

Teaser:
This review focuses on the latest developments in tau research for Alzheimer’s disease and other tauopathies, highlighting therapeutic approaches against antiphosphorylated tau using pharmacophore-based molecular and bioinformatics methods.
Phosphorylated tau (P-tau) has received much attention in the field of Alzheimer’s disease (AD), as a potential therapeutic target owing to its involvement with synaptic damage and neuronal dysfunction. The continuous failure of amyloid β (Aβ)-targeted therapeutics highlights the urgency to consider alternative therapeutic strategies for AD. The present review describes the latest developments in tau biology and function. It also explains abnormal interactions between P-tau with Aβ and the mitochondrial fission protein Drp1, leading to excessive mitochondrial fragmentation and synaptic damage in AD neurons. This article also addresses 3D pharmacophore-based drug models designed to treat patients with AD and other tauopathies.
Keywords: Alzheimer’s disease, tau protein, hyperphosphorylation, 3D pharmacophore, dynamin-related protein 1, tauopathies
Alzheimer’s disease and tau
Alzheimer’s disease (AD) is characterized by a collection of multiple, age-related mental conditions that include memory loss and other cognitive impairments. Currently, 50 million people, including 5.4 million Americans, suffer from AD [1]. With increased longevity in the general population, AD is a major health concern for society. Several years of intense research using postmortem human AD brains and brain specimens from AD mouse models have revealed physiological deregulation associated with AD disease progression, including loss of synapses and synaptic function, mitochondrial structural and functional abnormalities, inflammatory responses and neuronal loss, in addition to the presence of extracellular neuritic plaques and intracellular neurofibrillary tangles [2,3]. Although amyloid β (Aβ) and phosphorylated tau (P-tau) are involved in disease progression [4], mounting evidence suggests that the role of P-tau in the progression and pathogenesis of AD is largely through the impairment of axonal transport of subcellular organelles, including mitochondria, lysosomes, vesicles and proteins, to nerve terminals from cell soma [5]. In addition to its involvement in AD, P-tau is also reported to be involved in frontotemporal dementia and other tauopathies [6]. Cellular changes that are involved in disease progression of AD and other tauopathies are not completely understood. The purpose of this review is to highlight the recently discovered involvement of P-tau in AD and other tauopathies and to summarize the results from the latest studies of structure and function in healthy persons and in persons in diseased states. Our article also explains abnormal interactions between P-tau with Aβ and the mitochondrial fission protein dynamin-related protein (Drp)1, leading to excessive mitochondrial fragmentation and synaptic damage in AD neurons and how reduced P-tau and/or Drp1 protects from P-tau-induced toxicities in AD. This article also outlines pharmacophore-based models as therapeutic approaches in AD and other tauopathies.
Tau stabilizes microtubules
Tau is a cytoskeletal protein that stabilizes neuronal microtubules. These microtubules are abundant in nerve cells and present to a much lesser degree in oligodendrocytes and astrocytes [7]. The importance of tau in the assembly of microtubules was first elucidated in the 1970s [8–12]. Neuronal microtubules are the major component of nerve cells and are involved in nutrient transport and signal neurotransmission [13–15]. When tau becomes defective and fails to adequately stabilize microtubules, impaired axonal transport in neurons results [16]. Tau is an essential protein for signaling molecules that regulate gene transcription and cell cycle activity [17]. The cytoskeleton and plasma membrane, both of which act as scaffolding for a variety of signaling molecules, modulate the axonal transport process [18]. αβ-Tubulin proteins are the cytoskeleton proteins that are essential for the formation of the microtubule structure [19,20]. The microtubule-associated protein tau (MAPT) interacts with the αβ-tubulin dimers during structure formation and maintenance of the microtubules [21–23]. However, in neurodegenerative diseases, such as AD and frontotemporal dementia, excessive tau deposition disassembles and the structure of microtubules and neurofibrillary tangles forms [24,25]. Tau, in a nondiseased state, functions as a valuable protein in intracellular transport and in the development and maintenance of the cellular cytoskeleton. However, in AD, tau excessively aggregates in neurons, leading to abnormal tau phosphorylation and microtubule disintegration [26–28]. It has been hypothesized that, in disease states such as AD, aggregates of excessive tau disrupt microtubule formation and interfere with maintenance, impairing axonal transportation in the neuronal cells, resulting in synaptic starvation and neuronal death [29–31]. The human tau protein is encoded by a single gene known as MAPT, located on chromosome 17q21.
The structure of this gene has been hard to characterize owing to its alternative splicing and immature mRNA sequences [32]. MAPT comprises 16 exons. Although exons −1 and 14 are transcribed, they are not translated [33]. Six tau isoforms are produced with the full-length tau protein, which is 441 amino acid residues long. The major domains of the tau protein are the N-terminal acidic domain, the proline-rich domain and the C-terminal microtubule-binding repeat domain. The N-terminal acidic domain is composed of two inserts (exon 2 and exon 3), and the microtubule-binding domain is composed of the four inserts R1–R4, which are encoded by exons 9–12 in MAPT. In combination, these six tau isoforms contain either three (3R) or all four (4R) of the repeat inserts of the microtubule-binding domains [34,35]. The N-terminal part interacts with the neuronal plasma membrane and cytoskeletal elements, and the C-terminal microtubule-binding domain regulates the rate of microtubule polymerization and stabilization [36,37]. Either three (3R) or four (4R) repeat domains are responsible for tau interacting with microtubules [38]. There is a significant difference in the ratio of 4R to 3R in nondiseased and diseased brains. In microtubule-binding repeats of nondiseased human brains, 4R and 3R ratios are approximately equal (1:1) in contrast to their ratio in diseased states, such as in AD and tau pathologies, where the ratio is 2:1 (4R:3R) [39]. The tau protein promotes and regulates the assembly of microtubules and the stability of the cellular assembly [40] through the propagation of dendritic signals to particular axons.
Phosphorylated tau
Tau contains nearly 80 potential serine (Ser) and threonine (Thr) phosphorylation sites on the longest tau isoform [41]. Tau is hyperphosphorylated at specific sites in AD neurons [42] by kinases and phosphatases. In turn, P-tau plays a significant part in disrupting tau–microtubule interactions, impairing axonal transport, leading to defective neurite outgrowth [43]. Recent research also revealed that tau phosphorylation occurs owing to several factors, including Aβ-mediated caspase activation, Aβ-mediated oxidative stress, chronic oxidative stress, reduced insulin-like growth factor (IGF)1-mediated oxidative stress and mutations in the tau gene [44].
Interaction between tau and microtubules
Tau uses microtubule polymerization to regulate axonal stability and cell morphology, maintain cell structure, and regulate the transport of cargo across neurons. In the neuron, tau promotes the mobility of secretory vesicles, organelles and the organization of macromolecules, such as kinesin and dynein [44]. In several neurodegenerative diseases, including AD, P-tau has been abnormally associated with microtubules, leading to defective assembly of microtubules (Figure 1) [45]. This defective microtubule association can lead to impaired axonal transport of organelles and synaptic impairment; and could ultimately lead to neuronal dysfunction. In addition, in a diseased state, P-tau cannot bind to microtubules for stabilization, owing to binding vicinities of microtubule-associated proteins (MAPs) with P-tau. Studies have found that, in a diseased state, defective binding of P-tau with microtubules is associated with neuronal death in tauopathies [46–49]. In neurodegenerative diseases and tauopathies, the normal functioning of tau is disrupted, tau undergoes phosphorylation and tau is aberrantly cleaved. In AD, tau undergoes several aberrant changes that lead to neuronal damage including: (i) tau mislocalization; (ii) hyperphosphorylation [50,51]; and (iii) tau–Aβ interactions causing aggregation and/or fibrillation [52].
Figure 1.

Possible cascade of tau events. Normal tau maintains the stability of microtubules (a). Hyperphosphorylated tau can trigger the dissociation of radical tau from the microtubule surface, potentially destabilizing the microtubule structure (b). Changing the properties of the microtubule surface with dissociated microtubules could affect the transport of organelles and lead to the formation of paired helical filaments and (c) paired helical filaments in the Alzheimer’s disease (AD) state.
Mislocalization of tau
Aberrant tau, in particular P-tau, plays a crucial part in mediating tau mislocalization and subsequent synaptic impairments in AD. P-tau can cause abnormal interactions with other cytosolic proteins, such as MAPs, resulting in the inhibition and disruption of microtubules [53]. The mislocalization of P-tau in neurons in diseased states can also disrupt axonal transport, leading to the degeneration of synapses [54], in contrast to axonal transport in nondiseased states, where healthy neurons maintain the proper special gradient of tau higher in axons and where somato-dentrites exhibit comparatively low levels of healthy tau [55].
In the studies of tau using wild-type and AD mouse models, tau-mediated neurodegeneration was found to be widespread in the AD-affected brains and has been found to exhibit P-tau with an inverted gradient reported [56]. The mislocalization of P-tau in diseased states has been found to increase the proteasome components of tau oligomers, leading to increased synaptic damage in AD and tauopathies [57].
Tau phosphorylation and kinase regulations
Many kinases, such as proline-directed proteins, mitogen-activated proteins and cyclin-dependent kinases, are involved in the AD-associated phosphorylation of tau [58,59]. There is a counterbalance between kinase and phosphatase functions associated with stability and axonal growth maintained by normal tau. The phosphorylation of tau at different sites in tau protein impacts its function and pathogenic role differently [60]. When tau phosphorylation occurs at the sites of kinases, such as the cyclin-dependent kinases (Cdks), protein kinase A (PKA), protein kinase C (PKC) and calmodulin-dependent protein kinase (CaMK), tau functions abnormally. At Cdk sites, hyperphosphorylation of tau occurs at Ser235, Ser202 and Ser404, and tau promotes hyperphosphorylation and promotes self-aggregation of tau filaments. When tau phosphorylation occurs at Ser and Thr sites (e.g., Ser214, Ser324, Ser356, Ser409 and Ser416), tau targets PKA, resulting in the pathological state (Figure 2) [61]. All the Ser or Thr sites have been reported to participate in kinase-dependent tau hyperphosphorylation, leading to paired helical filaments (PHFs) of tau [62]. Phosphorylation sites of tau, whether at Ser, Thr or kinase sites, are affected by several proline-directed kinases, such as mitogen-activated protein kinase (MAPK), Cdk5 and glycogen synthase 3 (GSk3) [63]. The tau phosphorylation at kinase-dependent sites could have a role in the identification of diagnostic markers in AD and tauopathies.
Figure 2.
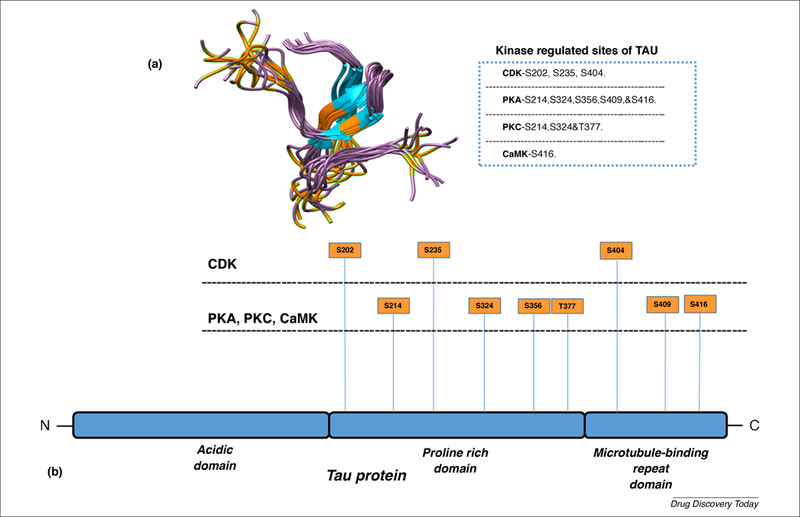
3D structure of the tau protein at serine and threonine sites. Tau regulated by four main kinase enzymes: cyclin-dependent kinase, protein kinase A, protein kinase C and calmodulin-dependent protein kinase. (a) The kinase-dependent regulated site of serine and threonine residue locations on a 3D structure of tau (highlighted in orange), and (b) the full-length tau protein consists of an acidic, proline-rich domain and a microtubule-binding repeat domain. See the four kinase-targeting locations of tau (highlighted on the protein model of tau).
Phosphorylated tau and Aβ interactions
Aβ is an important pathological protein found in the brains of individuals with AD [64]. Aβ is formed by successive cleavage of the Aβ precursor protein (APP), and the successive cascade of cellular events in the AD process [65]. Using postmortem brains from control subjects and AD patients at different stages of disease progression, and from AβPP, AβPPxPS1 and 3xTg-AD transgenic mouse models, researchers found that monomeric Aβ and oligomeric Aβ interact with P-tau and that these interactions progressively increased as AD progresses [66]. This increase in the interaction between P-tau and Aβ interaction was found to correlate with damaged neuronal structure, a condition that could lead to cognitive decline in AD patients. Tau aggregation and interactions, such as in hyperphosphorylation and mislocalization of tau, contribute to P-tau pathology, and apparent molecular mechanisms underlining P-tau-mediated neuronal loss acknowledged a potential therapeutic target in AD and tauopathies.
Phosphorylated tau and mitochondrial fission protein Drp1 interactions
Recent co-immunoprecipitation and co-localization studies from our laboratory revealed that the mitochondrial fission protein Drp1 interacted with P-tau, and this interaction increased as AD progressed [44]. Based on these findings, we hypothesize that a partial deficiency of Drp1 inhibits Drp1-phosphorylated tau interactions and protects neurons from P-tau-induced mitochondrial and synaptic toxicities; and maintains mitochondrial and neuronal functions in AD neurons. To test this hypothesis, Drp1 heterozygote knockout (+/−) mice were crossed with transgenic tau mice (P301L line) creating double-mutant (TauXDrp1+/−) mice [67]. Mitochondrial fission, fusion biogenesis and synaptic genes in brain tissues from 6-month-old Drp1+/−, tau, TauXDrp1+/− and wild-type mice were studied. Results showed mRNA and protein levels of fission genes and increased levels of fusion, biogenesis and synaptic genes in 6-month-old double-mutant (TauXDrp1+/−) mice relative to tau mice. P-tau was found to be reduced in double-mutant mice relative to tau mice. These findings suggest that a partial reduction of Drp1 decreases the production of P-tau, reduces mitochondrial dysfunction and maintains mitochondrial dynamics, enhancing mitochondrial biogenesis and synaptic activity in tau mice.
Loss of tau function
Increasing evidence suggests that tau is also involved in synaptic activity, and complete loss of tau triggers synaptic dysfunction and neuronal damage. To understand the links between loss of tau and synaptic and cognitive functions, Velazquez and colleagues [68] generated an adeno-associated virus (AAV) expressing a doxycycline-inducible short-hairpin (Sh)RNA targeted to tau, referred to as AAV-ShRNATau. Using bilateral stereotaxic injections in 7-month-old C57Bl6/SJL wild-type mice with either the AAV-ShRNATau or a control AAV, they acutely knocked-down tau in the adult hippocampus and found significantly impaired motor coordination and spatial memory. Blocking the expression of the AAV-ShRNATau, thereby allowing tau levels to return to control levels, restored motor coordination and spatial memory. Mechanistically, the reduced tau levels were associated with lower brain-derived neurotrophic factor (BDNF) levels, reduced levels of synaptic proteins associated with learning and decreased spine density. Their study findings suggest that tau is necessary for motor and cognitive function in the adult brain, indicating that tau loss-of-function could contribute to the clinical manifestations of many tauopathies [68].
Therapeutic strategies targeting P-tau
Our current understanding of tau function and dysfunction has been informed by pharmacophore-based strategies to discover new molecules that can target P-tau. P-tau-mediated therapeutic drug molecules can increase microtubule stability, inhibit P-tau production, as well as oligomerization and fibrillization, and activate phosphatase functions [69]. The inhibition of P-tau kinases and the activation of phosphatases in AD and other tauopathies could play a part in new molecular drugs that can target these kinases and phosphatases [70].
Tau reduction
Several tau mouse knockout studies have found that reduced neurotoxic functions of P-tau in tauopathies were linked to Aβ-dependent toxicity [70]. Other studies have found that an important therapeutic strategy in the treatment of neurodegenerative diseases might be to reduce tau expression [71].
Microtubule stabilization
Stabilizers of the microtubule structure have been found to be drug resistant and subject to toxicities that necessarily limit drug doses. These stabilizers include the antitumor drug taxol, which stabilizes the microtubule structure by stalling the cell cycle in its G1 or M phases [72]. However, adverse side effects have been associated with taxol and its second-generation analog docetaxel [73]. Some drugs, such as davunetide and epothilone D microtubule-stabilizing drugs, were tested in human clinical trials to determine their adverse and well-tolerated side effects [74–78]. All microtubule-stabilizing drugs showed no therapeutic benefits, and they did not significantly stabilize the microtubules.
Phosphatase activation and regulation
Tau is dephosphorylated by protein phosphatase (PP)-2A and, to a minor extent, by PP-1 and PP-2B (calcineurin). Recent studies revealed that the mRNA levels of PP-2A and PP-1 are reduced in AD patients compared with controls, indicating that P-tau influences the extent of phosphatase activation [79]. A decrease in phosphatase activity might result in impaired tau dephosphorylation, as well as enhanced tau phosphorylation because various tau-directed kinases are activated by hyperphosphorylation [80]. To date, there are no published papers that suggest pharmacological approaches have successfully activated PP-2A. Okadaic acid is an inhibitor of the Ser and Thr phosphatases PP-1 and PP-2A, each of which blocks the activation of extracellular, signal-regulated protein kinases [81]. However, phosphatase therapeutics might not serve as productive drugs in AD owing to their adverse side effects in clinical trials.
Drug development
Phosphorylation of tau could play a crucial part in the formation of neurofibrillary tangles. However, researchers have not yet been able to develop drugs to prevent P-tau propagation and aggregation.
Drug discovery
There have been significant advances in the development of drugs to treat AD and other tauopathies. Several small molecules have been identified that target tau-mediated AD and tauopathies. The current structure-based pharmacophore approach has accelerated pharmacokinetic studies and has focused on discovering small molecules capable of reducing various diseases, including AD and other tauopathies [82]. From the past decade to the present day, current therapeutics of AD and other tauopathies have been unsuccessful [83]. Such a lack of success could be caused, at least in part, by the lack of computational and bioinformatics methodologies in studies aiming to discover effective treatments for neurodegenerative diseases. One such computational methodology is pharmacophore molecular docking.
Molecular docking
Molecular docking is an effective methodology used to characterize the behavior of small molecules targeting binding sites of P-tau. In the ligand-based pharmacophore approach, molecular docking is designed to predict the primary binding modes of ligands within a 3D receptor molecule. Molecular docking explains how the ligands inhibit the target in a measurable manner. The interaction of residues and binding modes of ligands could help explain fundamental biochemical processes of a ligand. This molecular docking elucidates the binding behavior of lead molecules and the targets and is essential for rational drug discovery. In light of the significance of P-tau, 3D molecular docking interactions of ligands within the active core site are expected to provide the most effective inhibition of ligands for drug development. In studies of drug development for AD patients, small molecules have been reported to target and to bind at specific P-tau sites, resulting in a change in how P-tau functions [84].
Tau pharmacophore models
P-tau-based pharmacophore modeling procedures are expected to elucidate characteristics of tau aggregates and to identity hyperphosphorylated sites where binding occurs in AD and tauopathies. Structural-based pharmacophore models of P-tau have increased our knowledge of steric and electrostatic features of ligand atoms, which appear to be optimal for molecular docking and interacting with P-tau. 3D features of P-tau models are based on the actual biological features of molecular modeling. It is these actual features of pharmacophores that are responsible for the molecular behavior of the P-tau protein [85,86]. Further, based on these features, molecular modeling could effectively inhibit or reduce the phosphorylation of tau and reduce the interaction of P-tau with ligands. The P-tau pharmacophore models strengthen the phenomena with the key functional groups of atoms and pertained volume associated with the functional scaffold. As a pharmacophore, P-tau could target the supramolecular interactions between tau and ligands within the confined 3D structure of P-tau (Figure 3) [87].
Figure 3.

Pharmacophore models of P-tau. 3D structural architecture of P-tau and the drug discovery workflow with 3D structure of target (tau), pharmacophores, pharmacophore models, virtual screening, ligand sets, molecular docking and best leads.
Researchers of pharmacophore models are trying to develop drugs capable of inhibiting P-tau aggregation in AD and other tauopathies. These models are expected to mimic the features of tau kinases and to promote the interaction of tau and ligands [88] but their adverse side effects must prove to be minimal. They are also expected to facilitate HTS of drug targets. Ultimately, these models are expected to become the gold standard in drug delivery (Figure 4).
Figure 4.

Tau pharmacophore-based screening of drug targets. Shown are two procedures of pharmacophore development that could be encountered when starting a pharmacophore-based virtual screening of tau: (a) a ligand-based 3D pharmacophore with a known ligand structure and ligand fingerprints; and (b) a 3D pharmacophore model with a known protein structure. In both cases, pharmacophore query can be used to identify chemical features, such as H-bond acceptors and H-bond donors, and anionic (−), cationic (+), hydrophobic (H) and aromatic (R) groups for virtual screening. (c) Results are shown from virtual searches of chemical databases using preferential software algorithms to find the best reliable ligand sets with structure similarity targets for docking.
Concluding remarks and future perspectives
Tau plays an important part in the effective functioning of microtubules by promoting the structural integrity of microtubules and facilitating axonal transport. Results from studies of tau phosphorylation and neurofibrillary tangles have pointed to the importance of tau receiving more attention as a therapeutic target in AD and other tauopathies. Currently, Aβ-based therapeutics have failed patients with AD, forcing researchers and clinicians to consider other types of drugs, including P-tau-based therapeutics. Reduced P-tau is also considered as a therapeutic approach in AD. There is increasing interest in developing 3D, pharmacophore-based drug discovery systems that target P-tau for AD and related tauopathies.
Highlights:
Tau plays an important part in the formation of axonal microtubules
Phosphorylated tau (P-tau) plays an aberrant part in causing Alzheimer’s disease
P-tau is a potential therapeutic target in Alzheimer’s disease and tauopathies
P-tau-targeted 3D pharmacophore drug models are important for Alzheimer’s disease and tauopathies
Acknowledgments
This work was supported by NIH grants AG042178, AG47812 and NS105473; the Garrison Family Foundation; the CH Foundation; and a Sex and Gender Alzheimer’s Association (SAGA) grant (PHR). The present work is also supported by Alzheimer’s Association New Investigator Research Grant 2016-NIRG-39787 and Center of Excellence for Translational Neuroscience and Therapeutics grant number PN-CTNT20115-AR and Sex and Gender Alzheimer’s Association (SAGA) grant (to A.P.R.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Prince M et al. (2016) World Alzheimer’s Report. Improving healthcare for people living with dementia coverage, quality and costs now and in the future Available at: https://www.alz.co.uk/research/world-report-2016
- 2.Selkoe DJ (2001) Presenilin, notch, and the genesis and treatment of Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A 98, 11039–11041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.LaFerla FM et al. (2007) Intracellular amyloid-beta in Alzheimer’s disease. Nat. Rev. Neurosci 8, 499–509 [DOI] [PubMed] [Google Scholar]
- 4.Pascoal TA (2017) Synergistic interaction between amyloid and tau predicts the progression to dementia. Alzheimers Dement 13, 644–653 [DOI] [PubMed] [Google Scholar]
- 5.Reddy PH (2011) Abnormal tau, mitochondrial dysfunction, impaired axonal transport of mitochondria, and synaptic deprivation in Alzheimer’s disease. Brain Res 1415, 136–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Forner S et al. (2017) Synaptic impairment in Alzheimer’s disease: a dysregulated symphony. Trends Neurosci 40, 347–357 [DOI] [PubMed] [Google Scholar]
- 7.Orr ME et al. (2017) A brief overview of tauopathy: causes, consequences, and therapeutic strategies. Trends Pharmacol. Sci 38, 637–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weingarten MD et al. (1975) A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. U. S. A 72, 1858–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sabbagh JJ et al. (2016) The metamorphic nature of the tau protein: dynamic flexibility comes at a cost. Front. Neurosci 10, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahmadzadeh H et al. (2015) Mechanical effects of dynamic binding between tau proteins on microtubules during axonal injury. Biophys. J 109, 2328–2337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drubin D et al. (1985) Nerve growth factor induced neurite outgrowth in PC12 cells involves the coordinate induction of microtubule assembly and assembly-promoting factors. J. Cell Biol 101, 1790–1807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brandt R et al. (2016) Special issue on cytoskeletal proteins in health and neurodegenerative disease. Brain Res. Bull 126, 213–216 [DOI] [PubMed] [Google Scholar]
- 13.Dent EW et al. (2014) Microtubules in neurons as information carriers. J. Neurochem 129, 235–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van de Willige D et al. (2016) Microtubule plus-end tracking proteins in neuronal development. Cell. Mol. Life. Sci 73, 2053–2077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones SL et al. (2016) Axon initial segment cytoskeleton: architecture, development, and role in neuron polarity. Neural. Plast 2016, 6808293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spillantini MG et al. (2013) Tau pathology and neurodegeneration. Lancet Neurol 12, 609–622 [DOI] [PubMed] [Google Scholar]
- 17.Desdouets C et al. (1995) Cell cycle regulation of cyclin A gene expression by the cyclic AMP-responsive transcription factors CREB and CREM. Mol. Cell Biol 15, 3301–3309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kevenaar JT et al. (2005) The axonal cytoskeleton: from organization to function. Front. Mol. Neurosci 8, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siegel GJ et al. , eds (1999) Basic Neurochemistry: Molecular, Cellular and Medical Aspects (6th edn), Philadelphia: Lippincott-Raven [Google Scholar]
- 20.Penazzi L et al. (2016) Microtubule dynamics in neuronal development, plasticity, and neurodegeneration. Int. Rev. Cell. Mol. Biol 321, 89–169 [DOI] [PubMed] [Google Scholar]
- 21.Karki R et al. (2013) βIII-tubulin: biomarker of taxane resistance or drug target? Expert Opin. Ther. Targets 17, 461–472 [DOI] [PubMed] [Google Scholar]
- 22.Pilhofer M et al. (2011) Microtubules in bacteria: ancient tubulins build a five-protofilament homolog of the eukaryotic cytoskeleton. PLoS Biol 9, e1001213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schlieper D et al. (2005) Structure of bacterial tubulin BtubA/B: evidence for horizontal gene transfer. Proc. Natl. Acad. Sci. U. S. A 102, 9170–9175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brettschneider J et al. (2015) Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat. Rev. Neurosci 16, 109–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee SJ et al. (2011) Protein aggregate spreading in neurodegenerative diseases: problems and perspectives. Neurosci. Res 70, 339–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Campion D et al. (2016) Alzheimer disease: modeling an Aβ-centered biological network. Mol. Psychiatry 21 doi: 10.1038/mp.2016.38} [DOI] [PubMed] [Google Scholar]
- 27.Soto C (2003) Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci 4, 49–60 [DOI] [PubMed] [Google Scholar]
- 28.Puri R et al. (2009) Hyperphosphorylation and aggregation of tau in laforin-deficient mice, an animal model for Lafora disease. J. Biol. Chem 284, 22657–22663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Conde C et al. (2009) Microtubule assembly, organization, and dynamics in axons and dendrites. Nat. Rev. Neurosci 10, 319–332 [DOI] [PubMed] [Google Scholar]
- 30.Rajmohan R et al. (2017) Amyloid-beta and phosphorylated tau accumulations cause abnormalities at synapses of Alzheimer’s disease neurons. J. Alzheimers Dis. 57, 975–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pozueta J et al. (2013) Synaptic changes in Alzheimer’s disease and its models. Neuroscience 251, 51–65 [DOI] [PubMed] [Google Scholar]
- 32.Spillantini MG et al. (2015) Tau pathology and neurodegeneration. Lancet Neurol 12, 609–622 [DOI] [PubMed] [Google Scholar]
- 33.Caillet-Boudin ML et al. (2015) Regulation of human MAPT gene expression. Mol. Neurodegener 10, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.West RR et al. (1991) A model for microtubule-associated protein 4 structure. Domains defined by comparisons of human, mouse, and bovine sequences. J. Biol. Chem 266, 21886–21896 [PubMed] [Google Scholar]
- 35.Lapointe NE et al. (2009) Tau 6D and 6P isoforms inhibit polymerization of full-length tau in vitro. Biochemistry 48, 12290–12297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Csizmok V et al. (2016) Dynamic protein interaction networks and new structural paradigms in signaling. Chem. Rev 116, 6424–6462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levy SF et al. (2005) Three- and four-repeat tau regulates the dynamic instability of two distinct microtubule subpopulations in qualitatively different manners. Implications for neurodegeneration. J. Biol. Chem 280, 13520–13528 [DOI] [PubMed] [Google Scholar]
- 38.Morgan AA et al. (2013) Proline: the distribution, frequency, positioning, and common functional roles of proline and polyproline sequences in the human proteome. PLoS One 8, e53785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Espíndola SL et al. (2018) Modulation of tau isoform imbalance precludes tau pathology and cognitive decline in a mouse model of tauopathy. Cell Rep 23, 709–715 [DOI] [PubMed] [Google Scholar]
- 40.Hernandez F et al. (2007) Tauopathies. Cell Mol. Life Sci 64, 2219–2233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Despres C et al. (2017) Identification of the Tau phosphorylation pattern that drives its aggregation. Proc. Natl. Acad. Sci. U. S. A 114, 9080–9085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou L et al. (2018) Tau association with synaptic vesicles causes presynaptic dysfunction. Nat. Commun 8, 15295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brandt R and Bakota L (2017) Microtubule dynamics and the neurodegenerative triad of Alzheimer’s disease: the hidden connection. J. Neurochem 143, 409–417 [DOI] [PubMed] [Google Scholar]
- 44.Manczak M and Reddy PH (2012) Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 21, 2538–2547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leterrier C (2018) The axon initial segment: an updated viewpoint. J. Neurosci doi: 10.1523/JNEUROSCI.1922-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prezel E et al. (2018) Tau can switch microtubule network organizations: from random networks to dynamic and stable bundles. Mol. Biol. Cell 29, 154–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lathuiliere A et al. (2017) Motifs in the tau protein that control binding to microtubules and aggregation determine pathological effects. Sci. Rep 7, 13556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jebarupa B et al. (2018) Conformational heterogeneity of tau: implication on intrinsic disorder, acid stability and fibrillation in Alzheimer’s disease. Biophys. Chem 241, 27–37 [DOI] [PubMed] [Google Scholar]
- 49.Mandelkow E et al. (2007) Structural principles of tau and the paired helical filaments of Alzheimer’s disease. Brain Pathol 17, 83–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hoover BR et al. (2010) Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68, 1067–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller EC et al. (2014) Tau phosphorylation and tau mislocalization mediate soluble Aβ oligomer-induced AMPA glutamate receptor signaling deficits. Eur. J. Neurosci 39, 1214–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hefti F et al. (2013) The case for soluble Aβ oligomers as a drug target in Alzheimer’s disease. Trends Pharmacol. Sci 34, 261–266 [DOI] [PubMed] [Google Scholar]
- 53.Iqbal K et al. (2008) Cytosolic abnormally hyperphosphorylated tau but not paired helical filaments sequester normal MAPs and inhibit microtubule assembly. J. Alzheimers Dis 14, 365–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brandt R et al. (2005) Tau alteration and neuronal degeneration in tauopathies: mechanisms and models. Biochim. Biophys. Acta 1739, 331–354 [DOI] [PubMed] [Google Scholar]
- 55.Winters BD et al. (2017) Amplitude normalization of dendritic EPSPs at the soma of binaural coincidence detector neurons of the medial superior olive. J. Neurosci 37, 3138–3149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tracy TE and Gan L (2018) Tau-mediated synaptic and neuronal dysfunction in neurodegenerative disease. Curr. Opin. Neurobiol 51, 134–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ittner A and Ittner LM (2018) Dendritic tau in Alzheimer’s disease. Neuron 99, 13–27 [DOI] [PubMed] [Google Scholar]
- 58.Hosokawa T et al. (2010) Quantitative measurement of in vivo phosphorylation states of Cdk5 activator p35 by Phos-tag SDS-PAGE. Mol. Cell. Proteomics 9, 1133–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reddy PH et al. (2011) Mitochondria as a therapeutic target for aging and neurodegenerative diseases. Curr. Alzheimer. Res 8, 393–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Billingsley ML et al. (1997) Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem. J 323, 577–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Biernat J and Mandelkow EM (1999) The development of cell processes induced by tau protein requires phosphorylation of serine 262 and 356 in the repeat domain and is inhibited by phosphorylation in the proline-rich domains. Mol. Biol. Cell 10, 727–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rajmohan R and Reddy PH (2017) Amyloid-beta and phosphorylated tau accumulations cause abnormalities at synapses of Alzheimer’s disease neurons. J. Alzheimers Dis 57, 975–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee S et al. (2014) Divergent and convergent roles for kinases and phosphatases in neurofilament dynamics. J. Cell Sci 127, 4064–4077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bloom GS (2014) Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 71, 505–508 [DOI] [PubMed] [Google Scholar]
- 65.Norstrom E (2017) Metabolic processing of the amyloid precursor protein – new pieces of the Alzheimer’s puzzle. Discov. Med 23, 269–276 [PubMed] [Google Scholar]
- 66.Manczak M and Reddy PH (2013) Abnormal interaction of oligomeric amyloid-β with phosphorylated tau: implications to synaptic dysfunction and neuronal damage. J. Alzheimers Dis 36, 285–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kandimalla R et al. (2016) Reduced dynamin-related protein 1 protects against phosphorylated Tau-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet 25, 4881–4897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Velazquez R et al. (2018) Acute tau knockdown in the hippocampus of adult mice causes learning and memory deficits. Aging Cell doi: 10.1111/acel.12775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee VM et al. (2011) Developing therapeutic approaches to tau, selected kinases, and related neuronal protein targets. Cold Spring Harb. Perspect. Med 1, a006437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brunden KR et al. (2010) Tau-directed drug discovery for Alzheimer’s disease and related tauopathies: a focus on tau assembly inhibitors. Exp. Neurol 223, 304–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maina MB et al. (2018) The involvement of Aβ42 and tau in nucleolar and protein synthesis machinery dysfunction. Front. Cell Neurosci 12, 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Florenzano F et al. (2017) Extracellular truncated tau causes early presynaptic dysfunction associated with Alzheimer’s disease and other tauopathies. Oncotarget 8, 64745–64778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang CH and Horwitz SB (2017) Taxol®: the first microtubule stabilizing agent. Int. J. Mol. Sci 18, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sibaud V et al. (2017) Dermatological adverse events with taxane chemotherapy. Eur. J. Dermatol 26, 427–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Makani V et al. (2016) Evaluation of the brain-penetrant microtubule-stabilizing agent, dictyostatin, in the PS19 tau transgenic mouse model of tauopathy. Acta Neuropathol. Commun 4, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang B et al. (2012) The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. J. Neurosci 32, 3601–3611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.LaPointe NE et al. (2013) Effects of eribulin, vincristine, paclitaxel, and ixabepilone on fast axonal transport and kinesin-1 driven microtubule gliding: implications for chemotherapy-induced peripheral neuropathy. Neurotoxicology 37, 231–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gauthier S et al. (2016) Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: a randomised, controlled, double-blind, parallel-arm, Phase 3 trial. Lancet 388, 2873–2884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sontag JM et al. (2014) Altered protein phosphatase 2A methylation and Tau phosphorylation in the young and aged brain of methylenetetrahydrofolate reductase (MTHFR) deficient mice. Front. Aging Neurosci 6, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gong CX et al. (2000) Regulation of phosphorylation of neuronal microtubule-associated proteins MAP1b and MAP2 by protein phosphatase-2A and −2B in rat brain. Brain Res 853, 299–309 [DOI] [PubMed] [Google Scholar]
- 81.Garcia L et al. (2002) PP1/PP2A phosphatases inhibitors okadaic acid and calyculin A block ERK5 activation by growth factors and oxidative stress. FEBS Lett 523, 90–94 [DOI] [PubMed] [Google Scholar]
- 82.Shiri F et al. (2018) Dynamic structure based pharmacophore modeling of the acetylcholinesterase reveals several potential inhibitors. J. Biomol. Struct. Dyn 17, 1–13 [DOI] [PubMed] [Google Scholar]
- 83.Khanna MR et al. (2018) Therapeutic strategies for the treatment of tauopathies: hopes and challenges. Alzheimers Dement 12, 1051–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gribkoff VK et al. (2017) The need for new approaches in CNS drug discovery: why drugs have failed, and what can be done to improve outcomes. Neuropharmacology 120, 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.van Dun S et al. (2017) Supramolecular chemistry targeting proteins. J. Am. Chem. Soc 139, 13960–13968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hutton M et al. (2001) Analysis of tauopathies with transgenic mice. Trends Mol. Med 7, 467–470 [DOI] [PubMed] [Google Scholar]
- 87.Fichou Y et al. (2015) Molecular dynamics simulations of a powder model of the intrinsically disordered protein tau. J. Phys. Chem. B 119, 12580–12589 [DOI] [PubMed] [Google Scholar]
- 88.Wang B et al. (2013) Molecular recognition in a diverse set of protein−ligand interactions studied with molecular dynamics simulations and end-point free energy calculations. J. Chem. Inf. Model 53, 2659–2670 [DOI] [PMC free article] [PubMed] [Google Scholar]