

Abstract
Aim
Centrally‐acting acutely anxiolytic drugs, such as benzodiazepines, barbiturates and gabapentinoids, affect various central nervous system (CNS) functions, which reflects not only their anxiolytic effects but also neuropsychological side‐effects. To validate the pharmacodynamic biomarkers for GABA‐ergic anxiolytics, this study determined the pharmacodynamics of two anxiolytics and a nonanxiolytic control, and linked them to their anxiolytic and sedative effects, during an anxiety‐challenge study day.
Methods
Twenty healthy volunteers were randomized in this placebo‐controlled, double‐blind, four‐way cross‐over study with single‐dose alprazolam (1 mg), diphenhydramine (50 mg), pregabalin (200 mg) or placebo. The Neurocart was used between repeated fear‐potentiated startle assessments. Thus, the potential influence of anxiety on CNS pharmacodynamic markers could be examined.
Results
Compared to placebo, VAScalmness increased with alprazolam (2.0 mm) and pregabalin (2.5 mm) but not with diphenhydramine. Saccadic peak velocity (SPV) declined after alprazolam (−57 ° s–1) and pregabalin (−28 ° s–1), more than with diphenhydramine (−14 ° s–1); so did smooth pursuit. The average responses of SPV and smooth pursuit were significantly correlated with the drug‐induced increases in VAScalmness. The SPV‐relative responses of VASalertness, body‐sway and adaptive‐tracking also differed among alprazolam, pregabalin and diphenhydramine.
Conclusions
Compared with the antihistaminergic sedative diphenhydramine, alprazolam and pregabalin caused larger SPV reduction, which was correlated with simultaneous improvement of subjective calmness, during a study day in which anxiety was stimulated repeatedly. The different effect profiles of the three drugs are in line with their pharmacological distinctions. These findings corroborate the profiling of CNS effects to demonstrate pharmacological selectivity, and further support SPV as biomarker for anxiolysis involving GABA‐ergic neurons. The study also supports the use of prolonged mild threat to demonstrate anxiolytic effects in healthy volunteers.
Keywords: biomarker, pharmacodynamics, pharmacokinetics, psychopharmacology
What is Already Known about this Subject
Centrally acting acutely anxiolytic drugs, such as benzodiazepines, barbiturates and gabapentinoids, have an impact on a range of central nervous system functions, which reflects not only their anxiolytic effects but also side effects such as sedation, postural instability, and visuomotor and memory impairment.
The use of appropriate biomarkers may be especially useful for anxiety disorders, where therapeutic exploratory studies in patients can be difficult to achieve a clinically meaningful end‐point due to the nature of subjective assessments, the relatively large size and probability of placebo effect, and other ethical or practical issues.
Comparison of the effect‐profiles of putative anxiolytic and nonanxiolytic drugs may contribute to the validation of pharmacodynamic biomarkers for GABA‐ergic anxiolytics.
What this Study Adds
Compared with the antihistaminergic sedative diphenhydramine, alprazolam and pregabalin caused larger saccadic peak velocity reduction, which was correlated with simultaneous improvement of subjective calmness, during a study day in which anxiety was stimulated repeatedly.
The different effect profiles of the three drugs are in line with their pharmacological distinctions. These findings corroborate the profiling of central nervous system effects to demonstrate pharmacological selectivity, and further support saccadic peak velocity as a biomarker for anxiolysis involving GABA‐ergic neurons.
The study also supports the use of prolonged mild threat to demonstrate anxiolytic effects in healthy volunteers.
Tables of Links
| TARGETS | |
|---|---|
| Ligand gated‐ion channels 2 | GPCRs 4 |
| GABAA receptors | Histamine receptors |
| Other ion channels 3 | |
| Calcium activated chloride channel |
| LIGANDS | |
|---|---|
| alprazolam | diphenhydramine |
| pregabalin |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 1, and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 2, 3, 4.
Introduction
Centrally‐acting acute anxiolytic drugs, such as benzodiazepines (BZPs), barbiturates and gabapentinoids, have an impact on a range of central nervous system (CNS) functions, which reflects not only their anxiolytic effects but also side effects such as sedation, postural instability and visuomotor and memory impairment 5. It would be useful to identify the CNS activities for those compounds that are more closely linked to reduction of anxiety than to general CNS depression.
Pharmacodynamic (PD) approaches have been increasingly employed in early human pharmacology studies to obtain in vivo pharmacological information of different drugs acting on the central nervous system and of the systems with which the drugs interact. The general aim of these methodologies is to obtain information about the pharmacological characteristics of a drug (such as blood–brain barrier penetration, target engagement and mechanistically meaningful activity), which underlie its therapeutic effects 6, 7, 8. The use of appropriate biomarkers may be especially useful for anxiety disorders, where therapeutic exploratory studies in patients can make it difficult to achieve a clinically meaningful end‐point due to the nature of subjective assessments, the relatively large size and probability of placebo effect, and other ethical or practical issues 9, 10. A validated biomarker in early human pharmacology studies would certainly serve as a useful tool for the development of new therapeutic anxiolytics.
It has been well established that BZPs exert their pharmacological effects through positive allosteric modulation of the GABA‐A receptors. In recent years, the experiments on GABA‐A receptor subtype‐gene knock‐out mouse lines have greatly facilitated the identification of GABA‐A receptor subtypes that mediate BZPs‐induced sedation (α1 GABA‐A receptors), anxiolysis (α2 and α3 GABA‐A receptors), or memory impairment (α5 GABA‐A receptors) 11, 12, 13. To address the effects of BZPs in human pharmacological studies, a collection of PD measurements were employed and evaluated for their pharmacokinetic/PD relationship with BZPs, which include objective measures such as electroencephalography, semisubjective measures such as psychomotor performance, and subjective measures such as mood/sedation scales 14, 15, 16, 17. Despite of the acceptable sensitivity and the observed exposure–response relationship of these PD measurements for the effects of BZPs, as well as the potential involvement of eye movement in anxiety disorder and related neuropsychiatric disturbance, increasing attention has been paid to evaluate the relevance of these PD parameters to the pharmacological effects of established or novel anxiolytic drugs. The exact clinical relevance of quantitative electroencephalogram (EEG), for example, to the anxiolytic, anticonvulsant, sedative and hypnotic actions of BZPs, have not yet clearly been elucidated 18.
The Centre for Human Drug Research (CHDR; Leiden, The Netherlands) has developed a Neurocart battery of validated computerized tests for the assessments of various CNS functions. These tests have been shown to be sensitive to various aspects of sedation 19 and have been used in early studies of psychoactive drugs as pharmacodynamic biomarkers for postural (in)stability (body sway test), eye–hand cooperation (adaptive tracking test), subjective feelings of alertness, mood and calmness (visual analogue scale [VAS] Bond & Lader), and for neurophysiologicak functions (saccadic eye movement and smooth pursuit eye movement tests) 10. Our previous studies showed that the Neurocart battery presents distinct PD response‐patterns to different subtype‐selective partial GABAA‐agonists and nonselective BZP anxiolytics 20, 21, 22, 23, which may imply potential GABA‐A subtype specificity of these PD markers. Normally, this test battery does not provide any clear information about the specific anxiolytic properties of drugs, as measured by VAScalmness. BZPs or selective serotonin reuptake inhibitor, for instance, do not cause consistently significant increases of subjective calmness in healthy volunteers, when the measurement was performed in stress‐free experimental settings 9, 10. Such findings can be true for selective serotonin reuptake inhibitors that have a slow onset of action and can even worsen anxiety symptoms during initial treatment 24, but is not expected for fast‐acting anxiolytic drugs such as BZPs 25. We therefore combined the Neurocart test battery with a modified fear‐potentiated‐startle (FPS) paradigm 26. In this way, we could compare our more general CNS test battery with a specific anxiety test, which in some studies 27, 28, but not all 29, has been shown to be sensitive to anxiolytic drugs. To this end, we administered two sedating anxiolytic drugs (alprazolam and pregabalin) and a sedating nonanxiolytic (diphenhydramine) at therapeutic doses to healthy volunteers.
Methods
Ethics
The study was approved by the Medical Ethics Review Board of Leiden University Medical Centre, and was conducted according to the principles of the Helsinki Declaration and the International Conference on Harmonization/Good Clinical Practice.
Design
This was a single‐centre, randomized, placebo‐controlled, four‐way crossover, double‐blind study conducted in 20 healthy subjects. The scheme of this study included a screening period of maximally 14 days, four treatment periods separated by three washout periods of at least 3 days, and a telephone follow‐up.
Subjects
Ten men and 10 women, aged between 18 and 40 years, with a body mass index between 18 and 30 kg m−2, without any clinically significant abnormalities, were recruited. All volunteers provided written inform consent. Their eligibilities were evaluated before being randomized into the study. Subjects were instructed not to use alcoholic beverages from 24 h before admission until the next morning of each study day. No xanthine or tobacco containing products were allowed from 22:00 in the evening before each study day and during stay in the research unit. They were asked to keep a normal day/night pattern from 2 weeks before the first study day until the last study day.
Sample size determination
As was shown in Grillon et al. 27, the mean effect of the THREAT‐SAFE difference between unpredictable threat and a neutral context seen under placebo was about 15 ± 8.5 μV whereas the effect under 1 mg alprazolam was around 5 ± 8.5 μV (mean ± standard deviation). This leads to an alprazolam effect of 10 μV over placebo. Given that the within‐patient variability is normally not substantially greater than the between‐patient variability a residual standard deviation of 10 μV was assumed. Based on these assumptions, a sample size of 16 subjects was obtained to ensure a power of at least 80% with a two‐sided alpha level of 5%. For the Neurocart end points, using data from previous studies 21, 22, 23, the same sample size of 16 was determined to have ≥80% power to detect the mean differences of 1.244 in VAS alertness and 20.577 in saccadic peak velocity (SPV), respectively assuming standard deviations of 1.663 (VASalertness) and 27.429 (SPV) between placebo and lorazepam 2 mg using a paired t test with a 0.050 two‐sided significance level. Considering the possibility of drop‐out and the sample should be a multiple of four (to keep the study design balanced the sample size), a total sample size of 20 subjects was finally decided for the study.
Treatments
The study treatments were assigned according to a randomization schedule, which consisted of five blocks of the fully balanced 4 × 4 William Latin Squares. Each subject received a single oral dose of over‐capsulated pregabalin 200 mg, alprazolam 1 mg, diphenhydramine 50 mg or matching placebo in a fasted state at about 8–9 AM on each treatment period.
Safety
Adverse events (AEs), electrocardiograms (ECGs) and vital signs, as well as safety laboratory assays were frequently evaluated during the study. Twelve‐lead ECG recording was made using Nihon Kohden Cardiofax with Ecaps 12 software devices (Nihon Kohden, Tokyo, Japan). Vital signs (pulse rate and blood pressure) were taken using a Nihon Kohden BSM‐1101 K monitor or a Colin Pressmate bp 8800. All blood pressure, pulse rate, and ECG recordings were done after subject was resting in a supine position for at least 5 min. Safety laboratory tests on blood or urine samples were performed in the Central Clinical Laboratories of Leiden University Medical Centre.
Pharmacokinetic measurements
For the determination of drug concentrations, two venous blood samples of 5 and 2 ml were collected into ice‐bathed Li‐Hep tubes (Becton and Dickinson 367 684 and 368 200, respectively) within 0.5 h predose and at 0.5, 1.25, 1.75, 2.25, 3, 4, 6 and 8 h postdose. The samples were centrifuged (2000 × g, 15 min, 4°C). The obtained plasma was transferred into two polypropylene Sarstedt 2 ml tubes and stored at −20°C until analysis.
Plasma pregabalin concentrations were determined at AAI Pharma GmbH & Co KG, Neu‐Ulm, Germany, using liquid chromatography (LC)– tandem mass spectrometry (MS/MS) on a Finnigan LCQ system. A Phenomenex Gemini (50 × 3.0 mm i.d., 5 μm) was used as the high‐performance LC column. The quantification range was from 1.00 to 1000 μg l–1. The intra‐ and interassay variability was 2.1–10.5% and 0.9–6.6%, respectively. Plasma alprazolam and diphenhydramine concentrations were determined at the pharmacy of the Groningen University Medical Centre (Groningen, the Netherlands) using LC–MS/MS. All experiments were performed on a ThermoFisher (San Jose, CA, USA) triple quadrupole LC–MS/MS with a Finnigan Surveyor LC pump and a Finnigan Surveyor autosampler which was set at 20°C. Lower limit of quantification was 1.00 μg l–1 for alprazolam and 5.00 μg l–1 for diphenhydramine, respectively. Intra‐ and interassay variability were 2.1–7.2% and 0.0–3.3%, respectively, for alprazolam and 2.0–3.3% and 0.0–2.0%, respectively for diphenhydramine.
PD measurements
A training session of the PD tests (i.e. the Neurocart battery and the FPS paradigm) was performed during the screening. The purpose was to familiarize the subjects with the tests and prevent potential learning effect. In each study period, the FPS paradigm was carried out around 1 h after dosing; while the Neurocart battery was assessed at predose and 0.5, 1.25, 1.75, 2.25, 3, 4, 6 and 8 h postdose in the following sequence of tests: body sway, VAS Bond & Lader, saccadic eye movements, smooth pursuit eye movements, and adaptive tracking. At each assessment, one subject was assigned to a quiet room with ambient illumination.
Pharmaco‐EEG approach is currently widely used, and the empirical relation between this measure and other agonist effects of BZPs has been reported. However, the main purpose of this study was to compare the sensitivity and specificity of the Neurocart PD measurements vs. those of the FPS measurements to the effects of sedating, hypnotic and anxiolytic drugs. As the flowcharts of the study days were already quite busy with the combination of the non‐EEG PD tests and the FPS paradigm, and the device used for generation of electronic shocks in the FPS paradigm may interfere with the pharmaco‐EEG measurements, the EEG measures were omitted from the study design for the sake of smooth operation.
Body sway
Body sway was measured with an apparatus similar to the Wright ataxiameter 30, which integrates the amplitude of unidirectional body sway. The measurements were made in the antero–posterior direction with eyes closed for 2 min. The subject was asked to stand comfortably on a floor with their feet slightly apart. Body sway measures postural (in)stability. It has demonstrated considerable sensitivity to the effect of BZPs 31.
VAS of Bond & Lader
VASs, as originally described by Norris 32, were presented on a computer screen. Three composite factors were derived from the 16 items, corresponding to alertness, mood and calmness. These factors quantify subjective feelings and have been extensively used to delineate subjective effects of a variety of sedative agents 10.
Saccadic eye movements
Saccadic eye movements were evaluated using a computer‐based system composed of: (i) stimulus display and signal collection (Nihon Kohden Corporation, Tokyo, Japan); (ii) signal amplification (Grass‐Telefactor, An Astro‐Med, Inc. Product Group, Braintree, USA); (iii) data recording (Cambridge Electronics Design, Cambridge, UK); (iv) disposable silver–silver chloride electrodes (Medicotest N‐OO‐S, Olstykke, Denmark); and (v) the sampling and analysis scripts developed by CHDR (Leiden, the Netherlands). The parameters of this test were the average values of saccadic peak velocity (SPV, ° ms–1), reaction time (ms) and inaccuracy (%) of all artefact‐free saccades that were calculated on each session. Saccadic peak velocity appears to be the most sensitive measure for the sedative effect of BZPs 10 and has been found to be a promising biomarker for the anxiolytic component of BZPs and some newly developed compounds with potential anxiolytic effect 20, 21, 22, 23.
Smooth pursuit eye movements
The same system as used for saccadic eye movements was also used for measurement of smooth pursuit. For smooth pursuit eye movements, the target moved sinusoidally at frequencies ranging from 0.3 to 1.1 Hz, by steps of 0.1 Hz. The amplitude of target displacement corresponded to 22.5° eyeball rotation to both sides. Four cycles were recorded for each stimulus frequency. The method has been validated at the CHDR by Van Steveninck et al. 33 based on the work of Bittencourt et al. 34 and the original description of Baloh et al. 35. The time in which the eyes were in smooth pursuit of the target were calculated for each frequency and expressed as a percentage of stimulus duration. The average percentage of smooth pursuit for all stimulus frequencies were used as the parameter.
Adaptive tracking
The adaptive tracking test was performed as originally described by Borland and Nicholson 36, using customized equipment and software (Hobbs 2004, Hertfordshire, UK). After a 0.5‐min run‐in time without data‐recording, the average performance over the rest 3.0 min was scored and was used as the test parameter. Adaptive tracking is a pursuit‐tracking task. The subject was required to operate a joystick and try to keep a dot inside a circle moving randomly on the computer screen. If they succeeded, the speed of the moving circle increases, and vice versa.
FPS paradigm
The FPS paradigm is extensively described elsewhere 26. In brief, the test contained three contexts, which differed in the possibility of electronic shocks signalled by a computer displayed verbal instruction: “No shock” for the neutral (N) context, “Shock only during cue” for the predictable (P) context, and “Shock at any time” for the unpredictable (U) context. Duration of each context was 90–100 s, during which six startle probes were administered together with the assessment of startle response. Intervals between startle probes varied between 12 and 18 s (16 s on average). The FPS session consisted of two blocks with the following orders of contexts: (i) P‐N‐U‐N‐U‐N‐P and (ii) U‐N‐P‐N‐P‐N‐U. The order of these two blocks was counterbalanced across the subjects. A total of 12 shocks were administered during each FPS test session.
The shocks were delivered through two medal electrodes located on the inner side of one of the subjects' forearms. Shock stimuli were delivered using a Digitimer DS7A constant current stimulator (Digitimer Ltd, Hertfordshire, UK). Stimulation consists of short trains (total duration maximally 750 ms) of brief (2 ms) pulses. The maximum current intensity delivered during the study was 7 mA.
Statistical analysis
Pharmacokinetics
The plasma concentrations of pregabalin, diphenhydramine and alprazolam were summarized by time points, and graphically presented as mean concentration–time profiles. The error bars represent the standard deviation at each time point.
PD
Body sway values were log‐transformed prior to analysis to correct for the expected log‐normal distribution of the data 21, 22, 23. The effects of the four treatments on the PD measurements were compared with a mixed model analysis of variance. In this statistic model, treatment, period, time and treatment by time were set as fixed factors; and the random factors were subject, subject by treatment and subject by time; the baseline value was included as covariate, where baseline is defined as the average of the available measures obtained prior to dosing. The following contrasts were requested to demonstrate the effects of the active treatments: placebo–pregabalin, placebo–alprazolam and placebo–diphenhydramine.
A summary table of the analysis results was generated with estimates of the difference between each active treatment and placebo and a back‐transformed estimate of the difference in percentage for body sway, 95% confidence intervals (in percentage for body sway) and least square means (geometric means for body sway), and the P‐value of the contrasts. Least square means graphs were generated, with the least square means of the analysis of the data as change from baseline.
Previous studies suggested good sensitivity of SPV to the effect of BZPs 10 and α2,3 subtype‐selective GABA‐A receptor modulators 21, 22, 23, 37, 38, 39. There is a close association between the effect size of BZPs for SPV‐reduction and their administered doses 10. Based on the putative link between GABA‐A α2,3 receptors and anxiety 40, 41, this supports the consideration of SPV as a biomarker of clinical anxiolysis associated with GABA α2,3 activation 20, and the predictivity of SPV was supported by the selective SPV‐reduction caused by TPA023 21, combined with early clinical findings of this partial GABA α2,3 agonist 40. BZPs also affected body sway, VASalertness, adaptive tracking and VAScalmness, suggesting impairment of postural balance, subjective alertness, eye–hand coordination, and subjective calmness, respectively 21, 22, 23, 37, 38. Given the clinical relevance of these PD parameters, scatter plots of each PD measurement against simultaneously obtained SPV values were depicted to demonstrate SPV‐normalized effect profiles with the study treatments. Moreover, a regression analysis was performed using the mixed model with treatment as the fixed factor and SPV change from baseline and intercept as the random factors. Comparisons were made between each two active treatments with regards to the estimates of the slopes of the regression line obtained from each relative effect profile. The estimates of the slopes and their estimated difference were tabulated with the P‐values. The slopes of these regression lines can be regarded as a measure of pharmacological selectivity of the drugs in respect of their anxiolytic effect 20.
Results
Subjects
Twelve men and 10 women participated in the study. Ten subjects of each sex completed the study. The two drop‐outs withdrew for personal reasons unrelated to the study, and were replaced by male subjects who received the same order of study treatments. Subjects had an average age of 22 years (range 18–36), and body mass index of 23.3 kg m−2 (range 18.1–29.6). Data from all treated subjects were used in the analyses of safety and pharmacokinetics. Subjects who completed the study per protocol were included in the PD analysis.
Safety
No serious AEs were observed during the study. No subjects discontinued their study due to AEs. The most frequently reported AEs were somnolence, dizziness, fatigue and headache. Alprazolam was associated with the largest number of CNS‐related AEs (n = 21 in 14 out of 21 [66.7%] subjects), followed by diphenhydramine (n = 19 in 16 out of 21 [76.2%] subjects), pregabalin (n = 15 in 9 out of 20 [45.0%] subjects) and placebo (n = 14 in 11 out of 20 [55.0%] subjects). Most AEs were attributed to the CNS‐depressant effects of the study treatments. No ECG or laboratory abnormalities were judged clinically significant.
Pharmacokinetics
Sixty‐two concentration‐time profiles were obtained (20 for pregabalin, 21 for diphenhydramine and 21 for alprazolam). Following single‐dose oral administration, peak plasma concentrations of all three active treatments were reached at 2–3 h postdose. Mean (standard deviation) Cmax was 4.87 (0.94), 91.47 (29.85) and 15.17 (2.10) mg l–1 for pregabalin, diphenhydramin and alprazolam, respectively. Figure 1 shows the average concentration–time profiles of pregabalin, diphenhydramine and alprazolam.
Figure 1.
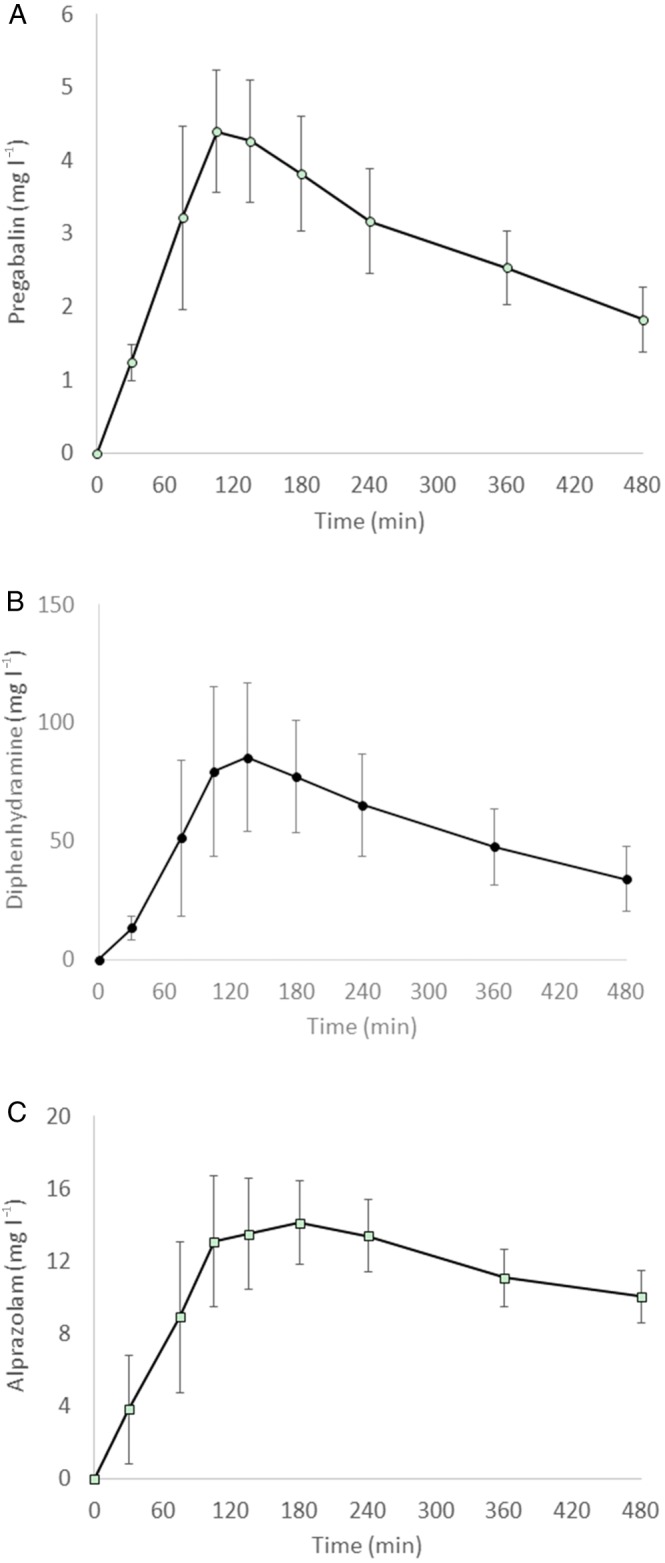
Mean plasma concentration‐time profiles after single oral administration of (A) pregabalin 200 mg, (B) diphenhydramine 50 mg or (C) alprazolam 1 mg in 20 healthy male volunteers. The error bars represent the standard deviation at each time point
PD
The profiles of the CNS PD 1parameters (Figures 2 and 3) show that peak effects of the study treatments were usually observed around the point of Tmax.
Figure 2.
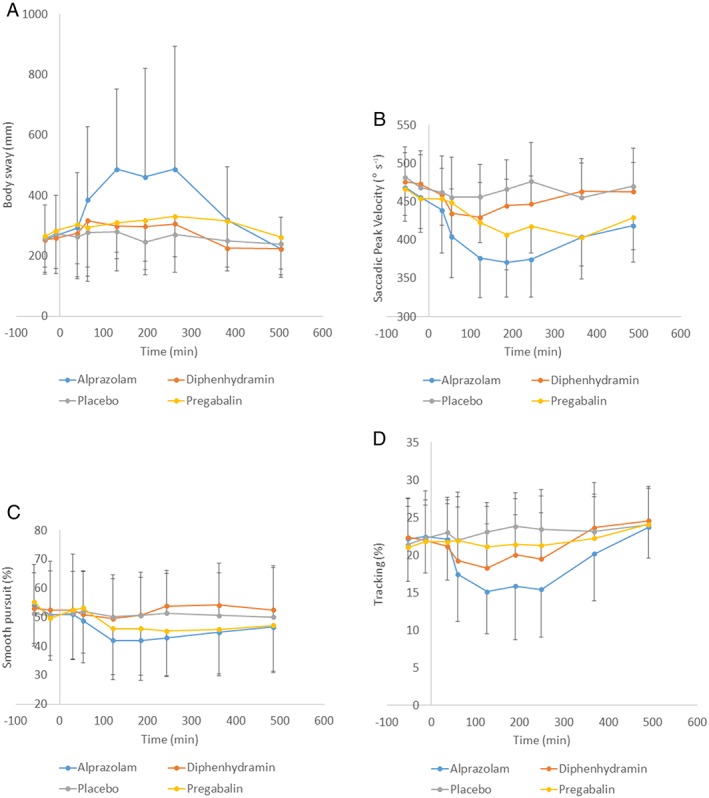
Graph of means of objective central nervous system–pharmacodynamic parameters with standard deviation as error bars. (A) Body sway; (B) saccadic peak velocity; (C) smooth pursuit; (D) adaptive tracking
Figure 3.
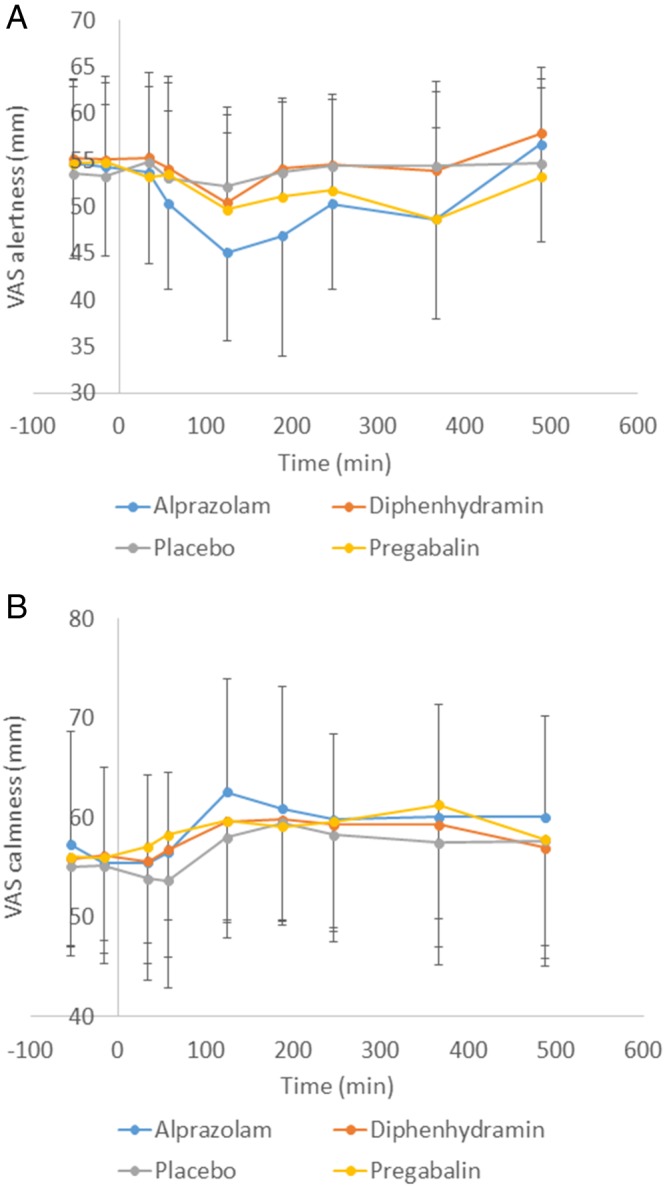
Graph of means of subjective central nervous system–pharmacodynamic parameters with standard deviation as error bars. (A) Visual analogue scale of alertness; (B) visual analogue scale of calmness
Table 1 summarizes the results of statistical comparisons between each active drug and placebo. Compared to placebo, VAScalmness increased statistically significantly with alprazolam (2.0 mm) and pregabalin (2.5 mm), but not with diphenhydramine (1.1 mm). In the meantime, saccadic peak velocity (SPV) declined after alprazolam (−57 ° s–1) and pregabalin (−28 ° s–1), more than by diphenhydramine (−14 ° s–1); so did smooth pursuit. The average responses of SPV were significantly correlated with the drug‐induced increases in VAScalmness.
Table 1.
Summary of the analysis results for central nervous system–pharmacodynamic parameters
| Parameter (unit) | Pregabalin vs. placebo | Alprazolam vs. placebo | Diphenhydramine vs. placebo |
|---|---|---|---|
| Body sway (‐) | 12.27% (−2.37%, 29.11%) P = 0.1026 | 34.43% (16.90%, 54.59%) P < 0.0001 | 12.25% (−2.35%, 29.03%) P = 0.1021 |
| Saccadic inaccuracy (%) | 0.4 (−0.2, 0.9) P = 0.1670 | 0.8 ( 0.3, 1.4) P = 0.0021 | 0.3 (−0.2, 0.8) P = 0.1827 |
| Saccadic peak velocity (° s–1) | –27.7 (−35.9, −19.5) P < 0.0001 | –56.9 (−65.0, −48.8) P < 0.0001 | –13.8 (−21.7, −5.9) P = 0.0010 |
| Saccadic reaction time (s) | 0.001 (−0.006, 0.009) P = 0.7032 | 0.010 (0.003, 0.017) P = 0.0082 | 0.002 (−0.005, 0.009) P = 0.6109 |
| Smooth pursuit (%) | –5.1 (−7.8, −2.5) P = 0.0003 | –6.8 (−9.5, −4.2) P < 0.0001 | –0.5 (−3.1,2.1) P = 0.7149 |
| Adaptive tracking (%) | –1.04 (−2.30, 0.22) P = 0.1039 | –5.04 (−6.30, −3.78) P < 0.0001 | –2.64 (−3.92, −1.36) P = 0.0001 |
| VASalertness (mm) | –2.3 (−5.7, 1.0) P = 0.1676 | –4.5 (−7.8, −1.1) P = 0.0096 | –1.0 (−4.4,2.3) P = 0.5377 |
| VAScalmness (mm) | 2.5 (0.4, 4.7) P = 0.0201 | 2.0 (−0.1, 4.1) P = 0.0606 | 1.1 (−1.0, 3.2) P = 0.3066 |
| VASmood (mm) | 0.7 (−0.5,2.0) P = 0.2483 | –0.1 (−1.4, 1.1) P = 0.8633 | 0.4 (−0.8, 1.7) P = 0.5059 |
The results are presented as the estimated differences between each active treatment and placebo in the least square mean change from baseline and the 95% confidence intervals of the differences. The results of body sway are presented as the differences of least square mean proportional change from baseline and their 95% confidence intervals
To further characterize the PD profiles of these compounds, various CNS PD effects were compared with the corresponding drug‐induced SPV reductions. According to the analyses about SPV‐relative effect profiles (Table 2), the SPV‐normalized impairment of adaptive tracking was higher after diphenhydramine and alprazolam, compared to that of pregabalin. The estimated slope for the regression line ΔSway/ΔSPV was rather flat with pregabalin and significantly smaller than alprazolam and diphenhydramine. The slope for the ΔVASalertness/ΔSPV relation was larger with pregabalin and alprazolam than with diphenhydramine. No significant difference was found among alprazolam, diphenhydramine, and pregabalin in the relative effect profiles of ΔVAScalmness vs. ΔSPV.
Table 2.
Summary of relative effect profile among the three active treatments
| Slope of the regression line | P‐value | |||||
|---|---|---|---|---|---|---|
| ALP | DPH | PRG | ALP‐DPH | ALP‐PRG | DPH‐PRG | |
| ΔSway/ΔSPV | −0.00208 | −0.00186 | −0.00106 | 0.5733 | 0.0055 | 0.0716 |
| ΔTracking/ΔSPV | 0.07785 | 0.06189 | 0.03056 | 0.1526 | <0.0001 | 0.0133 |
| ΔVASalertness/ΔSPV | 0.07227 | 0.01491 | 0.06061 | 0.0008 | 0.4540 | 0.0156 |
| ΔVAScalmness/ΔSPV | −0.03626 | −0.02776 | −0.05070 | 0.6564 | 0.4123 | 0.2834 |
ALP, alprazolam; DPH, diphenhydramine; PRG, pregabalin; SPV, saccadic peak velocity; VAS, visual analogue scale
The results are presented as least square mean estimates of the slope of regression line. The P‐values are presented for the comparisons of each two active treatments
The results of the FPS paradigm are reported in a separate article 26.
Discussion
In this study, a set of neuropsycho‐PD tests (i.e., the Neurocart battery) was performed to characterize the CNS profiles of three clinically anxiolytic and/or hypnotic drugs. Therapeutically relevant doses were administered as a single dose, because all drugs had a rapid onset of effects. The aim was to identify response patterns that are shared by fast‐acting anxiolytics (alprazolam and pregabalin) but differ from sedative effects (diphenhydramine).
For the assessment of FPS, none of the treatments reliably reduced either fear‐ or anxiety‐potentiated startle. Alprazolam and diphenhydramine reduced overall baseline startle. Pregabalin did not significantly affect any of the physiological measures 26. By contrast, as a full GABA‐A agonist, alprazolam induced robust effects on most CNS parameters. Such generalized CNS‐depressive PDs is similar to that of other BZPs 33, 37, 38 and can be explained by the nonselective modulation of alprazolam on different GABA‐A receptor subtypes, which constitute the most widely distributed inhibitory receptors in the CNS. Pregabalin and its congener gabapentin are more selective and affect the α2δ subunit of the voltage‐dependent calcium channel. Contrary to BZPs, ‘gabapentinoids’ do not bind to GABA‐receptors, but both drug classes lead to a decrease of the stimulatory neurotransmitters that are involved in anxiety, such as glutamate and the monoamines 42. In this study, pregabalin was associated with moderate reduction of SPV and smooth pursuit, as well as statistically significant increase of VAScalmness. Diphenhydramine, acting as an antagonist at the histamine H1 receptors, slightly reduced SPV, but it did not influence VAScalmness. As an indication that the 50 mg dose was functionally relevant, diphenhydramine showed a prominent effect on adaptive tracking.
An important finding of this study was the improvement of subjective calmness after a single dose of pregabalin and alprazolam. Moreover, the increase of VAScalmness was significantly correlated with SPV reductions. The literature is less clear about the subjective effects of anxiolytic drugs in healthy volunteers. In general, inconsistent changes of VAScalmness have been reported for single doses of lorazepam (2 mg) and some α2,3‐subtype selective GABA‐A agonists 21, 22, 23, 37, 38, even at dosages that are clinically more anxiolytic than the relatively low doses of alprazolam 1 mg or pregabalin 200 mg employed in the current study. These inconsistencies suggest that VAScalmness is a less reliable biomarker in studies where anxiety is not specifically stimulated. In such ‘normal’ drug studies, healthy subjects can experience different levels of anxiety, for instance depending on how familiar they are with these experiments, which may affect their sensitivity to anxiolytic drug effects. In the current study, subjects were repeatedly exposed to FPS tests, which include unpleasant electrical shocks. We assume that this has induced a mild anticipatory anxiety in the study subjects 43, which was suppressed by the anxiolytic drugs but not by the sedative antihistamine.
By contrast, the partial effect profiles of diphenhydramine and pregabalin and the more general CNS‐depression caused by alprazolam seems to match their pharmacological characteristics. Strictly speaking, a reliable comparison of pharmacological effect profiles is only justified across a wider dose range or at least at roughly equipotent dosages. Although it is difficult to establish dose equivalence across different drugs classes, all doses were in their therapeutic range. We tried to solve this further by looking at relative effect profiles across the entire profile of the plasma concentrations of the investigated drugs 20. With this approach, the concern regarding dose equivalence in PD comparisons is overcome by transforming from dose‐based PD‐effect relationship to exposure‐based PD‐effect relationship. SPV is one of the most sensitive PD biomarkers for anxiolytic doses of BZPs 10. Therefore, SPV was used to benchmark anxiolytic effects and was compared by linear regression with a second CNS biomarker to depict a drug effect on another CNS domain.
As can be seen in Tables 1 and 2, alprazolam and diphenhydramine lead to comparable impairments on body sway (measure of postural stability) relative to their effects on SPV. In contrast, the effect of pregabalin on body sway was less remarkable than SPV. The differential effects of pregabalin on these two PD parameters seem to be consistent with the clinical behaviour of this compound, which, compared to BZPs, shows a larger therapeutic window between anxiolysis and ataxia 44. The slopes of the ΔVASalertness/ΔSPV regression lines are comparable among the study treatments. This is different from our previous findings between selective and nonselective GABA‐A receptor agonists 20. As subjects were physically and mentally stressed by electronic shocks of the fear‐potentiated‐startle paradigm 26, this challenge probably increased the baseline level of VASalertness and hence reduced the responses to the investigated anxiolytic/hypnotic drugs. In addition, a distinct relationship was seen in the ΔSPV‐relative effect profiles of ΔTracking among the three compounds. The steeper slope of the ΔTracking/ΔSPV regression line after diphenhydramine reflects its minimal effect on SPV but substantial effect on tracking. Such a profile is linked to the clinical properties of diphenhydramine: it shows considerable hypnotic effects at the dose of 50 mg, but does not lead to anxiety relief. Known side‐effects of this compound, including drowsiness and motor impairment, are attributed to its inverse agonism at the histamine H1‐receptors distributed in the brain.
Taken together, the results of present study support the combination a physically stressful procedure to the subjective assessment of anxiolysis. Consistently, the simultaneous reduction of SPV and the correlation between these two PD measurements provide further confirmation for the use of these biomarkers for clinically relevant anxiolytic effects. The sensitivity of the experiment appears to have been increased by the constant mild anticipation of shock during repeated FPS testing. The different effect profiles of the three drugs are in line with their pharmacological distinctions. These findings corroborate the profiling of CNS effects to demonstrate pharmacological selectivity, optimize the previous use of EEG/psychomotor/subjective pharmacological assessments 45 to a more pharmacological mechanism‐based PD marker selection, and warrant the extension from a single, less reliable, subjective assessment to the combination of a stress‐challenged subjective measurement and a neurophysiological test for the evaluation and extrapolation of clinical anxiolysis.
Competing Interests
All authors have completed the Unified Competing Interest form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: X.C., F.B., M.d.K., J.B., A.C. and J.v.G. had no support from any organisation for the submitted work; no financial relationships with any organisations that might have an interest in the submitted work in the previous 3 years; no other relationships or activities that could appear to have influenced the submitted work.
We acknowledge all subjects for their contributions to the study. This study was funded by F. Hoffmann‐La Roche AG/Roche Global Development. This study was registered in Eudract with the number of ‘2006‐005707‐34’.
Contributors
The authors of this work are X.C., F.B., M.d.K., J.B., A.C. and J.v.G. X.C. was the primary medical writer of this paper. X.C. and M.d.K. performed the statistical analysis of the work. F.B. was responsible for the recruitment, informed consent, and medical care of subjects during the study, as well as study design, coordination and reporting. J.B., A.C. and J.v.G. were in charge of study design, study data analysis and study management. J.v.G. was the principle investigator of this study. All authors participated in study design, and reviewed and approved the submission of this manuscript. All authors met ICMJE authorship criteria and no person not named as an author qualified for authorship on this manuscript.
Chen, X. , Broeyer, F. , de Kam, M. , Baas, J. , Cohen, A. , and van Gerven, J. (2017) Pharmacodynamic response profiles of anxiolytic and sedative drugs. Br J Clin Pharmacol, 83: 1028–1038. doi: 10.1111/bcp.13204.
References
- 1. Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 2016; 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E, et al. The Concise Guide to PHARMACOLOGY 2015/16: Ligand gated‐ion channels. Br J Pharmacol 2015; 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E, et al. The Concise Guide to PHARMACOLOGY 2015/16: Other ion channels. Br J Pharmacol 2015; 172: 5942–5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alexander SPH, Fabbro D, Davenport AP, Kelly E, Marrion N, Peters JA, et al. The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 2015; 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Greenblatt DJ, Wright CE. Clinical pharmacokinetics of alprazolam. Therapeutic implications. Clin Pharmacokinet 1993; 24: 453–471. [DOI] [PubMed] [Google Scholar]
- 6. Cohen AF, Burggraaf J, Van Gerven JMA, Moerland M, Groeneveld GJ. The use of biomarkers in human pharmacology (Phase I) studies. Annu Rev Pharmacol Toxicol 2015; 55: 55–74. [DOI] [PubMed] [Google Scholar]
- 7. Cohen AF. Developing drug prototypes: pharmacology replaces safety and tolerability? Nat Rev Drug Discov 2010; 9: 856–865. [DOI] [PubMed] [Google Scholar]
- 8. Morgan P, Van Der Graaf PH, Arrowsmith J, Feltner DE, Drummond KS, Wegner CD, et al. Can the flow of medicines be improved? Fundamental pharmacokinetic and pharmacological principles toward improving Phase II survival. Drug Discov Today 2012; 17: 419–424. [DOI] [PubMed] [Google Scholar]
- 9. Dumont GJH, De Visser SJ, Cohen AF, Van Gerven JMA. Biomarkers for the effects of selective serotonin reuptake inhibitors (SSRIs) in healthy volunteers. Br J Clin Pharmacol 2005; 59: 495–510 (Review). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. De Visser SJ, Van der Post JP, De Waal PP, Cornet F, Cohen AF, Van Gerven JMA. Biomarkers for the effects of benzodiazepines in healthy volunteers. Br J Clin Pharmacol 2003; 55: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McKernan RM, Rosahl TW, Reynolds DS, Sur C, Wafford KA, Atack JR, et al. Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABA(A) receptor alpha1 subtype. Nat Neurosci 2000; 3: 587–592. [DOI] [PubMed] [Google Scholar]
- 12. Rudolph U, Crestani F, Benke D, Brünig I, Benson JA, Fritschy JM, et al. Benzodiazepine actions mediated by specific gamma‐aminobutyric acid(A) receptor subtypes. Nature 1999; 401: 796–800 .Erratum in: Nature 2000; 404: 629. [DOI] [PubMed] [Google Scholar]
- 13. Crestani F, Keist R, Fritschy JM, Benke D, Vogt K, Prut L, et al. Trace fear conditioning involves hippocampal alpha5 GABA(A) receptors. Proc Natl Acad Sci U S A 2002; 99: 8980–8985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Laurijssens BE, Greenblatt DJ. Pharmacokinetic‐pharmacodynamic relationships for benzodiazepines. Clin Pharmacokinet 1996; 30: 52–76. [DOI] [PubMed] [Google Scholar]
- 15. Besson M, Matthey A, Daali Y, Poncet A, Vuilleumier P, Curatolo M, et al. GABAergic modulation in central sensitization in humans: a randomized placebo‐controlled pharmacokinetic‐pharmacodynamic study comparing clobazam with clonazepam in healthy volunteers. Pain 2015; 156: 397–404. [DOI] [PubMed] [Google Scholar]
- 16. Tsunoda K, Uchida H, Suzuki T, Watanabe K, Yamashima T, Kashima H. Effects of discontinuing benzodiazepine‐derivative hypnotics on postural sway and cognitive functions in the elderly. Int J Geriatr Psychiatry 2010; 25: 1259–1265. [DOI] [PubMed] [Google Scholar]
- 17. Reilly JL, Lencer R, Bishop JR, Keedy S, Sweeney JA. Pharmacological treatment effects on eye movement control. Brain Cogn 2008; 68: 415–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mandema JW, Danhof M. Electroencephalogram effect measures and relationships between pharmacokinetics and pharmacodynamics of centrally acting drugs. Clin Pharmacokinet 1992; 23: 191–215. [DOI] [PubMed] [Google Scholar]
- 19. van Steveninck AL, van Berckel BN, Schoemaker RC, Breimer DD, van Gerven JM, Cohen AF. The sensitivity of pharmacodynamic tests for the central nervous system effects of drugs on the effects of sleep deprivation. J Psychopharmacol 1999; 13: 10–17. [DOI] [PubMed] [Google Scholar]
- 20. Chen X, de Haas S, de Kam M, van Gerven J. An overview of the CNS‐pharmacodynamic profiles of non‐selective and selective GABA receptor modulators. Adv Pharmacol Sci 2012; 2012: 134523. doi: 10.1155/2012/134523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. de Haas SL, de Visser SJ, van der Post JP, de Smet M, Schoemaker RC, Rijnbeek B, et al. Pharmacodynamic and pharmacokinetic effects of TPA023, a GABAA a2,3 subtype‐selective receptor modulator, compared to lorazepam and placebo in healthy volunteers. J Psychopharmacol 2007; 21: 374–383. [DOI] [PubMed] [Google Scholar]
- 22. de Haas SL, de Visser SJ, van der Post JP, Schoemaker RC, van Dyck K, Murphy MG, et al. Pharmacodynamic and pharmacokinetic effects of MK‐0343, a GABAA α2,3 subtype selective receptor modulator, compared to lorazepam and placebo in healthy male volunteers. J Psychopharmacol 2008; 22: 24–32. [DOI] [PubMed] [Google Scholar]
- 23. de Haas SL, Franson KL, Schmitt JA, Cohen AF, Fau JB, Dubruc C, et al. The pharmacokinetic and pharmacodynamic effects of SL65.1498, a GABAA 2,3 selective receptor modulator, in comparison with lorazepam in healthy volunteers. J Psychopharmacol 2009; 23: 625–632. [DOI] [PubMed] [Google Scholar]
- 24. Buoli M, Dell'osso B, Bosi MF, Altamura C. Slow vs standard up‐titration of paroxetine in the treatment of panic disorder: a prospective randomized trial. Psychiatry Clin Neurosci 2010; 64: 612–619. [DOI] [PubMed] [Google Scholar]
- 25. Blin O, Micallef J, Audebert C, Legangneux E. A double‐blind, placebo‐ and flurazepam‐controlled investigation of the residual psychomotor and cognitive effects of modified release zolpidem in young healthy volunteers. J Clin Psychopharmacol 2006; 26: 284–289. [DOI] [PubMed] [Google Scholar]
- 26. Baas JPM, Mol N, Kenemans JL, Prinssen EP, Niklson I, Chen X, et al. Validating a human model for anxiety using startle potentiated by cue and context: the effects of alprazolam, pregabalin, and diphenhydramine. Psychopharmacology (Berl) 2009; 205: 73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Grillon C, Baas JM, Pine DS, Lissek S, Lawley M, Ellis V, et al. The benzodiazepine alprazolam dissociates contextual fear from cued fear in humans as assessed by fear‐potentiated startle. Biol Psychiatry 2006; 60: 760–766. [DOI] [PubMed] [Google Scholar]
- 28. Hermans EJ, Putman P, Baas JM, Koppeschaar HP, van Honk J. A single administration of testosterone reduces fear‐potentiated startle in humans. Biol Psychiatry 2006; 59: 872–874. [DOI] [PubMed] [Google Scholar]
- 29. Baas JM, Grillon C, Böcker KB, Brack AA, Morgan CA 3rd, Kenemans JL, et al. Benzodiazepines have no effect on fear‐potentiated startle in humans. Psychopharmacology (Berl) 2002; 161: 233–247. [DOI] [PubMed] [Google Scholar]
- 30. Wright BM. A simple mechanical ataxia‐meter. J Physiol 1971; 218 (Suppl): 27P–28P. [PubMed] [Google Scholar]
- 31. van Steveninck AL, Gieschke R, Schoemaker RC, Roncari G, Tuk B, Pieters MS, et al. Pharmacokinetic and pharmacodynamic interactions of bretazenil and diazepam with alcohol. Br J Clin Pharmacol 1996; 41: 565–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Norris H. The action of sedatives on brain stem oculomotor systems in man. Neuropharmacology 1971; 10: 181–191. [DOI] [PubMed] [Google Scholar]
- 33. van Steveninck AL, Schoemaker HC, den Hartigh J, Pieters MS, Breimer DD, Cohen AF. Effects of intravenous temazepam. II. A study of the long‐term reproducibility of pharmacokinetics, pharmacodynamics, and concentration‐effect parameters. Clin Pharmacol Ther 1994; 55: 546–555. [DOI] [PubMed] [Google Scholar]
- 34. Bittencourt PR, Wade P, Smith AT, Richens A. The relationship between peak velocity of saccadic eye movements and serum benzodiazepine concentration. Br J Clin Pharmacol 1981; 12: 523–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baloh RW, Sills AW, Kumley WE, Honrubia V. Quantitative measurement of saccade amplitude, duration, and velocity. Neurology 1975; 25: 1065–1070. [DOI] [PubMed] [Google Scholar]
- 36. Borland RG, Nicholson AN. Visual motor co‐ordination and dynamic visual acuity. Br J Clin Pharmacol 1984; 18 (Suppl): 69S–72S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zuiker RG, Chen X, Østerberg O, Mirza NR, Muglia P, de Kam M, et al. NS11821, a partial subtype‐selective GABAA agonist, elicits selective effects on the central nervous system in randomized controlled trial with healthy subjects. J Psychopharmacol 2016; 30: 253–262. [DOI] [PubMed] [Google Scholar]
- 38. Chen X, Jacobs G, de Kam ML, Jaeger J, Lappalainen J, Maruff P, et al. AZD6280, a novel partial γ‐aminobutyric acid A receptor modulator, demonstrates a pharmacodynamically selective effect profile in healthy male volunteers. J Clin Psychopharmacol 2015; 35: 22–33. [DOI] [PubMed] [Google Scholar]
- 39. Chen X, Jacobs G, de Kam M, Jaeger J, Lappalainen J, Maruff P, et al. The central nervous system effects of the partial GABA‐Aα2,3 ‐selective receptor modulator AZD7325 in comparison with lorazepam in healthy males. Br J Clin Pharmacol 2014; 78: 1298–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Atack JR. GABA‐A receptor alpha2/alpha3 subtype‐selective modulators as potential nonsedating anxiolytics. Curr Top Behav Neurosci 2010; 2: 331–360. [DOI] [PubMed] [Google Scholar]
- 41. Engin E, Liu J, Rudolph U. α2‐containing GABA(A) receptors: a target for the development of novel treatment strategies for CNS disorders. Pharmacol Ther 2012; 136: 142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Micó JA, Prieto R. Elucidating the mechanism of action of pregabalin: α(2)δ as a therapeutic target in anxiety. CNS Drugs 2012; 26: 637–648. [DOI] [PubMed] [Google Scholar]
- 43. Robinson OJ, Vytal K, Cornwell BR, Grillon C. The impact of anxiety upon cognition: perspectives from human threat of shock studies. Front Hum Neurosci 2013; 7: 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gilron I. Gabapentin and pregabalin for chronic neuropathic and early postsurgical pain: current evidence and future directions. Curr Opin Anaesthesiol 2007; 20: 456–472. [DOI] [PubMed] [Google Scholar]
- 45. Laurijssens BE, Greenblatt DJ. Pharmacokinetic‐pharmacodynamic relationships for benzodiazepines. Clin Pharmacokinet 1996; 30: 52–76. [DOI] [PubMed] [Google Scholar]