji F, Bhang HE, Krishnamurthy Radhakrishna V, et al. (2016). Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med 22, 262–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeflich KP, O’Brien C, Boyd Z, Cavet G, Guerrero S, Jung K, Januario T, Savage H, Punnoose E, Truong T, et al. (2009). In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clinical Cancer Res 15, 4649–4664. [DOI] [PubMed] [Google Scholar]
- Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, and Westover KD (2015). Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res 13, 1325–1335. [DOI] [PubMed] [Google Scholar]
- Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, Chen Y, Babbar A, Firdaus SJ, Darjania L, et al. (2018). Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 172, 578–589e517. [DOI] [PubMed] [Google Scholar]
- Karoulia Z, Wu Y, Ahmed TA, Xin Q, Bollard J, Krepler C, Wu X, Zhang C, Bollag G, Herlyn M, et al. (2016). An integrated model of RAF inhibitor action predicts inhibitor activity against oncogenic BRAF signaling. Cancer Cell 30, 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karoulia Z, Gavathiotis E, and Poulikakos PI (2017). New perspectives for targeting RAF kinase in human cancer. Nat. Rev. Cancer 17, 676–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinghoffer RA, and Kazlauskas A (1995). Identification of a putative Syp substrate, the PDGF beta receptor. J. Biol. Chem 270, 22208–22217. [DOI] [PubMed] [Google Scholar]
- Kouhara H, Hadari YR, Spivak-Kroizman T, Schilling J, Bar-Sagi D, Lax I, and Schlessinger J (1997). A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell 89, 693–702. [DOI] [PubMed] [Google Scholar]
- Lito P, Pratilas CA, Joseph EW, Tadi M, Halilovic E, Zubrowski M, Huang A, Wong WL, Callahan MK, Merghoub T, et al. (2012). Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 22, 668–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lito P, Solomon M, Li LS, Hansen R, and Rosen N (2016). Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 351, 604–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainardi S, Mulero-Sánchez A, Prahallad A, Germano G, Bosma A, Krimpenfort P, Lieftink C, Steinberg JD, de Wit N, Gonçalves-Ribeiro S, et al. (2018). SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat. Med 24, 961–967. [DOI] [PubMed] [Google Scholar]
- Matalkah F, Martin E, Zhao H, and Agazie YM (2016). SHP2 acts both upstream and downstream of multiple receptor tyrosine kinases to promote basal-like and triple-negative breast cancer. Breast Cancer Res 18, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagner A, Yart A, Dance M, Perret B, Salles JP, and Raynal P (2005). A novel role for Gab1 and SHP2 in epidermal growth factor-induced Ras activation. J. Biol. Chem 280, 5350–5360. [DOI] [PubMed] [Google Scholar]
- Montero-Conde C, Ruiz-Llorente S, Dominguez JM, Knauf JA, Viale A, Sherman EJ, Ryder M, Ghossein RA, Rosen N, and Fagin JA (2013). Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov 3, 520–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols RJ, Haderk F, Stahlhut C, Schulze CJ, Hemmati G, Wildes D, Tzitzilonis C, Mordec K, Marquez A, Romero J, et al. (2018). RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat. Cell Biol 20, 1064–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly AM, and Neel BG (1998). Structural determinants of SHP-2 function and specificity in Xenopus mesoderm induction. Mol. Cell. Biol 18, 161–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrem JM, Peters U, Sos ML, Wells JA, and Shokat KM (2013). K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV, Chen Y, Kucharski JM, Feng J, Ely T, et al. (2016). Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov 6, 316–329. [DOI] [PubMed] [Google Scholar]
- Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, and Bernards R (2012). Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 483, 100–103. [DOI] [PubMed] [Google Scholar]
- Prahallad A, Heynen GJ, Germano G, Willems SM, Evers B, Vecchione L, Gambino V, Lieftink C, Beijersbergen RL, Di Nicolantonio F, et al. (2015). PTPN11 is a central node in intrinsic and acquired resistance to targeted cancer drugs. Cell Rep 12, 1978–1985. [DOI] [PubMed] [Google Scholar]
- Ruess DA, Heynen GJ, Ciecielski KJ, Ai J, Berninger A, Kabacaoglu D, Görgülü K, Dantes Z, Wörmann SM, Diakopoulos KN, et al. (2018). Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat. Med 24, 954–960. [DOI] [PubMed] [Google Scholar]
- Samatar AA, and Poulikakos PI (2014). Targeting RAS-ERK signalling in cancer: promises and challenges. Nat. Rev. Drug Discov 13, 928–942. [DOI] [PubMed] [Google Scholar]
- Shaffer SM, Dunagin MC, Torborg SR, Torre EA, Emert B, Krepler C, Beqiri M, Sproesser K, Brafford PA, Xiao M, et al. (2017). Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 546, 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, et al. (2010). A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141, 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MJ, Neel BG, and Ikura M (2013). NMR-based functional profiling of RASopathies and oncogenic RAS mutations. Proc. Natl. Acad. Sci. USA 110, 4574–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sos ML, Levin RS, Gordan JD, Oses-Prieto JA, Webber JT, Salt M, Hann B, Burlingame AL, McCormick F, Bandyopadhyay S, and Shokat KM (2014). Oncogene mimicry as a mechanism of primary resistance to BRAF inhibitors. Cell Rep. 8, 1037–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, and Bernards R (2014). Feedback and redundancy in receptor tyro-sine kinase signaling: relevance to cancer therapies. Trends Biochem. Sci 39, 465–474. [DOI] [PubMed] [Google Scholar]
- Sun C, Hobor S, Bertotti A, Zecchin D, Huang S, Galimi F, Cottino F, Prahallad A, Grernrum W, Tzani A, et al. (2014). Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep 7, 86–93. [DOI] [PubMed] [Google Scholar]
- Vogel W, and Ullrich A (1996). Multiple in vivo phosphorylated tyrosine phosphatase SHP-2 engages binding to Grb2 via tyrosine 584. Cell Growth Differ. 7, 1589–1597. [PubMed] [Google Scholar]
- Wong GS, Zhou J, Liu JB, Wu Z, Xu X, Li T, Xu D, Schumacher SE, Puschhof J, McFarland J, et al. (2018). Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat. Med 24, 468–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA sequencing data have been deposited in the Gene Expression Omnibus (GEO) under accession number GEO:GSE121117.