

Abstract
C–H bond activation at cationic [(η5-C5Me5)Ir(PMe2Ar′)] centers is described, where PMe2Ar′ are the terphenyl phosphine ligands PMe2ArXyl2 and PMe2ArDipp2. Different pathways are defined for the conversion of the five-coordinate complexes [(η5-C5Me5)IrCl(PMe2Ar′)]+, 2(Xyl)+ and 2(Dipp)+, into the corresponding pseudoallyls 3(Xyl)+ and 3(Dipp)+. In the absence of an external Brønsted base, electrophilic, remote ζ C–H activation takes place, for which the participation of dicationic species, [(η5-C5Me5)Ir(PMe2Ar′)]2+, is proposed. When NEt3 is present, the PMe2ArDipp2 system is shown to proceed via 4(Dipp)+ as an intermediate en route to the thermodynamic, isomeric product 3(Dipp)+. This complex interconversion involves a non-innocent C5Me5 ligand, which participates in C–H and C–C bond formation and cleavage. Remarkably, the conversion of 4(Dipp)+ to 3(Dipp)+ also proceeds in the solid state.
Cyclopentadienyls, C5R5, and tertiary phosphines, PR3, are unquestionably two of the most important classes of ligands in organometallic chemistry and catalysis.1 Although in most cases C5R5 and PR3 behave strictly as spectators, in some reactions they can also directly participate. As PR3 and C5R5 continue to be increasingly employed in homogeneous catalysis, knowledge of these unforeseen reactions is crucial because they might strongly influence catalytic outcomes2 or lead to catalyst deactivation.3 Certain aryl phosphines undergo facile cyclometalation,4,5 and recently, nickel- and palladium-mediated dearomatization of dialkylbiaryl phosphines has been reported.2,6 With cyclopentadienyl ligands, in particular C5Me5, ring methyl activation implying either deprotonation or hydride abstraction,7,8 as well as metal-to-ring hydride transfer,9,10 have all been documented.
Transition metal mediated C–H bond activation is a very important transformation with great potential for the functionalization of hydrocarbons. Decisive mechanistic advances have been made with the investigation of electrophilic C–H bond activation at (η5-C5Me5)Ir(III) centers,11 revealing, among other details, the influence of coligands, in particular their ability to act as a base to accept the generated proton.12 Here, we targeted the synthesis of cationic (η5-C5Me5)Ir(III) complexes of the terphenyl phosphines13 PMe2ArXyl2 and PMe2ArDipp2 (Scheme 1). In particular, we report that the five-coordinate complexes [(η5-C5Me5)IrCl(PR2Ar′)]+, 2(Xyl)+ and 2(Dipp)+, promote facile electrophilic C–H activation at remote ζ C–H bonds of the phosphine ligand to form 3(Xyl)+ and 3(Dipp)+. Moreover, for 2(Dipp)+, the observed ζ C–H activation in the presence of NEt3 occurs through a complex mechanism that implies reversible η5-C5Me5 deprotonation and reversible C–C bond formation between the resulting tetramethylfulvene terminal methylene group, and one of the flanking Dipp rings of the phosphine, that itself undergoes dearomatization.2,6 The resulting intermediate, 4(Dipp)+, contains a 10-membered phospha-iridacycle. Intriguingly, this complex transforms readily into the isomeric ζ C–H activation species, 3(Dipp)+, not only in solution, but also in the solid state.
Scheme 1. Dimethyl Terphenyl Phosphines and Corresponding Iridium(III) Cyclopentadienyl Complexes Reported in This Work.

Treatment of [(η5-C5Me5)IrCl2]2 with PMe2ArXyl2 in CH2Cl2 yielded the expected [(η5-C5Me5)IrCl2(PMe2ArXyl2)] product, 1(Xyl), in high yields (∼90%). Chloride abstraction by NaBArF was also straightforward and allowed isolation of the cationic complex [(η5-C5Me5)IrCl(PMe2ArXyl2)]+ (2(Xyl)+, Scheme 1) as its BArF salt, which appeared as a very dark red crystalline solid. Because of the high solution reactivity of this low-coordinate complex under ambient conditions, its synthesis and characterization were performed at −20 °C. Microanalytical and spectroscopic data (see the Supporting Information (SI)) were in agreement with the formulation indicated in Scheme 1, which was subsequently confirmed by X-ray crystallography (Figure 1, left). The short Ir–Cl bond length of 2.2785(9) Å (cf. the 2.396(1) Å average distance in 1(Xyl)), coupled with the distinct, intense dark color,14−16 suggests chloride acts as a π-donor in this formally 16e complex; similar Ru–Cl shortening was also reported in [(η5-C5Me5)RuCl(PiPr3)].17
Figure 1.

ORTEPs of the cations of complex [2(Xyl)]BArF and [3(Dipp)]BArF. Hydrogen atoms are excluded for clarity, and thermal ellipsoids are set at 50% probability. Gray lines represent Dipp iPr substituents.
At room temperature, dichloromethane solutions of 2(Xyl)+ underwent further chemical changes, as evidenced by a color change from the initial dark red to yellow-red. This process was accelerated by the presence of water and product crystallization from CH2Cl2/Et2O solvent yielded mixtures of a new iridium complex, 3(Xyl)+, along with [(η5-C5Me5)IrCl2]2 and [HPMe2ArXyl2]BArF. 3(Xyl)+ was unequivocally characterized as a pseudoallylic species formed via remote ζ C–H activation of a benzylic C–H bond of one of the Xyl substituents. It thus appears that the HCl released in the formation of 3(Xyl)+ decomposed unreacted 2(Xyl)+ to yield the above-mentioned side products.
Given that increased coligand steric demands often confer enhanced kinetic stability and hinder undesirable side reactions, (η5-C5Me5)Ir(III) complexes of the bulkier phosphine PMe2ArDipp2 (Scheme 1) were considered. Although the dichloride analogue of 1(Xyl) could not be generated, possibly because of steric hindrance, cationic 2(Dipp)+ formed rapidly when [(η5-C5Me5)IrCl2]2 and PMe2ArDipp2 were allowed to react in the presence of NaBArF. The similar properties of the two 2(PMe2Ar′)+ complexes, including the observation for 2(Dipp)+ of a 31P{1H} NMR singlet with a Δ(δ) shift relative to free PMe2ArDipp2 practically identical to the corresponding value for 2(Xyl)+, strongly supported a five-coordinate structure analogous to that of 2(Xyl)+. Notwithstanding the structural similarity, 2(Dipp)+ possesses much superior solution stability.
As the formation of cationic pseudoallyls, 3(PMe2Ar′)+, from the corresponding chlorides, 2(PMe2Ar′)+, implies electrophilic C–H activation and elimination of HCl, we considered it of interest to study (i) the generation of dicationic [(η5-C5Me5)Ir(PR2Ar′)]2+ species by chloride abstraction from 2(PMe2Ar′)+ with NaBArF and (ii) the use of an external Brønsted base such as NEt3 to facilitate HCl elimination. The first approach actually constitutes the best procedure for the high yield synthesis of complexes 3(Xyl)+ and 3(Dipp)+ (see Scheme 2). Focusing on the PMe2ArDipp2 analogues for additional solution reaction studies, it was found that the formation of 3(Dipp)+ promoted by NaBArF was very slow at room temperature, probably due to the absence of an effective base. Consistent with this hypothesis, reaction of PMe2ArDipp2 with [(η5-C5Me5)Ir(H2O)3](SO4)18 proceeded rapidly to afford 3(Dipp)+.
Scheme 2. Electrophilic ζ C–H Activation in Complexes 2+ To Give the Pseudoallylic Species 3+; S Represents a Solvent Molecule.

The BArF salts of the two pseudoallyl complexes 3(Xyl)+ and 3(Dipp)+ were fully characterized by microanalysis and multinuclear NMR spectroscopy. For 3(Xyl)+ distinct 1H NMR resonances corresponding to the anti and syn pseudoallylic protons are seen as multiplets at 3.14 and 1.04 ppm, with 2JHH = 3.9 and 3JHP = 1 and 14 Hz, respectively. The corresponding carbon atom gives a 13C{1H} signal at 26.3 ppm (2JCP = 4 Hz), whereas the Cortho and Cipso involved in the η3-bonded unit appear at 89.1 and 83.2 ppm, respectively. Single-crystals of [3(Dipp)]BArF were also investigated by X-ray crystallography (Figure 1, right) that confirms that a Dipp ring in 2(Dipp)+ has undergone ζ C–H activation to give a pseudoallylic product (Ir–CMe2 = 2.224(3), Ir–Cortho = 2.197(3) and Ir–Cipso = 2.257(3) Å).
The mechanism of the C–H bond activation to form the 3(PMe2Ar′)+ complexes was also investigated by DFT methods.19 The most accessible pathway involves initial Cl– dissociation to afford an ion-pair comprising dicationic [(η5-C5Me5)Ir(PMe2Ar′)]2+, in which the phosphine is bound in a κ-P, η3-Carene fashion (Figure S1), and Cl–, which resides in the outer coordination sphere. For 2(Xyl)+, this process entails a barrier of 18.4 kcal/mol and gives a species at +16.5 kcal/mol. Facile rearrangement then forms ζ C–H agostic intermediate at +19.3 kcal/mol (Scheme 3). The acidity of the agostic proton in this dicationic species promotes its facile abstraction by the Cl– ion via a transition state at +22.0 kcal/mol, this representing the overall barrier to the C–H activation process.20 In contrast, chloride-mediated deprotonation in 2(Dipp)+ does not occur at the agostic complex, but requires an additional C–H oxidative cleavage step to form an Ir(V) hydride, which is then deprotonated by Cl–. The overall barrier in this case is 24.7 kcal/mol, 2.7 kcal/mol higher than that in 2(Xyl)+ and so consistent with the observed enhanced solution stability of the former (see the SI for details). The formation of [HPMe2Ar′]BArF and [(η5-C5Me5)IrCl2]2 from 2+ and HCl seems to be the driving force of the reaction in both systems.
Scheme 3. Proposed Mechanism for the Electrophilic C–H Activation in 2(PMe2Ar′)+ Complexes (ΔG50°, kcal/mol, R = H, Me).

The addition of a slight excess of NEt3 to solutions of 2(Dipp)+ highlighted the remarkable chemical and structural changes that occur en route to 3(Dipp)+. The latter formed quantitatively by 1H NMR after stirring at room temperature for about 24 h. However, following the reaction by NMR demonstrated the formation of an intermediate, 4(Dipp)+, responsible for a 31P{1H} singlet resonance at −4.4 ppm, clearly distinguishable from those of 2(Dipp)+ and 3(Dipp)+ at 6.6 and 9.8 ppm, respectively. After careful NMR analysis of reaction temperature and time, we found that intermediate 4(Dipp)+ formed as the only observable product when 2(Dipp)+ and NEt3 were allowed to react at −20 °C for 2 h (Scheme 4).
Scheme 4. NEt3-Assisted Formation of Complex 4(Dipp)+ from 2(Dipp)+, and Solution and Solid-State Isomerization of 4(Dipp)+ to 3(Dipp)+.

BArF anions omitted for clarity.
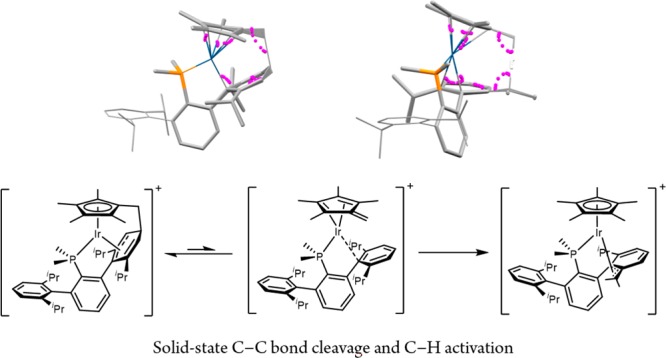
Although 3(Dipp)+ and 4(Dipp)+ are isomers, the latter exhibits a very different chemical constitution, for it contains a 10-membered metallacyclic unit resulting from deprotonation of the C5Me5 ring,7 followed by nucleophilic attack7a,7b at the para carbon atom of the coordinated Dipp ring, which is dearomatized.2,6 Unequivocal structural evidence was gained from variable temperature multinuclear NMR and X-ray studies (Figure 2). In solution, two degenerate pseudoallylic structures undergo fast exchange at room temperature, but reach the slow-exchange regime at −30 °C. At this temperature, the diastereotopic C5Me4CH2 protons resonate as doublets of doublets centered at 3.27 and 2.46 ppm, as a consequence of additional coupling to the adjacent para CH nucleus. The X-ray structure in Figure 2 reveals that, beyond the η5 coordination of the C5Me4CH2 moiety, the now activated phosphine ligand binds to iridium through the phosphorus atom and three adjacent carbon atoms of the dearomatized ring (Ir–C bond distances of 2.166(4) (to Cipso), 2.178(4) (Cortho), and 2.255(5) Å (Cmeta)), whereas the newly formed C–C bond has a length of 1.560(6) Å.
Figure 2.

ORTEP of the cation of complex [4(Dipp)]BArF. Hydrogen atoms are excluded for clarity, and thermal ellipsoids are set at 50% probability. Gray lines represent Dipp iPr substituents.
The isomerization of 4(Dipp)+ to 3(Dipp)+ required neither base (NEt3) nor acid (HNEt3+) catalysis. Instead, it occurred cleanly in CH2Cl2 solution (Scheme 4) following first-order kinetics (t1/2 ≈ 6 h; see the SI for details). It was, however, most notable to find that the 4(Dipp)+ to 3(Dipp)+ isomerization occurred also easily in the solid state (2 days, 30 °C).21,22 Periodical sampling and NMR monitoring disclosed no observable intermediates.
The conversion of 2(Dipp)+ into 3(Dipp)+ through 4(Dipp)+ was also studied computationally (Figure 3). Amine-mediated C5Me5 deprotonation (17.4 kcal/mol, TS2–A) led to the formation of a neutral, Ir(I) fulvene complex (12.0 kcal/mol, A). The thus generated triethylammonium cation then facilitates chloride release (20.2 kcal/mol, TSA–B) to yield intermediate B (1.0 kcal/mol). B is a cationic fulvene complex for which metal unsaturation is compensated by means of a π-arene interaction with one of the flanking aryl rings of the phosphine, and presents an appropriate geometry to undergo C–C bond formation via TSB–4 at 17.7 kcal/mol. We propose this ring dearomatization step proceeds with concomitant metal reoxidation to give Ir(III) complex 4(Dipp)+ at −2.1 kcal/mol. Isomerization of 4(Dipp)+ to 3(Dipp)+ involves the reversible formation of Ir(I) complex B via TSB–4. Attack of the fulvene moiety in B at the C–H of an isopropyl group of the proximate aryl ring (19.4 kcal/mol, TSB–C) reoxidizes the metal center to Ir(III) and gives the η1-allyl complex C (see the SI) at 7.6 kcal/mol. Isomerization to the corresponding η3-allyl occurs via TSC–3 (18.9 kcal/mol) and yields 3(Dipp)+ at −11.5 kcal/mol. It is striking that both the classically innocent ligands (C5Me5 and PR3) play a fundamental role in these transformations (C–H activation and reversible C–C bond formation), whereas the metal center participates by means of the Ir(I)–Ir(III) redox cycle (see the SI for details).
Figure 3.

ΔG50° profile for the conversion of 2(Dipp)+ into 3(Dipp)+ through 4(Dipp)+.
In conclusion, chloride abstraction from complexes 2(PMe2Ar′)+ (Ar′ = ArXyl2, ArDipp2) fosters electrophilic, remote C–H bond activation at dicationic intermediates [(η5-C5Me5)Ir(PMe2Ar′)]2+, to give the pseudoallyl products 3(PMe2Ar′)+ shown in Scheme 2. In the presence of NEt3, complex 2(Dipp)+ converts into the same C–H activation product 3(Dipp)+, though through an unforeseen intermediate, 4(Dipp)+. The latter participates in a complex reaction path involving a non-innocent C5Me5 ligand that undergoes reversible C–H and C–C bond formation and cleavage at one of the methyl termini. The 4(Dipp)+-to-3(Dipp)+ conversion occurs both in solution and in the solid state. The latter observation represents, we believe, a valuable contribution to the field of solid state organometallic chemistry, which, despite its importance as a bridge between molecular and solid-state chemistry, and hence between homogeneous and heterogeneous catalysis, is still underdeveloped.21a
Acknowledgments
This work has been supported by the Spanish Ministry of Economy and Competitiveness (Project CTQ2016-75193-P [AEI/FEDER, UE]) and the European Research Council (ERC Starting Grant, CoopCat, Project 756575). J.J.M. thanks the Universidad de Sevilla for a research grant and Heriot-Watt University for hosting an exchange visit. The use of computational facilities at the Supercomputing Center of Galicia (CESGA) and support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI) are acknowledged. This paper is dedicated to Professor Pablo Espinet in honor of his distinguished career and contributions to organometallic chemistry.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b11752.
All optimized geometries along with their SCF energies (electrophilic) (XYZ)
All optimized geometries along with their SCF energies (base promoted) (XYZ)
Crystallographic data for 2(Xyl)+ (CIF)
Crystallographic data for 3(Dipp)+ (CIF)
Crystallographic data for 4(Dipp)+ (CIF)
Experimental procedures, NMR spectra, full computational details and results and kinetic experiments (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Crabtree R. H. NHC ligands versus cyclopentadienyls and phosphines as spectator ligands in organometallic catalysis. J. Organomet. Chem. 2005, 690, 5451. 10.1016/j.jorganchem.2005.07.099. [DOI] [Google Scholar]
- a Maimone T. J.; Milner P. J.; Kinzel T.; Zhang Y.; Takase M. K.; Buchwald S. L. Evidence for in Situ Catalyst Modification during the Pd-Catalyzed Conversion of Aryl Triflates to Aryl Fluorides. J. Am. Chem. Soc. 2011, 133, 18106. 10.1021/ja208461k. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Milner P. J.; Maimone T. J.; Su M.; Chen J.; Müller P.; Buchwald S. L. Investigating the Dearomative Rearrangement of Biaryl Phosphine- Ligated Pd(II) Complexes. J. Am. Chem. Soc. 2012, 134, 19922. 10.1021/ja310351e. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Sather A. C.; Lee H. G.; De La Rosa V. Y.; Yang Y.; Müller P.; Buchwald S. L. A Fluorinated Ligand Enables Room-Temperature and Regioselective Pd-Catalyzed Fluorination of Aryl Triflates and Bromides. J. Am. Chem. Soc. 2015, 137, 13433. 10.1021/jacs.5b09308. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Sather A. C.; Buchwald S. L. The Evolution of Pd0/PdII-Catalyzed Aromatic Fluorination. Acc. Chem. Res. 2016, 49, 2146. 10.1021/acs.accounts.6b00247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree R. H. Deactivation in Homogeneous Transition Metal Catalysis: Causes, Avoidance, and Cure. Chem. Rev. 2015, 115, 127. 10.1021/cr5004375. [DOI] [PubMed] [Google Scholar]
- a Zhang S.; Chu X.; Li T.; Wang Z.; Zhu B. Synthesis, Structures, and Reactivity of Single and Double Cyclometalated Complexes Formed by Reactions of [Cp*MCl2]2 (M = Ir and Rh) with Dinaphthyl Phosphines. ACS Omega 2018, 3, 4522. 10.1021/acsomega.8b00193. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Guenther J.; Mallet-Ladeira S.; Estevez L.; Miqueu K.; Amgoune A.; Bourissou D. Activation of Aryl Halides at Gold(I): Practical Synthesis of (P,C) Cyclometalated Gold(III) Complexes. J. Am. Chem. Soc. 2014, 136, 1778. 10.1021/ja412432k. [DOI] [PubMed] [Google Scholar]; c Kiener C. A.; Shu C.; Incarvito C.; Hartwig J. F. Identification of an Activated Catalyst in the Iridium-Catalyzed Allylic Amination and Etherification. Increased Rates, Scope, and Selectivity. J. Am. Chem. Soc. 2003, 125, 14272. 10.1021/ja038319h. [DOI] [PubMed] [Google Scholar]
- a Montag M.; Leitus G.; Shimon L. J. W.; Ben-David Y.; Milstein D. Solvent-Dependent Interconversions between RhI, RhII, and RhIII Complexes of an Aryl–Monophosphine Ligand. Chem. - Eur. J. 2007, 13, 9043. 10.1002/chem.200700805. [DOI] [PubMed] [Google Scholar]; b Millard M. D.; Moore C. E.; Rheingold A. L.; Figueroa J. S. Four-Coordinate Iridium(I) Monohydrides: Reversible Dinitrogen Binding, Bond Activations, and Deprotonations. J. Am. Chem. Soc. 2010, 132, 8921. 10.1021/ja1037808. [DOI] [PubMed] [Google Scholar]; c Han Y.-F.; Jin G.-X. Cyclometalated [Cp*M(ĈX)] (M = Ir, Rh; X = N, C, O, P) complexes. Chem. Soc. Rev. 2014, 43, 2799. 10.1039/C3CS60343A. [DOI] [PubMed] [Google Scholar]
- a Nielsen D. K.; Doyle A. G. Nickel-Catalyzed Cross-Coupling of Styrenyl Epoxides with Boronic Acids. Angew. Chem., Int. Ed. 2011, 50, 6056. 10.1002/anie.201101191. [DOI] [PubMed] [Google Scholar]; b Allgeier A. M.; Shaw B. J.; Hwang T. L.; Milne J. E.; Tedrow J. S.; Wilde C. N. Characterization of Two Stable Degradants of Palladium tBuXPhos Catalyst and a Unique Dearomatization Reaction. Organometallics 2012, 31, 519. 10.1021/om200988f. [DOI] [Google Scholar]
- a Fan L.; Wei C.; Aigbirhio F. I.; Turner M. L.; Gusev O. V.; Morozova L. N.; Knowles D. R. T.; Maitlis P. M. Ring-Methyl Activation in Pentamethylcyclopentadienyl Complexes. 5.1 Syntheses and Structures of Tetramethylfulvene Complexes of Ruthenium(II). Organometallics 1996, 15, 98. 10.1021/om950462z. [DOI] [Google Scholar]; b Caldwell H.; Pregosin P. S. Intramolecular Allylation of a Ru-Cp* Methyl Group. Organometallics 2008, 27, 1591. 10.1021/om701248d. [DOI] [Google Scholar]; c Bernechea M.; Berenguer J. R.; Lalinde E.; Torroba J. Facile Single or Double C-H Bond Activation on a Cp* Ligand Promoted by the Presence of Alkynylphosphine Ligands. Organometallics 2009, 28, 312. 10.1021/om800867h. [DOI] [Google Scholar]; d Rais D.; Bergman R. G. Synthesis and Reactivity of the Monomeric Late-Transition-Metal Parent Amido Complex [Ir(Cp*)(PMe3)(Ph)(NH2)]. Chem. - Eur. J. 2004, 10, 3970. 10.1002/chem.200400151. [DOI] [PubMed] [Google Scholar]; e Glueck D. S.; Bergman R. G. Deprotonation of a Cp* Methyl Group by an Iridium Anilide: Formation, Structure, and Solution Dynamics of an η4-Tetramethylfulvene Complex. Organometallics 1990, 9, 2862. 10.1021/om00161a006. [DOI] [Google Scholar]; f Thomas H. P.; Marr A. C.; Morgan P. J.; Saunders G. C. Tethering of Pentamethylcyclopentadienyl and N-Heterocycle Stabilized Carbene Ligands by Intramolecular 1,4-Addition to a Polyfluorophenyl Substituent. Organometallics 2018, 37, 1339. 10.1021/acs.organomet.8b00164. [DOI] [Google Scholar]
- Meredith J. M.; Goldberg K. I.; Kaminsky W.; Heinekey D. M. η6-Tetramethylfulvene and μ-η3η3-Benzene Complexes of Iridium. Organometallics 2012, 31, 8459. 10.1021/om3010492. [DOI] [Google Scholar]
- a Paneque M.; Maitlis P. M. Loss of Pentamethylcyclopentadiene from Pentamethylcyclopentadienylrhodium Hydride Complexes. J. Chem. Soc., Chem. Commun. 1989, 105. 10.1039/c39890000105. [DOI] [Google Scholar]; b Jones W. D.; Kuykendall V. L.; Selmeczy A. D. Ring Migration Reactions of (C5Me5)Rh(PMe3)H2. Evidence for η3 Slippage and Metal-to-Ring Hydride Migration. Organometallics 1991, 10, 1577. 10.1021/om00051a057. [DOI] [Google Scholar]
- a Quintana L. M. A.; Johnson S. I.; Corona S. L.; Villatoro W.; Goddard W. A.; Takase M. K.; VanderVelde D. G.; Winkler J. R.; Gray H. B.; Blakemore J. D. Proton–hydride tautomerism in hydrogen evolution catalysis. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 6409. 10.1073/pnas.1606018113. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pitman C. L.; Finster O. N. L.; Miller A. J. M. Cyclopentadiene-mediated hydride transfer from rhodium complexes. Chem. Commun. 2016, 52, 9105. 10.1039/C6CC00575F. [DOI] [PubMed] [Google Scholar]; c Zamorano A.; Rendón N.; Valpuesta J. E. V.; Álvarez E.; Carmona E. Synthesis and Reactivity toward H2 of (η5-C5Me5)Rh(III) Complexes with Bulky Aminopyridinate Ligands. Inorg. Chem. 2015, 54, 6573. 10.1021/acs.inorgchem.5b00905. [DOI] [PubMed] [Google Scholar]
- a Labinger J. A.; Bercaw J. E. Understanding and exploiting C–H bond activation. Nature 2002, 417, 507. 10.1038/417507a. [DOI] [PubMed] [Google Scholar]; b Balcells D.; Clot E.; Eisenstein O. C–H Bond Activation in Transition Metal Species from a Computational Perspective. Chem. Rev. 2010, 110, 749. 10.1021/cr900315k. [DOI] [PubMed] [Google Scholar]; c Klei S. R.; Tilley T. D.; Bergman R. G. The Mechanism of Silicon-Hydrogen and Carbon-Hydrogen Bond Activation by Iridium(III): Production of a Silylene Complex and the First Direct Observation of Ir(III)/Ir(V) C-H Bond Oxidative Addition and Reductive Elimination. J. Am. Chem. Soc. 2000, 122, 1816. 10.1021/ja992954z. [DOI] [Google Scholar]; d Taw F. L.; Mellows H.; White P. S.; Hollander F. J.; Bergman R. G.; Brookhart M.; Heinekey D. M. Synthesis and Investigation of [Cp*(PMe3)Rh(H)(H2)]+ and Its Partially Deuterated and Tritiated Isotopomers: Evidence for a Hydride/Dihydrogen Structure. J. Am. Chem. Soc. 2002, 124, 5100. 10.1021/ja0165990. [DOI] [PubMed] [Google Scholar]; e Carlsen R.; Wohlgemuth N.; Carlson L.; Ess D. H. Dynamical Mechanism May Avoid High-Oxidation State Ir(V)–H Intermediate and Coordination Complex in Alkane and Arene C–H Activation by Cationic Ir(III) Phosphine. J. Am. Chem. Soc. 2018, 140, 11039. 10.1021/jacs.8b05238. [DOI] [PubMed] [Google Scholar]
- Davies D. L.; Donald S. M. A.; Al-Duaij O.; Macgregor S. A.; Pölleth M. Electrophilic C-H Activation at {Cp*Ir}: Ancillary-Ligand Control of the Mechanism of C-H Activation. J. Am. Chem. Soc. 2006, 128, 4210. 10.1021/ja060173+. [DOI] [PubMed] [Google Scholar]
- Ortega-Moreno L.; Fernández-Espada M.; Moreno J. J.; Navarro-Gilabert C.; Campos J.; Conejero S.; López-Serrano J.; Maya C.; Peloso R.; Carmona E. Synthesis, properties, and some rhodium, iridium, and platinum complexes of a series of bulky m-terphenylphosphine ligands. Polyhedron 2016, 116, 170. 10.1016/j.poly.2016.04.023. [DOI] [Google Scholar]
- a Johnson T. J.; Folting K.; Streib W. E.; Martin J. D.; Huffman J. C.; Jackson S. A.; Eisenstein O.; Caulton K. G. π-Stabilized, yet Reactive, Half-Sandwich Cp*Ru(PR3)X Compounds: Synthesis, Structure, and Bonding. Inorg. Chem. 1995, 34, 488. 10.1021/ic00106a010. [DOI] [Google Scholar]; b Heyn R. H.; Macgregor S. A.; Nadasdi T. T.; Ogasawara M.; Eisenstein O.; Caulton K. G. Is π-donation the only way? Unprecedented unsaturated Ru(II) species devoid of π-donor ligands. Inorg. Chim. Acta 1997, 259, 5. 10.1016/S0020-1693(97)05439-X. [DOI] [Google Scholar]
- a Blacker A. J.; Clot E.; Duckett S. B.; Eisenstein O.; Grace J.; Nova A.; Perutz R. N.; Taylor D. J.; Whitwood A. C. Synthesis and structure of “16-electron” rhodium(III) catalysts for transfer hydrogenation of a cyclic imine: mechanistic implications. Chem. Commun. 2009, 6801. 10.1039/b912943j. [DOI] [PubMed] [Google Scholar]; b Nova A.; Taylor D. J.; Blacker A. J.; Duckett S. B.; Perutz R. N.; Eisenstein O. Computational Studies Explain the Importance of Two Different Substituents on the Chelating Bis(amido) Ligand for Transfer Hydrogenation by Bifunctional Cp*Rh(III) Catalysts. Organometallics 2014, 33, 3433. 10.1021/om500356e. [DOI] [Google Scholar]; c Ishiwata K.; Kuwata S.; Ikariya T. Hydrogen- and Oxygen-Driven Interconversion between Imido-Bridged Dirhodium(III) and Amido-Bridged Dirhodium(II) Complexes. J. Am. Chem. Soc. 2009, 131, 5001. 10.1021/ja900650j. [DOI] [PubMed] [Google Scholar]; d Heiden Z. M.; Rauchfuss T. B. Homogeneous Catalytic Reduction of Dioxygen Using Transfer Hydrogenation Catalysts. J. Am. Chem. Soc. 2007, 129, 14303. 10.1021/ja073774p. [DOI] [PubMed] [Google Scholar]
- Zamorano A.; Rendón N.; López-Serrano J.; Valpuesta J. E. V.; Álvarez E.; Carmona E. Dihydrogen Catalysis of the Reversible Formation and Cleavage of C–H and N–H Bonds of Aminopyridinate Ligands Bound to (η5-C5Me5)IrIII. Chem. - Eur. J. 2015, 21, 2576. 10.1002/chem.201405340. [DOI] [PubMed] [Google Scholar]
- Campion B. K.; Heyn R. H.; Tilley T. D. Preparation and Reactivity of 16-Electron ’Half-Sandwich’ Ruthenium Complexes; X-Ray Crystal Structure of (η5-C5Me5)Ru(PPri3)Cl. J. Chem. Soc., Chem. Commun. 1988, 278. 10.1039/C39880000278. [DOI] [Google Scholar]
- Ogo S.; Makihara N.; Watanabe Y. pH-Dependent Transfer Hydrogenation of Water-Soluble Carbonyl Compounds with [Cp*IrIII(H2O)3]2+ (Cp* = η5-C5Me5) as a Catalyst Precursor and HCOONa as a Hydrogen Donor in Water. Organometallics 1999, 18, 5470. 10.1021/om9903689. [DOI] [Google Scholar]
- Calculations were performed with the Gaussian 09 program employing the hybrid functional PBE0. Geometry optimizations were carried out without geometry constraints and included solvent (dichloromethane) and dispersion effects (Grimme’s D3 parameter set). 50%-corrected free energy variations (ΔG50°) were employed to account for translational entropy overestimation.
- We also considered alternative C–H activation processes involving 16e 2(Xyl)+, but these proved to be not competitive (see SI).
- a Pike S. D.; Weller A. S. Organometallic synthesis, reactivity and catalysis in the solid state using well-defined single-site species. Philos. Trans. R. Soc., A 2015, 373, 20140187. 10.1098/rsta.2014.0187. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pike S. D.; Chadwick F. M.; Rees N. H.; Scott M. P.; Weller A. S.; Krämer T.; Macgregor S. A. Solid-State Synthesis and Characterization of σ-Alkane Complexes, [Rh(L2)(η2,η2-C7H12)][BArF4] (L2 = Bidentate Chelating Phosphine). J. Am. Chem. Soc. 2015, 137, 820. 10.1021/ja510437p. [DOI] [PubMed] [Google Scholar]; c Pike S. D.; Thompson A. L.; Algarra A. G.; Apperley D. C.; Macgregor S. A.; Weller A. S. Synthesis and Characterization of a Rhodium(I) σ-Alkane Complex in the Solid State. Science 2012, 337, 1648. 10.1126/science.1225028. [DOI] [PubMed] [Google Scholar]; d Chaplin A. B.; Green J. C.; Weller A. S. C–C Activation in the Solid State in an Organometallic σ-Complex. J. Am. Chem. Soc. 2011, 133, 13162. 10.1021/ja2047599. [DOI] [PubMed] [Google Scholar]
- a Huang Z.; White P. S.; Brookhart M. Ligand exchanges and selective catalytic hydrogenation in molecular single crystals. Nature 2010, 465, 598. 10.1038/nature09085. [DOI] [PubMed] [Google Scholar]; b Bianchini C.; Farnetti E.; Graziani M.; Kaspar J.; Vizza F. Molecular Solid-State Organometallic Chemistry of Tripodal (Polyphosphine)metal Complexes. Catalytic Hydrogenation of Ethylene at Iridium. J. Am. Chem. Soc. 1993, 115, 1753. 10.1021/ja00058a021. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.