

Abstract
The Dual-Specificity Tyrosine Phosphorylation-Regulated Kinase 1A (DYRK1A) is an enzyme that has been implicated as an important drug target in various therapeutic areas, including neurological disorders (Down syndrome, Alzheimer’s disease), oncology, and diabetes (pancreatic β-cell expansion). Current small molecule DYRK1A inhibitors are ATP-competitive inhibitors that bind to the kinase in an active conformation. As a result, these inhibitors are promiscuous, resulting in pharmacological side effects that limit their therapeutic applications. None are in clinical trials at this time. In order to identify a new DYRK1A inhibitor scaffold, we constructed a homology model of DYRK1A in an inactive, DFG-out conformation. Virtual screening of 2.2 million lead-like compounds from the ZINC database, followed by in vitro testing of selected 68 compounds revealed 8 hits representing 5 different chemical classes. We chose to focus on one of the hits from the computational screen, thiadiazine 1 which was found to inhibit DYRK1A with IC50 of 9.41 μM (Kd = 7.3 μM). Optimization of the hit compound 1, using structure-activity relationship (SAR) analysis and in vitro testing led to the identification of potent thiadiazine analogs with significantly improved binding as compared to the initial hit (Kd = 71–185 nM). Compound 3–5 induced human β-cell proliferation at 5 μM while showing selectivity for DYRK1A over DYRK1B and DYRK2 at 10 μM. This newly developed DYRK1A inhibitor scaffold with unique kinase selectivity profiles has potential to be further optimized as novel therapeutics for diabetes.
Keywords: Dual-specificity Tyrosine-Regulated Kinases (DYRKs), DYRK1A inhibitor, Structure Activity Relationship Study, Selectivity, Proliferation, Diabetes
Graphical Abstract
INTRODUCTION
The Dual-specificity Tyrosine-Regulated Kinases (DYRKs) belong to the CMCG family of eukaryotic protein kinases, which include the CDK-like kinases (CLKs), Glycogen Synthase Kinases 3 (GSK3s), Cyclin Dependent Kinases (CDKs) and Mitogen-Activated Protein Kinases (MAPKs). DYRK family proteins self-activate by auto-phosphorylation of the conserved tyrosine residue in the activation loop, then subsequently phosphorylate substrates only on serine and threonine residues.[1–3] The DYRK family consists of five subtypes including 1A, 1B, 2, 3 and 4. Among them, DYRK1A is the most extensively studied subtype. It is ubiquitously expressed and has been shown to play an important role in brain development and function,[4] neurodegenerative diseases,[5, 6] tumorigenesis, apoptosis,[7, 8] and human pancreatic β-cell proliferation[9–12].
Regulated expression of DYRK1A during fetal, postnatal life, as well as in adults is essential for normal neuronal development and brain function. DYRK1A is located in the Down Syndrome Critical region (DSCR) on human chromosome 21, a genomic region that has an important role in pathogenesis of Down Syndrome (DS), one of the most common and frequent human genetic disorders.[1, 13] Overexpression of DYRK1A in mouse and Drosophila models mimics the neurodevelopmental abnormalities associated with DS.[4, 5, 14, 15] Recent evidence has also implicated DYRK1A in the tau dysfunction and tau pathology of Alzheimer’s disease (AD), dementia with Lewy bodies, and Parkinson’s disease.[5, 6, 16] In addition, it has been reported that DYRK1A is overexpressed in various tumors such as, ovarian cancer, colon cancer, lung cancer and pancreatic cancer, signifying its role in tumorigenesis and uncontrolled cell proliferation.[7, 8] Inhibition of DYRK1A has been reported to promote EGFR degradation and reduce EGFR-dependent tumor growth in glioblastoma.[17] Furthermore, DYRK1A inhibition induces activation of caspase-9 which leads to massive apoptosis in specific cancer cell types.[18] Recently, DYRK1A has been shown to be involved in molecular pathways relevant to human pancreatic β-cell proliferation, making it a potential therapeutic target for β-cell regeneration in Type 1 and Type 2 diabetes.[9–12, 19]
DYRK1A has attracted increasing interest as a potential therapeutic target because it has been implicated, for example, in neurodegenerative disease, cancer and diabetes. A significant amount of research has been carried out to not only understand its underlying role in diseases, but also in identifying novel DYRK1A inhibitors.[1, 4, 6–10, 12] Several DYRK1A inhibitors have been identified and characterized from natural sources as well as from small molecule drug discovery programs. Among known DYRK1A inhibitors, harmine and its analogues (β-carbolines) are the most studied and remain the most potent and orally bioavailable class of inhibitors available.[1, 6] Apart from harmine, epigallocatechin (EGCG) and other flavan-3-ols,[20, 21] leucettines,[22, 23] quinalizarine,[24] peltogynoids Acanilol A and B,[25] benzocoumarins (dNBC),[26] indolocarbazoles (Starosporine, rebeccamycin and their analogues),[27] are other natural products that have been shown to inhibit DYRK1A and other kinases. Among the other scaffolds identified from small molecule drug discovery, GNF4877,[10] INDY,[15] DANDY,[28] and FINDY,[29] pyrazolidine-diones,[30, 31] amino-quinazolines,[32] meriolins,[33–35] pyridine and pyrazines,[36] chromenoindoles,[37] 11H-indolo[3,2-c]quinoline-6-carboxylic acids,[38] thiazolo[5,4-f]quinazolines (EHT 5372),[39, 40] CC-401[19] and 5-iodotubercidin[12, 41] showed potent DYRK1A activity with varying degrees of kinase selectivity.
Recently, using a phenotypic luciferase reporter high-throughput small-molecule screen (HTS), our group identified harmine as a new class of compounds that cause ~10–15-fold induction in human pancreatic β-cell proliferation, which is in the relevant range for therapeutic human β-cell expansion.[9] DYRK1A was defined as the likely target of harmine through genetic silencing and other studies, likely working through the Nuclear Factors of Activated T-cells (NFAT) family of transcription factors as mediators of human β-cell proliferation and differentiation.[9] Using three different mouse, rat and human pancreatic islet models, we have shown that harmine is able to induce β-cell proliferation and increase islet mass, and to improve glycemic control in mice transplanted with a marginal mass of human islets in vivo.[9]
Since DYRK1A and NFATs are widely expressed in other cell types, harmine analogs are known to hit off-targets that lead to pharmacological side effects, including CNS activity, thereby limiting its therapeutic utility and potential for pharmaceutical development for a chronic disease like diabetes. To counter these suboptimal selectivity and safety profiles, we were interested in identifying novel DYRK1A inhibitor scaffolds with the potential for a unique binding mode to DYRK1A other than the canonical ATP-competitive binding mode. As a starting point, we proposed to use a computational approach to model DYRK1A in an inactive conformation, the DFG-out conformation,[42],[43, 44] and to identify new inhibitors with the potential for improved selectivity and safety profile as compared to known DYRK1A type-I inhibitors.
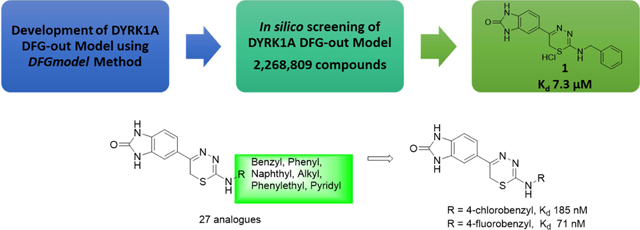
Herein, we report the identification of potent and novel DYRK1A inhibitor scaffolds, using an integrated computational modeling and medicinal chemistry approach (Figure 1A). We used DFGmodel,[42] a computational method for modeling the inactive kinase conformations, to model DYRK1A. Virtual screening of lead-like molecules (2,268,809 compounds) against the inactive DYRK1A models followed by binding and functional assays identified a novel DYRK1A inhibitor hit scaffold, 1 with IC50 of 9.41 μM (Kd = 7.3 μM). We also describe our hit-to-lead medicinal chemistry studies of this scaffold compound which led to the identification of a highly potent thiadiazine lead inhibitor 3–5 with 90-fold improved DYRK1A binding as compared to the initial hit, compound 1, and authentic human pancreatic β-cell proliferation with impressive selectivity for DYRK1A over the closely related DYRK1B and DYRK2.
Figure 1. DYRK1A DFG-out models.

(A) Strategy from computational modeling to experimental testing to identify DYRK1A inhibitor hit 1. (B) The DFG-in crystal structure and DFG-out model are shown in ribbon. The large-lobe of the two conformations is identical (gray); The small-lobe of the DFG-out structure and DFG-in model are shown in pink and cyan ribbons, respectively. (C) The conformational change between the DFG-in (cyan sticks) and DFG-out (pink sticks) states is shown. The type-I inhibitor harmine (cyan sticks) binds the ATP-binding site of DYRK1A in the DFG-in structure. (D) The electrostatic potential of the surface of the ligand-binding pocket in the DFG-out conformation is shown, with the DFG-pocket highlighted in blue ellipse.
RESULTS AND DISCUSSION
DYRK1A Kinase Modeling.
All currently known inhibitor-bound DYRK1A crystal structures adopt the DFG-in conformation, confirming these inhibitors as canonical type-I, ATP-competitive inhibitors.[15, 45–47] We anticipated that DYRK1A inhibitors that are identified from alternative kinase conformations could possess novel chemical scaffolds as well as exhibit different kinase selectivity and safety profiles as compared to type-I inhibitors. As a starting point, we constructed models of DYRK1A in the DFG-out conformation for virtual screening to identify hits with novel binding modes. Specifically, we applied DFGmodel, a homology modeling pipeline recently-developed in one of our labs (Figure 1B).[42] In brief, DFGmodel takes the amino acid sequence or known DFG-in structure of the target protein (if available) as input, and relies on a curated set of inactive kinase structures covering a range of unique conformations as modeling templates, to generate multiple DFG-out models. This method also models the rigid-body movement of the small-lobe observed in the DFG-out conformations of protein kinases. Fifty initial DFG-out DYRK1A models were generated with MODELLER[48] and evaluated and ranked with Z-DOPE, a normalized atomic distance-dependent statistical potential based on known protein structures[49]. The newly generated DYRK1A DFG-out models had a median Z-DOPE score of −0.93 ± 0.05, suggesting that they are reliable for further analysis.[42, 50]
Our model’s binding site comprises a portion of the ATP-binding pocket, which is partially blocked by the DFG-motif in the DFG-out conformation, and the DFG-pocket, which was previously occupied by the DFG-Phe (Phe308) side-chain of the known DFG-in structures (Figure 1C). Our protocol selects final models based on the size of the ligand-binding site.[42]
Notably, a key feature of DYRK1A is its gatekeeper residue Phe238, which is located at the narrow channel bridging the ATP-binding pocket and the DFG-pocket (Figure 1D). Several first-generation type-II kinase inhibitors such as imatinib and sorafenib have low affinity for kinases with large gatekeeper residues, such as Phe and Met, but have strong preference for kinases with small-sized gatekeeper residues (Thr and Val), which are over-represented in tyrosine kinases such as ABL and VEGFR2.[42, 43] Recent studies showed that kinases with large gatekeeper residues can be targeted with non-type-I kinase inhibitors (e.g., CDK8)[51, 52], suggesting that DYRK1A might be amenable to structure-based discovery of unique inhibitors with alternative binding modes.
Virtual screening of DYRK1A structural models.
We conducted virtual screening to each of the 10 DFG-out DYRK1A models using the ZINC12[53] lead-like small molecule library (2.2 million “available now”). We and others have shown that ligand enrichment for protein kinases is significantly improved when considering multiple conformations of homology models rather than a single structure.[42, 54, 55] The screen ranked molecules in the library according to their docking scores. To condense the results from these 10 models, we considered molecules that are in the 93rd percentile in 5 of 10 models (approximately 1,000 molecules). We clustered these molecules by their chemical similarity using the ECFP4 fingerprints with a Tanimoto coefficient (Tc) cutoff of 0.4 and visually inspected the predicted binding poses. We used the following considerations for selecting compounds for experimental testing: 1) the compound docks in the DFG-pocket; 2) potential for interaction with the amide backbone of the hinge-binding region, the αC-helix’s conserved glutamate residue (E203), or the DFG-Asp amide backbone (D307); 3) the compound samples unique chemical scaffolds not known as DYRK1A inhibitors; 4) scaffolds with drug-like properties[56]; and 5) the compound is synthetically accessible with few steps. Finally, we removed likely false positives, such as those predicted hits with unlikely tautomers or strained conformations. Although the compounds were predicted based on a DFG-out model, some of the compounds may dock well to DFG-in conformations, increasing our confidence in the prediction that they are DYRK1A inhibitors. For example, Harmine, a known type-I kinase inhibitor of DYRK1A docks well to DFG-out models with various docking programs (Figure 1C, D).
We purchased 68 commercially available compounds for testing of in vitro DYRK1A activity at 30 μM concentration using Life Technologies FRET-based LanthaScreen® Eu Kinase Binding Assay[57] (Figure 1A). Eight compounds showed inhibition of DYRK1A activity in 30–80% range at 30 μM (hit-rate of 11.7%). Of the 8 initial hits, we focused on compound 1 (CAS 931037-76-2), a 1,3,4-thiadiazine, with IC50 of 9.41 μM against DYRK1A. These data were further confirmed by a second, orthogonal assay KINOMEscan®[58] (DiscoverX) that quantifies DYRK1A binding through a sensitive qPCR method. The results obtained were consistent with those of the Life Technologies inhibition assay with the Kd of 7.3 μM for compound 1 in the DiscoverX assay.
Hit-to-lead optimization and SAR studies.
We confirmed the structure of the hit 1,3,4-thiadiazine 1 (HCl salt) by independent synthesis (Scheme 1) and comparison to the commercially purchased sample. NMR, LC-MS, and biological data confirmed the identity of the hit 1 in all aspects (Materials and Methods). The neutral analogue of compound 1 (3–1, non-salt) was found to have comparable DYRK1A activity with Kd of 4.5 μM (IC50 = 4.32 μM) (Table 1). With a confirmed scaffold in hand, we began a hit-to-lead SAR study to improve its DYRK1A binding potency and in vitro biological activity. We performed systematic structural modifications at the 2-amino position, keeping the rest of the molecule intact (Figure 2).
Scheme 1.

The Synthesis of 1,3,4-thiadiazines 3. i) Hydrazine monohydrate (2.0 eq.), Et2O, rt, 12 h, 39–99%; ii) Chloroacetyl chloride (2.0 eq.), AlCl3 (2.6 eq.), DCE, rt, 12 h, 99%; iii) 1–2 (1.1 eq.), DMF/AcOH, rt, 24 h, 23–90%; iv) K2CO3 (0.15 eq.), H2O2 (50% aq. Solution), DMSO, rt, 12 h, 42%.
Table 1.
Percent Inhibition at 30 μM and Kd of the Thiadiazine DYRK1A Inhibitors
 |
n.d.= not determined
‘% DYRK1A inhibition’ of tested compound at 30 μM, where higher values indicates stronger affinity
‘% DMSO Control’ of tested compound at 30 μM (n = 2), where lower values indicate stronger affinity
Kd values are determined using eleven serial three fold dilutions (in duplicate)
Value in parenthesis is IC50 determined at Life Technologies
Figure 2.

Summary of the SAR studies of compound 1.
The hit 1,3,4-thiadiazine compound 1 and its analogues were synthesized by the protocol outlined in Scheme 1.[59] Acylation of commercially available 2-benzimidazolone with chloroacetyl chloride in the presence of AlCl3 gave compound 2 in 99% crude yield.[60] Subsequently, the α-chloroketone 2 underwent smooth cyclo-condensation[59] with purchased or synthesized thiosemicarbazides (see Supporting Information) containing various R-groups to afford the desired thiadiazine analogues (27 compounds, Table 1) in range of 23–90% yield.
We first investigated the effect of various 2-substituted benzylamino groups on the DYRK1A binding activity as shown in Table 1. We found that DYRK1A binding activity is highly sensitive to the substitution pattern of the 2-benzylamino moiety. Notably, we observed that the introduction of fluorobenzylamino (3–2 to 3–4) and chlorobenzylamino (3–5 to 3–7) substituents at the 2-position of the thiadiazine improved the DYRK1A binding affinity of the hit 3–1 by 25- to 60-fold. Among these analogs, p-substituted benzylamino thiadiazines showed better DYRK1A binding as compared to their respective o- and m-substituted benzylamino analogues. Specifically, compounds 3–2 and 3–5 bearing p–chloro and p-fluorobenzylamino showed 24- to 60-fold improvement with Kd’s of 185 nM and 71 nM, respectively, as compared to the original hit 1 (Kd = 7300 nM) and 3–1 (Kd = 4500 nM). However, in case of trifluoromethyl-substituted benzylamino (3–8 to 3–10) and cyano-substituted benzylamino thiadiazine analogues (3–11 to 3–12), the improvement in binding activity was not as significant. Two exceptions are for m-trifluorobenzylamino thiadiazine (3–9) and m-cyanobenzylamino thiadiazine analogues (3–12) with Kd of 660 and 320 nM, respectively, a 7- to 15-fold improvement in DYRK1A binding as compared to original hit 3–1. For trifluoromethyl-substituted (3–8 to 3–10) and cyano substituents, the m-substituted benzyl thiadiazines were significantly more potent than corresponding o- and p-substituted analogs. These SAR differ from those of halogen substituents, which showed improved DYRK1A binding for the p-substituted benzyl group as compared to their respective o- and m-analogs.
We also introduced pyridylmethylamino substituents at the thiadiazine 2–position to study their effects on DYRK1A binding as well as to reduce the clogP of compound 3–1 (clogP = 2.83). Two pyridyl analogs (3–13 and 3–14) showed improved DYRK1A binding affinity compared to the hit 3–1 with reduce lipophilicity (3–13 clogP = 1.41). For example, compound 3–13 with 2-(pyridine-3-yl)methylamino substituent had binding affinity Kd of 860 nM, a 5-fold increase over compound 3–1. In addition, compound 3–15 bearing more polar m-carboxamide substituted benzyl group was synthesized by hydrolysis of the corresponding cyano derivative (3–12) using basic hydrogen peroxide solution (Scheme 1).[61] This compound was found to have improved DYRK1A binding (Kd = 440 nM) as compared to hit compound 3–1 with ~10-fold increase in Kd.
We next examined the effect of introducing methyl[62] at the α-carbon of the benzylaminothiadiazine 3–1. The 2-(α-methyl)benzylamino thiadiazine compound (3–16) was well tolerated, providing 4-fold improvement in the DYRK1A binding assay (Kd = 1100 nM). Since 4-fluorobenzylamino substituent at the 2-position improved DYRK1A binding, we synthesized 2-(4-fluoro(α-methyl))benzylamino thiadiazine (3–17). Surprisingly, introduction of fluorine substituent reduced the DYRK1A binding by half with Kd of 2300 nM. We continued to study the effects of combinations of benzyl substituents to further develop our binding models and SAR studies.
Last, we investigated the effect of changing the carbon chain length between the 2-amino and the phenyl group. Since m-trifluoromethylbenzylamino and m-cyanobenzylamino substituents were important to improve binding affinity, we synthesized m-phenylamino substituted thiadiazines (3–19 to 3–21) that are commonly found in DFG-out specific kinase inhibitors.[63] The 2-phenyl substituted thiadiazines did not show any improvement in binding affinity as compared to their superior benzylic analogs, except for compound 3–21 (Kd = 3800 nM). Increasing the carbon chain length between the amino and the phenyl group by one carbon atom, resulted in improvement in binding affinity for 2-(2-phenylethyl)amino thiadiazine (3–22) (Kd = 1600 nM), a 3-fold increase in activity as compared to the compound 3–1. Introduction of methyl β to 2-amino (3–23), further improved the binding affinity of the compound by 5-fold. Corresponding N-(2-(pyridine-2-yl)ethyl) (3–24) and N-(2-(pyridine-3-yl)ethyl) (3–25) thiadiazine analogues of compound 3–22 did not show any improvement in DYRK1A binding affinity as compared to their phenyl analogues. However, all of these analogues had 4- to 5-fold more potent Kd’s as compared to the hit compound 3–1.
Bulky substituents, including naphthylamino groups (3–26 to 3–28) at the thiadiazine 2-position did not improve binding affinity. Among all the analogues prepared, 4–chloro (3–2) and 4-fluoro (3–5) benzylamino thiadiazines showed superior affinity to DYRK1A for the analogues prepared.
Human β-cell proliferation assay:
As mentioned earlier, DYRK1A inhibition has been reported to drive human pancreatic β-cell proliferation,[9, 10, 11, 19] thus making it a potential therapeutic target for both Type 1 and Type 2 diabetes. Therefore, the effect of the most active DYRK1A inhibitor 3–5 on human β-cell proliferation was investigated using our reported assay protocols.[9] Compound 3–5 induced human β-cell proliferation at 5 μM, as compared to vehicle-treated cells, as quantified by increased KI67 labeling of insulin-labeled cells (Figure 3A, 3B). These data indicate that this 1,3,4-thiadiazine analogue induces β-cell proliferation similar to the positive control harmine. This confirms that our 1,3,4-thiadiazine scaffold have the potential for further improvement as therapeutic agents for diabetes treatment.
Figure 3: Analysis of DYRK1A inhibitors on human β-cell proliferation.

(A) Human β-cell proliferation assay using 5 μM of compound 3–5 and DMSO as negative control. (B) A representative example from A of a KI67. Error bars indicate sem *P < 0.05. A minimum of 1,000 beta cells was counted from each of 4 different human islet donors.
Kinase selectivity profile against DYRK family kinases:
In order to assess the kinase selectivity profile of these inhibitors, we carried out kinase binding assays of compound 3–5 and harmine (CAS 442-51-3) on the closely related DYRK1A, DYRK1B, and DYRK2 at 10 μM (Table 2) using the DiscoverX assay technology. Harmine inhibited DYRK1B and DYRK2 (<20% activity remaining) in addition to DYRK1A (< 1%) at 10 μM, exhibiting poor selectivity as previously reported. In contrast, the thiadiazine scaffold lead compound 3–5 exhibited an impressive selectivity for DYRK1A (4.3% remaining) as compared to DYRK1B (31% remaining) and DYRK2 (55% remaining). These data indicate the possibility that the selectivity is a result of a different binding mode as compared to canonical ATP-competitive inhibitors for DYRK1A, like harmine, which show reduced selectivity for closely related kinases.
Table 2.
Selectivity Profile of Harmine and Thiadiazine 3–5 against the DYRK-family of Protein Kinasesa
| Target |
 Harmine |
 3–5 |
|---|---|---|
| DYRK1A | 0 | 4.3 |
| DYRK1B | 6.1 | 31 |
| DYRK2 | 3.2 | 55 |
Compounds were screened at 10 μM using DiscoverX KINOMEscan®. Results for primary screen binding interactions are reported as ‘% DMSO Ctrl’, where lower values indicate stronger inhibition.
CONCLUSIONS
We have described the identification of a novel DYRK1A inhibitor scaffold using an integrated computational modeling and medicinal chemistry optimization approach. We developed a homology model of DYRK1A in the inactive conformation using a newly developed computational protocol. This DYRK1A structural model revealed a flexible and hydrophobic inhibitor-binding site with a weakly negative electrostatic potential. Virtual screening of this binding site for lead-like compounds from the ZINC12 database, followed by chemical biology and medicinal chemistry evaluation led to the selection of 68 compounds for in vitro assay of DYRK1A binding. 8 compounds were identified with DYRK1A binding in the range of 30–80% at a concentration of 30 μM, exhibiting a high hit-rate for a discovery campaign using a homology model. From these initial hits, compound 1 was titrated and found to inhibit DYRK1A with IC50 of 9.41 μM (Kd = 7.3 μM), exhibiting a unique scaffold for DYRK1A kinase inhibitors. The preliminary hit-to-lead medicinal chemistry was performed on 2-benzylamino-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4-thiadiazine (3–1), to optimize the 2-benzylamino substituents through systematic modifications. We observed that the substitution pattern of the 2-benzylamino group substantially impacts the DYRK1A binding activity of this lead series. The majority of thiadiazine analogues bearing fluoro, chloro, cyano and trifluoromethylbenzylamino substituents at the thiadiazine 2-position showed significantly improved DYRK1A binding affinity as compared to unsubstituted benzyl group of hit 1. Importantly, p–chloro (3–2) and p-fluoro (3–5) benzylamino thiadiazine were the most potent analogs identified with Kd of 185 nM and 71 nM, respectively. Similarly, introduction of polar groups at the thiadiazine 2-position with 3-pyridylmethylamino, 3-cyanobenzyl and 3-carboxyaminobenzyl groups, enhanced the DYRK1A binding affinity in the range of 5- to 15-fold as compared to 3–1. Increasing the carbon chain length between the 2-aminothiadiazine and the phenyl subsituent from 1 to 2 carbon atoms (3–22 to 3–25) improved the DYRK1A binding affinity by 4- to 5-fold. However, decreasing the chain length (i.e. 2-phenylamino substituted thiadiazines, 3–18 to 3–21), provided no noticeable improvement in DYRK1A binding as compared to 3–1. These data suggest that both benzylic and phenylethyl chain lengths are well tolerated for potent DYRK1A binding of this lead series (Figure 4). The bulkiness of substituents at the 2-position beyond phenyl, for example with naphthylamino substituents, negatively impacted DYRK1A binding.
Since DYRK1A inhibition has been proposed to drive β-cell proliferation, the effect of the most active DYRK1A inhibitor 3–5 on human β-cell proliferation was investigated. Compound 3–5 induced human β-cell proliferation at 5 μM as compared to vehicle treated cells, while also exhibiting impressive selectivity for DYRK1A over closely related kinases, DYRK1B and DYRK2 at 10 μM.
Further improvements in DYRK1A binding affinity as well pharmacological studies beyond DYRK1A binding of this lead series will be required to advance these translational drug discovery efforts. While the initial hit compound was identified via docking to the DFG-out model, several analogues designed based on this DYRK1A conformational model did not correlate with DYRK1A binding activity. Therefore, DYRK1A co-crystallization studies and other biophysical studies with our most potent thiadiazine analogues are in progress to understand the binding modes of these novel thiadiazine DYRK1A inhibitor scaffolds, which will be useful to guide further optimization to development candidate DYRK1A inhibitors. The results of our on-going SAR for this lead scaffold and, DYRK1A biophysical studies will be reported in due course.
MATERIAL AND METHODS
1H and 13C NMR spectra were acquired on a Bruker DRX-600 spectrometer at 600 MHz for 1H and 150 MHz for 13C. TLC was performed on silica coated aluminum sheets (thickness 200 μm) or alumina coated (thickness 200 μm) aluminum sheets supplied by Sorbent Technologies and column chromatography was carried out on Teledyne ISCO combiflash equipped with a variable wavelength detector and a fraction collector using a RediSep Rf high performance silica flash columns by Teledyne ISCO. LCMS and purity analysis was conducted on an Agilent Technologies G1969A high-resolution API-TOF mass spectrometer attached to an Agilent Technologies 1200 HPLC system. Samples were ionized by electrospray ionization (ESI) in positive mode. Chromatography was performed on a 2.1 × 150 mm Zorbax 300SB-C18 5-μm column with water containing 0.1% formic acid as solvent A and acetonitrile containing 0.1% formic acid as solvent B at a flow rate of 0.4 mL/min. The gradient program was as follows: 1% B (0−1 min), 1−99% B (1− 4 min), and 99% B (4−8 min). The temperature of the column was held at 50 °C for the entire analysis. The chemicals and reagents were purchased from Aldrich Co., Alfa Aesar, Enamine, TCI USA, eMolecules. All solvents were purchased in anhydrous from Acros Organics and used without further purification. Harmine CAS No. 442-51-3, Thiadiazine 1 CAS No. 931037-76-2.
All active compounds were synthesized independently in analytically pure form. They were chemically stable and exhibited a dose dependent DYRK1A binding without showing any erroneous/misleading readouts due to any aggregation, and decomposition, prevalent among known classes of Pan Assay Interference compounds (PAINS).
Generation of DFG-out models.
Models of DYRK1A in the inactive DFG-out conformation were generated using DFGmodel[42]. Briefly, DFGmodel takes the amino acid sequence of the target kinase domain as input. Generation of DFG-out models is based on a manually curated set of experimentally determined structures, sampling a range of DFG-out conformations adopted by kinases. DFGmodel uses the structure-based sequence alignment function of T_COFFEE/Expresso[64] v11.00.8 to align the target kinase to the template structures, and builds multi-template homology models with MODELLER[48] v9.14. The crystal structure of DYRK1A in the DFG-in conformation (PDBID: 3ANR[15]) was used as the input structure for DFGmodel. 50 models were built and ranked based on the size of the inhibitor-binding site calculated by POVME[65] v2.0. 10 models with largest binding site volume were selected for virtual screening.
Optimizing docking parameters.
Molecular docking was performed using FRED[42, 66] and Glide (Schrödinger 2015–3[67]; shown in Figures 3 and 4). We optimized the scaling parameters used in Glide’s protein grid generation for docking inhibitors targeting DFG-out conformations. The OPLS3 force field was used to parameterize both protein and ligands.[68] Aromatic C-H hydrogens were considered as hydrogen-bond donors. The SP docking accuracy mode was used. Several combinations of the scaling of van der Waals (vdW) radius for atoms with partial charge exceeding a cutoff were examined: the default, 1.00× scaling of vdW for partial charge > ± 0.25; 0.75× scaling of vdW for partial charge > ± 0.20; and 0.75× scaling of vdW for partial charge > ± 0.15. We adapted the performance test set of kinases with known type-II and non-type-II kinase inhibitors as described in our previous study.[42] Specifically, we used the enrichment plot to derive the values of the area-under-curve (AUC) and the logarithmic scale of the plot (logAUC).[54, 69] Glide docking was performed on all 50 DFG-out models of the test kinases. Docking results from the 10 DFG-out models with largest binding site volume were processed with the open-source cheminformatics toolkit RDKit (www.rdkit.org) and combined to generate a consensus ranking, represented by the best-scoring pose that can be used in the enrichment plot. We favor the logAUC since it emphasizes the early detection of known type-II inhibitors. Using a 0.75× scaling of vdW for atoms with partial charge > ± 0.15 yields the highest performance in both AUC and logAUC (Tables S1, S2).
Virtual screening.
10 DYRK1A models with the largest binding site were screened against a lead-like small molecule library (~2.2 million “available now” compounds, ZINC[53] database downloaded in 2013). The docking results were averaged for 10 models, as described by Ung et al.[42] We selected molecules that ranked above the 93 percentiles in at least 5 of 10 models, represented by the best-scoring pose, resulting in ~1,000 top-ranking molecules for manual inspection. Results were checked for unfavorable docking pose, unmatched ionization states and tautomers, or strained conformations. 68 compounds, with an average molecular weight of 330, were purchased and tested by in vitro activity testing.
DYRK1A Binding Assays.
Compounds were tested for DYRK1A binding activity at two different commercial kinase profiling services, Life Technologies and DiscoverX. Life Technologies uses the FRET-based LanthaScreen® Eu Kinase Binding Assay[57] whereas DiscoverX uses proprietary qPCR-based KINOMEscan® Assay[58, 70]. Compounds were screened for DYRK1A activity at single concentration of 30 μM in duplicates. Similarly, the dissociation constant Kd of the hit compounds from the initial screening was determined at DiscoverX using their proprietary KINOMEscan® Assay[58]. Kd values are determined using eleven serial three fold dilutions with highest concentration of 60 μM.
β-cell proliferation assay.
Human pancreatic islets were obtained from the NIH/NIDDK-supported Integrated Islet Distribution Program (IIDP) (https://iidp.coh.org). Islets were first dispersed with Accutase (Sigma, St. Louis, MO) onto coverslips as described earlier.[9] After 2 hours, dispersed human islet cells were treated with compound in RPMI1640 complete medium for 96 hours. Then the cells were fixed and immunolabeled for insulin and Ki67.[9] Total insulin-positive cells and double Ki67-plus insulin-positive cells were imaged and counted. At least 1000 cells were counted per human islet donor.
Kinome Scan Profile.
Compounds were screened against DYRK1A, DYRK1B, and DYRK2 at single concentration of 10 μM in duplicates at DiscoverX using their proprietary KINOMEscan® Assay.[58] The results for primary screen binding interactions are reported as ‘% DMSO Ctrl’, where lower values indicate stronger affinity.
Synthetic Procedures.
5-(2-Chloroacetyl)-1H-benzo[d]imidazol-2(3H)- one (2)[60]
To a suspension of aluminium chloride (5.16 g, 38.76 mmol) in DCE (20 mL) was added 2-chloroacetyl chloride (2.34 mL, 29.8 mmol) dropwise at 0°C under Argon atmosphere and stirred for 30 min. A solution of 2-hydroxybenzimidazole 1 (2 g, 14.9 mmol) in dichloroethane (DCE) (5 mL) was added slowly to the above solution and stirred at 50°C for 2 hours and then, overnight at room temperature. Upon completion of reaction monitored by LC/MS, the mixture was poured onto ice to obtain the product as white precipitate which was filtered and washed with water and ethyl acetate (EtOAc). Consequently, the compound was dried under high vacuum to provide the desired product 5-(2-chloroacetyl)-1H-benzo[d]imidazol-2(3H)- one 2 (3.1 g, 99 %) as white solid. 1H-NMR (600 MHz, DMSO-d6): δ 11.13 (s, 1H), 10.98 (s, 1H), 7.69–7.67 (d, 1H), 7.49 (s, 1H), 7.05–7.04 (d, 1H), 5.13 (s, 2H); MS (ESI) m/z 211.02 (M+H)+.
N-benzyl-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine hydrochloride salt (1)[59]
A solution of 5-(2-chloroacetyl)-1H-benzo[d]imidazol-2(3H)-one 2 (0.15 g, 0.714 mmol, 1 eq.) and N-benzylhydrazinecarbothioamide (0.15 g, 1.1 eq.) in N,N-dimethylformamide (DMF)/acetic acid (AcOH) (2 mL/0.2 mL) was stirred at room temperature for 12 h. Upon completion of reaction monitored by LC/MS, the mixture was concentrated under high vacuum to remove solvent, the residue was triturated with CH2Cl2, the precipitate was wash with CH2Cl2 and dried under high vacuum to give product desired product 1 (0.22 g, 90%) as white solid. 1H-NMR (600 MHz, DMSO-d6): δ 11.01 (s, 1H), 10.94 (s, 1H), 7.54–7.53 (d, 1H), 7.48 (s, 1H), 7.44–7.39 (m, 4H), 7.37–7.35 (m, 1H), 7.07–7.05 (d, 1H), 4.71 (s, 2H), 4.22 (s, 2H); 13C-NMR (150 MHz, DMSO-d6): δ 158.8, 155.4, 152.1, 135.7, 132.8, 130.3, 128.7, 127.9, 125.1, 120.7, 108.5, 106.6, 47.3, 22.2; HRMS (ESI): m/z [M + H]+ calcd for C17H16N5OS+ : 338.1070, found: 338.1070; HPLC: t = 3.45 min, Purity >95%
General procedure for the synthesis of thiadiazine analogs 3[59]
A solution of 5-(2-chloroacetyl)-1H-benzo[d]imidazol-2(3H)-one 2 (0.47 mmol, 1 eq.) and N-alkyl thiosemicarbazides (1.1 eq.) in DMF/AcOH (2 mL/0.2 mL) was stirred at room temperature for 12 h. Upon completion of reaction monitored by LC/MS, the mixture was concentrated under high vacuum to remove solvent and aqueous ammonia solution was added to it. The resulting precipitate was filtered, washed with water and dried under high vacuum to give the desired 1,3,4-thiadiazines 3.
N-benzyl-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–1)
Yellow solid. Yield 90%. 1H-NMR (600 MHz, DMSO-d6): δ 10.76 (s, 1 H), 10.75 (s, 1 H), 7.51 (s, 1 H), 7.45 (m, 2 H), 7.34 (m, 3 H), 7.24 (m, 1 H), 6.95 (d, 1 H, J = 7.8 Hz), 4.56 (s, 2 H), 3.65 (s, 2 H); 13C-NMR (150 MHz, DMSO-d6): δ 155.4, 147.3, 139.8, 130.9, 130.1, 128.7, 128.2, 127.4, 126.6, 119.6, 108.1, 106.1, 45.4, 21.7; HRMS (ESI): m/z [M + H]+ calcd for C17H16N5OS+ : 338.1070, found: 338.1063; HPLC: t = 3.48 min, Purity >95%.
N-(4-chlorobenzyl)-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–2)
Yellow solid. Yield 53%. 1H-NMR (600 MHz, DMSO-d6): δ 10.77 (s, 1 H), 10.76 (s, 1H), 7.51 (s, 1 H), 7.45 (m, 2 H), 7.38 (m, 3 H), 6.95 (d, 1 H, J = 8.4 Hz), 4.53 (s, 2 H), 3.65 (s, 1 H); 13C-NMR (150 MHz, DMSO-d6): δ 155.4, 147.5, 138.8, 131.2, 131.0, 130.1, 129.3, 128.5, 128.1, 127.9, 119.6, 108.1, 106.1, 45.4, 21.8; HRMS (ESI): m/z [M + H]+ calcd for C17H15ClN5OS+ : 372.0680, found: 372.0662; HPLC: t = 3.73 min, Purity >95%
N-(3-chlorobenzyl)-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–3)
Yellow solid. Yield 49%. 1H-NMR (600 MHz, DMSO-d6): δ 10.79 (s, 1 H), 10.77 (s, 1H), 7.46 (d, 2 H, J = 7.2 Hz), 7.41 (s, 1 H), 7.36 (d, 1 H, J = 7.8 Hz), 7.33 (d, 2 H, J = 6.6 Hz), 6.96 (d, 1 H, J = 8.4 Hz), 4.56 (s, 2 H), 3.72 (s, 2 H); 13C-NMR (150 MHz, DMSO-d6): δ 155.4, 147.7, 142.3, 132.9, 131.1, 130.1, 128.3, 127.2, 126.7, 126.1, 119.7, 108.1, 106.1, 45.4, 21.8; HRMS (ESI): m/z [M + H]+ calcd for C17H15ClN5OS+ : 372.0680, found: 372.0667; HPLC: t = 3.75 min, Purity >95%
N-(2-chlorobenzyl)-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–4)
Yellow solid. Yield 57%. 1H-NMR (600 MHz, DMSO-d6): δ 10.79 (s, 1 H), 10.77 (s, 1H), 7.46 (m, 3 H), 7.32 (m, 3 H), 6.96 (d, 1 H, J = 9.0 Hz), 4.61 (s, 2 H), 3.73 (s, 2 H); 13C-NMR (150 MHz, DMSO-d6): δ 155.4, 148.1, 136.2, 132.0, 131.3, 130.1, 129.1, 128.6, 128.0, 127.5, 127.1, 119.8, 108.1, 106.2, 44.7, 21.9; HRMS (ESI): m/z [M + H]+ calcd for C17H15ClN5OS+ : 372.0680, found: 372.0668; HPLC: t = 3.62 min, Purity >95%
N-(4-fluorobenzyl)-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–5)
Yellow solid. Yield 36%. 1H-NMR (600 MHz, DMSO-d6): δ 10.77 (s, 1 H), 10.76 (s, 1H), 7.45 (m, 2 H), 7.39 (m, 2 H), 7.15 (t, 3 H, J = 9.0 Hz), 6.94 (d, 1 H, J = 8.4 Hz), 4.53 (s, 2 H), 3.67 (s, 2 H); 13C-NMR (150 MHz, DMSO-d6): δ 161.9, 160.3, 155.4, 148.0, 147.4, 136.0, 131.0, 130.1, 129.8, 129.4, 129.3, 128.6, 119.6, 114.9, 114.8, 108.1, 106.1, 45.4, 21.8; HRMS (ESI): m/z [M + H]+ calcd for C17H15FN5OS+ : 356.0976, found: 356.0983; HPLC: t = 3.57 min, Purity >95%.
N-(3-fluorobenzyl)-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–6)
Yellow solid. Yield 62%. 1H-NMR (600 MHz, DMSO-d6): δ 10.78 (s, 1 H), 10.77 (s, 1H), 7.46 (d, 2 H, J = 7.2 Hz), 7.38 (m, 1 H), 7.17 (m, 2 H), 7.08 (m, 1 H), 6.96 (d, 1 H, J = 7.8 Hz), 4.56 (s, 2 H), 3.70 (s, 2 H); 13C-NMR (150 MHz, DMSO-d6): δ 163.0, 161.4, 155.4, 147.8, 142.7, 131.1, 130.1, 128.3, 123.4, 119.7, 114.1, 113.5, 106.1, 45.3, 21.8; HRMS (ESI): m/z [M + H]+ calcd for C17H15FN5OS+ : 356.0976, found: 356.0975; HPLC: t = 3.54 min, Purity >95%.
N-(2-fluorobenzyl)-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–7)
Yellow solid. Yield 44%. 1H-NMR (600 MHz, DMSO-d6): δ 10.81 (s, 1 H), 10.79 (s, 1H), 7.46 (m, 3 H), 7.33 (m, 1 H), 7.20 (m, 2 H), 6.96 (d, 1 H, J = 8.4 Hz), 4.61 (s, 2 H), 3.76 (s, 2 H); 13C-NMR (150 MHz, DMSO-d6): δ 160.9, 159.3, 155.4, 131.4, 130.1, 129.8, 129.1, 124.3, 119.8, 115.1, 108.1, 106.2, 21.8; HRMS (ESI): m/z [M + H]+ calcd for C17H15FN5OS+: 356.0976, found: 356.0989; HPLC: t = 3.48 min, Purity >95%.
N-(4-trifluoromethylbenzyl)-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–8)
Yellow solid. Yield 33%. 1H-NMR (600 MHz, DMSO-d6): δ 10.77 (s, 1 H), 10.76 (s, 1 H), 7.71 (d, 2 H, J = 7.8 Hz), 7.56 (d, 2 H, J = 7.8 Hz), 7.46 (d, 2 H, J = 7.8 Hz), 6.95 (d, 1 H, J = 9.0 Hz), 4.63 (s, 2 H), 3.66 (s, 2 H); 13C-NMR (150 MHz, DMSO-d6): δ 155.4, 147.5, 144.9, 131.0, 130.0, 128.5, 128.0, 127.4, 125.5, 125.0, 124.6, 123.4, 119.6, 108.1, 106.1, 45.5, 21.8; HRMS (ESI): m/z [M + H]+ calcd for C18H15F3N5OS+ : 406.0944, found: 406.0934; HPLC: t = 3.91 min, Purity >95%.
N-(3-trifluoromethylbenzyl)-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–9)
Yellow solid. Yield 54%. 1H-NMR (600 MHz, DMSO-d6): δ 10.78 (s, 1 H), 10.77 (s, 1 H), 7.70 (m, 1 H), 7.68 (d, 2 H, J = 7.2 Hz), 7.61 (m, 2 H), 7.46 (m 2 H), 6.95 (d, 1 H, J = 8.4 Hz), 4.64 (s, 2 H), 3.68 (s, 2 H); 13C-NMR (150 MHz, DMSO-d6): δ 155.4, 147.5, 141.4, 131.5, 131.0, 130.1, 129.3, 128.5, 123.8, 123.4, 119.6, 108.2, 106.1, 44.9, 21.8; HRMS (ESI): m/z [M + H]+ calcd for C18H15F3N5OS+ : 406.0944, found: 406.0924; HPLC: t = 3.87 min, Purity >95%.
N-(2-trifluoromethylbenzyl)-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–10)
Yellow solid. Yield 23%. 1H-NMR (600 MHz, DMSO-d6): δ 10.78 (s, 1 H), 10.77 (s, 1 H), 7.73 (d, 1 H, J = 7.8 Hz), 7.67 (d, 2 H, J = 7.2 Hz), 7.61 (m, 1 H), 7.48 (m 2 H), 6.95 (d, 1 H, J = 8.4 Hz), 4.75 (s, 2 H), 3.72 (s, 2 H); 13C-NMR (150 MHz, DMSO-d6): δ 155.4, 147.7, 138.0, 132.6, 131.0, 130.1, 128.7, 128.5, 127.2, 126.1, 125.9, 123.6, 119.7, 108.1, 106.1, 41.9, 21.8; HRMS (ESI): m/z [M + H]+ calcd for C18H15F3N5OS+: 406.0944, found: 406.0927; HPLC: t = 3.80 min, Purity >95%.
N-(4-cyanobenzyl)-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–11)
Yellow solid. Yield 45%. 1H-NMR (600 MHz, DMSO-d6): δ 10.77 (s, 1 H), 10.76 (s, 1H), 7.81 (d, 2 H, J = 8.4 Hz), 7.65 (s, 1 H), 7.54 (d, 2 H, J = 7.8 Hz), 7.46 (m, 2 H), 6.94 (d, 1 H, J = 8.4 Hz), 4.62 (s, 2 H), 3.67 (s, 2 H); 13C-NMR (150 MHz, DMSO-d6): δ 155.4, 147.5, 146.0, 132.6, 132.2, 131.7, 131.0, 130.1, 128.5, 128.1, 127.7, 119.6, 118.9, 109.3, 108.1, 106.1, 45.1, 21.8; HRMS (ESI): m/z [M + H]+ calcd for C18H15N6OS+: 363.1023, found: 363.1006; HPLC: t = 3.30 min, Purity >95%.
N-(3-cyanobenzyl)-5-(benzo[]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–12)
Yellow solid. Yield 45%. 1H-NMR (600 MHz, DMSO-d6): δ 10.77 (s, 1 H), 10.74 (s, 1H), 7.79 (s, 1 H), 7.72 (m, 2 H), 7.56 (t,1 H, J = 7.8 Hz), 7.46 (m, 2 H), 6.96 (d, 1 H, J = 8.4 Hz), 4.59 (s, 2 H), 3.71 (s, 2 H); 13C-NMR (150 MHz, DMSO-d6): δ 155.4, 147.6, 141.7, 132.4, 131.0, 130.9, 130.4, 130.1, 129.4, 128.5, 119.7, 118.9, 111.1, 108.1, 106.1, 44.9, 21.8; HRMS (ESI): m/z [M + H]+ calcd for C18H15N6OS+: 363.1023, found: 363.1015; HPLC: t = 3.34 min, Purity >95%.
N-(pyridine-3yl)methyl-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–13)
Yellow solid. Yield 43%. 1H-NMR (600 MHz, DMSO-d6): δ 10.77 (s, 1 H), 10.76 (s, 1H), 8.57 (s, 1 H), 8.45 (s, 1 H), 7.76 (d, 1 H, J =6.6 Hz), 7.57 (s, 1 H), 7.46 (m, 1 H), 7.36 (m, 1 H), 6.96 (d, 1 H, J = 8.4 Hz), 4.57 (s, 2 H), 3.66 (s, 2 H); 13C-NMR (150 MHz, DMSO-d6): δ 155.4, 149.4, 148.9, 148.4, 148.0, 147.5, 135.2, 131.0, 130.1, 128.6, 123.4, 119.6, 108.1, 106.1, 43.3, 21.8; HRMS (ESI): m/z [M + H]+ calcd for C16H15N6OS+: 339.1023, found: 339.1020; HPLC: t= 1.25 min, Purity >95%.
N-(pyridine-4yl)methyl-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–14)
Yellow solid. Yield 34%. 1H-NMR (600 MHz, DMSO-d6): δ 10.77 (s, 1 H), 10.76 (s, 1H), 8.51 (m, 2 H), 7.46 (m, 2 H), 7.34 (m, 2 H), 6.96 (d, 1 H, J = 9 Hz), 4.56 (s, 2 H), 3.70 (s, 2 H); 13C-NMR (150 MHz, DMSO-d6): δ 155.4, 150.2, 149.5, 149.0, 131.0, 130.1, 128.5, 122.8, 122.2, 119.7, 108.2, 106.1, 45.2, 21.8; HRMS (ESI): m/z [M + H]+ calcd for C16H15N6OS+ : 339.1023, found: 339.1004; HPLC: t = 1.23 min, Purity >95%.
N-(3-carboxyaminobenzyl)-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–15)[61]
To a solution of 3.12 (0.091 mmol) in DMSO (1 mL) was added 50 % hydrogen peroxide solution (0.013 mL) at 0 °C followed by potassium carbonate (0.015 eq.). The reaction mixture was allowed to warm up to room temperature and stirred overnight. Upon the completion of reaction, the reaction mixture was vacuum dried and purified using flash chromatography with mixture of methanol/dichloromethane/ammonia(10:89:1) as eluent to get the final product as yellow solid. Yield 42%. 1H-NMR (600 MHz, DMSO-d6): δ 10.77 (s, 1 H), 10.76 (s, 1H), 7.95 (m, 2 H), 7.86 (m, 1 H), 7.75 (m,1 H), 7.51 (m, 1 H), 7.47 (m, 2 H), 7.40 (m, 1 H), 7.34 (m, 1 H), 6.96 (d, 1 H, J = 7.8 Hz), 4.59 (s, 2 H), 3.68 (s, 2 H); 13C-NMR (150 MHz, DMSO-d6): 167.9, 155.4, 147.3, 139.9, 134.2, 131.0, 130.3, 130.0, 128.6, 128.0, 126.3, 124.2, 119.6, 108.2, 106.1, 45.9, 21.7; HRMS (ESI): m/z [M + H]+ calcd for C18H17N6O2S+ : 381.1128, found: 381.1110; HPLC: t = 1.80 min, Purity >95%.
N-(1-phenylethyl)-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–16)
Yellow solid. Yield 41%. 1H-NMR (600 MHz, DMSO-d6): δ 10.76 (s, 1 H), 10.75 (s, 1H), 7.42 (m, 2 H), 7.36 (d, 2 H, J = 7.2 Hz), 7.32 (m, 2 H), 7.21 (m,1 H), 6.94 (d, 1 H, J = 7.8 Hz), 5.20 (s, 1 H), 3.72 (s, 1 H), 3.52 (s, 1 H), 1.43 (d, 3 H, J = 7.2 Hz); 13C-NMR (150 MHz, DMSO-d6): δ 155.4, 147.1, 145.4, 130.9, 130.0, 128.6, 128.1, 126.4, 126.0, 118.5, 108.0, 106.0, 52.1, 23.3, 21.7; HRMS (ESI): m/z [M + H]+ calcd for C18H18N5OS+: 3352.1227, found: 352.1235; HPLC: t = 3.63 min, Purity 95%.
N-(1-(4-fluorophenyl)ethyl)-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–17)
Yellow solid. Yield 41%. 1H-NMR (600 MHz, DMSO-d6): δ 10.82 (s, 1 H), 10.80 (s, 1H), 7.43 (m, 4 H), 7.17 (m, 2 Hz), 6.96 (d, 1 H, J = 8.4 Hz), 5.15 (s, 1 H), 3.81 (m, 1 H), 3.67 (m, 1 H), 1.44 (d, 3 H, J = 6.6 Hz); 13C-NMR (150 MHz, DMSO-d6): δ 161.8, 160.2, 155.4, 148.3, 140.7, 131.4, 130.1, 128.0, 119.8, 115.0, 114.9, 108.1, 106.2, 51.7, 23.1, 21.8 HRMS (ESI): m/z [M + H]+ calcd for C18H17FN5OS+: 370.1132, found: 370.1120; HPLC: t = 3.70 min, Purity >95%.
N-phenyl-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–18)
Yellow solid. Yield 66%. 1H-NMR (600 MHz, DMSO-d6): δ 10.82 (s, 1 H), 10.80 (s, 1 H), 7.39 (m, 2 H), 7.30 (m, 2 H), 7.02 (m, 2 H), 6.96 (m, 1 H), 6.83 (m, 2 H), 3.89 (s, 2 H); 13C-NMR (150 MHz, DMSO-d6): δ 155.1, 131.6, 130.1, 129.6, 128.5, 123.7, 122.4, 120.4, 119.4, 109.1, 108.0, 22.5; HRMS (ESI): m/z [M + H]+ calcd for C16H14N5OS+: 324.0914, found: 324.0900; HPLC: t = 3.43 min, Purity 95%.
N-(3-fluorophenyl)-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–19)
Yellow solid. Yield 69%. 1H-NMR (600 MHz, DMSO-d6): δ 10.83 (s, 1 H), 10.81 (s, 1 H), 7.49 (m, 2 H), 7.30 (m,2 H), 7.36 (m, 2 H), 7.05 (m, 1 H), 6.98 (d, 1 H, J = 7.2 Hz), 6.66 (m, 1 H), 3.91 (s, 2 H); 13C-NMR (150 MHz, DMSO-d6): δ 163.1, 161.5, 155.4, 131.3, 130.1, 127.6, 119.5, 109.0, 108.2, 107.9, 105.9, 22.3; HRMS (ESI): m/z [M + H]+ calcd for C16H13FN5OS+: 342.0819, found: 342.0812; HPLC: t = 3.82 min, Purity >95%.
N-(3-trifluoromethylphenyl)-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–20)
Yellow solid. Yield 74%. 1H-NMR (600 MHz, DMSO-d6): δ 10.84 (s, 1 H), 10.82 (s, 1 H), 7.54 (m,2 H), 7.36 (m, 2 H), 7.15 (m, 2 H), 6.98 (d, 1 H, J = 7.8 Hz), 3.92 (s, 2 H); 13C-NMR (150 MHz, DMSO-d6): δ 155.4, 131.4, 130.2, 129.8, 129.6, 129.4, 127.4, 125.0, 123.2, 119.7, 119.0, 108.2, 106.1, 22.4; HRMS (ESI): m/z [M + H]+ calcd for C17H13F3N5OS+ : 392.0787, found: 392.0795; HPLC: t = 4.29 min, Purity >95%.
N-(3-cyanophenyl)-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–21)
Yellow solid. Yield 68%. 1H-NMR (600 MHz, DMSO-d6): δ 10.84 (s, 1 H), 10.81 (s, 1 H), 7.51 (m,3 H), 7.41 (m, 2 H), 7.17 (m, 2 H), 6.98 (d, 1 H, J = 7.2 Hz), 3.94 (s, 2 H); 13C-NMR (150 MHz, DMSO-d6): δ 155.4, 131.3, 130.1, 130.0, 127.54 119.5, 118.8, 111.5, 108.2, 105.8, 22.4; HRMS (ESI): m/z [M + H]+ calcd for C17H13N6OS+ : 349.0866, found: 349.0866; HPLC: t = 3.79 min, Purity >95%.
N-(2-phenylethyl)-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–22)
Yellow solid. Yield 60%. 1H-NMR (600 MHz, DMSO-d6): δ 10.78 (s, 1 H), 10.77 (s, 1 H), 7.48 (m, 2 H), 7.30 (m, 2 H), 7.26 (m, 2 H), 7.21 (m, 1 H), 6.96 (d, 1 H, J = 8.4 Hz), 3.69 (s, 2 H), 3.56 (m, 2 H), 2.89 (m, 2 H); 13C-NMR (150 MHz, DMSO-d6): δ 155.4, 147.3, 139.5, 131.0, 130.1, 129.0, 128.6, 128.6, 126.0, 119.6, 108.1, 106.1, 44.2, 35.2, 21.7; HRMS (ESI): m/z [M + H]+ calcd for C18H18N5OS+ : 352.1227, found: 3352.1217; HPLC: t = 3.57 min, Purity >95%.
N-(3-phenylpropyl)-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–23)
Yellow solid. Yield 41%. 1H-NMR (600 MHz, DMSO-d6): δ 11.03 (s, 1 H), 10.98 (s, 1H), 7.51 (d, 1 H, J = 8.4 Hz), 7.46 (s, 1 H), 7.32 (m, 3 H), 7.24 (m,1 H), 7.05 (d, 1 H, J = 8.4 Hz), 4.15 (s, 2 H), 3.68 (s, 2 H), 3.16 (m, 1 H), 1.28 (d, 3 H, J = 6.6 Hz); 13C-NMR (150 MHz, DMSO-d6): δ 155.3, 151.7, 143.2, 132.6, 130.3, 128.4, 127.3, 126.8, 126.6, 125.3, 120.5, 108.4, 106.5, 50.1, 48.5, 22.1, 19.0; HRMS (ESI): m/z [M + H]+ calcd for C19H20N5OS+ : 366.1383, found: 366.1389; HPLC: t = 3.72 min, Purity >95%.
N-(2-(pyridine-2-yl)ethyl)-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–24)
Yellow solid. Yield 44%. 1H-NMR (600 MHz, DMSO-d6): δ 10.77 (s, 1 H), 10.76 (s, 1H), 8.51 (d, 1 H, J = 4.8 Hz), 7.71 (m, 1 H), 7.47 (m, 2 H), 7.22 (t, 1 H, J = 4.8 Hz), 6.94 (d, 1 H, J = 8.4 Hz), 3.65 (m, 2 H), 3.04 (t, 2 H, J = 7.2 Hz); 13C-NMR (150 MHz, DMSO-d6): δ 159.2, 155.4, 149.0, 147.3, 136.4, 131.0, 130.1, 128.6, 123.2, 121.4, 119.6, 108.1, 106.1, 42.6, 37.4, 21.7; HRMS (ESI): m/z [M + H]+ calcd for C17H17N6OS+ : 353.1179, found: 353.1166; HPLC: t = 1.46 min, Purity >95%.
N-(2-(pyridine-3-yl)ethyl)-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–25)
Yellow solid. Yield 25%. 1H-NMR (600 MHz, DMSO-d6): δ 10.75 (s, 2 H), 8.46 (s, 1 H), 8.42 (m, 1 H), 7.68 (d, 1 H, J = 7.2 Hz), 7.46 (m, 2 H), 7.33 (m, 1 H), 6.94 (d, 1 H, J = 8.4 Hz), 3.65 (m, 2 H), 3.58 (m, 2 H), 2.91 (t, 2 H, J = 7.2 Hz); 13C-NMR (150 MHz, DMSO-d6): δ 155.4, 150.6, 147.6, 136.7, 135.1, 130.9, 130.1, 128.7, 123.4, 119.6, 108.2, 106.1, 43.4, 32.3, 21.6; HRMS (ESI): m/z [M + H]+ calcd for C17H17N6OS+ : 353.1166, found: 353.1166; HPLC: t = 1.33 min, Purity >95%.
N-(2-naphthylmethy)-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–26)
Yellow solid. Yield 39%. 1H-NMR (600 MHz, DMSO-d6): δ 10.78 (s, 1 H), 10.76 (s, 1 H), 7.88 (m, 3 H), 7.84 (m, 1 H), 7.49 (m, 3 H), 7.48 (m, 5 H), 6.96 (d, 1 H, J = 8.4 Hz), 4.73 (s, 2 H), 3.70 (s, 2 H); 13C-NMR (150 MHz, DMSO-d6): δ 155.4, 147.4, 137.4, 132.9, 132.1, 131.0, 130.1, 128.6, 127.6, 126.1, 125.5, 119.6, 108.1, 106.1, 45.6, 21.8; HRMS (ESI): m/z [M + H]+ calcd for C21H18N5OS+: 388.1227, found: 388.1204; HPLC: t = 3.84 min, Purity >95%.
N-(1-naphthylmethy)-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–27)
Yellow solid. Yield 75%. 1H-NMR (600 MHz, DMSO-d6): δ 10.78 (s, 1 H), 10.76 (s, 1 H), 8.11 (d, 1 H, J = 8.4 Hz), 7.95 (m, 1 H), 7.86 (d, 1 H, J = 7.8 Hz), 7.56 (m, 3 H), 7.48 (m, 3 H), 6.96 (d, 1 H, J = 7.8 Hz), 5.03 (s, 2 H), 3.70 (s, 2 H); 13C-NMR (150 MHz, DMSO-d6): δ 155.4, 147.3, 134.8, 133.2, 131.0, 130.1, 128.6, 126.1, 125.7, 125.5, 125.4, 119.6, 108.1, 106.1, 43.5, 21.8; HRMS (ESI): m/z [M + H]+ calcd for C21H18N5OS+: 388.1227, found: 388.1205; HPLC: t = 3.88 min, Purity >95%.
N-(1-naphthyl)-5-(benzo[d]imidazol-2(3H)-one)-6H-1,3,4- thiadiazin-2-amine (3–28)
Yellow solid. Yield 56%. 1H-NMR (600 MHz, Methanol-d4): δ 11.52 (s, 1 H), 10.82 (s, 1 H), 10.79 (s, 1 H), 8.00 (d, 1 H, J = 7.8 Hz), 7.89 (d, 1 H, J = 7.8 Hz), 7.61 (d, 1 H, J = 7.8 Hz), 7.49 (m, 2 H), 7.42 (m, 3 H), 6.96 (d, 1 H, J = 7.8 Hz), 6.82 (s, 1 H), 3.91 (s, 2 H); 13C-NMR (150 MHz, DMSO-d6): δ 155.4, 153.0, 146.0, 145.3, 133.8, 131.1, 130.1, 127.6, 126.1, 125.7, 125.2, 123.0, 119.2, 117.2, 108.2, 105.6, 22.7; HRMS (ESI): m/z [M + H]+ calcd for C20H16N5OS+: 374.1070, found: 374.1055; HPLC: t = 4.04 min, Purity >95%.
Supplementary Material
Homology model of DYRK1A in the inactive, DFG-out conformation was developed using DFGmodel protocol.
Virtual screen of the ZINC database and in vitro testing of 68 compounds revealed inhibitor hit thiadiazine 1 with DYRK1A Kd = 7.3 μM.
Hit-to-lead SAR study was carried out by systematic structural modifications at the 2-amino position to improve hit DYRK1A binding potency.
27 analogues synthesized to identify two compounds with significantly improved binding to DYRK1A (Kd = 71–185 nM).
New lead compound 3–5 induced human β-cell proliferation at 5 μM and improved selectivity for closely related kinases.
Acknowledgements
K.K., P.W., R.J.D. and A.F.S. were supported in part by NIH grant by NIDDK R01DK015015–01-A.
P.W. and A.F.S. were also supported by NIH grants UC4 DK104211, P-30 DK 020541 and JDRF grant 2-SRA-2015–62
K.K., H.W., H. L., M.K.A. and R.J.D. were supported in part by Icahn School of Medicine Seed Fund 0285–3980.
A.S. and P.M.U.U were supported in part by NIH grants R01-GM108911 and U54-OD020353 Partial support for a postdoctoral fellowship to Dr. Hailing Li from the Swiss National Science Foundation is gratefully acknowledged. M.K.A. gratefully acknowledges a summer research intern fellowship from Dartmouth University. Finally, we thank the NIH/NIDDK-supported Diabetes Research Centers (DRC) and Human Islet Research Network (HIRN) for their support.
ABBREVIATIONS
- DYRK1A
The Dual-Specificity Tyrosine Phosphorylation-Regulated Kinase 1A
- DS
Down syndrome
- AD
Alzheimer’s disease
- DFG
Aspartate-Phenylalanine-Glycine
- CLKs
CDK-like kinases
- GSK3s
Glycogen Synthase Kinases 3
- CDKs
Cyclin Dependent Kinases
- MAPKs
Mitogen-Activated Protein Kinases
- DSCR
Down Syndrome Critical region
- EGFR
epidermal growth factor receptor
- NFAT
nuclear factor of activated T cells
- EGCG
Epigallocatechin gallate
- INDY
inhibitor of DYRK1A
- FINDY
Folding intermediate inhibitor of DYRK1A
- DANDY
diaryl azaindole inhibitor of DYRK1A
- CNS
central nervous system
- PDB
Protein data bank
- RMSD
root mean square deviation
- FRET
Fluorescence resonance energy transfer
- qPCR
quantitative polymerase chain reaction
- SAR
structure activity relationship
- TLC
thin layer chromatography
- DCE
Dichloroethane
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting Information.
Chemical synthesis of alkylthiosemicarbazides (1–2), Performance of glide parameters, DYRK1A binding curves and spectral data (PDF) are available as supplementary data.
REFERENCES:
- [1].Becker W, Sippl W, Activation, regulation, and inhibition of DYRK1A, FEBS J, 278 (2011) 246–256. [DOI] [PubMed] [Google Scholar]
- [2].Lochhead PA, Sibbet G, Morrice N, Cleghon V, Activation-loop autophosphorylation is mediated by a novel transitional intermediate form of DYRKs, Cell (Cambridge, MA, U. S.), 121 (2005) 925–936. [DOI] [PubMed] [Google Scholar]
- [3].Walte A, Rueben K, Birner-Gruenberger R, Preisinger C, Bamberg-Lemper S, Hilz N, Bracher F, Becker W, Mechanism of dual specificity kinase activity of DYRK1A, FEBS J, 280 (2013) 4495–4511. [DOI] [PubMed] [Google Scholar]
- [4].Becker W, Soppa U, Tejedor FJ, DYRK1A: A Potential Drug Target for Multiple Down Syndrome Neuropathologies, CNS Neurol. Disord.: Drug Targets, 13 (2014) 26–33. [DOI] [PubMed] [Google Scholar]
- [5].Wegiel J, Gong C-X, Hwang Y-W, The role of DYRK1A in neurodegenerative diseases, FEBS J, 278 (2011) 236–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Smith B, Medda F, Gokhale V, Dunckley T, Hulme C, Recent Advances in the Design, Synthesis, and Biological Evaluation of Selective DYRK1A Inhibitors: A New Avenue for a Disease Modifying Treatment of Alzheimer’s?, ACS Chem. Neurosci, 3 (2012) 857–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ionescu A, Dufrasne F, Gelbcke M, Jabin I, Kiss R, Lamoral-Theys D, DYRK1A kinase inhibitors with emphasis on cancer, Mini-Rev. Med. Chem, 12 (2012) 1315–1329. [DOI] [PubMed] [Google Scholar]
- [8].Fernandez-Martinez P, Zahonero C, Sanchez-Gomez P, DYRK1A: the double-edged kinase as a protagonist in cell growth and tumorigenesis, Mol. Cell. Oncol, (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wang P, Alvarez-Perez J-C, Felsenfeld DP, Liu H, Sivendran S, Bender A, Kumar A, Sanchez R, Scott DK, Garcia-Ocana A, Stewart AF, A high-throughput chemical screen reveals that harmine-mediated inhibition of DYRK1A increases human pancreatic beta cell replication, Nat. Med. (N. Y., NY, U. S.), 21 (2015) 383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Shen W, Taylor B, Jin Q, Nguyen-Tran V, Meeusen S, Zhang Y-Q, Kamireddy A, Swafford A, Powers AF, Walker J, Lamb J, Bursalaya B, DiDonato M, Harb G, Qiu M, Filippi CM, Deaton L, Turk CN, Suarez-Pinzon WL, Liu Y, Hao X, Mo T, Yan S, Li J, Herman AE, Hering BJ, Wu T, Seidel HM, McNamara P, Glynne R, Laffitte B, Inhibition of DYRK1A and GSK3B induces human β-cell proliferation, Nat. Commun, 6 (2015) 8372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rachdi L, Kariyawasam D, Aiello V, Herault Y, Janel N, Delabar J-M, Polak M, Scharfmann R, Dyrk1A induces pancreatic β cell mass expansion and improves glucose tolerance, Cell Cycle, 13 (2014) 2221–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dirice E, Walpita D, Vetere A, Meier BC, Kahraman S, Hu J, Dancik V, Burns SM, Gilbert TJ, Olson DE, Clemons PA, Kulkarni RN, Wagner BK, Inhibition of DYRK1A stimulates human beta-cell proliferation, Diabetes, 65 (2016) 1660–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Becker W, Joost H-G, Structural and functional characteristics of Dyrk, a novel subfamily of protein kinases with dual specificity, Prog. Nucleic Acid Res. Mol. Biol, 62 (1999) 1–17. [DOI] [PubMed] [Google Scholar]
- [14].Park J, Song W-J, Chung KC, Function and regulation of Dyrk1A: towards understanding Down syndrome, Cell. Mol. Life Sci, 66 (2009) 3235–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ogawa Y, Nonaka Y, Goto T, Ohnishi E, Hiramatsu T, Kii I, Yoshida M, Ikura T, Onogi H, Shibuya H, Hosoya T, Ito N, Hagiwara M, Development of a novel selective inhibitor of the Down syndrome-related kinase Dyrk1A, Nat. Commun, 1 (2010) [DOI] [PubMed] [Google Scholar]
- [16].Stotani S, Giordanetto F, Medda F, DYRK1A inhibition as potential treatment for Alzheimer’s disease, Future Med. Chem, 8 (2016) 681–696. [DOI] [PubMed] [Google Scholar]
- [17].Pozo N, Zahonero C, Fernandez P, Linares JM, Ayuso A, Hagiwara M, Perez A, Ricoy JR, Hernandez-Lain A, Sepulveda JM, Sanchez-Gomez P, Inhibition of DYRK1A destabilizes EGFR and reduces EGFR-dependent glioblastoma growth, J. Clin. Invest, 123 (2013) 2475–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Seifert A, Allan LA, Clarke PR, DYRK1A phosphorylates caspase 9 at an inhibitory site and is potently inhibited in human cells by harmine, FEBS J, 275 (2008) 6268–6280. [DOI] [PubMed] [Google Scholar]
- [19].Abdolazimi Y, Lee S, Xu H, Allegretti P, Horton TM, Yeh B, Moeller HP, McCutcheon D, Armstrong NA, Annes JP, Allegretti P, Horton TM, McCutcheon D, Smith M, Annes JP, Horton TM, Nichols RJ, Shalizi A, Smith M, CC-401 Promotes β-Cell Replication via Pleiotropic Consequences of DYRK1A/B Inhibition, Endocrinology, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Guedj F, Sebrie C, Rivals I, Ledru A, Paly E, Bizot JC, Smith D, Rubin E, Gillet B, Arbones M, Delabar JM, Green tea polyphenols rescue of brain defects induced by overexpression of DYRK1A, PLoS One, 4 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bain J, McLauchlan H, Elliott M, Cohen P, The specificities of protein kinase inhibitors: an update, Biochem. J, 371 (2003) 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tahtouh T, Elkins JM, Filippakopoulos P, Soundararajan M, Burgy G, Durieu E, Cochet C, Schmid RS, Lo DC, Delhommel F, Oberholzer AE, Pearl LH, Carreaux F, Bazureau J-P, Knapp S, Meijer L, Selectivity, Cocrystal Structures, and Neuroprotective Properties of Leucettines, a Family of Protein Kinase Inhibitors Derived from the Marine Sponge Alkaloid Leucettamine B, J. Med. Chem, 55 (2012) 9312–9330. [DOI] [PubMed] [Google Scholar]
- [23].Naert G, Ferre V, Meunier J, Keller E, Malmstrom S, Givalois L, Carreaux F, Bazureau J-P, Maurice T, Leucettine L41, a DYRK1A-preferential DYRKs/CLKs inhibitor, prevents memory impairments and neurotoxicity induced by oligomeric Aβ25–35 peptide administration in mice, Eur. Neuropsychopharmacol, 25 (2015) 2170–2182. [DOI] [PubMed] [Google Scholar]
- [24].Cozza G, Mazzorana M, Papinutto E, Bain J, Elliott M, di Maira G, Gianoncelli A, Pagano MA, Sarno S, Ruzzene M, Battistutta R, Meggio F, Moro S, Zagotto G, Pinna LA, Quinalizarin as a potent, selective and cell-permeable inhibitor of protein kinase CK2, Biochem. J, 421 (2009) 387–395. [DOI] [PubMed] [Google Scholar]
- [25].Ahmadu A, Abdulkarim A, Grougnet R, Myrianthopoulos V, Tillequin F, Magiatis P, Skaltsounis A-L, Two new peltogynoids from Acacia nilotica Delile with kinase inhibitory activity, Planta Med, 76 (2010) 458–460. [DOI] [PubMed] [Google Scholar]
- [26].Sarno S, Mazzorana M, Traynor R, Ruzzene M, Cozza G, Pagano MA, Meggio F, Zagotto G, Battistutta R, Pinna LA, Structural features underlying the selectivity of the kinase inhibitors NBC and dNBC: role of a nitro group that discriminates between CK2 and DYRK1A, Cell. Mol. Life Sci, 69 (2012) 449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sanchez C, Salas AP, Brana AF, Palomino M, Pineda-Lucena A, Carbajo RJ, Mendez C, Moris F, Salas JA, Generation of potent and selective kinase inhibitors by combinatorial biosynthesis of glycosylated indolocarbazoles, Chem. Commun. (Cambridge, U. K.), (2009) 4118–4120. [DOI] [PubMed] [Google Scholar]
- [28].Gourdain S, Dairou J, Denhez C, Bui LC, Rodrigues-Lima F, Janel N, Delabar JM, Cariou K, Dodd RH, Development of DANDYs, New 3,5-Diaryl-7-azaindoles Demonstrating Potent DYRK1A Kinase Inhibitory Activity, J. Med. Chem, 56 (2013) 9569–9585. [DOI] [PubMed] [Google Scholar]
- [29].Kii I, Sumida Y, Goto T, Sonamoto R, Okuno Y, Yoshida S, Kato-Sumida T, Koike Y, Abe M, Nonaka Y, Ikura T, Ito N, Shibuya H, Hosoya T, Hagiwara M, Selective inhibition of the kinase DYRK1A by targeting its folding process, Nat. Commun, 7 (2016) 11391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Koo KA, Kim ND, Chon YS, Jung M-S, Lee B-J, Kim JH, Song W-J, QSAR analysis of pyrazolidine-3,5-diones derivatives as Dyrk1A inhibitors, Bioorg. Med. Chem. Lett, 19 (2009) 2324–2328. [DOI] [PubMed] [Google Scholar]
- [31].Kim ND, Yoon J, Kim JH, Lee JT, Chon YS, Hwang M-K, Ha I, Song W-J, Putative therapeutic agents for the learning and memory deficits of people with Down syndrome, Bioorg. Med. Chem. Lett, 16 (2006) 3772–3776. [DOI] [PubMed] [Google Scholar]
- [32].Rosenthal AS, Tanega C, Shen M, Mott BT, Bougie JM, Nguyen D-T, Misteli T, Auld DS, Maloney DJ, Thomas CJ, Potent and selective small molecule inhibitors of specific isoforms of Cdc2-like kinases (Clk) and dual specificity tyrosine-phosphorylation-regulated kinases (Dyrk), Bioorg. Med. Chem. Lett, 21 (2011) 3152–3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Giraud F, Alves G, Debiton E, Nauton L, Thery V, Durieu E, Ferandin Y, Lozach O, Meijer L, Anizon F, Pereira E, Moreau P, Synthesis, Protein Kinase Inhibitory Potencies, and in Vitro Antiproliferative Activities of Meridianin Derivatives, J. Med. Chem, 54 (2011) 4474–4489. [DOI] [PubMed] [Google Scholar]
- [34].Echalier A, Bettayeb K, Ferandin Y, Lozach O, Clelment M, Valette A, Liger F, Marquet B, Morris JC, Endicott JA, Joseph B, Meijer L, Meriolins (3-(Pyrimidin-4-yl)-7-azaindoles): Synthesis, Kinase Inhibitory Activity, Cellular Effects, and Structure of a CDK2/Cyclin A/Meriolin Complex, J. Med. Chem, 51 (2008) 737–751. [DOI] [PubMed] [Google Scholar]
- [35].Akue-Gedu R, Debiton E, Ferandin Y, Meijer L, Prudhomme M, Anizon F, Moreau P, Synthesis and biological activities of aminopyrimidyl-indoles structurally related to meridianins, Bioorg. Med. Chem, 17 (2009) 4420–4424. [DOI] [PubMed] [Google Scholar]
- [36].Kassis P, Brzeszcz J, Beneteau V, Lozach O, Meijer L, Le Guevel R, Guillouzo C, Lewinski K, Bourg S, Colliandre L, Routier S, Merour J-Y, Synthesis and biological evaluation of new 3-(6-hydroxyindol-2-yl)-5-(Phenyl) pyridine or pyrazine V-Shaped molecules as kinase inhibitors and cytotoxic agents, Eur. J. Med. Chem, 46 (2011) 5416–5434. [DOI] [PubMed] [Google Scholar]
- [37].Neagoie C, Vedrenne E, Buron F, Merour J-Y, Rosca S, Bourg S, Lozach O, Meijer L, Baldeyrou B, Lansiaux A, Routier S, Synthesis of chromeno[3,4-b]indoles as Lamellarin D analogues : A novel DYRK1A inhibitor class, Eur. J. Med. Chem, 49 (2012) 379–396. [DOI] [PubMed] [Google Scholar]
- [38].Falke H, Chaikuad A, Becker A, Loaec N, Lozach O, Abu Jhaisha S, Becker W, Jones PG, Preu L, Baumann K, Knapp S, Meijer L, Kunick C, 10-Iodo-11H-indolo[3,2-c]quinoline-6-carboxylic Acids Are Selective Inhibitors of DYRK1A, J. Med. Chem, 58 (2015) 3131–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Foucourt A, Heou D, Dubouilh-Benard C, Desire L, Casagrande A-S, Leblond B, Loaec N, Meijer L, Besson T, Design and synthesis of thiazolo[5,4-f]quinazolines as DYRK1A inhibitors, part I, Molecules, 19 (2014) 15546–15571, 15526 pp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Coutadeur S, Benyamine H, Delalonde L, de Oliveira C, Leblond B, Foucourt A, Besson T, Casagrande A-S, Taverne T, Girard A, Pando MP, Desire L, A novel DYRK1A (Dual specificity tyrosine phosphorylation-regulated kinase 1A) inhibitor for the treatment of Alzheimer’s disease: effect on Tau and amyloid pathologies in vitro, J. Neurochem, 133 (2015) 440–451. [DOI] [PubMed] [Google Scholar]
- [41].Annes JP, Ryu JH, Lam K, Carolan PJ, Utz K, Hollister-Lock J, Arvanites AC, Rubin LL, Weir G, Melton DA, Adenosine kinase inhibition selectively promotes rodent and porcine islet β-cell replication, Proc. Natl. Acad. Sci. U. S. A, 109 (2012) 3915–3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ung PM-U, Schlessinger A, DFGmodel: Predicting Protein Kinase Structures in Inactive States for Structure-Based Discovery of Type-II Inhibitors, ACS Chem. Biol, 10 (2015) 269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhao Z, Wu H, Wang L, Liu Y, Knapp S, Liu Q, Gray NS, Exploration of type II binding mode: A privileged approach for kinase inhibitor focused drug discovery?, ACS Chem Biol, 9 (2014) 1230–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Roskoski R Jr., Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes, Pharmacol Res, 103 (2016) 26–48. [DOI] [PubMed] [Google Scholar]
- [45].Anderson K, Chen Y, Chen Z, Dominique R, Glenn K, He Y, Janson C, Luk KC, Lukacs C, Polonskaia A, Qiao Q, Railkar A, Rossman P, Sun H, Xiang Q, Vilenchik M, Wovkulich P, Zhang X, Pyrido[2,3-d]pyrimidines: discovery and preliminary SAR of a novel series of DYRK1B and DYRK1A inhibitors, Bioorg Med Chem Lett, 23 (2013) 6610–6615. [DOI] [PubMed] [Google Scholar]
- [46].Falke H, Chaikuad A, Becker A, Loaec N, Lozach O, Abu Jhaisha S, Becker W, Jones PG, Preu L, Baumann K, Knapp S, Meijer L, Kunick C, 10-iodo-11H-indolo[3,2-c]quinoline-6-carboxylic acids are selective inhibitors of DYRK1A, J Med Chem, 58 (2015) 3131–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Rothweiler U, Stensen W, Brandsdal BO, Isaksson J, Leeson FA, Engh RA, Svendsen JS, Probing the ATP-Binding Pocket of Protein Kinase DYRK1A with Benzothiazole Fragment Molecules, J Med Chem, 59 (2016) 9814–9824. [DOI] [PubMed] [Google Scholar]
- [48].Sali A, Blundell TL, Comparative protein modelling by satisfaction of spatial restraints, J Mol Biol, 234 (1993) 779–815. [DOI] [PubMed] [Google Scholar]
- [49].Shen MY, Sali A, Statistical potential for assessment and prediction of protein structures, Protein Sci, 15 (2006) 2507–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Eramian D, Eswar N, Shen MY, Sali A, How well can the accuracy of comparative protein structure models be predicted?, Protein Sci, 17 (2008) 1881–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Bergeron P, Koehler MF, Blackwood EM, Bowman K, Clark K, Firestein R, Kiefer JR, Maskos K, McCleland ML, Orren L, Ramaswamy S, Salphati L, Schmidt S, Schneider EV, Wu J, Beresini M, Design and Development of a Series of Potent and Selective Type II Inhibitors of CDK8, ACS Med Chem Lett, 7 (2016) 595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Schneider EV, Bottcher J, Blaesse M, Neumann L, Huber R, Maskos K, The structure of CDK8/CycC implicates specificity in the CDK/cyclin family and reveals interaction with a deep pocket binder, J Mol Biol, 412 (2011) 251–266. [DOI] [PubMed] [Google Scholar]
- [53].Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG, ZINC: A Free Tool to Discover Chemistry for Biology, J. Chem. Inf. Model, 52 (2012) 1757–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Fan H, Irwin JJ, Webb BM, Klebe G, Shoichet BK, Sali A, Molecular docking screens using comparative models of proteins, J Chem Inf Model, 49 (2009) 2512–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Amaro RE, Li WW, Emerging methods for ensemble-based virtual screening, Curr Top Med Chem, 10 (2010) 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Arrowsmith CH, Audia JE, Austin C, Baell J, Bennett J, Blagg J, Bountra C, Brennan PE, Brown PJ, Bunnage ME, Buser-Doepner C, Campbell RM, Carter AJ, Cohen P, Copeland RA, Cravatt B, Dahlin JL, Dhanak D, Edwards AM, Frye SV, Gray N, Grimshaw CE, Hepworth D, Howe T, Huber KVM, Jin J, Knapp S, Kotz JD, Kruger RG, Lowe D, Mader MM, Marsden B, Mueller-Fahrnow A, Muller S, O’Hagan RC, Overington JP, Owen DR, Rosenberg SH, Roth B, Ross R, Schapira M, Schreiber SL, Shoichet B, Sundstrom M, Superti-Furga G, Taunton J, Toledo-Sherman L, Walpole C, Walters MA, Willson TM, Workman P, Young RN, Zuercher WJ, The promise and peril of chemical probes, Nat. Chem. Biol, 11 (2015) 536–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].LanthaScreen® Eu Kinase Binding Assay - Customer Protocol and Assay Conditions documents located at. https://www.thermofisher.com/us/en/home/industrial/pharmabiopharma/drug-discovery-development/target-and-lead-identification-and-validation/kinasebiology/kinase-activity-assays/lanthascreentm-eu-kinase-binding-assay.html.
- [58].Fabian MA, Biggs WH, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, Ford JM, Galvin M, Gerlach JL, Grotzfeld RM, Herrgard S, Insko DE, Insko MA, Lai AG, Lelias J-M, Mehta SA, Milanov ZV, Velasco AM, Wodicka LM, Patel HK, Zarrinkar PP, Lockhart DJ, A small molecule-kinase interaction map for clinical kinase inhibitors, Nat. Biotechnol, 23 (2005) 329–336. [DOI] [PubMed] [Google Scholar]
- [59].Pfeiffer W-D, Ahlers K-D, Saghyan AS, Villinger A, Langer P, Unexpected Ring Enlargement of 2-Hydrazono-2,3-dihydro-1,3-thiazoles to 1,3,4-Thiadiazines, Helv. Chim. Acta, 97 (2014) 76–87. [Google Scholar]
- [60].Kornberg BE, Lewthwaite RA, Manning DD, Nikam SS, Scott IL, Preparation of piperidine derivatives as subtype selective n-methyl-d-aspartate antagonists useful in the treatment of cerebral vascular disorders, in, Warner-Lambert Company, USA: 2002, pp. 154 pp. [Google Scholar]
- [61].Katritzky AR, Pilarski B, Urogdi L, Efficient conversion of nitriles to amides with basic hydrogen peroxide in dimethyl sulfoxide, Synthesis, (1989) 949–950. [Google Scholar]
- [62].Schoenherr H, Cernak T, Profound Methyl Effects in Drug Discovery and a Call for New C-H Methylation Reactions, Angew. Chem., Int. Ed, 52 (2013) 12256–12267. [DOI] [PubMed] [Google Scholar]
- [63].Zhao Z, Wu H, Wang L, Liu Y, Knapp S, Liu Q, Gray NS, Exploration of Type II Binding Mode: A Privileged Approach for Kinase Inhibitor Focused Drug Discovery?, ACS Chem. Biol, 9 (2014) 1230–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Notredame C, Higgins DG, Heringa J, T-Coffee: A Novel Method for Fast and Accurate Multiple Sequence Alignment, J. Mol. Biol, 302 (2000) 205–217. [DOI] [PubMed] [Google Scholar]
- [65].Durrant JD, Votapka L, Soerensen J, Amaro RE, POVME 2.0: An Enhanced Tool for Determining Pocket Shape and Volume Characteristics, J. Chem. Theory Comput, 10 (2014) 5047–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].McGann M, FRED pose prediction and virtual screening accuracy, J Chem Inf Model, 51 (2011) 578–596. [DOI] [PubMed] [Google Scholar]
- [67].G. Schrödinger Release 2015–3: Maestro, LigPrep, Schrödinger, LLC, New York, NY, 2017., in. [Google Scholar]
- [68].Harder E, Damm W, Maple J, Wu C, Reboul M, Xiang JY, Wang L, Lupyan D, Dahlgren MK, Knight JL, Kaus JW, Cerutti DS, Krilov G, Jorgensen WL, Abel R, Friesner RA, OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins, J Chem Theory Comput, 12 (2016) 281–296. [DOI] [PubMed] [Google Scholar]
- [69].Schlessinger A, Geier E, Fan H, Irwin JJ, Shoichet BK, Giacomini KM, Sali A, Structure-based discovery of prescription drugs that interact with the norepinephrine transporter, NET, Proc Natl Acad Sci U S A, 108 (2011) 15810–15815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].KINOMEscan® Assay-Customer Protocol and Assay Conditions documents located at. https://www.discoverx.com/technologies-platforms/competitive-binding-technology/kinomescan-technology-platform.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.