

Abstract
Voltage-gated Kv1.1 potassium channel α-subunits, encoded by the Kcna1 gene, have traditionally been regarded as neural-specific with no expression or function in the heart. However, recent data revealed that Kv1.1 subunits are expressed in atria where they may have an overlooked role in controlling repolarization and arrhythmia susceptibility independent of the nervous system. To explore this concept in more detail and to identify functional and molecular effects of Kv1.1 channel impairment in the heart, atrial cardiomyocyte patch-clamp electrophysiology and gene expression analyses were performed using Kcna1 knockout (Kcna1−/−) mice. Specifically, we hypothesized that Kv1.1 subunits contribute to outward repolarizing K+ currents in mouse atria and that their absence prolongs cardiac action potentials. In voltage-clamp experiments, dendrotoxin-K (DTX-K), a Kv1.1-specific inhibitor, significantly reduced peak outward K+ currents in wild-type (WT) atrial cells but not Kcna1−/− cells, demonstrating an important contribution by Kv1.1-containing channels to mouse atrial repolarizing currents. In current-clamp recordings, Kcna1−/− atrial myocytes exhibited significant action potential prolongation which was exacerbated in right atria, effects that were partially recapitulated in WT cells by application of DTX-K. Quantitative RT-PCR measurements showed mRNA expression remodeling in Kcna1−/− atria for several ion channel genes that contribute to the atrial action potential including the Kcna5, Kcnh2, and Kcnj2 potassium channel genes and the Scn5a sodium channel gene. This study demonstrates a previously undescribed heart-intrinsic role for Kv1.1 subunits in mediating atrial repolarization, thereby adding a new member to the already diverse collection of known K+ channels in the heart.
Keywords: action potential, atria, channel remodeling, dendrotoxin-K, Kv1.1
INTRODUCTION
Cardiac action potentials are shaped by the coordinated activation and inactivation of inward depolarizing (Na+ and Ca2+) currents and outward repolarizing (K+) currents carried by ion channels (25). Whereas only a few voltage-gated Na+ (Nav) and Ca2+ (Cav) channels are responsible for the depolarizing cardiac currents, multiple types of functionally and molecularly diverse K+ channels participate in myocardial repolarization (24). In mammalian hearts, the prominent repolarizing K+ currents are categorized into several broad classes based on their gating, kinetics, and voltage-dependence. These include the transient outward (Ito) and delayed rectifier (IK) currents, which are encoded by voltage-gated K+ (Kv) channels, and the inwardly rectifying currents, which are encoded by non-voltage-gated inwardly rectifying K+ (Kir) channels (24). Changes in the functional expression of cardiac K+ channels due to gene mutations or heart disease can alter cardiac repolarization, and thereby action potential waveforms, propagation, and rhythmicity, leading to susceptibility to deleterious arrhythmias (25).
Recently, a potential role has emerged for voltage-gated Kv1.1 K+ channel α-subunits in the regulation of atrial repolarization (5). Kv1.1 channels, which are encoded by the Kcna1 gene, are best known for their roles in the brain and peripheral nervous system, where they are expressed at high levels and where they act to dampen axonal excitability by regulating action potential firing properties (12, 13, 31). In humans, KCNA1 mutations cause neurological diseases associated with neuronal hyperexcitability, including episodic ataxia type 1, epilepsy, and myokymia (3, 14, 36). Kcna1−/− mice, which lack Kv1.1 due to gene knockout, also exhibit neuronal hyperexcitability defects which manifest as severe epilepsy and neurogenic cardiac dysfunction (5, 6, 20, 21, 30). Although traditionally thought to be absent in heart, Kv1.1 subunits were recently detected at the mRNA and protein levels in both mouse and human atria where they could impact cardiac function intrinsically (5). In addition, patch-clamp recordings of isolated human atrial cardiomyocytes revealed significant outward K+ current components that were sensitive to dendrotoxin-K (DTX-K), a selective blocker of Kv1.1 subunits, suggesting a possible contribution by Kv1.1 to atrial repolarization (5). However, this previous study of the role of Kv1.1 in atrial electrophysiology was limited in scope. For example, the recordings performed in that study were restricted to human cells, which constrained the evaluation of Kv1.1 function to pharmacological methods without the ability to test the effects of KCNA1 gene mutation. In addition, those human cardiomyocyte recordings were obtained only in the voltage-clamp configuration and only for a single voltage-step. Thus, the effect of Kv1.1-mediated currents on action potential morphology, which can influence arrhythmia susceptibility, remains unknown.
Therefore, this work seeks to more clearly define the role of Kv1.1 subunits in the atria. Specifically, this research examines the hypothesis that Kv1.1 subunits contribute to outward repolarizing K+ currents in mouse atria and that their absence prolongs cardiac action potentials. To test this, voltage- and current-clamp recordings were obtained from isolated atrial myocytes in wild-type (WT) and Kcna1−/− mice, utilizing DTX-K to selectively inhibit Kv1.1 channel components. In addition, levels of mRNA for the other predominant cardiac ion channels that contribute to the cardiac action potential were measured to determine whether compensatory expression changes in other channels underlie the atrial electrophysiological phenotype of Kcna1−/− mice. The results of this work provide the first detailed evidence that Kv1.1 subunits contribute to repolarizing currents in atria.
MATERIALS AND METHODS
Animals and genotyping.
Kcna1−/− knockout (KO) mice carry null alleles of the Kcna1 gene (chromosome 6) resulting from targeted deletion of the open reading frame, as previously described (30). The mice were bred and maintained on a Black Swiss (Tac:N:NIHS-BC) genetic background. Animals were housed at 22°C, fed ad libitum, and maintained on a 12:12-h light-dark cycle. All procedures were performed in accordance with the guidelines of the National Institutes of Health (NIH), as approved by the Institutional Animal Care and Use Committee of the Louisiana State University Health Sciences Center- Shreveport.
For genotyping, genomic DNA was isolated by enzymatic digestion of tail clips using Direct-PCR Lysis Reagent (Viagen Biotech, Los Angeles, CA). The genotypes of Kcna1 mice were determined by performing PCR amplification of genomic DNA using allele-specific primers: a mutant specific primer (5′-CCTTCTATCGCCTTCTTGACG-3′), a WT-specific primer (5′-GCCTCTGACAGTGACCTCAGC-3′), and a common primer (5′-GCTTCAGGTTCGCCACTCCCC-3′). The PCR yielded amplicons of ∼337 bp for the wild-type allele and ∼475 bp for the null allele.
Isolation of mouse atrial myocytes.
Atrial myocytes were enzymatically isolated from hearts of male and female mice (ages 6–8 wk). Briefly, mice were intraperitoneally injected with 5000 U/kg heparin (Sigma-Aldrich, St. Louis, MO) and euthanized by cervical dislocation. The heart was quickly removed and mounted on a Langendorff apparatus followed by a 3-min retrograde perfusion with oxygenated (100% O2) Ca2+-free Tyrode’s solution containing (in mmol/l): 140 NaCl, 5.4 KCl, 0.5 MgCl2, 10 glucose, and 10 HEPES (pH 7.4; 37°C). Hearts were then perfused with the same Tyrode’s solution but containing Liberase TH enzymes (0.025 mg/ml; Sigma-Aldrich) and bovine serum albumin (BSA; 1 mg/ml; Sigma-Aldrich). Left and right atrial tissue was then removed and minced, and atrial myocytes were dispersed in KB solution containing (in mmol/l): 80 KOH, 40 KCl, 25 KH2PO4, 3 MgSO4, 50 glutamic acid, 20 taurine, 1 EGTA, 10 glucose, and 10 HEPES (pH 7.2 with KOH; 20–22°C). Cells were stored at room temperature (20–22°C) for at least 1 h before use. All chemicals used to make the solutions for cell isolations were obtained from Sigma-Aldrich.
Whole cell patch-clamp recordings.
Whole cell patch-clamp recordings were performed at 37°C using a mixture of cardiomyocytes pooled from left and right atria unless otherwise indicated. Borosilicate glass pipette (Warner Instruments, Hamden, CT) microelectrodes were used with tip resistances of 2–3 MΩ when filled with pipette solution. Electrodes were connected to a MultiClamp 700B microelectrode amplifier equipped with a CV-7B head stage (Axon Instruments, Molecular Devices, San Jose, CA). Electrical signals were sampled at 4 kHz and digitized with an Axon analog/digital converter (Digidata 1440A). Data acquisition and analysis were performed using Clampfit software (version 10.3, Axon Instruments, Molecular Devices). Dendrotoxin-K (10 nM; Sigma-Aldrich) was used to selectively block Kv1.1 channels, as done previously (5). All chemicals used to make the bath and pipette solutions for recordings were obtained from Sigma-Aldrich.
For voltage-clamp recordings, seal resistances were 4–8 GΩ. Whole cell capacitance was compensated. The bath solution contained (in mmol/l): 132 NaCl, 4 KCl, 1.8 CaCl2, 1.2 MgCl2, 10 HEPES, and 5 glucose (pH 7.4 with NaOH). L-type Ca2+ currents were blocked by adding nimodipine (1 μM) to the bath solution. The pipette solution contained (in mmol/l): 125 K-aspartate, 20 KCl, 1 Mg-ATP, 3 GTP-Na, 10 EGTA, 1 MgCl2, and 10 HEPES (pH = 7.2 with KOH). To control for myocyte-size variability, currents were expressed as densities (pA/pF). Outward currents were measured as the peak current density evoked by 1-s depolarizing voltage steps of +10 mV increments from a holding potential of –50 mV. Activation curves were obtained by plotting peak current amplitude against test potential voltage.
For current-clamp recordings, action potentials were evoked by electrical stimulation with 1-ms, 2-nA current pulses at a frequency of 1 Hz. The bath solution contained (in mmol/l): 126 NaCl, 5.4 KCl, 1.8 CaCl2, 1.0 MgCl2, 20 HEPES, and 11 glucose (pH = 7.4 with NaOH). The pipette solution contained (in mmol/l): 90 K-aspartate, 30 KCl, 10 NaCl, 5.5. glucose, 1.0 MgCl2, 10 EGTA, 4.0 Na-GTP, and 10 HEPES (pH = 7.2 with KOH).
Quantitative PCR analyses.
Following euthanasia by cervical dislocation, the left atria of WT and Kcna1−/− mice (ages 6–8 wk; n = 3 males and 3 females per genotype) were quickly harvested and homogenized in ice-cold TRI reagent (Zymo Research, Irvine, CA). Left atria were used to avoid possible contamination by sinoatrial pacemaker cells in the right atria. Total RNA was extracted using PureLink RNA Mini Kit (Thermo Fisher, Waltham, MA). Genomic DNA was eliminated using DNA-free DNA Removal Kit (Thermo Fisher). The quantity of total RNA was measured using a NanoDrop 1000 spectrophotometer (Thermo Fisher) and quality was confirmed using an RNA ScreenTape assay on a 2200 TapeStation system (Agilent, Santa Clara, CA). The resulting RNA integrity number was used to estimate total RNA integrity and only samples with scores ≥8.0 were used for experiments. The RNA samples (375 ng) were converted to first-strand cDNA using the iScript Advanced cDNA Kit for reverse transcriptase (RT)-quantitative PCR (qPCR) with oligo(dT) primers (Bio-Rad, Hercules, CA). qPCR of first-strand cDNA was performed with TaqMan gene expression assays (Thermo Fisher) that were designed and preoptimized by Thermo Fisher for the detection of Kcna4 (Mm00445241_s1), Kcna5 (Mm00524346_s1), Kcnb1 (Mm00492791_m1), Kcnd2 (Mm01161732_m1), Kcnh2 (Mm00465377_mH), Kcnj2 (Mm00434616_m1), Kcnj3 (Mm00434618_m1), Kcnq1 (Mm00434640_m1), Scn5a (Mm01342518_m1), Cacna1c (Mm01188822_m1), Kcnab1 (Mm00440018_m1), Kcnab2 (Mm01260263_m1), and Kcnab3 (Mm01337143_m1). qPCR experiments were performed using a 1:15 cDNA dilution, which was determined to be the optimal concentration by analyzing qPCR amplification across a five-point cDNA dilution series to generate a standard curve. Individual PCR reactions were performed in triplicate using cDNA and TaqMan Gene Expression Master Mix (Thermo Fisher) on a CFX96 Fast Real-Time PCR System (Bio-Rad). No template and no reverse transcriptase (–RT) reactions were included as negative controls to verify the absence of contamination leading to unwanted PCR amplification and detection. Reactions were normalized to the geometric mean of the amplification threshold cycle (CT) of the housekeeping genes hypoxanthine phosphoribosyltransferase 1 (Hprt1; Mm03024075_m1) and β-actin (Actb; Mm02619580_g1). Relative mRNA expression was calculated as normalized values by use of the 2−ΔΔCT formula. The expression levels of Kcnab3 frequently fell below the limit of reliable detection, so those data were excluded from analysis.
Statistical analysis.
All data are expressed as means ± SE. For electrophysiology data, the sample sizes (n) indicate the number of cells recorded per mouse (i.e., total number of cells recorded/total number of mice used). Statistical analyses were performed using Prism for Windows (version 6; GraphPad Software, La Jolla, CA) and OriginPro (version 7.5, OriginLab, Northampton, MA). For comparisons involving two groups, either paired or unpaired two-tailed Student’s t-tests were employed as appropriate.
RESULTS
Atrial myocytes exhibit Kv1.1-associated currents.
To determine whether Kv1.1 subunits contribute to cardiac currents in mouse atria, whole cell voltage-clamp recordings of outward currents were obtained from cardiomyocytes of WT mice. In recordings from WT atrial myocytes, outward currents were routinely evoked by a voltage-step protocol composed of 1-s depolarizing voltage steps to potentials between –40 mV and +70 mV from a holding potential of –50 mV (Fig. 1A). To measure the specific contribution of Kv1.1-containing channels to the outward currents in WT atrial myocytes, Kv1.1 subunits were selectively inhibited by application of DTX-K (10 nM), as done previously (5). Kv1.1 channel blockade with DTX-K significantly attenuated the outward K+ currents, reducing the peak current density by ∼40% (Fig. 1, B–D). This large, DTX-K-sensitive component suggests a substantial contribution by Kv1.1-containing channels to the repolarizing currents in mouse atria (Fig. 1C).
Fig. 1.
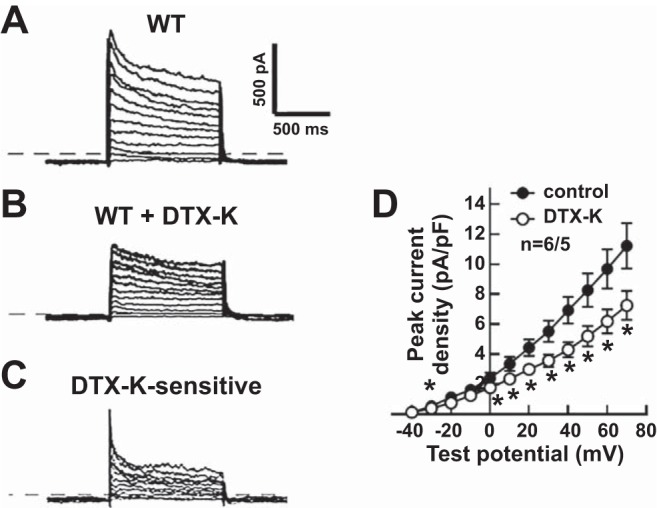
Dendrotoxin-K (DTX-K) reduced outward K+ currents in wild-type (WT) mouse atrial myocytes. A–C: representative raw current traces in a WT cell in response to 1-s depolarizing voltage steps of +10 mV from a holding potential of −50 mV to +70 mV: at baseline (A); with application of 10 nM DTX-K (B); and following subtraction of the current difference to isolate the DTX-K-sensitive component (C). D: quantification of the peak current densities before (control) and after (DTX-K) application of DTX-K in WT atrial myocytes. The dotted line in the traces indicates 0 pA. Sample numbers (n) indicate myocytes per mouse. *P ≤ 0.05 (2-tailed paired Student’s t-test).
To verify the specificity of DTX-K for Kv1.1 channels, voltage-clamp recordings were repeated in atrial myocytes from Kcna1−/− mice which lack Kv1.1 subunits. Depolarizing voltage steps regularly evoked outward K+ currents in Kcna1−/− atrial myocytes (Fig. 2A). However, application of DTX-K (10 nM) had no significant effect on peak outward K+ currents in Kv1.1-deficient cells, indicating elimination of the DTX-K-sensitive component (Fig. 2, B–D). Thus, the lack of DTX-sensitive currents in Kcna1−/− atrial myocytes demonstrates the drug’s specificity for Kv1.1 subunits and confirms that the DTX-K-sensitive currents in WT cells are due to the contribution of Kv1.1-containing channels.
Fig. 2.
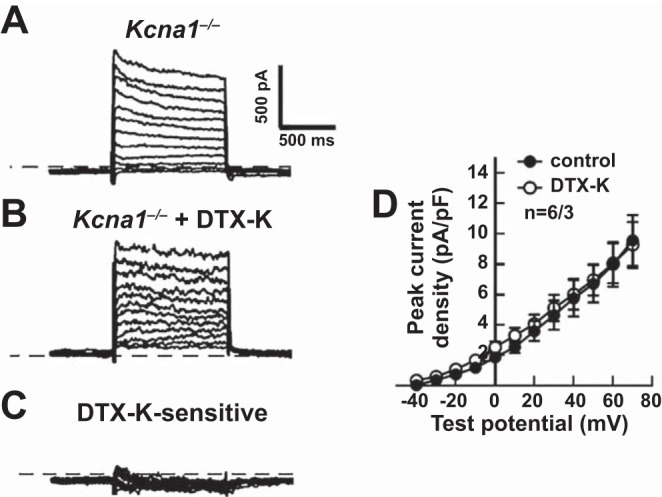
Dendrotoxin-K (DTX-K) had no significant effect on outward K+ currents in Kcna1−/− atrial myocytes. A–C: representative raw current traces in a Kcna1−/− cell in response to 1-s depolarizing voltage steps of +10 mV from a holding potential of −50 mV to +70 mV: at baseline (A); with application of 10 nM DTX-K (B); and following subtraction of the current difference to isolate the DTX-K-sensitive component (C). D: quantification of the peak current densities before (control) and after (DTX-K) application of DTX-K in Kcna1−/− atrial myocytes. The dotted line in the traces indicates 0 pA. Sample numbers (n) indicate myocytes per mouse.
Action potentials are prolonged in atrial myocytes with genetic or pharmacological Kv1.1 inhibition.
To test whether the absence of Kv1.1 subunits alters action potential waveforms, whole cell current-clamp recordings were performed at physiologically relevant temperature (37°C) in atrial myocytes isolated from WT and Kcna1−/− mice. In WT cells, action potentials were typically very brief, lasting <50 ms; however, in Kcna1−/− cells, action potentials were drastically broadened, with durations exceeding 100 ms (Fig. 3A). Measurement of action potential durations at 30, 50, and 90% repolarization (APD30, APD50, and APD90) revealed significant prolongation in Kcna1−/− cells compared with WT cells at APD50 (P ≤ 0.05) and APD90 (P ≤ 0.0001). APD30, APD50, and APD90 were 4.7 ± 0.3 ms, 6.7 ± 0.4 ms, and 44.2 ± 1.6 ms in WT (n = 29/16) and 5.6 ± 0.3 ms, 8.5 ± 0.7 ms, and 96.8 ± 7.5 ms in Kcna1−/− atrial myocytes (n = 19/10), respectively (Fig. 3B). In contrast, resting membrane potentials and action potential amplitudes were not significantly different (Fig. 3, C and D).
Fig. 3.
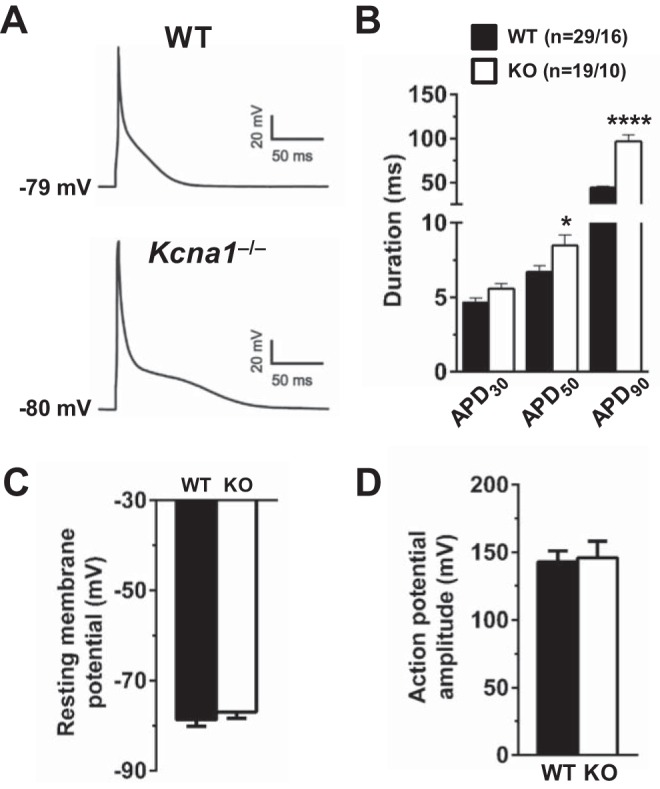
Kcna1−/− atrial myocytes exhibit action potential duration (APD) prolongation. A: representative atrial action potentials recorded in a wild-type (WT) cell and a Kcna1−/− cell. The resting membrane potential is indicated next to each waveform. B: average APD in WT and Kcna1−/− cells at 30, 50, and 90% repolarization (APD30, APD50, and APD90). C and D: average resting membrane potential (C) and action potential amplitude (D) in WT (n = 29/16) and Kcna1−/− cells (n = 19/10). Sample numbers (n) indicate myocytes per mouse. *P ≤ 0.05; ****P ≤ 0.0001 (2-tailed unpaired Student’s t-test). KO, knockout.
Next, the effects of pharmacological Kv1.1 channel inhibition on APD were investigated in WT and Kcna1−/− cells. Application of DTX-K (10 nM) to selectively block Kv1.1 subunits caused substantial prolongation of action potentials in WT atrial myocytes, resembling the effects of genetic Kv1.1 ablation, but to a lesser degree (Fig. 4A). APD90 was significantly (P = 0.0001) prolonged by 58% in WT cells (n = 15/12) following DTX-K administration (45.1 ± 2.1 ms before drug; 71.2 ± 3.2 ms after drug); however, APD30 and APD50 were unchanged (Fig. 4B). To further confirm the specificity of DTX-K for Kv1.1 subunits, its effects on action potential morphology were reexamined in Kcna1−/− cardiomyocytes. Exposure of Kcna1−/− cells to DTX-K (10 nM) failed to significantly alter APD at any time point, providing additional evidence of the toxin’s specificity for Kv1.1 (Fig. 4, C and D).
Fig. 4.
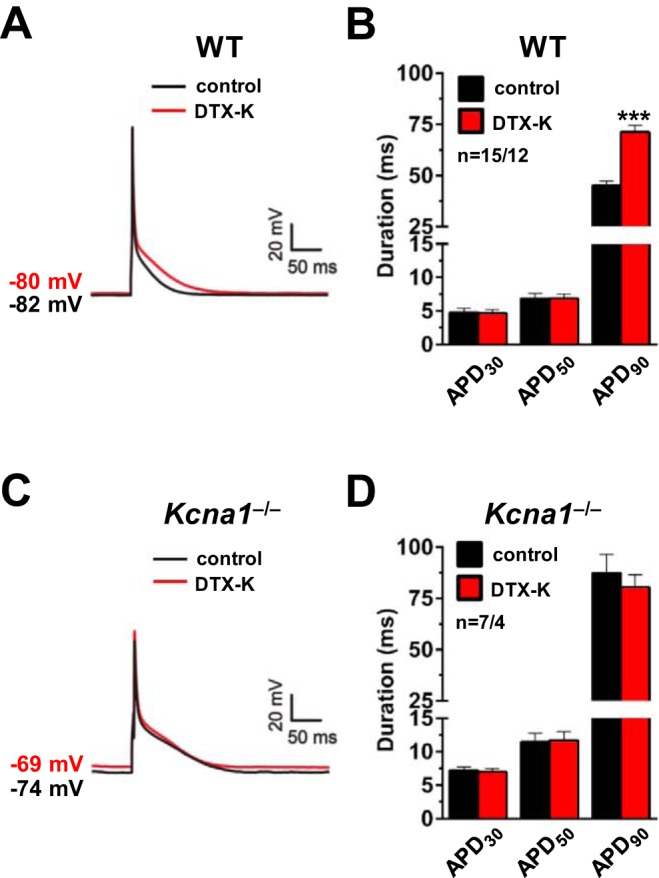
Dendrotoxin-K (DTX-K) prolongs action potentials in wild-type (WT) atrial myocytes. A: a representative action potential recording in a WT cell at baseline (black line; labeled control) overlaid with the resulting action potential after application of 10 nM DTX-K (red line; labeled DTX-K). B: average action potential duration (APD) in WT cells at 30, 50, and 90% repolarization (APD30, APD50, and APD90) before (control) and after (DTX-K) application of 10 nM DTX-K. C: a representative action potential recording in a Kcna1−/− cell at baseline (black line; labeled control) overlaid with the resulting action potential after application of 10 nM DTX-K (red line; labeled DTX-K). D: average APD in Kcna1−/− cells at APD30, APD50, and APD90 before (control) and after (DTX-K) application of 10 nM DTX-K. The resting membrane potential before and after DTX-K is indicated next to each waveform. Sample numbers (n) indicate myocytes per mouse. ***P ≤ 0.001 (2-tailed paired Student’s t-test).
In mice and other mammalian species, action potential duration can vary between the left and right atria due to differences in repolarizing currents (19, 22). However, the above experiments were performed using a mixture of cells isolated together from left and right atria, preventing analysis of interatrial differences. Therefore, to determine whether the absence of Kv1.1 subunits affects action potentials differently in left and right atria, cardiomyocytes were separately isolated from the left and right atria and recorded to measure APD in WT and Kcna1−/− mice. In WT mice, APD was similar for cells from the left and right sides (Table 1 and Fig. 5, A and B). In contrast, in Kcna1−/− mice, APD90 was significantly longer (P < 0.01) in cells from the right atria compared with cells from the left atria (Table 1 and Fig. 5, C and D). In addition, the APD90 of left and right atrial myocytes was significantly prolonged in Kcna1−/− cells compared with WT cells from the same side (P < 0.01; Table 1), consistent with our observations above using mixtures of left and right cells (Fig. 3). Application of DTX-K (10 nM) to left and right WT atrial myocytes led to significant prolongation of APD90 by ~50% on each side (P < 0.05; Table 2 and Fig. 5, A and B). As expected, DTX-K had no significant effect on APD in Kcna1−/− cells regardless of side (Table 2 and Fig. 5, C and D). Thus, genetic ablation or pharmacological inhibition of Kv1.1 subunits prolongs action potentials in cardiomyocytes from both left and right atria.
Table 1.
Action potential durations of left and right atrial myocytes by genotype
| WT |
Kcna1−/− |
|||
|---|---|---|---|---|
| Left Atrium (n = 8/4) | Right Atrium (n = 8/5) | Left Atrium (n = 6/4) | Right Atrium (n = 4/3) | |
| APD30, ms | 5.6 ± 0.8 | 5.2 ± 0.6 | 6.3 ± 0.8 | 6.2 ± 0.7 |
| APD50, ms | 8.0 ± 1.1 | 7.9 ± 0.7 | 11.0 ± 1.8 | 8.6 ± 1.0 |
| APD90, ms | 49.9 ± 2.6 | 46.7 ± 2.9 | 70.1 ± 3.1* | 102.6 ± 11.1†,‡ |
Values are means ± SE. APD30, APD50, and APD90, action potential duration at 30%, 50%, and 90% of repolarization, respectively. Sample numbers (n) indicate myocytes per mouse. WT, wild type.
P < 0.01 vs. WT left atrium (Student’s unpaired t-test);
P < 0.01 vs. WT right atrium (Student’s unpaired t-test);
P < 0.01 vs. Kcna1−/− left atrium (Student’s unpaired t-test).
Fig. 5.
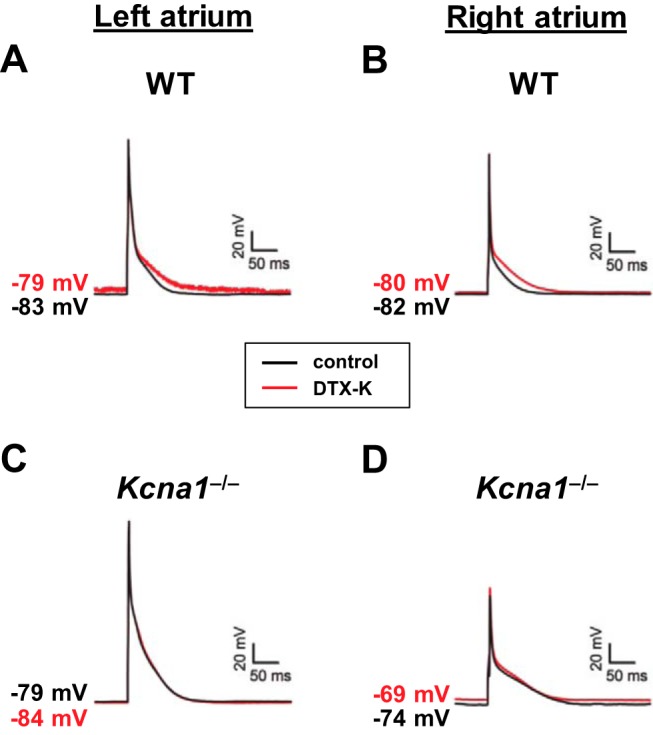
Action potential prolongation in Kcna1−/− cardiomyocytes is exacerbated in right atria. A–D: representative action potential recordings before (black line; labeled control) and after (red line; labeled DTX-K) application of 10 nM DTX-K in wild-type (WT) cells from left (A) and right atria (B) and in Kcna1−/− cells from left (C) and right atria (D).
Table 2.
Action potential durations of left and right atrial myocytes by genotype before and after DTX-K (10 nM)
| Left Atrium (WT n = 4/3; KO n = 4/3) |
Right Atrium (WT n = 4/4; KO n = 3/3) |
|||
|---|---|---|---|---|
| Control | DTX-K | Control | DTX-K | |
| WT | ||||
| APD30, ms | 6.1 ± 1.6 | 5.4 ± 0.7 | 5.4 ± 1.3 | 5.7 ± 1.5 |
| APD50, ms | 8.8 ± 2.0 | 8.2 ± 1.2 | 7.5 ± 1.4 | 8.3 ± 1.7 |
| APD90, ms | 50.8 ± 4.9 | 73.7 ± 6.4* | 43.5 ± 4.1 | 65.7 ± 5.0* |
| Kcna1−/− | ||||
| APD30, ms | 7.3 ± 0.8 | 7.0 ± 0.8 | 6.8 ± 0.5 | 7.0 ± 0.2 |
| APD50, ms | 13.0 ± 1.9 | 13.3 ± 2.0 | 9.4 ± 0.7 | 9.6 ± 0.2 |
| APD90, ms | 72.1 ± 3.9 | 71.6 ± 8.7 | 107.7 ± 14.0 | 92.1 ± 1.4 |
Values are means ± SE. APD30, APD50, and APD90, action potential duration at 30%, 50%, and 90% of repolarization, respectively. Sample numbers (n) indicate myocytes per mouse. DTX-K, dendrotoxin-K; KO, knockout; WT, wild-type.
P < 0.05 vs. control (Student’s paired t-test).
Kv1.1 channel deficiency leads to potassium channel remodeling in atria.
To test whether the absence of Kv1.1 subunits leads to changes in the expression of other ion channels which could contribute to the observed electrophysiological phenotypes, quantitative RT-PCR (qPCR) was performed to compare cardiac ion channel mRNA expression levels in WT and KO atria. Previously, mRNA levels of the related Kv1.x α-subunits (Kcna2–Kcna7) and β-subunits (Kcnab1–Kcnab3) were measured in adult WT and Kcna1−/− atria from 4-mo-old mice revealing minimal compensatory transcriptional remodeling (5); however, other cardiac channels were not investigated. Here, qPCR was performed using 6- to 8-wk-old mice (corresponding to the ages of animals in the patch-clamp electrophysiology experiments) to measure mRNA levels of the main ion channel genes underlying atrial action potential properties and repolarizing currents. These genes (with their associated protein and current) examined included: Kcna4 (Kv1.4; Ito); Kcna5 (Kv1.5; IKur); Kcnb1 (Kv2.1; Iss); Kcnd2 (Kv4.2; Ito,f); Kcnh2 (Kv11.1/mERG; IKr); Kcnj2 (Kir2.1; IK1); Kcnj3 (Kir3.1; IK,ACh); Scn5a (Nav1.5; INa); and Cacna1c (Cav1.2; ICa,L). In addition, the following Kv1-associated β-subunits were measured: Kcnab1 and Kcnab2. qPCR revealed that transcript levels were significantly increased for the genes Kcnh2 (+118%, P = 0.027), Kcnj2 (+59%, P = 0.014), and Kcna5 (+47%, P = 0.043), suggesting possible compensatory upregulation by K+ channel subunits (Fig. 6). In addition, the abundance of Scn5a mRNA was also significantly elevated (+63%, P = 0.007; Fig. 6), suggesting a possible increase in the contribution of depolarizing currents. Neither of the β-subunit genes exhibited significant expression differences between WT and Kcna1−/− atria (Fig. 6).
Fig. 6.
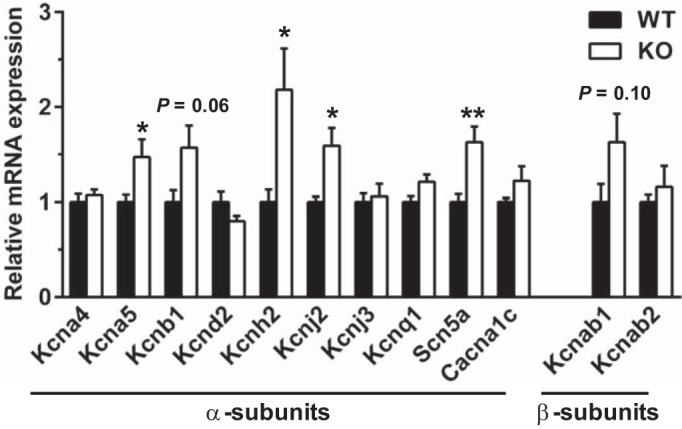
Kcna1−/− atria exhibit evidence of ion channel gene expression remodeling. qPCR was used to measure relative differences in atrial mRNA levels of the primary cardiac ion channel α-subunit genes associated with the atrial action potential, as well as the main Kv1-associated β-subunit genes (n = 6 per genotype). *P ≤ 0.05; **P ≤ 0.01 (2-tailed unpaired Student’s t-test). KO, knockout; WT, wild type.
DISCUSSION
In this study, whole cell patch-clamp recordings were used to demonstrate the first evidence of functional expression of Kv1.1 subunits in mouse atrial cardiomyocytes, as well as the electrophysiological and molecular consequences of their absence. Voltage-clamp recordings revealed that WT cells exhibit significant DTX-K-sensitive outward K+ currents, indicating a contribution by Kv1.1-containing potassium channels to atrial repolarization. In current-clamp recordings, the absence of Kv1.1 subunits due to Kcna1 gene deletion led to drastically prolonged atrial action potentials, which were exacerbated in right atria. Gene expression analysis suggests that Kv1.1 deficiency leads to complex changes in other cardiac ion channels, which could partially compensate for or exacerbate the loss of Kv1.1-mediated repolarization.
The ability of DTX-K to significantly decrease peak outward K+ currents and prolong action potentials in WT atrial cardiomyocytes, but not in Kcna1−/− cells, demonstrates the specificity of DTX-K for Kv1.1 subunits. To our knowledge, this study is the first to report the effects of any type of dendrotoxin on outward currents in mouse atrial cardiomyocytes. The dendrotoxins constitute a family of structurally similar peptides that were originally isolated from the venom of green (DTX-K and DTX-I) and black mamba snakes (α-DTX, β-DTX, δ-DTX, and γ-DTX) and found to have varying degrees of potency in blocking voltage-gated K+ channels (7, 8, 26). DTX-K is unique among the dendrotoxins for its specificity. Whereas other dendrotoxins block multiple targets, DTX-K has a high binding affinity only for Kv1.1 subunits and not for other members of the Kv1 family, including the structurally similar Kv1.2 (29, 34). The presence of a single Kv1.1 subunit in a K+ channel tetramer is sufficient for binding to DTX-K, but the presence of other partners in the tetramer reduces this affinity (34, 35). Furthermore, unlike most other dendrotoxins, DTX-K dissociates very slowly from the channel once bound, rendering its effects relatively irreversible and resistant to washout (26). DTX-K is also unique among dendrotoxins for its potency. In studies of its ability to inhibit mouse Kv1.1 currents in cell lines, DTX-K was ~20 times more potent (in terms of EC50 values) than any other dendrotoxin, including ≥300 times more potent than the commonly used α-DTX, which also blocks both Kv1.1- and Kv1.2-mediated currents with similar efficacy (7, 26). Thus, the DTX-K-sensitive outward currents described here suggest that mouse atrial myocytes possess functional Kv1.1-containing channels that contribute to cardiac repolarizing currents, the specific identity of which remains to be determined.
The electrophysiological recordings performed in this work provide additional insight into a possible role for Kv1.1 subunits in the pathomechanisms of atrial fibrillation (AF). Previously, Kcna1−/− mice were found to exhibit increased susceptibility to AF in response to intracardiac burst pacing stimulation (5). In addition, patients with chronic AF showed an increase in Kv1.1 protein levels with a corresponding increase in DTX-K-sensitive outward K+ currents in atrial myocytes (5). The two principal electrophysiological mechanisms believed to underlie AF initiation and maintenance are focal ectopic firing (i.e., afterdepolarizations or automaticity) and reentry (i.e., abnormal impulse propagation) (23). Spontaneous AF with underlying atrial APD prolongation has been described in at least six mouse models: Gαq-transgenic (TG); F1759A-Nav1.5-dTG; CREM-IbΔC-X; TG-junctate-1; TG-junctin; and Dct−/− (9–11, 16, 18, 33). Most of these mouse mutants also exhibited atrial remodeling in the form of fibrosis or enlargement which could promote reentry. However, in Dct−/− mice, the APD prolongation was associated with AF-promoting early afterdepolarizations (EADs) in the absence of obvious structural remodeling (16). In our previous work, we did not find a significant change in atrial fibrosis in Kcna1−/− hearts despite increased levels of collagen, type VI, α1 (col6a1) mRNA, which encodes a protein component of fibrotic tissue (5). Future studies will be needed to directly test whether the APD prolongation in Kcna1−/− cells increases their propensity for AF-promoting afterdepolarizations.
The significantly longer APDs in Kcna1−/− myocytes isolated from the right atria versus the left atria suggest that Kv1.1 subunits can contribute to interatrial differences in repolarization. In mice and other mammals, left atrial cells typically exhibit shorter action potentials than right atrial cells as a means to maintain synchronous firing between atria and normal right-to-left atrial electrical conduction (17, 19, 22). In contrast to previous studies, our findings showed no significant left-versus-right differences in WT APDs (19, 22). One explanation for the lack of left-right differences in WT APD in our study could be the slightly younger age of the animals used (6–8 wk) compared with the other studies, which used 8- to 10-wk-old animals (19, 22). The atrial repolarizing currents that shape the action potential have been shown to change and develop over time during postnatal development (32). In addition, mouse strain differences could also contribute to the lack of left-right differences in our study since our mice are on a Black Swiss genetic background, whereas previous studies utilized the C57BL/6 strain (19, 22). Mouse genetic background has been shown to influence the expression of cardiac ion channels, including the main K+ channel genes, which could lead to strain differences in repolarization affecting action potential morphology (4). Although the APD prolongation in Kcna1−/− mice was more pronounced in right atrial myocytes, pharmacological inhibition of Kv1.1 with DTX-K in WT cells produced a similar degree of APD prolongation in left and right atrial cells. Furthermore, the right atrial APDs in Kcna1−/− myocytes were significantly longer than the APDs in WT right atrial cells that were treated with DTX-K. Thus, the significantly longer APDs in right atrial Kcna1−/− myocytes may reflect complex changes due to the functional absence of Kv1.1 subunits, as well as expression remodeling of other channels.
Gene transcript analyses of Kcna1−/− atria identified significant changes in the mRNA levels of several ion channel genes that contribute to the atrial action potential (Kcnh2, Kcnj2, Kcna5, and Scn5a), indicating the possibility of compensatory electrical remodeling. These changes could be a direct consequence of the absence of cardiac Kv1.1 subunits or they could represent an adaptive response of the heart to repeated seizures, such as occur in Kcna1−/− mice. The increased expression of K+ channel subunits could represent a compensatory mechanism by the atria to attempt to maintain proper repolarization in the absence of Kv1.1 subunits. In contrast, the increase in Scn5a mRNA levels may signify a maladaptive response that could increase depolarizing currents promoting prolongation of the action potential. Interestingly, Kcna1−/− cells showed significantly prolonged action potentials at both the APD50 and APD90 time points, whereas in WT cells, DTX-K exposure led to APD prolongation only at APD90. This difference could be partially due to upregulation of Scn5a in Kcna1−/− cells which can increase APD during both early and late phases of the action potential (1). The existence of seizures in Kcna1−/− mice adds another layer of complexity in trying to understand the cause of channel expression remodeling in the atria of these animals. In rats, genetic and chemically induced epilepsy leads to changes (increases and decreases) in cardiac ion channel expression at the mRNA and/or protein levels, as has been observed for the genes Kcnd2 (decrease), Kcnd3 (decrease), Hcn2 (decrease), Hcn4 (decrease), Scn5a (increase), and Scn1a (increase) (1, 2, 15, 27). Of note, Kcnd2 expression decreased by 20% in Kcna1−/− atria (P = 0.14), similar to changes in the rat epilepsy models (2, 15). However, one significant caveat is that most of the channel expression remodeling studies in rat epilepsy models have been restricted to the ventricles rather than the atria. In addition, an important limitation of our study is that expression analysis was confined to mRNA levels, which do not always correlate with protein abundance.
In summary, this work identifies a novel cardiac role for Kv1.1 channels in mediating atrial repolarization, thereby adding a new member to the already diverse collection of known K+ channels in the heart. In mice, Kv1.1 expression predominates in the atria compared with the ventricles (5). Therefore, assuming a similar expression pattern in humans, drug targeting of Kv1.1 channels could represent a novel atrial-selective clinical therapeutic strategy for control of atrial fibrillation that would potentially avoid severe negative effects on ventricular repolarization; however, care would have to be taken not to provoke seizures (28). The identification of Kv1.1-mediated currents in humans and now mice shows that Kv1.1 subunits make a previously unexpected but significant contribution to intrinsic cardiac function, especially in the atria, and that the Kcna1 gene should be included in molecular genetic screens for arrhythmogenic mutations in patients.
GRANTS
This study was supported by National Institutes of Health Grants R00 HL-107641, R01 NS-100954, and R01 NS-099188 (to E. Glasscock) and by Malcolm Feist Postdoctoral (to M. Si) and Predoctoral (to K. Trosclair) Fellowships from the Center for Cardiovascular Diseases and Stroke at the Louisiana State University Health Sciences Center-Shreveport.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
E.G. conceived and designed research; M.S. and K.T. performed experiments; M.S., K.T., and K.A.H. analyzed data; M.S., K.T., K.A.H., and E.G. interpreted results of experiments; M.S., K.T., and E.G. prepared figures; M.S., K.T., and E.G. drafted manuscript; M.S., K.T., K.A.H., and E.G. edited and revised manuscript; M.S., K.T., K.A.H., and E.G. approved final version of manuscript.
REFERENCES
- 1.Biet M, Morin N, Lessard-Beaudoin M, Graham RK, Duss S, Gagné J, Sanon NT, Carmant L, Dumaine R. Prolongation of action potential duration and QT interval during epilepsy linked to increased contribution of neuronal sodium channels to cardiac late Na+ current: potential mechanism for sudden death in epilepsy. Circ Arrhythm Electrophysiol 8: 912–920, 2015. doi: 10.1161/CIRCEP.114.002693. [DOI] [PubMed] [Google Scholar]
- 2.Brewster AL, Marzec K, Hairston A, Ho M, Anderson AE, Lai YC. Early cardiac electrographic and molecular remodeling in a model of status epilepticus and acquired epilepsy. Epilepsia 57: 1907–1915, 2016. doi: 10.1111/epi.13516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Browne DL, Gancher ST, Nutt JG, Brunt ER, Smith EA, Kramer P, Litt M. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet 8: 136–140, 1994. doi: 10.1038/ng1094-136. [DOI] [PubMed] [Google Scholar]
- 4.Demolombe S, Marionneau C, Le Bouter S, Charpentier F, Escande D. Functional genomics of cardiac ion channel genes. Cardiovasc Res 67: 438–447, 2005. doi: 10.1016/j.cardiores.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 5.Glasscock E, Voigt N, McCauley MD, Sun Q, Li N, Chiang DY, Zhou XB, Molina CE, Thomas D, Schmidt C, Skapura DG, Noebels JL, Dobrev D, Wehrens XH. Expression and function of Kv1.1 potassium channels in human atria from patients with atrial fibrillation. Basic Res Cardiol 110: 505, 2015. doi: 10.1007/s00395-015-0505-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glasscock E, Yoo JW, Chen TT, Klassen TL, Noebels JL. Kv1.1 potassium channel deficiency reveals brain-driven cardiac dysfunction as a candidate mechanism for sudden unexplained death in epilepsy. J Neurosci 30: 5167–5175, 2010. doi: 10.1523/JNEUROSCI.5591-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grissmer S, Nguyen AN, Aiyar J, Hanson DC, Mather RJ, Gutman GA, Karmilowicz MJ, Auperin DD, Chandy KG. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol Pharmacol 45: 1227–1234, 1994. [PubMed] [Google Scholar]
- 8.Harvey AL. Twenty years of dendrotoxins. Toxicon 39: 15–26, 2001. doi: 10.1016/S0041-0101(00)00162-8. [DOI] [PubMed] [Google Scholar]
- 9.Hirose M, Takeishi Y, Niizeki T, Shimojo H, Nakada T, Kubota I, Nakayama J, Mende U, Yamada M. Diacylglycerol kinase ζ inhibits Gαq-induced atrial remodeling in transgenic mice. Heart Rhythm 6: 78–84, 2009. doi: 10.1016/j.hrthm.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 10.Hong CS, Cho MC, Kwak YG, Song CH, Lee YH, Lim JS, Kwon YK, Chae SW, Kim DH. Cardiac remodeling and atrial fibrillation in transgenic mice overexpressing junctin. FASEB J 16: 1310–1312, 2002. doi: 10.1096/fj.01-0908fje. [DOI] [PubMed] [Google Scholar]
- 11.Hong CS, Kwon SJ, Cho MC, Kwak YG, Ha KC, Hong B, Li H, Chae SW, Chai OH, Song CH, Li Y, Kim JC, Woo SH, Lee SY, Lee CO, Kim DH. Overexpression of junctate induces cardiac hypertrophy and arrhythmia via altered calcium handling. J Mol Cell Cardiol 44: 672–682, 2008. doi: 10.1016/j.yjmcc.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 12.Jan LY, Jan YN. Voltage-gated potassium channels and the diversity of electrical signalling. J Physiol 590: 2591–2599, 2012. doi: 10.1113/jphysiol.2011.224212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kamb A, Iverson LE, Tanouye MA. Molecular characterization of Shaker, a Drosophila gene that encodes a potassium channel. Cell 50: 405–413, 1987. doi: 10.1016/0092-8674(87)90494-6. [DOI] [PubMed] [Google Scholar]
- 14.Kinali M, Jungbluth H, Eunson LH, Sewry CA, Manzur AY, Mercuri E, Hanna MG, Muntoni F. Expanding the phenotype of potassium channelopathy: severe neuromyotonia and skeletal deformities without prominent episodic ataxia. Neuromuscul Disord 14: 689–693, 2004. doi: 10.1016/j.nmd.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 15.Lai YC, Li N, Lawrence W, Wang S, Levine A, Burchhardt DM, Pautler RG, Valderrábano M, Wehrens XH, Anderson AE. Myocardial remodeling and susceptibility to ventricular tachycardia in a model of chronic epilepsy. Epilepsia Open 3: 213–223, 2018. doi: 10.1002/epi4.12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levin MD, Lu MM, Petrenko NB, Hawkins BJ, Gupta TH, Lang D, Buckley PT, Jochems J, Liu F, Spurney CF, Yuan LJ, Jacobson JT, Brown CB, Huang L, Beermann F, Margulies KB, Madesh M, Eberwine JH, Epstein JA, Patel VV. Melanocyte-like cells in the heart and pulmonary veins contribute to atrial arrhythmia triggers. J Clin Invest 119: 3420–3436, 2009. doi: 10.1172/JCI39109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li D, Zhang L, Kneller J, Nattel S. Potential ionic mechanism for repolarization differences between canine right and left atrium. Circ Res 88: 1168–1175, 2001. doi: 10.1161/hh1101.091266. [DOI] [PubMed] [Google Scholar]
- 18.Li N, Chiang DY, Wang S, Wang Q, Sun L, Voigt N, Respress JL, Ather S, Skapura DG, Jordan VK, Horrigan FT, Schmitz W, Müller FU, Valderrabano M, Nattel S, Dobrev D, Wehrens XH. Ryanodine receptor-mediated calcium leak drives progressive development of an atrial fibrillation substrate in a transgenic mouse model. Circulation 129: 1276–1285, 2014. doi: 10.1161/CIRCULATIONAHA.113.006611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lomax AE, Kondo CS, Giles WR. Comparison of time- and voltage-dependent K+ currents in myocytes from left and right atria of adult mice. Am J Physiol Heart Circ Physiol 285: H1837–H1848, 2003. doi: 10.1152/ajpheart.00386.2003. [DOI] [PubMed] [Google Scholar]
- 20.Mishra V, Karumuri BK, Gautier NM, Liu R, Hutson TN, Vanhoof-Villalba SL, Vlachos I, Iasemidis L, Glasscock E. Scn2a deletion improves survival and brain-heart dynamics in the Kcna1-null mouse model of sudden unexpected death in epilepsy (SUDEP). Hum Mol Genet 26: 2091–2103, 2017. doi: 10.1093/hmg/ddx104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moore BM, Jerry Jou C, Tatalovic M, Kaufman ES, Kline DD, Kunze DL. The Kv1.1 null mouse, a model of sudden unexpected death in epilepsy (SUDEP). Epilepsia 55: 1808–1816, 2014. doi: 10.1111/epi.12793. [DOI] [PubMed] [Google Scholar]
- 22.Nakamura H, Ding W-G, Sanada M, Maeda K, Kawai H, Maegawa H, Matsuura H. Presence and functional role of the rapidly activating delayed rectifier K+ current in left and right atria of adult mice. Eur J Pharmacol 649: 14–22, 2010. doi: 10.1016/j.ejphar.2010.08.025. [DOI] [PubMed] [Google Scholar]
- 23.Nattel S, Dobrev D. Electrophysiological and molecular mechanisms of paroxysmal atrial fibrillation. Nat Rev Cardiol 13: 575–590, 2016. doi: 10.1038/nrcardio.2016.118. [DOI] [PubMed] [Google Scholar]
- 24.Nerbonne JM. Molecular basis of functional myocardial potassium channel diversity. Card Electrophysiol Clin 8: 257–273, 2016. doi: 10.1016/j.ccep.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev 85: 1205–1253, 2005. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- 26.Owen DG, Hall A, Stephens G, Stow J, Robertson B. The relative potencies of dendrotoxins as blockers of the cloned voltage-gated K+ channel, mKv1.1 (MK-1), when stably expressed in Chinese hamster ovary cells. Br J Pharmacol 120: 1029–1034, 1997. doi: 10.1038/sj.bjp.0701004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Powell KL, Jones NC, Kennard JT, Ng C, Urmaliya V, Lau S, Tran A, Zheng T, Ozturk E, Dezsi G, Megatia I, Delbridge LM, Pinault D, Reid CA, White PJ, O’Brien TJ. HCN channelopathy and cardiac electrophysiologic dysfunction in genetic and acquired rat epilepsy models. Epilepsia 55: 609–620, 2014. doi: 10.1111/epi.12563. [DOI] [PubMed] [Google Scholar]
- 28.Ravens U, Odening KE. Atrial fibrillation: therapeutic potential of atrial K+ channel blockers. Pharmacol Ther 176: 13–21, 2017. doi: 10.1016/j.pharmthera.2016.10.003. [DOI] [PubMed] [Google Scholar]
- 29.Robertson B, Owen D, Stow J, Butler C, Newland C. Novel effects of dendrotoxin homologues on subtypes of mammalian Kv1 potassium channels expressed in Xenopus oocytes. FEBS Lett 383: 26–30, 1996. doi: 10.1016/0014-5793(96)00211-6. [DOI] [PubMed] [Google Scholar]
- 30.Smart SL, Lopantsev V, Zhang CL, Robbins CA, Wang H, Chiu SY, Schwartzkroin PA, Messing A, Tempel BL. Deletion of the KV1.1 potassium channel causes epilepsy in mice. Neuron 20: 809–819, 1998. doi: 10.1016/S0896-6273(00)81018-1. [DOI] [PubMed] [Google Scholar]
- 31.Tempel BL, Jan YN, Jan LY. Cloning of a probable potassium channel gene from mouse brain. Nature 332: 837–839, 1988. doi: 10.1038/332837a0. [DOI] [PubMed] [Google Scholar]
- 32.Trépanier-Boulay V, Lupien MA, St-Michel C, Fiset C. Postnatal development of atrial repolarization in the mouse. Cardiovasc Res 64: 84–93, 2004. doi: 10.1016/j.cardiores.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 33.Wan E, Abrams J, Weinberg RL, Katchman AN, Bayne J, Zakharov SI, Yang L, Morrow JP, Garan H, Marx SO. Aberrant sodium influx causes cardiomyopathy and atrial fibrillation in mice. J Clin Invest 126: 112–122, 2016. doi: 10.1172/JCI84669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang FC, Bell N, Reid P, Smith LA, McIntosh P, Robertson B, Dolly JO. Identification of residues in dendrotoxin K responsible for its discrimination between neuronal K+ channels containing Kv1.1 and 1.2 alpha subunits. Eur J Biochem 263: 222–229, 1999. doi: 10.1046/j.1432-1327.1999.00494.x. [DOI] [PubMed] [Google Scholar]
- 35.Wang FC, Parcej DN, Dolly JO. α Subunit compositions of Kv1.1-containing K+ channel subtypes fractionated from rat brain using dendrotoxins. Eur J Biochem 263: 230–237, 1999. doi: 10.1046/j.1432-1327.1999.00493.x. [DOI] [PubMed] [Google Scholar]
- 36.Zuberi SM, Eunson LH, Spauschus A, De Silva R, Tolmie J, Wood NW, McWilliam RC, Stephenson JB, Kullmann DM, Hanna MG. A novel mutation in the human voltage-gated potassium channel gene (Kv1.1) associates with episodic ataxia type 1 and sometimes with partial epilepsy. Brain 122: 817–825, 1999. doi: 10.1093/brain/122.5.817. [DOI] [PubMed] [Google Scholar]