

Abstract
Objective
To assess whether neurodegenerative pathologies are differentially related to trajectories of change in different cognitive abilities.
Methods
At annual intervals for up to 21 years, 915 older participants in a longitudinal clinical-pathologic cohort study completed a battery of 15 tests from which previously established composite measures of episodic memory, semantic memory, working memory, and perceptual speed were derived. At death, they underwent a neuropathologic examination to quantify Alzheimer disease pathology, Lewy bodies, transactive response DNA-binding protein 43 (TDP-43) pathology, and hippocampal sclerosis plus multiple markers of cerebrovascular disease. Time-varying effect models were used to assess change over time in the relation of neuropathologic markers to cognitive trajectories.
Results
Controlling for pathology, decline in perceptual speed was evident about 15 years before death; modest decline in semantic and working memory occurred later; and there was little change in episodic memory. Each neurodegenerative marker was associated with lower episodic memory function beginning about 10 to 16 years before death. As time before death decreased, Alzheimer disease pathology, Lewy bodies, and hippocampal sclerosis were associated with impairment in other cognitive domains but the association of TDP-43 pathology with cognition continued to be mainly confined to episodic memory.
Conclusions
The results suggest that episodic memory impairment is an early sign of multiple neurodegenerative conditions, which primarily differ in their associations with other cognitive systems.
Despite widespread awareness that neurodegenerative lesions are frequently observed in the brains of older persons and are important determinants of cognitive decline, little is known about how their deleterious effects on cognition unravel over the course of aging. This gap in knowledge is in part driven by the frequent co-occurrence of age-related neurodegenerative lesions,1–3 many of which cannot currently be measured in vivo. Moreover, neurodegenerative lesions have different patterns of accumulation, may preferentially affect specific cognitive systems, and their effects may change over time. We recently employed a novel time-varying effect model to examine whether the associations of common age-related neuropathologies with global cognition changed over the course of aging and reported that neurodegenerative lesions were associated with increasingly deleterious effects on cognition over time; by contrast, vascular diseases exerted relatively mild and stable effects on cognition.4
In this study, we expand on prior work by examining the effects of common age-related neuropathologies on change in 4 specific cognitive systems. We used time-varying effect models to flexibly assess the shape of cognitive trajectories over time and to capture change in the association of neuropathologic markers with cognition over time. Participants in this study were 915 deceased older persons from 2 longitudinal clinical-pathologic cohort studies of aging. All were without dementia at baseline, completed a minimum of 2 annual cognitive evaluations (maximum = 21), died, and underwent uniform neuropathologic examinations for identification of age-related neuropathologies.
Methods
Participants
Analyses are based on deceased participants in 2 ongoing clinical-pathologic studies. The Religious Orders Study began in 1994. It involves older Catholic priests, monks, and nuns recruited from groups across the United States.5,6 The Rush Memory and Aging Project began in 1997. It involves older lay persons recruited from the Chicago metropolitan region.7,8 In each study, eligibility requires agreement to annual clinical evaluations plus brain autopsy and neuropathologic examination in the event of death. The clinical and neuropathologic assessments in the 2 studies are identical in essential details.
Eligibility for these analyses required the absence of dementia at the study baseline, a completed cognitive assessment at baseline and at least one follow-up evaluation, and a complete neuropathologic examination. At the time of these analyses (February 2016), 3,124 people had enrolled in the parent studies and completed the baseline evaluation; 194 persons with dementia were excluded. Of the remaining 2,930 individuals, 100 died before the first follow-up and 141 had been in the study less than 1 year. This left 2,689 eligible for follow-up and 2,448 (91%) had cognitive follow-up data. Among those with follow-up, there were 1,278 deaths, and a brain autopsy was done on 1,121 (87.7%); of these, the neuropathologic examination was completed in 915 individuals who formed the final analytic group. Their mean age was 80.1 (SD = 6.9) years at baseline and 89.5 (SD = 6.6) years at death with a mean of 8.1 (SD = 4.5) years of follow-up. They had completed a mean of 16.2 (SD = 3.6) years of education; 284 (31.0%) were men.
Standard protocol approvals, registrations, and patient consents
Participants signed informed consent forms after thorough discussion with the study staff. The institutional review board of Rush University Medical Center approved each study.
Clinical classification
Each annual clinical evaluation included a medical history, cognitive assessment, and neurologic examination. On the basis of this evaluation, an experienced clinician diagnosed dementia following the criteria of the joint working group of the National Institute of Neurological and Communicative Disorders and Stroke/Alzheimer's Disease and Related Disorders Association,9 which require a history of cognitive decline and impairment in 2 or more cognitive domains. Further information on the implementation of these criteria in the parent studies is published elsewhere.6,8,10
Cognitive assessment
Each annual clinical evaluation includes administration of a battery of cognitive tests in an approximately 1-hour session. A set of 15 tests is used to assess 4 cognitive domains. Episodic memory is assessed with Word List Memory, Word List Recall, and Word List Recognition11 plus immediate and delayed recall of Logical Memory Story A12 and the East Boston Story.13,14 Semantic memory is assessed with a Boston Naming Test15 short form,11 category fluency test,11,14 and a brief word reading test.14 Working memory is assessed with Digit Span Forward and Digit Span Backward12 plus a modified form14 of Digit Ordering.16 Modified versions14 of the Symbol Digit Modalities Test17 and Number Comparison18 were used to assess perceptual speed.
In analyses, we used composite cognitive measures based on 2 or more individual tests. Composite measures of this sort can accommodate a wider range of performance than individual tests thereby helping to minimize floor and ceiling artifacts and other forms of measurement error. Supported by factor analyses in these14,19,20 and other21 cohorts, individual tests were assigned to 4 cognitive domains: episodic memory (7 tests), semantic memory (3 tests), working memory (3 tests), and perceptual speed (2 tests). Raw scores on each test were converted to z scores, using the baseline mean and SD in the combined parent cohorts, and the z scores of component tests in each domain were averaged to yield the composite measure of that domain. Further information on the individual tests and cognitive domain measures is contained in earlier publications.14,19,20
Neuropathologic examination
The neuropathologic examination was designed to yield quantitative markers of common conditions associated with late-life loss of cognition. Because previous research in these cohorts has shown that regional measures of a given pathologic condition are robustly intercorrelated,22,23 in the interests of parsimony, the present analyses are based on composite pathologic measures averaged across brain regions. Depending on their distributions, pathologic markers were either treated as continuous measures ranging from no pathology to higher levels (Alzheimer disease [AD] pathology, transactive response DNA-binding protein 43 [TDP-43] pathology, cerebral amyloid angiopathy) or as dichotomous measures that were either present or absent (hippocampal sclerosis, Lewy bodies, gross infarcts, microinfarcts, moderate/severe atherosclerosis, moderate/severe arteriolar sclerosis).
Brain removal and sectioning and preservation of the tissue followed a standard protocol.24 The hemispheres were cut coronally into 1-cm slabs and examined for gross cerebral infarcts. We used hematoxylin & eosin stain to identify microinfarcts in 9 regions from 1 hemisphere (6 cortical, 2 subcortical, 1 midbrain).25 Cerebral β-amyloid angiopathy was assessed with β-amyloid immunostaining in 4 regions (midfrontal cortex, inferior temporal cortex, angular cortex, calcarine cortex). β-Amyloid deposition in the meningeal and parenchymal vessels of each region was rated on a 5-point scale (no deposition, scattered segmental but no circumferential deposition, circumferential deposition up to 10 vessels, circumferential deposition up to 75% of region, circumferential deposition over 75% of region) and the mean of the regional scores was used in analyses.26 Atherosclerosis was based on visual inspection of vessels in the circle of Willis; arteriolar sclerosis was assessed histologically using hematoxylin & eosin–stained sections of the anterior basal ganglia.27 In analyses, both conditions were treated as present if the grade was moderate/severe or absent if findings were less severe.
We used a modified Bielschowsky silver stain to identify neuritic plaques, diffuse plaques, and neurofibrillary tangles in 5 brain regions. Regional scores of each pathology were scaled (raw counts of each type of pathology in each region were divided by the standard deviation for that pathology in that region) and averaged to yield a continuous measure of AD pathology.22 Monoclonal antibodies to phosphorylated TDP-43 (p5409/410; 1:100)28 were used to detect TDP-43 cytoplasmic inclusions in 6 brain regions; inclusion density was rated on a 6-point scale; and regional inclusion density ratings were averaged to yield a composite measure of TDP-43 pathology.29,30 Severe neuronal loss and gliosis in the hippocampus or subiculum (based on hematoxylin & eosin staining) was classified as hippocampal sclerosis.30,31 Lewy bodies were identified with a monoclonal antibody to α-synuclein in 6 brain regions (substantia nigra, anterior cingulate cortex, entorhinal cortex, midfrontal cortex, superior or middle temporal cortex, inferior parietal cortex).32
Statistical analysis
We assessed the dynamic associations of the postmortem pathologic markers with the longitudinal cognitive measures using time-varying effect models.33,34 This modeling approach has 2 advantages: it makes no assumption about the shape of cognitive trajectories over time and it allows the association of covariates with cognition to vary over time. Thus, the model can not only accommodate complex nonlinear cognitive change but it can also assess the relation of a given pathologic index to cognition at each annual observation, helping to inform how each neuropathologic condition contributes to the shape of cognitive aging trajectories. Each model included terms for age at death, years of education, sex, and the 9 postmortem neuropathologic variables, with all covariates permitted to have time-varying effects. Age at death and years of education were centered at the mean. The continuous neuropathologic markers (AD pathology, TDP-43 pathology, and cerebral amyloid angiopathy) were centered at the 10th percentile to represent a light neuropathologic burden.
To assess cognitive decline, we estimated the mean level of cognition over time with the corresponding 95% confidence bands after adjusting for demographics and common neuropathologies. We compared the mean level of cognition during follow-up with the baseline level. If the confidence interval of the coefficient during the follow-up did not cover the baseline, we concluded that cognition changed (figure 1). To assess associations of neuropathologies with cognition, we estimated the coefficients of association over time with the corresponding 95% confidence bands and examined whether the coefficients were different from 0. If the confidence interval of the coefficient did not cover 0, we concluded that the cognitive-pathologic association was statistically significant (figures 2 and 3).
Figure 1. Residual change in cognitive domains after adjustment for cerebrovascular and neurodegenerative conditions.

The time-varying effect model adjusted for age at death, education, sex, AD pathology, gross infarcts, microinfarcts, TDP-43 pathology, hippocampal sclerosis, Lewy bodies, cerebral amyloid angiopathy, atherosclerosis, and arteriolar sclerosis. The data represent the estimated mean cognition for a woman with average age at death (88.9 years), average years of education (16.4 years), low levels (10th percentile) of AD pathology (0.044), TDP-43 pathology (0), and cerebral amyloid angiopathy (0), and no other neuropathologies, with the black dotted line showing the initial estimated cognitive level and the red dotted line (with blue shading indicating the 95% confidence intervals) showing change in the estimated cognitive level as a function of years before death. AD = Alzheimer disease; TDP-43 = transactive response DNA-binding protein 43.
Figure 2. Neurodegenerative disease and change in cognitive domains (A) AD pathology, (B) Lewy bodies, (C) TDP-43 pathology, and (D) hippocampal sclerosis.

The association of markers of neurodegenerative disease with change in different cognitive domains as a function of years before death, from time-varying effect models adjusted for age at death, education, sex, AD pathology, gross infarcts, microinfarcts, TDP-43 pathology, hippocampal sclerosis, Lewy bodies, cerebral amyloid angiopathy, atherosclerosis, and arteriolar sclerosis. The black dotted lines show the initial cognitive-pathologic association and the red dotted lines (with blue shading indicating the 95% confidence intervals) show change in the cognitive-pathologic associations as a function of years before death. AD = Alzheimer disease; TDP-43 = transactive response DNA-binding protein 43.
Figure 3. Cerebrovascular disease and change in cognitive domains (A) gross infarcts, (B) microinfarcts, (C) cerebral amyloid angiopathy, (D) atherosclerosis, and (E) arteriolar sclerosis.

The association of markers of cerebrovascular disease with change in different cognitive domains as a function of years before death, from time-varying effect models adjusted for age at death, education, sex, AD pathology, gross infarcts, microinfarcts, TDP-43 pathology, hippocampal sclerosis, Lewy bodies, cerebral amyloid angiopathy, atherosclerosis, and arteriolar sclerosis. The black dotted lines show the initial cognitive-pathologic association and the red dotted lines (with blue shading indicating the 95% confidence intervals) show change in the cognitive-pathologic associations as a function of years before death. AD = Alzheimer disease; TDP-43 = transactive response DNA-binding protein 43.
We used cubic B-splines to estimate the coefficient functions. There are 3 criteria for assessing time-varying effect model fit: log likelihood, Akaike information criterion, and Bayesian information criterion. In the analyses of each cognitive system, we let the number of knots vary from 1 to 10. Because of the large number of covariates included in the time-varying effect models, it was not feasible to explore all possible combinations of number of knots for each variable. Therefore, we assumed an equal number of knots for each variable. A model with 1 knot gave the best fit by all 3 criteria (data available on request). There was little difference in model fit when the number of knots changed from 1 to 5, and the fitted curves gave similar estimated coefficient functions, with more knots resulting in slightly more wiggly curves.
We conducted 2 sets of additional analyses. First, we used the difference between pairs of cognitive domains as alternate outcomes. Second, we repeated the core model for episodic memory by adding, separately, the interactions of AD pathology with Lewy bodies and of AD pathology with hippocampal sclerosis to the time-varying effect model.
Data availability
All data in these analyses (and descriptions of the studies and variables) are available through the Rush Alzheimer's Disease Center Research Resource Sharing Hub at radc.rush.edu. After logging in, qualified users can request deidentified data.
Results
Cognitive and neuropathologic data
Table 1 provides psychometric information on the composite measures of cognitive domains. At baseline, each measure had a mean of approximately 0. The standard deviations are less than one because the component tests making up each composite measure are correlated. Higher performance on each measure was associated with younger age and higher educational attainment.
Table 1.
Descriptive information on cognitive domain measures at baseline

Death occurred a median of 7.6 months after the last clinical evaluation (interquartile range: 4.0–11.2) with a median postmortem interval of 6.7 hours (interquartile range: 5.0–9.8). The composite measure of AD pathology ranged from 0.00 (indicating no AD pathology) to 2.81 (mean = 0.71, SD = 0.60) and ratings of TDP-43 pathology ranged from 0.00 (indicating no TDP-43 pathology) to 4.50 (mean = 0.61, SD = 0.94). Lewy bodies were present in 220 persons (24.0%) and hippocampal sclerosis was present in 76 persons (8.3%). With respect to cerebrovascular disease, 316 (34.5%) had one or more gross infarcts, 263 (28.7%) had one or more microinfarcts, 270 (29.5%) had at least moderate atherosclerosis, 303 (33.1%) had at least moderate arteriolar sclerosis, and the rating of cerebral β-amyloid angiopathy ranged from 0 (indicating no evidence of pathology) to 4 (mean = 1.08, SD = 1.05). Consistent with prior research in these cohorts,35 most participants had multiple pathologic conditions (data available from Dryad, supplemental table 1, doi.org/10.5061/dryad.mf7hk2q).
Cognitive trajectories controlling for neurodegeneration
We used time-varying effect models to estimate trajectories of change in each cognitive system during up to 21 years of observation (mean = 8.1, SD = 4.5). Each analysis included 3 demographic variables (age at death, education, sex) and 9 neuropathologic variables consisting of 4 neurodegenerative markers (AD pathology, Lewy bodies, TDP-43 pathology, hippocampal sclerosis) and 5 cerebrovascular markers (gross infarcts, microscopic infarcts, cerebral amyloid angiopathy, atherosclerosis, arteriolar sclerosis), with all variables treated as having time-varying effects.
Figure 1 shows the mean level of cognition in each domain at baseline (black dotted line) and over time (red dotted line with 95% confidence intervals in blue) predicted by the model for a woman with average age at death (88.9 years), average years of education (16.4 years), low levels (10th percentile) of AD pathology (0.044), TDP-43 pathology (0), and cerebral amyloid angiopathy (0), and no other neuropathologies. The trajectories range from about 17.6 years before death to 1.5 years before death because 95% of cognitive data were collected in this time frame. The figure suggests different patterns of healthy aging across cognitive domains. Decline in perceptual speed is evident beginning about 15 years before death. Modest decline occurs later in semantic and working memory. By contrast, episodic memory shows little change across the observation period.
Relation of neurodegeneration to cognitive trajectories
The associations of AD pathology with lower episodic memory became evident about 16 years before death (figure 2), when every 1 additional unit of AD pathology was associated with a 0.086-SD decrease in episodic memory (SE = 0.043; p = 0.046); this effect doubled about 14 years before death and tripled about 12 years before death (range of estimates: −0.773 to −0.041, SD of estimates = 0.196). The association of AD pathology with semantic memory impairment became evident about 12 years before death, when every 1 additional unit of AD pathology was associated with a 0.060-SD decrease in semantic memory (SE = 0.030; p = 0.044); this effect doubled about 9 years and tripled about 7 years before death (range of estimates: −0.534 to 0.011, SD = 0.150). The associations of AD pathology with lower working memory were evident early (p < 0.025) but were relatively stable until about 4 years before death (range of estimates: −0.175 to −0.108, SD = 0.020) when a progressive effect was observed (range of estimates: −0.303 to −0.173, SD = 0.040).
The associations of AD pathology with lower perceptual speed were evident about 16 years before death, when every 1 additional unit of pathology was associated with a 0.101-SD decrease in perceptual speed (SE = 0.049; p = 0.041); this effect doubled about 7 years and tripled about 3 years before death (range of estimates: −0.049 to −0.037, SD = 0.075).
The associations of Lewy bodies with episodic memory were evident about 12 years before death (figure 2), when their presence was associated with a 0.070-SD decrease in episodic memory (SE = 0.034; p = 0.042); this effect doubled about 9 years and tripled about 2 years before death (range of estimates: −0.233 to 0.057, SD = 0.079). The associations of Lewy bodies with semantic memory were evident about 10 years before death, when their presence was associated with a 0.052-SD decrease in semantic memory (SE = 0.025; p = 0.040); this effect was mildly progressive (range of estimates: −0.240 to 0.080, SD = 0.083). The results for working memory were unexpected, with Lewy bodies associated with slightly better function in the domain many years prior to death. The associations of Lewy bodies with perceptual speed were evident about 7 years before death, with a 0.073-SD decrease in perceptual speed (SE = 0.036; p = 0.042); this effect doubled about 4 years and tripled about 3 years before death (range of estimates: −0.312 to 0.164, SD = 0.134). The associations of hippocampal sclerosis with episodic memory were evident about 14 years before death (figure 2), with a 0.155-SD decrease in episodic memory (SE = 0.076; p = 0.041); this effect doubled about 11 years and tripled about 7 years before death (range of estimates: −0.498 to 0.099, SD = 0.186). The effects of hippocampal sclerosis on semantic memory were evident about 12 years before death and were associated with a 0.104-SD decrease in semantic memory (SE = 0.052; p = 0.047); this effect doubled about 8 years and tripled about 6 years before death (range of estimates: −0.440 to 0.040, SD = 0.132). The effects of hippocampal sclerosis on working memory were evident about 14 years before death, with a 0.173-SD decrease in working memory (SE = 0.086; p = 0.045). The effects were relatively stable (range of estimates: −0.202 to −0.018, SD = 0.042). Hippocampal sclerosis was associated with perceptual speed about 8 years before death, with a 0.117-SD decrease in perceptual speed (SE = 0.058; p = 0.045); this effect doubled about 5 years before death and gradually increased thereafter (range of estimates: −0.273 to 0.114, SD = 0.146).
The associations of TDP-43 pathology with episodic memory were evident about 9 years before death (figure 2), with a 0.040-SD decrease in episodic memory (SE = 0.019; p = 0.039); this effect doubled about 7 years and tripled about 6 years before death (range of estimates: −0.282 to 0.041, SD = 0.094). The effects of TDP-43 pathology on semantic memory and perceptual speed were not evident until a few years before death when a very mild effect was observed. The effect of TDP-43 pathology on working memory was evident about 16 years before death and was small but positive; this finding was unexpected and likely the result of having relatively limited data so many years before death.
Relation of cerebrovascular disease to cognitive trajectories
The associations of postmortem markers of cerebrovascular disease with decline in cognitive systems are graphically displayed in figure 3. Microinfarcts, cerebral amyloid angiopathy, and arteriolar sclerosis had little association with cognitive function. Gross infarcts were associated with decline in multiple cognitive domains. Atherosclerosis was associated with lower level of function in multiple cognitive domains, particularly perceptual speed.
Additional analyses
To confirm the robustness of findings from the primary analyses examining the associations of the pathologic indices with each cognitive domain in separate models, we conducted sensitivity analyses using difference scores between the cognitive domains (e.g., episodic memory–semantic memory) as alternate outcomes. Results were supportive of findings from the primary modeling approach.
Clinical-pathologic studies have shown that mild cognitive impairment and dementia are usually associated with multiple pathologic conditions.1–3 To explore copathologic effects without greatly increasing model complexity, we added the interaction of AD pathology with hippocampal sclerosis and Lewy bodies in separate models with the composite measure of episodic memory as the outcome. For a given level of AD pathology, there was a more deleterious effect on episodic memory if hippocampal sclerosis was also present (figure 4). The effect was stable over time from about 17 years before death to about 5 years before death. The addition of Lewy bodies to AD pathology had a similar stable (but slightly smaller) deleterious effect on episodic memory from about 17 years before death to about 8 years before death (figure 5).
Figure 4. Interaction of AD pathology and hippocampal sclerosis with episodic memory.
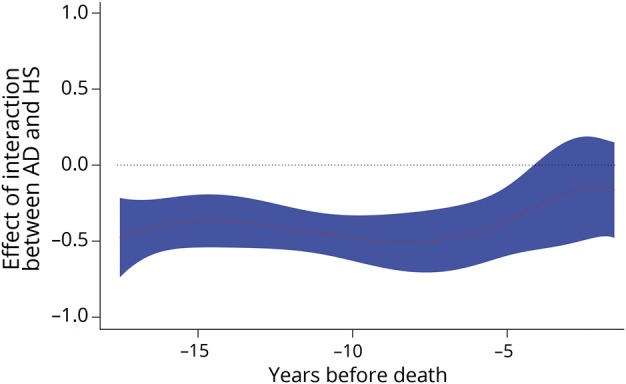
The association of AD pathology and hippocampal sclerosis with episodic memory as a function of years before death from a time-varying effect model adjusted for age at death, education, sex, gross infarcts, microinfarcts, TDP-43 pathology, Lewy bodies, cerebral amyloid angiopathy, atherosclerosis, and arteriolar sclerosis. The black dotted line shows the initial cognitive-pathologic association and the red dotted line (with blue shading indicating the 95% confidence interval) shows the cognitive-pathologic association as a function of years before death. AD = Alzheimer disease; HS = hippocampal sclerosis; TDP-43 = transactive response DNA-binding protein 43.
Figure 5. Interaction of AD pathology and Lewy bodies with episodic memory.
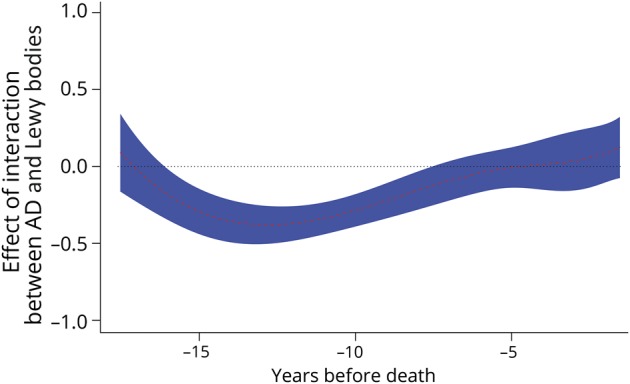
The association of AD pathology and Lewy bodies with episodic memory as a function of years before death from a time-varying effect model adjusted for age at death, education, sex, gross infarcts, microinfarcts, TDP-43 pathology, hippocampal sclerosis, cerebral amyloid angiopathy, atherosclerosis, and arteriolar sclerosis. The black dotted line shows the initial cognitive-pathologic association and the red dotted line (with blue shading indicating the 95% confidence interval) shows the cognitive-pathologic association as a function of years before death. AD = Alzheimer disease; TDP-43 = transactive response DNA-binding protein 43.
Discussion
We annually assessed multiple domains of cognitive function in more than 900 older individuals for up to 21 years. Upon death, there was a uniform neuropathologic examination to quantify common neurodegenerative and cerebrovascular conditions. Each form of neurodegeneration was associated with impairment in episodic memory, but the timing of their effects varied and the different forms were differentially related to impairment in other cognitive systems. The results suggest that different neurodegenerative conditions have relatively distinctive associations with cognitive systems in old age.
Because the brains of older persons often contain a mixture of pathologies,1–3 most of which are difficult to identify antemortem, knowledge about the behavioral manifestations of specific conditions is mainly based on clinical-pathologic research. The present study builds on past clinical-pathologic research in 2 ways. First, we assessed multiple domains of cognition. Second, the time-varying effect model allowed us to characterize nonlinearity in cognitive trajectories and capture change in pathologic correlations with cognition over time. The most striking findings involved episodic memory. Each of the 4 neurodegenerative conditions was associated with episodic memory impairment about 10 to 16 years before death. More proximal to death, AD, Lewy bodies, and hippocampal sclerosis were related to impairment in other cognitive domains whereas, as previously reported with linear modeling,29 TDP-43 pathology was more selectively associated with episodic memory. A related observation is that absent pathology, no decline in episodic memory was evident. These findings provide empirical support for the centrality of episodic memory loss to late-life dementia.
As time to death diminished, the association of the neurodegenerative conditions became less selective. For example, AD was associated with impairment in all cognitive domains in the last 10 years of life, hippocampal sclerosis was associated with decline in semantic memory and perceptual speed in addition to episodic memory in the last 5 to 10 years of life, and Lewy bodies were associated with impairment in all domains in the last few years of life. These data support the idea that as death approaches, widespread neurobiologic changes are impairing all cognitive systems so that the decline in different cognitive abilities becomes more correlated and distinct cognitive domains become dedifferentiated.36
Recognition that late-life dementia is preceded by many years of gradually accelerating cognitive decline37,38 and that these prodromal cognitive symptoms are probably preceded by many years of pathologic changes in the brain39 has blurred the distinction between normal and pathologic cognitive aging. With multiple neurodegenerative and cerebrovascular pathologies statistically controlled, the present analyses provide a view of cognitive aging in the absence of pathologies known to be associated with dementia. Little change was evident in episodic memory. By contrast, there was substantial decline in perceptual speed (nearly 1.0 unit during the observational period) beginning more than 15 years before death. Although neurodegenerative and cerebrovascular pathologies were related to decline in perceptual speed, factors unrelated to these pathologies are also apparently contributing to late-life change in perceptual speed. There was also moderate decline in working memory and semantic memory after controlling for pathologies related to dementia. Understanding the factors accounting for residual variability in perceptual speed, working memory, and semantic memory after adjustment for these pathologies might suggest novel strategies for maintaining cognitive abilities in old age.
In some instances, neurodegenerative markers were associated with better cognitive function. Thus, Lewy bodies were associated with better working memory about a decade before death with similar associations observed between Lewy bodies and perceptual speed and between TDP-43 pathology and working memory (figure 2). We are not aware of evidence linking Lewy bodies or TDP-43 pathology to higher premorbid cognitive ability. However, exploratory examination of the data suggested that individuals with Lewy bodies did have higher cognition at baseline, which may account for this finding. Further investigation of this issue is warranted.
This study has notable strengths. The high rates of participation in the follow-up clinical evaluations and brain autopsy make it less likely that selective attrition biased results. The availability of metrically sound measures of multiple cognitive domains and neuropathologic conditions plus use of time-varying effect models allowed us to flexibly characterize nonlinear change in different abilities and to assess how the relation of neuropathologic conditions to cognitive trajectories changes over time.
An important limitation of the clinical-pathologic approach used here is that assessment of pathology necessarily occurs after clinical assessments, possibly introducing bias into estimates of cognitive-pathologic associations. Further development of antemortem biomarkers may help address this issue. Another limitation is that analyses are based on a selected group and so the generalizability of the findings remains to be determined. Despite our attempt to select the optimal number of knots by maximizing model fit, we cannot rule out the possibility of overfitting the data, which could also affect the generalizability of the findings. A longer observation period might identify earlier cognitive-neuropathologic correlations.
Acknowledgment
The authors thank the many Catholic nuns, priests, and monks who participated in the Religious Orders Study and the many Illinois residents who have participated in the Rush Memory and Aging Project; Traci Colvin, MPH, for coordination of the clinical data collection; Karen Skish, MS, for coordination of the pathologic data collection; and John Gibbons, MS, and Greg Klein, MS, for data management.
Glossary
- AD
Alzheimer disease
- TDP-43
transactive response DNA-binding protein 43
Author contributions
Robert S. Wilson: study concept and design, analysis and interpretation of data, critical revision of manuscript for intellectual content. Jingyun Yang: analysis and interpretation of data, critical revision of manuscript for intellectual content. Lei Yu: analysis and interpretation of data, critical revision of manuscript for intellectual content. Sue E. Leurgans: analysis and interpretation of data, critical revision of manuscript for intellectual content. Ana W. Capuano: analysis and interpretation of data, critical revision of manuscript for intellectual content. Julie A. Schneider: critical revision of manuscript for intellectual content. David A. Bennett: critical revision of manuscript for intellectual content. Patricia A. Boyle: study concept and design, analysis and interpretation of data, critical revision of manuscript for intellectual content.
Study funding
Study funded by NIA (R01AG17917, P30AG10161, R01AG15819, R01AG34374) and the Illinois Department of Public Health. The funding organizations had no role in the design or conduct of the study; the collection, analysis, or interpretation of the data; or the writing of the report or the decision to submit it for publication.
Disclosure
R. Wilson is an associate editor for Neuropsychology, Psychology and Aging, and Aging, Neuropsychology, and Cognition and receives research support from NIH grants RF1AG022018, P30AG010161, RF1AG015819, R01AG017917, U01AG046152-02S1, R01AG054058, R01AG034374, R01AG033678, R01AG020048, U01AG029824, U24AG056270, R01AG051635, RF1AG057532, and R01NS093870. J. Yang receives research support from NIH grants RF1AG015819, RF1AG052476, R01AG054058, and U01AG046152. L. Yu receives research support from NIH grants R01AG017917, R01AG050631, RF1AG048056, RF1AG015819, RF1AG036042, U01AG046152, R01AG033678, R01AG054058, R01AG034374, R01AG053446, R01DK099269, and R01AG052488. S. Leurgans is an associate editor for Biostatistics for Neurology and receives research support from NIH grants R01AG054476, P30AG010161, RF1AG054057, UH2NS100599, RF1AG051641, R01AG054058, R01AG042210, P20MD006886, R01AG034374, R01AG047976, RF1AG015819, U01AG046152-02S1, and R01AG033570. A. Capuano receives research support from NIH grants RF1AG022018, R01AG054058, P30AG010161, R01AG040039, and R01NS084965. J. Schneider receives research support from NIH grants R01AG042210, UH2NS100599, P30AG010161, R01NS084965, R01AG054058, R01AG017917, R01AG033678, R01AG047976, RF1AG015819, RF1AG022018, U01AG046152, R01AG057911, R01AG034374, R01AG056352, UL1TR000430, R01AG043379, RF1AG054057, R01AG054476, R01DK099269, U01AG046161, R01AG033570, P01AG014449, R01AG048108, and R01NS089674. D. Bennett is on the editorial board of Neurology® and receives research support from NIH grants P30AG010161, RF1AG015819, R01AG017917, RF1AG036042, U01AG046152, U01AG046152-02S1, R01AG054058, RF1AG052476, RF1AG057473, RF1AG057471, R01AG053446, RF1AG051641, R01AG057457, R01DK099269, R01AG057911, R01AG055430, R01AG048108, R01AG057914, R01AG033570, U01AG046161, R01AG057912, R01AG052488, R01NS086736, U24AG056270, RF1AG054057, R01AG054476, P01AG014449, R01AG050431, R01NS093870, R01AG050631, R01AG056533, UM1HG009443, RF1AG048056, R01AG046174, and R01AG057907. P. Boyle receives research support from NIH grants R01AG033678, R01AG034374, P30AG010161, R01AG056533, R01AG033570, UH2NS100599, and R01AG055430. Go to Neurology.org/N for full disclosures.
References
- 1.Matthews FE, Brayne C, Lowe J, Wharton SB, Ince P. Epidemiological pathology of dementia: attributable-risks at death in the Medical Research Council Cognitive Function and Ageing Study. PLoS Med 2009;6:e1000180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stephan BC, Matthews FE, Hunter S, et al. ; Medical Research Council Cognitive Function and Aging Study. Neuropathological profile of mild cognitive impairment from a population perspective. Alzheimer Dis Assoc Discord 2012;26:205–212. [DOI] [PubMed] [Google Scholar]
- 3.Abner EL, Kryscio RJ, Schmitt FA, et al. Outcomes after diagnosis of mild cognitive impairment in a large autopsy series. Ann Neurol 2017;81:549–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boyle PA, Yang J, Yu L, et al. Varied effects of age-related neuropathologies on the trajectory of late life cognitive decline. Brain 2017;140:804–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilson RS, Bienias JL, Evans DA, Bennett DA. Religious Orders Study: overview and change in cognitive and motor speed. Aging Neuropsychol Cogn 2004;11:280–303. [Google Scholar]
- 6.Bennett DA, Schneider JA, Arvanitakis Z, Wilson RS. Overview and findings from the Religious Orders Study. Curr Alzheimer Res 2012;9:628–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bennett DA, Schneider JA, Buchman AS, Mendes de Leon CF, Wilson RS. The Rush Memory and Aging Project: study design and baseline characteristics of the study cohort. Neuroepidemiology 2005;25:163–175. [DOI] [PubMed] [Google Scholar]
- 8.Bennett DA, Schneider JA, Buchman AS, Barnes LL, Boyle PA, Wilson RS. Overview and findings from the Rush Memory and Aging Project. Curr Alzheimer Res 2012;9:646–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 10.Bennett DA, Schneider JA, Aggarwal NT, et al. Decision rules guiding the clinical diagnosis of Alzheimer's disease in two community-based cohort studies compared to standard practice in a clinic-based cohort study. Neuroepidemiology 2006;27:169–176. [DOI] [PubMed] [Google Scholar]
- 11.Welsh KA, Butters N, Mohs RC, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD), part V: a normative study of the neuropsychological battery. Neurology 1994;44:609–614. [DOI] [PubMed] [Google Scholar]
- 12.Wechsler D. Wechsler Memory Scale–revised: manual. San Antonio: Psychological Corporation; 1987. [Google Scholar]
- 13.Albert M, Scherr P, Taylor J, Evans D, Funkenstein H. Use of brief cognitive tests to identify individuals in the community with clinically diagnosed Alzheimer's disease. Int J Neurosci 1991;57:167–178. [DOI] [PubMed] [Google Scholar]
- 14.Wilson RS, Beckett LA, Barnes LL, et al. Individual differences in rates of change in cognitive abilities of older persons. Psychol Aging 2002;17:179–193. [PubMed] [Google Scholar]
- 15.Kaplan EF, Goodglass H, Weintraub S. The Boston Naming Test. Philadelphia: Lea & Febiger; 1983. [Google Scholar]
- 16.Cooper JA, Sagar HJ, Jordan N, Harvey NS, Sullivan EV. Cognitive impairment in early, untreated Parkinson's disease and its relationship to motor disability. Brain 1991;114:2095–2122. [DOI] [PubMed] [Google Scholar]
- 17.Smith A. Symbol Digit Modalities Test. Manual (Revised). Los Angeles: Western Psychological Services; 1982. [Google Scholar]
- 18.Ekstrom RB, French JW, Harman HH, Kermen D. Manual for Kit of Factor-Referenced Cognitive Tests. Princeton: Educational Testing Service; 1976. [Google Scholar]
- 19.Wilson RS, Barnes LL, Bennett DA. Assessment of lifetime participation in cognitively stimulating activities. J Clin Exp Neuropsychol 2003;25:634–642. [DOI] [PubMed] [Google Scholar]
- 20.Wilson RS, Barnes LL, Krueger KR, Hoganson G, Bienias JL, Bennett DA. Early and late life cognitive activity and cognitive systems in old age. J Int Neuropsychol Soc 2005;11:400–407. [PubMed] [Google Scholar]
- 21.Wilson RS, Aggarwal NT, Barnes LL, Bienias JL, Mendes de Leon CF, Evans DA. Biracial population study of mortality in mild cognitive impairment and Alzheimer's disease. Arch Neurol 2009;66:767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE. Neurofibrillary tangles mediate the association of amyloid with clinical AD and level of cognitive function. Arch Neurol 2004;61:348–384. [DOI] [PubMed] [Google Scholar]
- 23.Wilson RS, Nag S, Boyle PA, et al. Neural reserve, neuronal density in the locus coeruleus, and cognitive decline. Neurology 2013;80:1202–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol 2009;66:200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arvanitakis Z, Leurgans SE, Barnes LL, Bennett DA, Schneider JA. Microinfarct pathology, dementia, and cognitive systems. Stroke 2011;42:722–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boyle PA, Yu L, Nag S, et al. Cerebral amyloid angiopathy, Alzheimer’s dementia and cognitive decline in community based older persons. Neurology 2015;85:1930–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arvanitakis Z, Capuano AW, Leurgans SE, Bennett DA, Schneider JA. Relation of cerebral vessel disease to Alzheimer’s disease dementia and cognitive function in elderly people: a cross-sectional study. Lancet Neurol 2016;15:934–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neumann M, Kwong LK, Lee EB, et al. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol 2009;117:137–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilson RS, Yu L, Trojanowski JQ, et al. TDP-43 pathology, cognitive decline, and dementia in old age. JAMA Neurol 2013;70:1418–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nag S, Yu L, Capuano AW, et al. Hippocampal sclerosis and TDP-43 pathology in aging and Alzheimer's disease. Ann Neurol 2015;77:942–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nag S, Yu L, Wilson RS, Chen EY, Bennett DA, Schneider JA. TDP-43 pathology and memory impairment in elders without the pathological diagnoses of AD or FTLD. Neurology 2017;88:653–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schneider JA, Li JL, Li Y, Wilson RS, Kordower JH, Bennett DA. Neurofibrillary tangles in the substantia nigra are related to gait impairment in older persons. Ann Neurol 2006;59:166–173. [DOI] [PubMed] [Google Scholar]
- 33.Tan X, Shiyko MP, Li R, Li Y, Dierrker L. A time-varying effect model for intensive longitudinal data. Psychol Methods 2012;17:61–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shiyko MP, Lanza ST, Tan X, Li R, Shiffman S. Using the time-varying effect model (TVEM) to examine dynamic associations between negative affect and self confidence on smoking urges: differences between successful quitters and relapsers. Prev Sci 2012;13:288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boyle PA, Yu L, Wilson RS, Leurgans SE, Schneider JA, Bennett DA. Person-specific contribution of neuropathologies to cognitive loss in old age. Ann Neurol 2018;83:74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilson RS, Segawa E, Hizel LP, Boyle PA, Bennett DA. Terminal dedifferentiation of cognitive abilities. Neurology 2012;78:1116–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Amieva H, Le Goff M, Millett X, et al. Prodromal Alzheimer’s disease: successive emergence of the clinical symptoms. Ann Neurol 2008;64:492–498. [DOI] [PubMed] [Google Scholar]
- 38.Wilson RS, Leurgans SE, Boyle PA, Bennett DA. Cognitive decline in prodromal Alzheimer disease and mild cognitive impairment. Arch Neurol 2011;68:351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aisen PS, Cummings J, Jack CR Jr, et al. On the path to 2025: understanding the Alzheimer's disease continuum. Alzheimers Res Ther 2017;9:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data in these analyses (and descriptions of the studies and variables) are available through the Rush Alzheimer's Disease Center Research Resource Sharing Hub at radc.rush.edu. After logging in, qualified users can request deidentified data.