

Abstract
Bpifa1 (BPI fold-containing group A member 1) is an airway host-protective protein with immunomodulatory properties that binds to LPS and is regulated by infectious and inflammatory signals. Differential expression of Bpifa1 has been widely reported in lung disease, yet the biological significance of this observation is unclear. We sought to understand the role of Bpifa1 fluctuations in modulating lung inflammation. We treated wild-type (WT) and Bpifa1−/− mice with intranasal LPS and performed immunological and transcriptomic analyses of lung tissue to determine the immune effects of Bpifa1 deficiency. We show that neutrophil (polymorphonuclear cells, PMNs) lung recruitment and transmigration to the airways in response to LPS is impaired in Bpifa1−/− mice. Transcriptomic analysis revealed a signature of 379 genes that differentiated Bpifa1−/− from WT mice. During acute lung inflammation, the most downregulated genes in Bpifa1−/− mice were Cxcl9 and Cxcl10. Bpifa1−/− mice had lower bronchoalveolar lavage concentrations of C-X-C motif chemokine ligand 10 (Cxcl10) and Cxcl9, interferon-inducible PMN chemokines. This was consistent with lower expression of IFNγ, IFNλ, downstream IFN-stimulated genes, and IFN-regulatory factors, which are important for the innate immune response. Administration of Cxcl10 before LPS treatment restored the inflammatory response in Bpifa1−/− mice. Our results identify a novel role for Bpifa1 in the regulation of Cxcl10-mediated PMN recruitment to the lungs via IFNγ and -λ signaling during acute inflammation.
INTRODUCTION
The airway epithelium plays an important role in the innate immune response to pathogens. This complex response includes the expression of cytokines and chemokines, antimicrobial proteins, and immune mediators and epithelial interactions with inflammatory cells. Bpifa1 (BPI fold-containing group A member 1), is an airway-secreted protein that regulates multiple aspects of epithelial host defense through its antimicrobial, ion transport, and immunomodulatory properties (16).
Bpifa1, also known as Splunc1 (short palate lung and nasal epithelium clone 1), is a member of the bactericidal/permeability-increasing protein fold-containing (Bpif) superfamily, a heterogeneous group of proteins sharing the structure of bactericidal/permeability-increasing protein (BPI), a lipopolysaccharide (LPS)-binding protein with antimicrobial and immunomodulatory properties (1, 4, 7, 10, 13, 19). Genomic and proteomic studies have reported differential expression of Bpifa1 in lung disease, yet the biological significance of this observation remains unclear (15, 28, 31, 48, 49).
Bpifa1 is expressed primarily in the respiratory tract, a site of high exposure to environmental pathogens and irritants (9, 17, 22). Its distribution and abundance make it readily available to serve as an antimicrobial and early regulator of innate immune responses. Bpifa1 is known to inhibit bacterial and viral proliferation, regulate ion transport, and work as a surfactant (1, 2, 12, 16, 27, 50). These functions act in synergy to promote mucociliary clearance and maintain airway epithelial integrity. Furthermore, Bpifa1 modulates inflammation in models of gram-negative infection and noninfectious airway inflammation (3, 29, 30, 37, 56, 57). Like other members of the BPIF family, its binding to LPS may be one mechanism by which Bpifa1 exerts its immunomodulatory role (11, 42, 54).
Our previous studies focused on Bpifa1 fluctuations in response to infection and inflammation. Under basal conditions, Bpifa1 is highly concentrated in the respiratory tract and rapidly decreases in response to pathogen-associated molecular patterns (PAMP) and cytokines. In addition to infectious stimuli, we identified interferon-γ (IFNγ) as a rapid and potent inhibitor of Bpifa1 expression (17). In this previous work, we proposed that decreased Bpifa1 during infection serves as a sensor of pathogen exposure that regulates local immunity to maintain homeostasis.
Two patterns of immunomodulatory effects have been described in models of Bpifa1 dysregulation. In the absence of Bpifa1, Bpifa1−/− mice infected with gram-negative bacteria had increased inflammation, possibly due to the absence of antimicrobial effects of Bpifa1, leading to increased pathogen burden (20, 21, 26, 32, 37–39, 47, 56). However, in a noninfectious model using inhaled carbon nanotubes as an irritant, mice that overexpressed Bpifa1 had increased inflammation (23, 51). Thus, in one model Bpifa1 promoted inflammation, and in another it protected against it. Here, we sought to understand the role of Bpifa1 in modulating acute lung inflammation triggered by LPS derived from Pseudomonas aeruginosa, rather than P. aeruginosa itself, therefore excluding contributions from active bacterial growth. These studies show that Bpifa1 serves an important function in regulating neutrophilic responses to gram-negative bacterial products and suggest that Bpifa1 is important to IFN-inducible responses to pathogens in the airways.
METHODS
Mice, LPS model of lung inflammation and Cxcl10 reconstitution.
C57BL/6 Bpifa1−/− mice were generated from C57BL/6 ES cells containing a gene-ablating deletion of the Bpifa1 gene in chromosome 2. Six- to twelve-week-old Bpifa1−/− mice and age- and sex-matched wild-type (WT) C57BL/6 littermate controls were used in all experiments. Mice received LPS from P. aeruginosa, strain PAO1, administered intranasally (5 μg per mouse; Sigma-Aldrich, St. Louis, MO). Animals were euthanized at time points as indicated. For experiments reconstituting C-X-C motif chemokine ligand 10 (Cxcl10), we administered recombinant mouse Cxcl10 intraperitoneally (0.6 μg per mouse; BioLegend, San Diego, CA) 30 min before intranasal LPS. All animal experiments were performed according to guidelines of and approved by Yale Animal Care and Use Committee.
Assessment of lung inflammation.
Bronchoalveolar lavage (BAL) was performed by cannulation of the trachea and instillation of 1 ml of PBS. BAL was collected for ELISA and measurement of inflammatory mediators. Cells were counted as previously described (43). To count lung leukocytes, we performed lung tissue digestion with collagenase type IV at 150 U/ml (Worthington Biochemical, Lakewood, NJ) and DNase at 10 U/ml (Sigma-Aldrich) for 1 h at 37°C and passed tissue through a wire mesh to dissociate the cells. Cytospin preparations of BAL and lung cells were stained with Diff-Quik (Baxter Healthcare, Deerfield, IL), and differentials were performed on 200 cells on the basis of morphology and staining characteristics.
Lung histology, immunohistochemistry, and immunofluorescence.
Lung tissue was dissected, fixed in 4% PFA, and incubated overnight at 4°C. The next day, tissue was washed twice with PBS and placed in 70% ethanol before paraffin embedding. Lung tissue sections were deparaffinized with xylene, rehydrated with graded alcohol solutions, and washed with deionized water. A hematoxylin-eosin stain was used to assess overall inflammation and the distribution of lung infiltrating cells (peribronchial and alveolar) in 10 random fields at ×20 magnification for each sample. The number of cells in alveolar or peribronchial distribution in each field was quantified using an Inflammatory Infiltration Histology Score (modified from Dubin et al.) (6, 24). To detect differences in inducible expression of Cxcl10 by infiltrating leukocytes and other lung cell types during inflammation, we used immunohistochemistry (IHC) and immunofluorescence (IF) stains, colocalizing Cxcl10 protein and specific cell/tissue markers. Heat-induced antigen retrieval with target retrieval solution (pH 6.0; Dako, Carpinteria, CA) was performed. Sections were blocked with serum-free protein blocking buffer (Dako) and incubated with the following primary antibodies overnight at 4°C: 1) F4:80 rat anti mouse (11-4801-81; Invitrogen, Carlsbad, CA), 2) Ly6G rat anti-mouse (MCA-771EL; Bio-Rad, Hercules, CA), 3) Cxcl10 rabbit anti-mouse (701225; ThermoFisher, Waltham, MA), 4) pan-cytokeratin rabbit anti-cow (Dako), and 5) von Willebrand factor rabbit anti-human (A0082, Dako). For IHC, samples were washed by immersion in PBS and incubated with the following secondary antibodies: 1) alkaline phosphatase-conjugated goat anti-rabbit (GR602 H; Biocare Medical, Pacheco, CA), 2) horseradish peroxidase (HRP)-conjugated goat anti-rabbit (K3467, Dako), and 3) alkaline phosphatase-conjugated goat anti-rat (GAP515, Biocare Medical). For IF, samples were washed by immersion in PBS and incubated with the following secondary antibodies: Dylight 549 anti-rabbit (DI-1549; Vector Laboratories, Burlingame, CA), Alexa fluor 488 conjugate anti-rat (4416; Cell Signaling Tech, Danvers, MA). Finally, samples were washed again by immersion in PBS and mounted with Prolong Gold mounting medium with DAPI (Life Technologies, Carlsbad, CA). For IHC staining, after chromogen was applied, slides were counterstained with hematoxylin and mounted with mounting medium. Rabbit IgG (ThermoFisher Scientific) was used as an isotype control. The entire lung was surveyed for each mouse, and four to six images were taken from each lung. Images were acquired with Nikon Eclipse Ti (IF) and Nikon Eclipse Ni (IHC) microscopes (Nikon, Tokyo, Japan).
RNA extraction, real-time PCR, and analysis of microarray data.
RNA was isolated from whole lung tissue homogenates (TRIzol, Invitrogen) and used as a template to generate cDNA (iScript kit, Bio-Rad). RNA quantity was determined by NanoDrop at 260 nm. Labeling was performed using the Agilent Technologies Low Input Quick Amp Labeling kit (Agilent Technologies, Santa Clara, CA). After purification and fragmentation, aliquots of each sample were hybridized to SurePrint G3 Mouse Gene Expression v.2 8 × 60K microarrays (Agilent Technologies). Each array was sequentially washed and scanned with an Agilent Technologies microarray scanner. Arrays were visually inspected for hybridization defects, and quality control procedures were applied. Intensity information from captured array images and the annotation information from the microarray experiments were determined using Agilent Feature Extraction 12.0.0 software. The Agilent microarray processed signals were normalized using Quantile normalization as previously described (5, 14). Interquartile normalization was applied to normalize the gene expression signals by BRB-ArrayTools v.4.5.0 (https://brb.nci.nih.gov/BRB-ArrayTools/). We selected only transcripts with annotated Entrez ID. Given that gene expression microarrays contain multiple replicated probes for the same transcript, we selected the transcripts with the highest interquartile range (IQR) variation. Data were visualized by generating heat maps with Java TreeView. The microarray data were deposited at the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=yvctemmwrtyttgj&acc=GSE84901) under accession number GSE84901. Microarray data analyses were performed using GeneSpring (Agilent Technologies). A multivariate permutation t-test (1,000 permutations) for unpaired samples was used to identify differentially expressed transcripts between WT and Bpifa1−/− mice at baseline. A two-way ANOVA was used to identify gene expression differences in WT vs. Bpifa1−/− mice after differences in expression changes were accounted for over time at 0, 8, and 24 h. Results of the two-way ANOVA were corrected for multiple hypothesis testing using the Benjamini & Hochberg false discovery rate (FDR) approach. This correction works by ranking the P value of each gene from smallest to largest, multiplying the rank number by the false discovery rate selected, and then dividing by the total number of tests performed (8). If the value obtained is less than 0.05, it is considered statistically significant. This is done sequentially for every value on the rank list.
Pathway enrichment analysis was performed using GeneSpring. Statistical significance was defined as a FDR < 0.05, and we focused on genes with a statistically significant fold change (FC) greater than 1.5. Changes in mRNA expression are represented as FC in Bpifa1−/− mice relative to WT. Pathway analysis was performed with Ingenuity Pathway Analysis software (IPA, Ingenuity Systems, Redwood City, CA). The IPA software package (http://www.ingenuity.com) uses a curated database containing gene expression and gene annotation and information from multiple sources (52). IPA calculates single P values for the enrichment of each gene category by using Fisher’s exact test, taking into consideration the total number of molecules from the analyzed data set and the total number of molecules linked to the same gene. The software generates a Z-score that represents an approximate interaction between the focus molecule and each network (34, 46, 52).
For real-time PCR, RNA expression was measured by quantitative PCR reaction (SYBR FAST; KAPA Biosystems, Woburn, MA). Murine PCR primers used for amplification are detailed in Table 1. For each set of primers, validation experiments showed a linear dependency of threshold cycle values at different RNA concentrations. The data were analyzed after mouse genes were normalized to gapdh or tubbulin-5 (tubb5). Statistical analyses were performed using SAS v. 9.4 (SAS, Cary, NC), GraphPad Prism v. 7.0 (GraphPad Software, La Jolla, CA), and Data Assist software, v. 2.0 (Applied Biosystems, Carlsbad, CA). Changes in mRNA expression are represented as fold change (FC) in Bpifa1−/− relative to WT mice.
Table 1.
Real-time PCR mouse primers used to assess gene expression in this study
| Gene | Forward | Reverse |
|---|---|---|
| Bpifa1 | GTCCACCCTTGCCACTGAACCA | CACCGCTGAGAGCATCTGTGAA |
| Tubb-5 | CGGTGCTAAGTTCTGGGAGGTGATA | TGAGCGAACGGAGTCCATAGTC |
| Ifna2 | GATGAGGAGGCTCCCCTTTCT | GATGGCTTGAGCCTTCTGGAT |
| Ifnb1 | AGCTCCAAGAAAGGACGAACA | GCCCTGTAGGTGAGGTTGAT |
| Ifng | ATGAACGCTACACACTGCATC | CCATCCTTTTGCCAGTTCCTC |
| Ifnl2/3 | ACCCTGAAGGTCTGGGAGAA | AAGCTGTGTACAGGTCTGC |
| Cxcl10 | TCCATATCGATGACGGGCCA | TTCATCGTGGCAATGATCTCAAC |
| Cxcl9 | TGCGACTTCACTCCAACACAG | AGGGTTCCTCGAACTCCACAC |
| Irf7 | TGCTCTGCCCACACAGGTTC | GGTTCCTCGTAAACACGGTCT |
| Oas1g | CCTTTGATGTCCTGGGTCATGG | CAGATGAGGATGGTGTAGATTAAGG |
| Ccl12 | ATTTCCACACTTCTATGCCTCCT | ATCCAGTATGGTCCTGAAGATCA |
| Ccl7 | TCCCTGGGAAGCTGTTATCTTCAA | AAGGCTTTGGAGTTGGGGTT |
| Mx1 | GACCATAGGGGTCTTGACCAA | AGACTTGCTCTTTCTGAAAAGCC |
| Cxcl1 | CCGAAGTCATAGCCACACTC | TCCGTTACTTGGGGACACCT |
| Tnfa | TAGCCCACGTCGTAGCAAAC | ACAAGGTACAACCCATCGGC |
| Ccl2 | CACTCACCTGCTGCTACTCA | GCTTGGTGACAAAAACTACAGC |
| Il6 | CCGGAGAGGAGACTTCACAG | TTGCCATTGCACAACTCTTTT |
| Ifitm3 | CCCCCAAACTACGAAAGAATCA | ACCATCTTCCGATCCCTAGAC |
| Isg15 | GGTGTCCGTGACTAACTCCAT | TGGAAAGGGTAAGACCGTCCT |
Bpifa1 ELISA and quantitative analysis of cytokines, chemokines, and leukotriene B4 in BAL.
Plates were coated with BAL, followed by polyclonal sheep anti-mouse Bpifa1 antibody (R&D, Minneapolis, MN), followed by anti-sheep HRP (Millipore, Billerica, MA). Reactions were developed as previously described (17) and were measured at optical densities of 450 and 550 nm. Total protein (BCA protein assay; Abcam, Cambridge, MA) and LDH activity (colorimetric assay; Thermo Scientific, Rockford, IL) were quantified in BAL according to the manufacturer’s specifications. Leukotriene B4 was measured in BAL by ELISA (Enzo Life Sciences, Farmingdale, NY). Cytokines and chemokines in BAL fluid were assessed using a 25-multiplexed Luminex assay (Milliplex MAP Mouse Cytokine/Chemokine Kit, Millipore) and read on a Bio-plex 200 system.
Neutrophil isolation and chemotaxis assays.
Heparinized mouse blood was obtained by flushing bone marrow from the long bones of WT and Bpifa1−/− mice. Polymorphonuclear cells (PMN) were isolated by negative selection (EasySep Mouse Neutrophil Enrichment Kit, StemCell Technologies, Vancouver, BC, Canada). Chemotaxis assays were performed using a modified Boyden chamber technique (ECM515, Millipore). A solution containing 105 PMN in 100 μl was placed in the upper chamber, and PMN were stimulated with keratinocyte chemoattractant (KC; 50 ng/ml, Peprotech), N-formyl-methionyl-leucyl-phenylalanine (fMLP; 105 M, Sigma-Aldrich), and recombinant mouse Cxcl9 (50 ng/ml, R&D Systems) and Cxcl10 (100 ng/ml, BioLegend). Cells were lysed and collected, and cell migration was estimated based on relative fluorescence units (RFU) after 2 h at 37°C in a 5% CO2 incubator.
Statistical analysis.
Data were analyzed using GraphPad Prism v. 7.0 and SAS v. 9.4. Results are reported as mean ± SE unless otherwise specified. Statistical significance was determined using the Mann-Whitney test for data with a nonnormal distribution. In experiments with repeated measures and multiple time points, we used a generalized linear mixed models (GLIMMIX) procedure in SAS statistical software. This approach fitted statistical models to data with nonconstant variability and with nonnormal distributions while accounting for repeated measures of the same sample, variability within samples, and multiple comparisons. Statistical significance was defined as a P value <0.05.
RESULTS
Bpifa1 deficiency impairs airway neutrophilic inflammation in response to LPS.
We administered LPS intranasally to Bpifa1−/− and WT control mice to characterize the immunomodulatory effects of Bpifa1. BAL concentrations of Bpifa1 decreased in response to LPS and reached nadir at 24 h (Fig. 1A, gray triangular markers). Neutrophil counts were significantly lower in the airways and lungs of Bpifa1−/− compared with WT mice after LPS administration (Fig. 1, A and B). Bpifa1−/− mice had 31% fewer airway PMN at 8 h (3.62 vs. 6.5 × 105/ml, P = 0.0455) and 66% fewer airway PMN at 24 h (Fig. 1A, 7.65 vs. 22.42 × 105/ml, P = 0.0317). In whole lungs, PMN counts were also lower in Bpifa1−/− compared with WT mice at 8 and 24 h (Fig. 1B; 8 h, 2.98 vs. 4.92 × 106/ml, 39% reduction, P = 0.03; 24 h, 4.87 vs. 8.11 × 106/ml, 32% reduction, P = 0.009). In addition to PMN differences, BAL macrophage counts were modestly increased in Bpifa1−/− mice at 24 h (Fig. 1C). Although the total number of macrophages increased throughout the experiment in WT and Bpifa1−/− mice, there were no differences in lymphocyte or eosinophil counts throughout the experiment (Fig. 1, C and D). There were no significant differences in PMN counts in the lungs or airways of untreated Bpifa1−/− and WT mice. These data show that, in Bpifa1−/− mice, the response to LPS is characterized by reduced airway and lung PMN infiltration at 8 and 24 h. These findings suggest that the role of Bpifa1 in LPS-induced inflammation involves promoting PMN accumulation in the lungs.
Fig. 1.

BPI fold-containing group A member 1-deficient (Bpifa1−/−) mice have impaired lung neutrophilic inflammation after lipopolysaccharide (LPS). A: neutrophils (black lines) in bronchoalvolar lavage (BAL) fluid from Bpifa1+/+ [wild-type (WT), ●], Bpifa1−/− mice (○), and Bpifa1 protein in BAL (▲) after intranasal LPS (5 μg/mouse, from Pseudomonas aeruginosa). B: neutrophils in lung digests after intranasal LPS. C: total BAL cell counts in WT (●) and Bpifa1−/− mice (○). D: total lung cell counts in WT and Bpifa1−/− mice. O.D., optical density; PMN, polymorphonuclear neutrophils. Mann-Whitney test: *P < 0.05. Horizontal lines indicate mean values. Data represent 3 experiments; n = 4–7 mice/group.
Bpifa1-deficient mice have limited peribronchial and alveolar inflammation in response to LPS.
To determine whether impaired PMN inflammation was associated with a specific histological distribution within the lungs, we assessed the distribution of inflammatory cells in the peribronchial and alveolar compartments after LPS (6, 24). This analysis demonstrated a significant decrease in inflammatory infiltration in the alveolar and peribronchial compartments of Bpifa1−/− mice relative to WT controls (Fig. 2, A–C). These findings suggested that Bpifa1 controls inflammation by modulating PMN ability to infiltrate the peribronchial and alveolar compartments.
Fig. 2.

BPI fold-containing group A member 1-deficient (Bpifa1−/−) mice have limited peribronchial and alveolar inflammation in response to lipopolysaccharide (LPS). A: tissue sections of paraffin-embedded lung tissue stained with hematoxylin-eosin from wild-type (WT; left) and Bpifa1−/− mice (right) after treatment with LPS (LPS 8 h, LPS 24 h) or PBS (Control). Images obtained with optical microscope at ×20 magnification. B: assessment of peribronchial inflammation in WT (●) and Bpifa1−/− mice (○) based on histology score. C: assessment of alveolar inflammation in WT and Bpifa1−/− mice based on histology score. Each data point represents the mean inflammation score of 10 random fields scored within the same section. Black horizontal lines indicate mean values. Generalized linear mixed model: *P < 0.05. Data represent 2 experiments; n = 4–7 mice/group.
Bpifa1 regulates expression of IFN-driven pathways.
To identify mechanisms driving impaired PMN accumulation in the lungs of Bpifa1−/− mice, we analyzed gene expression microarrays in LPS-treated mice at 0, 8, and 24 h and found differential expression of 379 genes (Fig. 3, A and B). Cxcl10, Cxcl9, and IFN-regulatory factor 7 (Irf7) were the lowest expressed genes at 8 and 24 h in LPS-treated Bpifa1−/− compared with WT mice (Tables 2 and 4). Cxcr3, a receptor for these chemokines, was also reduced at all time points. Many genes involved in IFNγ signaling pathways that affect PMN recruitment and function were also reduced (Tables 2 and 4). Pathway analysis showed that “IFN signaling” and “activation of IRFs by pathogen recognition receptors (PRRs)” were the most strongly impaired pathways in Bpifa1−/− mice (Fig. 3C). Our gene expression analysis also highlighted decreased expression of genes important to granulocyte adhesion and diapedesis, including C-X-C motif receptor 3 (cxcr3), integrin β2-like (Itgb2l), cadherin 2 (cdh2), lymphocyte selectin (sell), and platelet-ligand selectin (selplg) (Table 2). These genes may further contribute to abnormal adhesion, accumulation, and transmigration of inflammatory cells in our model. In untreated Bpifa1−/− compared with WT mice, there were significant differences in 10 genes (Table 3). The most strongly downregulated genes included schlafen 4 (slfn4, an IFNγ-inducible molecule that disrupts myelopoiesis), plasminogen activator urokinase (plau, involved in thrombolysis and extracellular matrix degradation), and Z-DNA-binding protein-1 (zbp1, a protein that stimulates type I and -II IFN and IRF expression in response to viral DNA). Consistent with these findings, our ANOVA also uncovered significant impairments in the expression of the downstream IFN-stimulated gene (ISG) response (Table 4). In summary, we identified broad defects in IFN-inducible genes that regulate host defense and cell recruitment, adhesion, and transmigration to the lungs.
Fig. 3.

Gene expression profile of BPI fold-containing group A member 1-deficient (Bpifa1−/−) and wild-type (WT) mice during acute lung inflammation. A: whole lung microarray heatmap showing differential gene expression in WT (Bpifa1+/+) and Bpifa1−/− mice after intranasal instillation of lipopolysaccharide (LPS) [2-way ANOVA, multiple hypothesis testing corrected using Benjamini-Hochberg false discovery rate (FDR) set at <0.05]. Adjacent color scale in log base-2 scale. Yellow denotes increase over the geometric mean of samples; purple denotes decrease. B: microarray heatmap showing the top 20 most up/downregulated genes in Bpifa1−/− mice after intranasal instillation of LPS (2-way ANOVA, multiple-hypothesis testing corrected using Benjamini-Hochberg FDR set at <0.05). Adjacent color scale in log base-2 scale: yellow denotes increase over the geometric mean of samples; purple denotes decrease relative to WT expression. C: activated canonical pathways based on gene expression profile. IRF, interferon regulatory factors. Z-scores (bars) represent the strength of the association between the regulated genes within the canonical pathway. Negative Z-score represents statistically significant downregulation of the genes within the pathway relative to WT expression (P < 0.05); positive Z-score represents upregulation (P < 0.05). Ratio (yellow line) represents the fraction of the genes associated with the pathway that are significantly regulated in Bpifa1−/− mice; n = 5–6 mice/group.
Table 2.
Immune response and host defense gene expression differences in Bpifa1−/− mice during acute inflammation
| Symbol | 0 h | 8 h | 24 h | P | |
|---|---|---|---|---|---|
| Chemokines and cytokines | |||||
| Chemokine (C-X-C motif) ligand 9 | Cxcl9 | −1.38 | −1.69 | −5.42 | 0.049 |
| Chemokine (C-X-C motif) ligand 10 | Cxcl10 | −1.06 | −1.99 | −4.56 | 0.036 |
| Chemokine (C-C motif) ligand 7 | Ccl7 | −1.12 | −1.79 | −3.83 | 0.021 |
| Chemokine (C-C motif) ligand 12 | Ccl12 | −1.12 | −1.47 | −3.80 | 0.018 |
| Interleukin 10 | Il10 | −1.01 | −2.70 | −2.79 | 0.018 |
| Chemokine (C-X-C motif) ligand 16 | Cxcl16 | −1.16 | −1.09 | −1.61 | 0.010 |
| Chemokine (C-X-C motif) ligand 15 | Cxcl15 | 1.22 | 1.22 | 1.50 | 0.029 |
| Cytokine receptors | |||||
| Chemokine (C-C motif) receptor 5 | Ccr5 | −1.31 | −1.59 | −2.24 | 0.036 |
| Interleukin 12 receptor, β1 | Il12rb1 | 1.07 | −1.77 | −2.09 | 0.036 |
| Interleukin 2 receptor, β-chain | Il2rb | −1.10 | −1.95 | −1.83 | 0.015 |
| Chemokine (C-X-C motif) receptor 3 | Cxcr3 | −1.16 | −1.56 | −1.66 | 0.015 |
| Interleukin 2 receptor, γ-chain | Il2rg | −1.10 | −1.44 | −1.60 | 0.015 |
| Chemokine (C-C motif) receptor 2 | Ccr2 | −1.50 | −1.84 | −1.19 | 0.049 |
| Involved in cell migration | |||||
| Integrin β2-like | Itgb2l | 1.23 | −3.33 | −2.65 | 0.028 |
| Cadherin, EGF LAG 7-pass G-type receptor 3 | Celsr3 | −1.13 | −1.73 | −1.52 | 0.046 |
| Selectin, lymphocyte | Sell | −1.15 | −1.64 | −1.44 | 0.049 |
| Selectin, platelet (p-selectin) ligand | Selplg | −1.05 | −1.51 | −1.43 | 0.019 |
| Cadherin 2 | Cdh2 | 1.12 | 1.49 | 1.53 | 0.039 |
| AMP and PRR signaling | |||||
| Granzyme B | Gzmb | −1.15 | −1.48 | −2.30 | 0.042 |
| Secretin | Sct | 1.18 | −1.43 | −2.24 | 0.016 |
| Toll-like receptor 3 | Tlr3 | −1.14 | −1.32 | −1.41 | 0.044 |
| Myeloid differentiation primary response gene 88 | Myd88 | −1.00 | −1.34 | −1.57 | 0.041 |
| Surfactant-associated protein B | Sftpb | 1.23 | 1.40 | 1.60 | 0.013 |
| Class I histocompatibility antigen superfamily | |||||
| Histocompatibility 2, T region locus 9 | H2-T9 | −1.08 | −1.42 | −1.86 | 0.010 |
| Histocompatibility 2, Q region locus 8 | H2-Q8 | −1.25 | −1.18 | −1.85 | 0.031 |
| Histocompatibility 2, T region locus 10 | H2-T10 | −1.09 | −1.20 | −1.79 | 0.020 |
| Histocompatibility 2, M region locus 11 | H2-M11 | −1.16 | −1.05 | −1.66 | 0.032 |
| Histocompatibility 2, T region locus 24 | H2-T24 | −1.10 | −1.28 | −1.60 | 0.031 |
| Histocompatibility 2, T region locus 23 | H2-T23 | −1.08 | −1.08 | −1.59 | 0.028 |
| Histocompatibility 2, M region locus 3 | H2-M3 | −1.11 | −1.27 | −1.53 | 0.007 |
Whole lung microarray results showing differential gene expression in untreated mice (0 h) and at 8 and 24 h (8 h, 24 h) after intranasal instillation of lipopolysaccharide (LPS). Data are represented as fold change (FC) from wild-type (WT) gene expression. Negative FC values denote lower gene expression relative to WT. Two-way ANOVA, false discovery rate < 0.05, at least one time point with FC >1.5. Bpifa1−/−, BPI fold-containing group A member 1 deficient; AMP, antimicrobial proteins, PRR, pathogen recognition receptors.
Table 4.
IFN regulatory and IFN-inducible gene expression in Bpifa1−/− mice during acute inflammation
| Symbol | 0 h | 8 h | 24 h | P | |
|---|---|---|---|---|---|
| Interferon signaling | |||||
| Suppressor of cytokine signaling 1 | Socs1 | −1.49 | −1.72 | −2.71 | 0.010 |
| Signal transducer and activator of transcription 2 | Stat2 | −1.08 | −1.34 | −1.95 | 0.010 |
| Janus kinase 2 | Jak2 | −1.07 | −1.19 | −1.31 | 0.025 |
| Interferon regulatory factors (IRF) | |||||
| Interferon regulatory factor 7 | Irf7 | −1.10 | −1.39 | −4.71 | 0.018 |
| Interferon regulatory factor 1 | Irf1 | −1.09 | −1.21 | −1.76 | 0.015 |
| Interferon regulatory factor 5 | Irf5 | −1.06 | −1.65 | −1.68 | 0.021 |
| Interferon regulatory factor 9 | Irf9 | −1.05 | −1.11 | −1.52 | 0.009 |
| Interferon-stimulated genes (ISG) | |||||
| 2′–5′-Oligoadenylate synthetase 1G | Oas1g | 1.11 | −1.93 | −4.24 | 0.037 |
| Interferon-inducible GTPase 1B | Iigp1b | −1.40 | −2.92 | −4.11 | 0.029 |
| ISG15 ubiquitin-like modifier | Isg15 | −1.31 | −1.47 | −3.72 | 0.018 |
| 2′–5′-Oligoadenylate synthetase-like 1 | Oasl1 | −1.18 | −1.60 | −3.72 | 0.012 |
| Interferon-induced protein with tetratricopeptide repeats 2 | Ifit2 | −1.27 | −1.73 | −3.67 | 0.015 |
| Myxovirus (influenza virus) resistance 1 | Mx1 | −1.03 | −1.56 | −3.64 | 0.013 |
| Interferon-inducible GTPase 1 | Iigp1 | −1.30 | −1.60 | −3.54 | 0.049 |
| Interferon γ-induced GTPase | Igtp | −1.15 | −1.75 | −3.50 | 0.014 |
| Interferon-activated gene 204 | Ifi204 | −1.08 | −1.48 | −3.34 | 0.036 |
| Interferon-induced protein with tetratricopeptide repeats 3 | Ifit3 | −1.44 | −1.58 | −2.99 | 0.004 |
| 2′–5′-Oligoadenylate synthetase 1A | Oas1a | −1.15 | −1.56 | −2.92 | 0.009 |
| Interferon-induced protein 44 | Ifi44 | −1.15 | −1.82 | −2.90 | 0.021 |
| Interferon-induced protein with tetratricopeptide repeats 1 | Ifit1 | −1.11 | −1.48 | −2.84 | 0.036 |
| 2′–5′-Oligoadenylate synthetase-like 2 | Oasl2 | −1.20 | −1.14 | −2.80 | 0.013 |
| Myxovirus (influenza virus) resistance 2 | Mx2 | −1.13 | −1.79 | −2.73 | 0.014 |
| 2′–5′-Oligoadenylate synthetase 1F | Oas1f | −1.11 | −1.38 | −2.42 | 0.021 |
| Interferon-induced transmembrane protein 3 | Ifitm3 | −1.14 | −1.51 | −2.08 | 0.014 |
| Interferon-activated gene 203 | Ifi203 | −1.17 | −1.62 | −2.01 | 0.015 |
| Interferon-stimulated protein | Isg20 | −1.00 | −1.62 | −1.94 | 0.009 |
| Interferon-induced protein 35 | Ifi35 | 1.03 | −1.26 | −1.65 | 0.037 |
| Interferon-induced transmembrane protein 6 | Ifitm6 | −1.66 | −2.13 | −1.34 | 0.022 |
Whole lung microarray results showing differential gene expression in untreated mice (0 h), and at 8 and 24 h after intranasal instillation of lipopolysaccharide (LPS). Data are represented as fold change (FC) from wild-type (WT) gene expression. Negative FC values denote lower gene expression relative to WT. Two-way ANOVA, false discovery rate < 0.05, at least one time point with FC > 1.5.
Table 3.
Basal gene expression differences in untreated Bpifa1−/− mice
| Genes | Symbol | FC | P |
|---|---|---|---|
| Schlafen 4 | Slfn4 | −2.41 | 0.034 |
| Pyrin domain-containing 4 | Pydc4 | −2.05 | 0.010 |
| Plasminogen activator, urokinase | Plau | −1.77 | 0.015 |
| Interferon-induced transmembrane protein 6 | Ifitm6 | −1.66 | 0.022 |
| Z-DNA binding protein 1 | Zbp1 | −1.61 | 0.005 |
| Apolipoprotein L 9a | Apol9a | −1.61 | 0.005 |
| Pyrin domain containing 3 | Pydc3 | −1.54 | 0.008 |
| Chemokine (C-C motif) receptor 2 | Ccr2 | −1.50 | 0.049 |
| Major facilitator superfamily domain-containing 2A | Mfsd2a | 1.64 | 0.023 |
| Fatty acid-binding protein-12 | Fabp12 | 4.79 | 0.039 |
Whole-lung microarray results showing differential gene expression in untreated wild-type (WT) and Bpifa1−/−, BPI fold-containing group A member 1 deficient (Bpifa1−/−) mice [2-way ANOVA, false discovery rate < 0.05; fold change (FC) >1.5]. Negative FC values denote lower gene expression relative to WT.
Cxcl10 is decreased in Bpifa1−/− airways during early LPS-induced inflammation.
To evaluate the impact of decreased chemokine gene expression on airway cytokine concentrations and PMN lung recruitment, we quantified canonical and noncanonical PMN chemoattractants in Bpifa1−/− BAL (Fig. 4). IFN-inducible Cxcl10 was the only cytokine significantly decreased in Bpifa1−/− compared with WT mice at 8 h. This time point is important for the development of inflammation, because early changes in gene expression and cytokine expression will drive PMN recruitment at later time points, even though differences in PMN accumulation are not yet detectable (40, 53). At 8 h, there were no significant differences in granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin-6 (IL-6), KC, tumor necrosis factor-α (TNFα), macrophage inflammatory protein-2 (MIP2), IL-1β, or leukotriene B4 in BAL fluid (Fig. 4, top row).
Fig. 4.

C-X-C motif chemokine ligand 10 (CXCL10) is decreased in BPI fold-containing group A member 1-deficient (Bpifa1−/−) airways during early lung inflammation. Bronchoalveolar lavage from Bpifa1+/+ [wild-type (WT), ●] and Bpifa1−/− mice (○) was collected 8 h (top row) and 24 h (bottom row) after intranasal lipopolysaccharide (LPS). Cytokine concentrations measured by multiplexed Luminex assay. Additional cytokines tested without significant difference: interferon (IFN)γ, interleukin (IL)-1α, IL-1β, IL-2, IL-4, IL-5, IL-7, IL-9, IL-10, IL-15, IL-17, C-C motif chemokine ligand 5 (CCL5), macrophage inflammatory protein (MIP)-1α, MIP-1β. Leukotriene B4 (LTB4) was not significantly different. TNFα, tumor necrosis factor-α; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; KC, keratinocyte chemoattractant; n = 4–7 mice/group. Mann-Whitney test: *P < 0.05. Horizontal lines indicate mean value. Data represent 2 experiments.
Twenty-four hours after LPS administration, significant reductions in IFNγ-inducing Ccl2, and IFNγ-inducible Cxcl2 appeared in Bpifa1−/− mice. These IFN-associated cytokines have a role in myeloid cell activation, adhesion, and recruitment to mucosal surfaces. IFNγ, a potent inducer of Cxcl10 and inhibitor of Bpifa1 expression, was below the level of detection in BAL (data not shown). There were no differences in canonical PMN-recruiting cytokines at 24 h in Bpifa1−/− BAL (Fig. 4, bottom row). There were no differences in total protein and LDH activity (indirect markers of acute lung injury, not shown).
To determine whether Bpifa1−/− PMN had intrinsic defects in chemotactic response, we performed chemotaxis assays using WT and Bpifa1−/− PMN. There was no difference in migration of Bpifa1−/− PMN compared with WT PMN in response to chemoattractants, including fMLP and KC, nor in response to the IFN-inducible cytokines Cxcl10, Cxcl9, or their combination (Fig. 5). In summary, these data show that Bpifa1 regulates the production of Cxcl10 early after exposure to PAMP, and this may serve to amplify PMN recruitment without specific effects on canonical PMN chemoattractants. They also show that Bpifa1−/− PMN do not have impaired chemotactic responses that contribute to this effect.
Fig. 5.
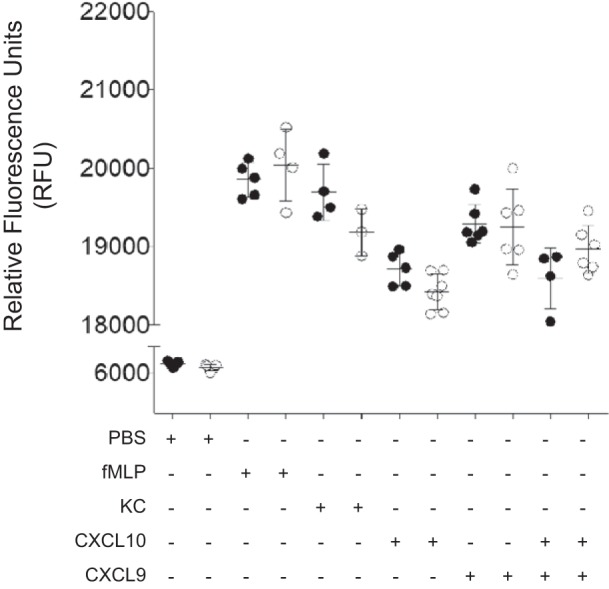
BPI fold-containing group A member 1-deficient (Bpifa1−/−) polymorphonuclear neutrophils (PMN) do not have impaired chemotaxis response. PMN from wild-type (WT) and Bpifa1−/− mice were isolated from bone marrrow. Chemotaxis assays were performed using a modified Boyden technique. WT and Bpifa1−/− PMN were stimulated with PMN chemoattractant N-formyl-methionyl-leucyl-phenylalanine (fMLP; 105 M), keratinocyte chemoattractant (KC; 50 ng/ml), C-X-C motif chemokine ligand 10 (Cxcl10; 100 ng/ml), Cxcl9 (50 ng/ml), and combination of Cxcl10 and Cxcl9. Total PMN migration represented as relative fluorescence units (RFU). ●, WT PMN; ○, Bpifa1−/− PMN; n = 4–6 wells/group. Generalized linear mixed model: no significant differences observed. Horizontal bars indicate mean values. Data represent 2 experiments.
IFNγ and IFNλ expression is impaired in Bpifa1−/− mice during acute inflammation.
Since IFN levels were not detectable in BAL, we performed targeted RT-PCR for type I (ifnα2, ifnα4, ifnβ1), type II (ifnγ), and type III IFN (ifnλ2/3) in lungs from LPS-treated Bpifa1−/− and WT mice at 0, 8, and 24 h. IFNγ and IFNλ mRNA expressions were significantly lower at 24 h in Bpifa1−/− compared with WT mice (Fig. 6A). Type I IFN, ifnα2, ifnα4, and ifnβ1, showed a trend toward lower expression. We also confirmed decreased expression of IFN-stimulated cytokines and IFN-stimulated genes (ISG) by using this methodology (Fig. 6, B and C). Our findings suggest that lower expression of IFNγ and IFNλ in Bpifa1−/− mice reduces PMN accumulation in the lungs through downregulation of IFN-stimulated cytokines and ISG.
Fig. 6.

Interferon (IFN) and IFN-stimulated gene (ISG) expression in BPI fold-containing group A member 1-deficient (Bpifa1−/−) lungs during acute inflammation. RNA was extracted from whole lung lysates from wild-type (WT) and Bpifa1−/− naive mice (0 h) and treated mice 8 and 24 h after lipopolysaccharide (LPS; 8 h, 24 h). Differences in gene expression were assessed by RT-PCR. A: time course of IFN mRNA expression in Bpifa1−/− mice. Relative expression (RQ) in Bpifa1−/− mice (◇) vs. WT gene expression (WT expression = 1, marked with horizontal dashed line). RQ values lower than 1 denote lower gene expression relative to WT. B: IFN-inducible cytokine gene expression at 24 h: relative expression of cytokine gene mRNA in Bpifa1−/− lungs (◇) relative to WT (◆). Cxcl, C-X-C motif chemokine ligand; Ccl, C-C motif chemokine ligand; tubb5, tubulin-β5. C: IFN-stimulated gene (ISG) expression at 24 h: relative expression of ISG mRNA in Bpifa1−/− lungs (◇) relative to WT (◆). Data represent 2 experiments. Each data point represents the mean mRNA expression of Bpifa1−/− mice within each group. Generalized linear mixed model: n = 4–6 mice/group, *P < 0.05.
Cxcl10 upregulation is impaired in Bpifa1−/−-infiltrating myeloid cells during acute inflammation.
We hypothesized that PMN and other infiltrating cells would be the primary contributors to the acute inflammation gene signature. To test this, we costained lung sections for Cxcl10 and Ly6G (for PMN) or F4:80 (for macrophages). Twenty-four hours after intranasal LPS, Cxcl10 staining was lower in PMN and macrophages from Bpifa1−/− lung sections compared with WT mice (Fig. 7). These observations suggest that Bpifa1 regulates the induction of Cxcl10 in myeloid inflammatory cells (PMN and macrophages) to enhance PMN recruitment in response to LPS.
Fig. 7.

C-X-C motif chemokine ligand 10 (Cxcl10) induction in myeloid cells during inflammation is impaired in BPI fold-containing group A member 1-deficient (Bpifa1−/−) mice. Top: lung sections from wild-type (WT) and Bpifa1−/− mice 24 h after intranasal instillation of lipopolysaccharide (LPS). Tissue sections were stained with antibodies against Cxcl10 (red) and F4:80, a marker for macrophages, or Ly6G, a marker that primarily stains polymorphonuclear neutrophils (PMN; both in green). Cell nuclei were stained with DAPI (blue); colocalization of Cxcl10 and cell markers is shown in yellow (white arowheads). Bottom: ratios of Cxcl10+ stained cells to total Ly6G+, F4:80+, or combined Ly6G+ plus F4:80+ cells in tissue sections from mice in these experiments. Each data point represents the average number of cells per field in 10 random fields at ×20 magnification. ●, WT values; ○, Bpifa1−/− values; n = 3 mice/group. Generalized linear mixed model: *P < 0.05. Data represent 2 experiments.
Reconstituting Cxcl10 in Bpifa1−/− mice restores lung PMN inflammation.
To determine whether Cxcl10 deficiency was critical to impaired airway PMN inflammation in Bpifa1−/− mice, we administered recombinant Cxcl10 intraperitoneally to WT and Bpifa1−/− mice before treatment with LPS and monitored their inflammatory response. PMN infiltration into the lungs was equivalent in LPS-treated WT and Cxcl10 pretreated Bpifa1−/− mice (Fig. 8), indicating that reconstitution of Cxcl10, in the absence of Bpifa1, can restore lung neutrophilic inflammation.
Fig. 8.
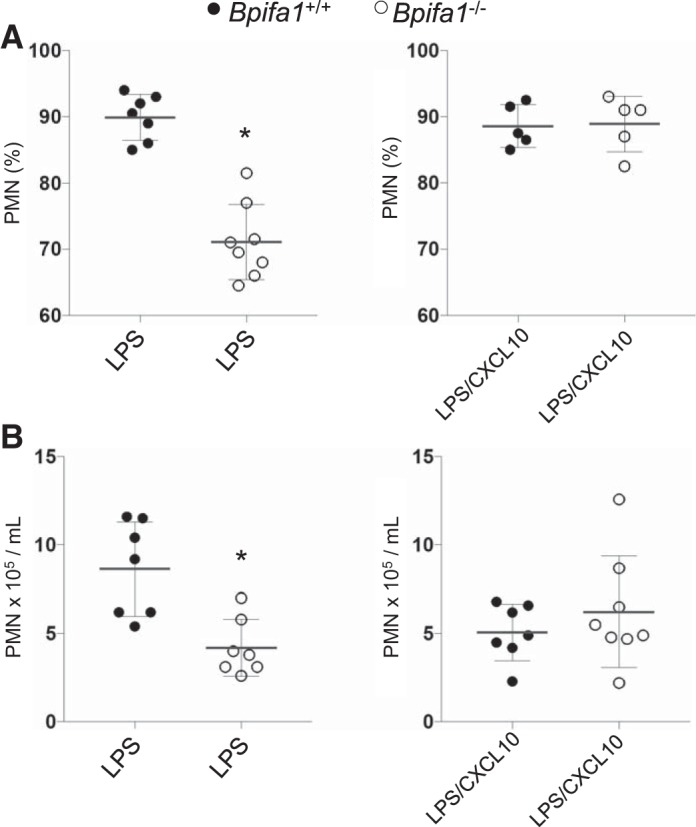
Reconstitution of C-X-C motif chemokine ligand 10 (Cxcl10) in BPI fold-containing group A member 1-deficient (Bpifa1−/−) mice restores lung polymorphonuclear neutrophils (PMN). Wild-type (WT) and Bpifa1−/− mice received intraperitoneal Cxcl10 before treatment with lipopolysaccharide (LPS). PMN bronchoalveolar lavage (BAL) counts were assessed at 24 h. A: %PMN isolated in BAL fluid (BALF). B: absolute PMN count in BALF after LPS. Bpifa1+/+, ●; Bpifa1−/−, ○; LPS, 5 μg/mouse, from Pseudomonas aeruginosa PAO1; Cxcl10, 0.6 μg/mouse; n = 5–8 mice/group. Mann-Whitney test: *P < 0.05. Horizontal lines indicate mean values. Data represent 2 experiments.
DISCUSSION
These studies show that Bpifa1 regulates PMN accumulation in the lung and reveal a novel immunomodulatory function of this protein. After LPS, Bpifa1−/− mice have fewer PMN in their lungs and impaired expression of IFN-stimulated genes. Decreased Cxcl10 expression, as we show here, plays an essential role in PMN recruitment in our model. Our previous studies showed that Bpifa1 is expressed at very high levels under basal conditions in untreated mice and that its expression quickly decreases in response to inflammatory signals. To isolate these immunomodulatory properties, we sought to exclude Bpifa1’s antimicrobial effects using LPS, rather than whole bacteria, whose growth could be inhibited by Bpifa1. It is important to note that, although LPS was derived from P. aeruginosa, this PAMP does not fully simulate the broad immune activation and responses triggered by the pathogen itself. We used Bpifa1−/− mice to simulate Bpifa1 downregulation after exposure to LPS, where Bpifa1 typically reaches nadir at less than 20% of its basal level. In the absence of Bpifa1, neutrophilia was impaired. Therefore we hypothesized that the downregulation of Bpifa1 in response to PAMP blunts PMN recruitment through its effects on IFN signaling pathways. These studies suggest that Bpifa1 fluctuation modulates inflammation, possibly to counterregulate excessive lung neutrophilia in response to inflammatory or infectious stimuli.
Bpifa1 is expressed abundantly, but not exclusively, in the proximal respiratory tract, where exposure to pathogens and irritants is high. This localization is consistent with its ability to inhibit bacterial growth and limit microbial invasion (2). The rapid downregulation of Bpifa1 within hours after some infections suggests that the antimicrobial effects of Bpifa1 may be limited in the hours following initial exposure to a pathogen; yet, others have shown that, even at low concentrations, Bpifa1 is important in protecting the airway epithelium from microbial invasion (17). Notably, there are other antimicrobials in the respiratory tract, not downregulated during infection, which also serve as immediate barriers against pathogen growth while Bpifa1 levels are dropping. This distinctive reduction in Bpifa1 suggests a role for Bpifa1 fluctuations in regulating the immune response to PAMPs. It is important to note that our findings contrast with studies using P. aeruginosa rather than LPS (37). In those studies, the authors showed that Bpifa1 deficiency resulted in increased bacterial loads and biofilm formation. This would likely increase immune activation and PMN infiltration. Since we focused on understanding the immunomodulatory properties of Bpifa1 in response to PAMP, we purposefully excluded bacterial organisms in order to study a discrete immune signaling cascade and limit the antimicrobial effects of Bpifa1. We propose that the inflammation differences observed between our studies reflect antimicrobial effects of Bpifa1 on bacterial growth and subsequent inflammation. Our studies provide further understanding of the seemingly paradoxical downregulation of Bpifa1 and contribute the first report of a link among this protein, IFN signaling, and PMN responses in the lung.
In Bpifa1−/− mice we observed fewer PMN in lungs and airways 8 h after LPS administration. Yet, none of the canonical PMN chemotaxins, such as IL-6, KC, TNFα, G-CSF, or GM-CSF were decreased early on to suggest hat they drove this defect. Others had shown that Bpifa1 was itself a chemotaxin (47). We tested this mechanism with chemotaxis assays, but we did not observe significant differences in PMN response to Bpifa1−/− and WT BAL obtained during acute inflammation (not shown).
To broadly define the mechanisms of impaired lung neutrophilia, we examined lung gene expression of LPS-treated WT and Bpifa1−/− mice. There were significant differences in the expression of Cxcl10, Cxcl9, and numerous IFN-responsive genes induced by IFNγ and IFNλ. Furthermore, we showed that Cxcl10 expression is reduced in both neutrophils and macrophages in Bpifa1−/− mice. Taken together, these data suggest that the downregulation of Bpifa1 preferentially affects inflammatory cells. We hypothesize that under basal conditions Bpifa1 modulates LPS-PRR interactions to enhance downstream IFN signaling and expression of IRF, Cxcl10, and Cxcl9.
To better understand the cellular source of this signature, we performed IHC using antibodies against Cxcl10 and cell-specific markers. This demonstrated that a larger number of WT myeloid inflammatory cells upregulated Cxcl10 expression after 24 h of acute inflammation compared with Bpifa1−/− myeloid cells. These findings support the hypothesis that the primary mechanism for impaired PMN inflammation in these mice is through defects in cytokine expression by inflammatory cells.
LPS-binding proteins (LBPs) modulate immune responses to pathogens by forming LPS-protein-CD14 complexes that bind to Toll-like receptor 4 (TLR4) (35, 44, 45). LBP, a proinflammatory LPS-binding molecule, enhances LPS-induced inflammation through the activation of TIR domain-containing adapter-inducing INFβ (TRIF)/TRIF-related adaptor molecule (TRAM) signaling, leading to IFN production (11, 35, 41, 55). Bpifa1 binds LPS, and it is likely to modulate IFN signaling through this mechanism (20). Bpifa1 may modulate TLR4 signaling by activating TRIF/TRAM pathways, explaining the defects observed in the IFN pathways and Cxcl10 in LPS-stimulated Bpifa1−/− mice (Fig. 9). IFNγ and IFNλ can exert pro- and anti-inflammatory effects during acute neutrophilic inflammation in the lungs and the intestinal tract (18, 25). Furthermore, the critical role of Cxcl10-Cxcr3 interactions in promoting PMN recruitment to the lung after acute lung injury and infection has been well characterized (33, 36). In our model, we observed significantly decreased expression of IFNγ (type II) and IFNλ (type III), potent inducers of Cxcl10. IFNα and IFNβ (type I) expression also decreased, but not as robustly as types II and III IFN. Importantly, we show that supplementing Cxcl10 restores the Bpifa1−/− phenotype to a WT-like response, indicating that this IFN-inducible cytokine is a primary mechanism responsible for impaired PMN recruitment. Cxcl9 expression was also decreased in our gene expression analysis, but the restoration of a WT-like phenotype in Bpifa1−/− mice with Cxcl10 suggests that Cxcl9 is less likely to be a major contributor to the limited inflammatory response.
Fig. 9.

Diagram: BPI fold-containing group A member 1 (Bpifa1) regulation of noncanonical Toll-like receptor 4 (TLR4) and interferon (IFN) signaling. Left: Bpifa1 is a lipopolysaccharide (LPS)-binding molecule and is likely to influence inflammatory signaling by modulating LPS-TLR4 interactions. In the absence of Bpifa1, impaired noncanonical TLR4 signaling through TIR domain-containing adapter-inducing INFβ1 (TRIF) results in decreased interferon regulatory factor 7 (IRF7) expression, leading to decreased IFNλ expression. Canonical LPS-TLR4 activation and cytokine expression is not affected by Bpifa1 deficiency. Middle: Bpifa1 binding to LPS facilitates immune signaling through TNF receptor associated factors (TRAF), which in turn activates IRF5 to induce interleukin (IL)-12 expression, inducing IFNγ. During Bpifa1 deficiency, key components of this pathway are downregulated (IL-12rb1, Jak2, Irf5), resulting in decreased IFNγ expression. Right: decreased expression of IFNγ and IFNλ during Bpifa1 deficiency causes downstream downregulation of Irf9, Irf7, Cxcl10, Cxcl9 and IFN-stimulated genes (ISG). Shaded boxes denote lower gene expression in our model. Dashed arrows represent impaired signaling pathways in our model. AEC, airway epithelial cell; APC, antigen-presenting cell; AL, airway lumen; NF-κB, nuclear factor-κB; TNFα, tumor necrosis factor-α; JAK, Janus kinase; STAT, signal transducer and activator of transcription; CXCL, C-X-C motif chemokine ligand.
Our work shows a novel mechanism of immune modulation that involves PAMP sensing by Bpifa1, leading to a Bpifa1-deficient state that suppresses IFNγ and IFNλ signaling. Decreased IFN signaling after Bpifa1 downregulation limits PMN recruitment and may serve as a protective mechanism to limit exaggerated PMN infiltration.
In summary, we show that Bpifa1 is necessary for normal neutrophilic responses to LPS. In the absence of Bpifa1, and likely during normal downward fluctuations triggered by environmental stimuli, decreased IFN signaling limits IFN-mediated cytokine induction and decreases PMN recruitment through effects on Cxcl10. These and our prior studies show that Bpifa1 and type II/III interferons work in a coordinated program to sense PAMP and modulate inflammatory responses to pathogen signals in the lung.
GRANTS
This work was supported by National Institutes of Health Grants R01-HL081160 and R21-AI-083475 (to L. E. Cohn) and T32-HL-007778 and K01-HL-125514-01 (to C. J. Britto) and Cystic Fibrosis Foundation through its Fifth Year Clinical Fellowship Award (to C. J. Britto).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
C.J.B. and L.E.C. conceived and designed research; C.J.B., N.N., S.K., L.H., J.H.-M., M.S., L.S., and C.S.D.C. performed experiments; C.J.B., N.N., S.K., L.H., J.H.-M., A.T., M.S., M.D.S., L.S., C.S.D.C., N.K., and L.E.C. analyzed data; C.J.B., N.N., S.K., L.H., J.H.-M., A.T., M.S., M.D.S., L.S., C.S.D.C., N.K., and L.E.C. interpreted results of experiments; C.J.B., S.K., J.H.-M., A.T., M.S., and C.S.D.C. prepared figures; C.J.B. and L.E.C. drafted manuscript; C.J.B., L.H., J.H.-M., A.T., M.S., M.D.S., L.S., C.S.D.C., N.K., and L.E.C. edited and revised manuscript; C.J.B., N.N., S.K., L.H., J.H.-M., A.T., M.S., M.D.S., L.S., C.S.D.C., N.K., and L.E.C. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Marie Egan from the Department of Cell Biology and Molecular Physiology at Yale University for thoughtful review and contributions in the development of this manuscript.
REFERENCES
- 1.Aichele D, Schnare M, Saake M, Röllinghoff M, Gessner A. Expression and antimicrobial function of bactericidal permeability-increasing protein in cystic fibrosis patients. Infect Immun 74: 4708–4714, 2006. doi: 10.1128/IAI.02066-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akram KM, Moyo NA, Leeming GH, Bingle L, Jasim S, Hussain S, Schorlemmer A, Kipar A, Digard P, Tripp RA, Shohet RV, Bingle CD, Stewart JP. An innate defense peptide BPIFA1/SPLUNC1 restricts influenza A virus infection. Mucosal Immunol 11: 71–81, 2018. doi: 10.1038/mi.2017.45. [DOI] [PubMed] [Google Scholar]
- 3.Allard JB, Poynter ME, Marr KA, Cohn L, Rincon M, Whittaker LA. Aspergillus fumigatus generates an enhanced Th2-biased immune response in mice with defective cystic fibrosis transmembrane conductance regulator. J Immunol 177: 5186–5194, 2006. doi: 10.4049/jimmunol.177.8.5186. [DOI] [PubMed] [Google Scholar]
- 4.Barnes FA, Bingle L, Bingle CD. Pulmonary genomics, proteomics, and PLUNCs. Am J Respir Cell Mol Biol 38: 377–379, 2008. doi: 10.1165/rcmb.2007-0388TR. [DOI] [PubMed] [Google Scholar]
- 5.Bauer Y, Tedrow J, de Bernard S, Birker-Robaczewska M, Gibson KF, Guardela BJ, Hess P, Klenk A, Lindell KO, Poirey S, Renault B, Rey M, Weber E, Nayler O, Kaminski N. A novel genomic signature with translational significance for human idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol 52: 217–231, 2015. doi: 10.1165/rcmb.2013-0310OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bayes HK, Ritchie N, Irvine S, Evans TJ. A murine model of early Pseudomonas aeruginosa lung disease with transition to chronic infection. Sci Rep 6: 35838, 2016. doi: 10.1038/srep35838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beamer LJ, Carroll SF, Eisenberg D. Crystal structure of human BPI and two bound phospholipids at 2.4 angstrom resolution. Science 276: 1861–1864, 1997. doi: 10.1126/science.276.5320.1861. [DOI] [PubMed] [Google Scholar]
- 8.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57: 289–300, 1995. doi: 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
- 9.Bingle CD, Bingle L. Characterisation of the human plunc gene, a gene product with an upper airways and nasopharyngeal restricted expression pattern. Biochim Biophys Acta 1493: 363–367, 2000. doi: 10.1016/S0167-4781(00)00196-2. [DOI] [PubMed] [Google Scholar]
- 10.Bingle CD, Bingle L, Craven CJ. Distant cousins: genomic and sequence diversity within the BPI fold-containing (BPIF)/PLUNC protein family. Biochem Soc Trans 39: 961–965, 2011. doi: 10.1042/BST0390961. [DOI] [PubMed] [Google Scholar]
- 11.Bingle CD, Craven CJ. Meet the relatives: a family of BPI- and LBP-related proteins. Trends Immunol 25: 53–55, 2004. doi: 10.1016/j.it.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 12.Bingle CD, Gorr SU. Host defense in oral and airway epithelia: chromosome 20 contributes a new protein family. Int J Biochem Cell Biol 36: 2144–2152, 2004. doi: 10.1016/j.biocel.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 13.Bingle CD, Seal RL, Craven CJ. Systematic nomenclature for the PLUNC/PSP/BSP30/SMGB proteins as a subfamily of the BPI fold-containing superfamily. Biochem Soc Trans 39: 977–983, 2011. doi: 10.1042/BST0390977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19: 185–193, 2003. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 15.Boon K, Bailey NW, Yang J, Steel MP, Groshong S, Kervitsky D, Brown KK, Schwarz MI, Schwartz DA. Molecular phenotypes distinguish patients with relatively stable from progressive idiopathic pulmonary fibrosis (IPF). PLoS One 4: e5134, 2009. doi: 10.1371/journal.pone.0005134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Britto CJ, Cohn L. Bactericidal/permeability-increasing protein fold-containing family member A1 in airway host protection and respiratory disease. Am J Respir Cell Mol Biol 52: 525–534, 2015. doi: 10.1165/rcmb.2014-0297RT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Britto CJ, Liu Q, Curran DR, Patham B, Dela Cruz CS, Cohn L. Short palate, lung, and nasal epithelial clone-1 is a tightly regulated airway sensor in innate and adaptive immunity. Am J Respir Cell Mol Biol 48: 717–724, 2013. doi: 10.1165/rcmb.2012-0072OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Broggi A, Tan Y, Granucci F, Zanoni I. IFN-λ suppresses intestinal inflammation by non-translational regulation of neutrophil function. Nat Immunol 18: 1084–1093, 2017. doi: 10.1038/ni.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Canny G, Levy O, Furuta GT, Narravula-Alipati S, Sisson RB, Serhan CN, Colgan SP. Lipid mediator-induced expression of bactericidal/permeability-increasing protein (BPI) in human mucosal epithelia. Proc Natl Acad Sci USA 99: 3902–3907, 2002. doi: 10.1073/pnas.052533799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chu HW, Thaikoottathil J, Rino JG, Zhang G, Wu Q, Moss T, Refaeli Y, Bowler R, Wenzel SE, Chen Z, Zdunek J, Breed R, Young R, Allaire E, Martin RJ. Function and regulation of SPLUNC1 protein in Mycoplasma infection and allergic inflammation. J Immunol 179: 3995–4002, 2007. doi: 10.4049/jimmunol.179.6.3995. [DOI] [PubMed] [Google Scholar]
- 21.Di YP. Functional roles of SPLUNC1 in the innate immune response against Gram-negative bacteria. Biochem Soc Trans 39: 1051–1055, 2011. doi: 10.1042/BST0391051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Di YP, Harper R, Zhao Y, Pahlavan N, Finkbeiner W, Wu R. Molecular cloning and characterization of spurt, a human novel gene that is retinoic acid-inducible and encodes a secretory protein specific in upper respiratory tracts. J Biol Chem 278: 1165–1173, 2003. doi: 10.1074/jbc.M210523200. [DOI] [PubMed] [Google Scholar]
- 23.Di YP, Tkach AV, Yanamala N, Stanley S, Gao S, Shurin MR, Kisin ER, Kagan VE, Shvedova A. Dual acute proinflammatory and antifibrotic pulmonary effects of short palate, lung, and nasal epithelium clone-1 after exposure to carbon nanotubes. Am J Respir Cell Mol Biol 49: 759–767, 2013. doi: 10.1165/rcmb.2012-0435OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dubin PJ, Kolls JK. IL-23 mediates inflammatory responses to mucoid Pseudomonas aeruginosa lung infection in mice. Am J Physiol Lung Cell Mol Physiol 292: L519–L528, 2007. doi: 10.1152/ajplung.00312.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Espinosa V, Dutta O, McElrath C, Du P, Chang YJ, Cicciarelli B, Pitler A, Whitehead I, Obar JJ, Durbin JE, Kotenko SV, Rivera A. Type III interferon is a critical regulator of innate antifungal immunity. Sci Immunol 2: eaan5357, 2017. doi: 10.1126/sciimmunol.aan5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gally F, Di YP, Smith SK, Minor MN, Liu Y, Bratton DL, Frasch SC, Michels NM, Case SR, Chu HW. SPLUNC1 promotes lung innate defense against Mycoplasma pneumoniae infection in mice. Am J Pathol 178: 2159–2167, 2011. doi: 10.1016/j.ajpath.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia-Caballero A, Rasmussen JE, Gaillard E, Watson MJ, Olsen JC, Donaldson SH, Stutts MJ, Tarran R. SPLUNC1 regulates airway surface liquid volume by protecting ENaC from proteolytic cleavage. Proc Natl Acad Sci USA 106: 11,412–11,417, 2009. [Erratum in Proc Natl Acad Sci USA 106: 15091, 2009.] doi: 10.1073/pnas.0903609106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Genter MB, Van Veldhoven PP, Jegga AG, Sakthivel B, Kong S, Stanley K, Witte DP, Ebert CL, Aronow BJ. Microarray-based discovery of highly expressed olfactory mucosal genes: potential roles in the various functions of the olfactory system. Physiol Genomics 16: 67–81, 2003. doi: 10.1152/physiolgenomics.00117.2003. [DOI] [PubMed] [Google Scholar]
- 29.Ghafouri B, Kihlström E, Ståhlbom B, Tagesson C, Lindahl M. PLUNC (palate, lung and nasal epithelial clone) proteins in human nasal lavage fluid. Biochem Soc Trans 31: 810–814, 2003. doi: 10.1042/bst0310810. [DOI] [PubMed] [Google Scholar]
- 30.Ghafouri B, Kihlström E, Tagesson C, Lindahl M. PLUNC in human nasal lavage fluid: multiple isoforms that bind to lipopolysaccharide. Biochim Biophys Acta 1699: 57–63, 2004. doi: 10.1016/S1570-9639(04)00003-2. [DOI] [PubMed] [Google Scholar]
- 31.Ghafouri B, Ståhlbom B, Tagesson C, Lindahl M. Newly identified proteins in human nasal lavage fluid from non-smokers and smokers using two-dimensional gel electrophoresis and peptide mass fingerprinting. Proteomics 2: 112–120, 2002. doi: 10.1002/1615-9861(200201)2:1<112:AID-PROT112>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 32.Gross CA, Bowler RP, Green RM, Weinberger AR, Schnell C, Chu HW. beta2-agonists promote host defense against bacterial infection in primary human bronchial epithelial cells. BMC Pulm Med 10: 30, 2010. doi: 10.1186/1471-2466-10-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ichikawa A, Kuba K, Morita M, Chida S, Tezuka H, Hara H, Sasaki T, Ohteki T, Ranieri VM, dos Santos CC, Kawaoka Y, Akira S, Luster AD, Lu B, Penninger JM, Uhlig S, Slutsky AS, Imai Y. CXCL10-CXCR3 enhances the development of neutrophil-mediated fulminant lung injury of viral and nonviral origin. Am J Respir Crit Care Med 187: 65–77, 2013. doi: 10.1164/rccm.201203-0508OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krämer A, Green J, Pollard J Jr, Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30: 523–530, 2014. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krasity BC, Troll JV, Weiss JP, McFall-Ngai MJ. LBP/BPI proteins and their relatives: conservation over evolution and roles in mutualism. Biochem Soc Trans 39: 1039–1044, 2011. doi: 10.1042/BST0391039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lang S, Li L, Wang X, Sun J, Xue X, Xiao Y, Zhang M, Ao T, Wang J. CXCL10/IP-10 neutralization can ameliorate lipopolysaccharide-induced acute respiratory distress syndrome in rats. PLoS One 12: e0169100, 2017. doi: 10.1371/journal.pone.0169100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y, Di ME, Chu HW, Liu X, Wang L, Wenzel S, Di YP. Increased susceptibility to pulmonary Pseudomonas infection in Splunc1 knockout mice. J Immunol 191: 4259–4268, 2013. doi: 10.4049/jimmunol.1202340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lukinskiene L, Liu Y, Reynolds SD, Steele C, Stripp BR, Leikauf GD, Kolls JK, Di YP. Antimicrobial activity of PLUNC protects against Pseudomonas aeruginosa infection. J Immunol 187: 382–390, 2011. doi: 10.4049/jimmunol.1001769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McGillivary G, Bakaletz LO. The multifunctional host defense peptide SPLUNC1 is critical for homeostasis of the mammalian upper airway. PLoS One 5: e13224, 2010. doi: 10.1371/journal.pone.0013224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mukherjee S, Chen LY, Papadimos TJ, Huang S, Zuraw BL, Pan ZK. Lipopolysaccharide-driven Th2 cytokine production in macrophages is regulated by both MyD88 and TRAM. J Biol Chem 284: 29391–29398, 2009. doi: 10.1074/jbc.M109.005272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mulero JJ, Boyle BJ, Bradley S, Bright JM, Nelken ST, Ho TT, Mize NK, Childs JD, Ballinger DG, Ford JE, Rupp F. Three new human members of the lipid transfer/lipopolysaccharide binding protein family (LT/LBP). Immunogenetics 54: 293–300, 2002. doi: 10.1007/s00251-002-0467-3. [DOI] [PubMed] [Google Scholar]
- 42.Ning F, Wang C, Berry KZ, Kandasamy P, Liu H, Murphy RC, Voelker DR, Nho CW, Pan CH, Dai S, Niu L, Chu HW, Zhang G. Structural characterization of the pulmonary innate immune protein SPLUNC1 and identification of lipid ligands. FASEB J 28: 5349–5360, 2014. doi: 10.1096/fj.14-259291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Niu N, Laufer T, Homer RJ, Cohn L. Cutting edge: Limiting MHC class II expression to dendritic cells alters the ability to develop Th2- dependent allergic airway inflammation. J Immunol 183: 1523–1527, 2009. doi: 10.4049/jimmunol.0901349. [DOI] [PubMed] [Google Scholar]
- 44.Noppert SJ, Fitzgerald KA, Hertzog PJ. The role of type I interferons in TLR responses. Immunol Cell Biol 85: 446–457, 2007. doi: 10.1038/sj.icb.7100099. [DOI] [PubMed] [Google Scholar]
- 45.Pålsson-McDermott EM, O’Neill LA. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology 113: 153–162, 2004. doi: 10.1111/j.1365-2567.2004.01976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Romero R, Friel LA, Velez Edwards DR, Kusanovic JP, Hassan SS, Mazaki-Tovi S, Vaisbuch E, Kim CJ, Erez O, Chaiworapongsa T, Pearce BD, Bartlett J, Salisbury BA, Anant MK, Vovis GF, Lee MS, Gomez R, Behnke E, Oyarzun E, Tromp G, Williams SM, Menon R. A genetic association study of maternal and fetal candidate genes that predispose to preterm prelabor rupture of membranes (PROM). Am J Obstet Gynecol 203: 361.e1–361.e30, 2010. doi: 10.1016/j.ajog.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sayeed S, Nistico L, St Croix C, Di YP. Multifunctional role of human SPLUNC1 in Pseudomonas aeruginosa infection. Infect Immun 81: 285–291, 2013. doi: 10.1128/IAI.00500-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scheetz TE, Zabner J, Welsh MJ, Coco J, Eyestone MF, Bonaldo M, Kucaba T, Casavant TL, Soares MB, McCray PB Jr. Large-scale gene discovery in human airway epithelia reveals novel transcripts. Physiol Genomics 17: 69–77, 2004. doi: 10.1152/physiolgenomics.00188.2003. [DOI] [PubMed] [Google Scholar]
- 49.Shultz MA, Zhang L, Gu YZ, Baker GL, Fannuchi MV, Padua AM, Gurske WA, Morin D, Penn SG, Jovanovich SB, Plopper CG, Buckpitt AR. Gene expression analysis in response to lung toxicants. I. Sequencing and microarray development. Am J Respir Cell Mol Biol 30: 296–310, 2004. doi: 10.1165/rcmb.2003-0214OC. [DOI] [PubMed] [Google Scholar]
- 50.Tarran R, Redinbo MR. Mammalian short palate lung and nasal epithelial clone 1 (SPLUNC1) in pH-dependent airway hydration. Int J Biochem Cell Biol 52: 130–135, 2014. doi: 10.1016/j.biocel.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Teeguarden JG, Webb-Robertson BJ, Waters KM, Murray AR, Kisin ER, Varnum SM, Jacobs JM, Pounds JG, Zanger RC, Shvedova AA. Comparative proteomics and pulmonary toxicity of instilled single-walled carbon nanotubes, crocidolite asbestos, and ultrafine carbon black in mice. Toxicol Sci 120: 123–135, 2011. doi: 10.1093/toxsci/kfq363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thomas S, Bonchev D. A survey of current software for network analysis in molecular biology. Hum Genomics 4: 353–360, 2010. doi: 10.1186/1479-7364-4-5-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ulich TR, Guo K, Yin S, del Castillo J, Yi ES, Thompson RC, Eisenberg SP. Endotoxin-induced cytokine gene expression in vivo. IV. Expression of interleukin-1 alpha/beta and interleukin-1 receptor antagonist mRNA during endotoxemia and during endotoxin-initiated local acute inflammation. Am J Pathol 141: 61–68, 1992. [PMC free article] [PubMed] [Google Scholar]
- 54.Walton WG, Ahmad S, Little MS, Kim CS, Tyrrell J, Lin Q, Di YP, Tarran R, Redinbo MR. Structural features essential to the antimicrobial functions of Human SPLUNC1. Biochemistry 55: 2979–2991, 2016. doi: 10.1021/acs.biochem.6b00271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weiss J. Bactericidal/permeability-increasing protein (BPI) and lipopolysaccharide-binding protein (LBP): structure, function and regulation in host defence against Gram-negative bacteria. Biochem Soc Trans 31: 785–790, 2003. doi: 10.1042/bst0310785. [DOI] [PubMed] [Google Scholar]
- 56.Zhou HD, Li XL, Li GY, Zhou M, Liu HY, Yang YX, Deng T, Ma J, Sheng SR. Effect of SPLUNC1 protein on the Pseudomonas aeruginosa and Epstein-Barr virus. Mol Cell Biochem 309: 191–197, 2008. doi: 10.1007/s11010-007-9659-3. [DOI] [PubMed] [Google Scholar]
- 57.Zhou HD, Wu MH, Shi L, Zhou M, Yang YX, Zhao J, Deng T, Li XL, Sheng SR, Li GY. [Effect of growth inhibition of the secretory protein SPLUNC1 on Pseudomonas aeruginosa]. Zhong Nan Da Xue Xue Bao Yi Xue Ban 31: 464–469, 2006. [PubMed] [Google Scholar]