

Abstract
Hypertonicity increases water permeability, independently of vasopressin, in the inner medullary collecting duct (IMCD) by increasing aquaporin-2 (AQP2) membrane accumulation. We investigated whether protein kinase C (PKC) and adenosine monophosphate kinase (AMPK) are involved in hypertonicity-regulated water permeability. Increasing perfusate osmolality from 150 to 290 mosmol/kgH2O and bath osmolality from 290 to 430 mosmol/kgH2O significantly stimulated osmotic water permeability. The PKC inhibitors chelerythrine (10 µM) and rottlerin (50 µM) significantly reversed the increase in osmotic water permeability stimulated by hypertonicity in perfused rat terminal IMCDs. Chelerythrine significantly increased phosphorylation of AQP2 at S261 but not at S256. Previous studies show that AMPK is stimulated by osmotic stress. We tested AMPK phosphorylation under hypertonic conditions. Hypertonicity significantly increased AMPK phosphorylation in inner medullary tissues. Blockade of AMPK with Compound C decreased hypertonicity-stimulated water permeability but did not alter phosphorylation of AQP2 at S256 and S261. AICAR, an AMPK stimulator, caused a transient increase in osmotic water permeability and increased phosphorylation of AQP2 at S256. When inner medullary tissue was treated with the PKC activator phorbol dibutyrate (PDBu), the AMPK activator metformin, or both, AQP2 phosphorylation at S261 was decreased with PDBu or metformin alone, but there was no additive effect on phosphorylation with PDBu and metformin together. In conclusion, hypertonicity regulates water reabsorption by activating PKC. Hypertonicity-stimulated water reabsorption by PKC may be related to the decrease in endocytosis of AQP2. AMPK activation promotes water reabsorption, but the mechanism remains to be determined. PKC and AMPK do not appear to act synergistically to regulate water reabsorption.
Keywords: AMPK, hypertonicity, nephrogenic diabetes insipidus, PKC, water transport
INTRODUCTION
Renal water excretion is important for the maintenance of water balance in the body. Vasopressin is the major hormone regulating water permeability in the kidney (30). Vasopressin binds to the V2 receptors on the basolateral plasma membrane of the inner medullary collecting duct (IMCD), which results in stimulation of adenylyl cyclase and generation of cAMP. cAMP stimulates protein kinase A, which phosphorylates key transporters and channels resulting in an increase in water permeability (5). Vasopressin stimulation of water permeability is due in large part to stimulation of the aquaporin 2 (AQP2) water channel in the IMCD (7, 20). AQP2, located at the apical plasma membrane and subapical vesicles in the principal cells of the collecting duct, is regulated by vasopressin-stimulated increases in both its phosphorylation and apical plasma membrane accumulation (16).
However, there are situations where vasopressin or its receptor is either absent or nonfunctional. Nephrogenic diabetes insipidus (NDI) is such a condition. In X-linked congenital NDI, a mutation exists in the gene encoding the V2 receptor that results in greatly diminished or absent ability to respond to vasopressin (23, 28). The result is that cAMP does not get stimulated. PKA is therefore not activated, and AQP2 phosphorylation does not occur when needed. In NDI, very large quantities of dilute urine are produced due to an inability of the kidney to respond to vasopressin (2). It is therefore important to look for an alternative kinase that can mediate water transport independent of vasopressin to provide a therapy for NDI. Luckily, there is evidence showing that a vasopressin-independent activator, hypertonicity, regulates water transport in the inner medulla. For example, hypertonicity increases ADH (antidiuretic hormone, vasopressin)-stimulated water permeability in rat IMCDs (25) and increases hydraulic conductivity in the absence and presence of vasopressin in rat IMCDs (22). Acute hypertonicity alters AQP2 trafficking at the plasma membrane of renal epithelial cells (13), but direct evidence that hypertonicity alone stimulates water permeability in IMCDs independently of vasopressin has not been obtained. Whether hypertonicity regulates water permeability by phosphorylation of AQP2 in IMCDs needs to be determined.
Previous studies (10) show that hypertonicity increases intracellular calcium but does not change cAMP accumulation. These findings suggest that hypertonicity as an independent activator of water transport acts through intracellular calcium instead of cAMP. Our previous data show that hypertonicity increases urea permeability in rat IMCDs through a PKC signaling pathway initiated by intracellular calcium (32). This is important because urea accumulation in the interstitium causes hyperosmolality and thus enhances water absorption. Given that both urea and water permeability increase under hypertonic conditions, it is likely that hypertonicity may also stimulate water permeability through a PKC signaling pathway.
Adenosine monophosphate-activated kinase (AMPK) is an energy-sensing kinase that can be stimulated by osmotic stress, hypoxia, and exercise (27), conditions that cause a high AMP or low ATP level. The renal inner medulla is normally hypertonic and hypoxic. Hypertonicity increases AMPKα phosphorylation at threonine 172 in cultured IMCD cells (12). Our previous study shows that AMPK is present in the kidney inner medulla (18) and that metformin, an AMPK stimulator, increases osmotic water permeability in rat IMCDs (18). These findings suggest that AMPK may have an important role in regulation of water reabsorption in a hypertonic inner medulla.
Since both PKC and AMPK can be activated by hypertonicity, the purpose of the present study was to determine whether PKC, AMPK, or both are involved in hypertonicity-stimulated water transport and AQP2 water channel activity.
METHODS
Animals.
All animal protocols were approved by the Emory University Institutional Animal Care and Use Committee. These studies used both male and female rats from Charles River Laboratories (Wilmington, MA). There were no apparent differences between the responses of males or females in either osmotic water permeability or protein phosphorylation in these studies. To measure osmotic water permeability (Pf), rats weighing 50–75 g were killed by decapitation to avoid any anesthesia effect, and the kidneys were quickly dissected to remove the inner medullas (IMs). To test AQP2 phosphorylation, rats weighing 100–140 g were killed, and the IMs were homogenized on ice.
Tubule perfusion.
IMs were transferred to a dissection dish and the terminal IMCDs microdissected in a dissection buffer at 17°C. The dissecting solution contained the following (in mM): 125 NaCl, 25 NaHCO3, 2 CaCl2, 2.5 K2HPO4, 1.2 MgSO4, 5.5 glucose, and 5 raffinose. The osmolality of the solution was adjusted to 430 mosmol/kgH2O with NaCl only for the purpose of dissecting tubules (25). The perfusion and bath solutions were identical to the dissection medium, except that an additional 70 mM NaCl was added to the bath. Thus, a bath-to-lumen osmolality gradient of ~140 mosmol/kgH2O was established. This solution was gassed continuously with 95% air and 5% CO2 before and during the dissection and perfusion.
Single IMCDs were dissected, mounted on glass pipettes, and perfused as described previously (29, 30). In general, 45 min after warming the tubules to 37°C in 2 ml of bath solution, two 2~3-min collections of 6–8 nl/min were made. To measure the response to the treatments, the bath solution was changed or the treatments were added to the bath, and tubules were allowed to equilibrate for 10–15 min, then 2 further collections were made. To measure Pf, collected solutions were assayed for raffinose content by ultramicrofluorometry (19). Raffinose was used as a volume marker for the measurement of osmotic water permeability. Pf was calculated as previously described (30). When IMCDs were perfused, the paracelluar leak was checked by monitoring the osmotic water permeability in response to the flow rate. If the inhibitors and activators change paracellular water permeability, the water permeability in the IMCD would be sufficiently high to reach equilibrium in a short (within 2–5 min) time and would not change with altered flow rate.
To subject the tubules to hypertonic conditions, the tubules were first perfused under isotonic conditions with a hypotonic perfusate (150 mosmol/kgH2O) and an isotonic bath (290 mosmol/kgH2O) fluid. Then, the osmolality of the perfusate was increased from 150 to 290 mosmol/kgH2O (isotonic perfusate) and the osmolality of the bath was increased from 290 to 430 mosmol/kgH2O (hypertonic bath) by adding 70 mM NaCl; thus, the same osmotic gradient was maintained between the isotonic conditions and hypertonic conditions.
To assess the contribution of PKC to hypertonicity-stimulated osmotic water permeability, a cell-permeable PKC inhibitor (10 µM chelerythrine or 50 µM rottlerin) was added to the hypertonic bath after the osmotic water permeability was measured under hypertonic conditions.
To assess the contribution of AMPK, a cell-permeable AMPK inhibitor (10 µM Compound C) was added to the hypertonic bath. We also tested AICAR-stimulated osmotic water permeability. AICAR is an AMPK stimulator. The tubules were first perfused under isotonic conditions with a hypotonic perfusate (150 mosmol/kgH2O) and an isotonic bath (290 mosmol/kgH2O) fluid. Then, 1 mM AICAR was added to the bath, and the osmotic water permeability was measured.
Tissue incubation.
To measure the phosphorylation of AQP2, inner medullary (IM) tissues were first incubated at 37°C for 15 min in isotonic Hanks’ balanced salt solution (HBSS) to establish baseline resting conditions and then stimulated for 15 min with NaCl-enriched hypertonic HBSS (690 mosmol/kgH2O), hypertonic HBSS in the presence of chelerythrine (10 µM), or hypertonic HBSS with Compound C (10 µM) or 30 min with isotonic HBSS in the presence of phorbol dibutyrate (PDBu) (4 µM), metformin (400 µM), or both. The tissues were homogenized and analyzed for AQP2 phosphorylation with Western blotting.
To measure the phosphorylation of AMPK, IM tissues were first incubated at 37°C for 15 min in isotonic Dulbecco’s modified Eagle’s medium (DMEM) to establish baseline resting conditions and then stimulated for 45 min with NaCl-enriched hypertonic DMEM (690 mosmol/kgH2O). The tissues were homogenized and analyzed for phosphorylation of AMPK with Western blotting.
Western blot analysis.
Samples of whole IM protein lysate (20 µg/lane), one rat per lane, were size-separated by SDS-PAGE on 12.5% gels then electroblotted to polyvinylidene difluoride (PVDF) membranes (Immobilon, Millipore, Bedford, MA). Blots were blocked with 5% nonfat dry milk in Tris-buffered saline (TBS; 20 mM Tris·HCl, 0.5 M NaCl, pH 7.5) then incubated with primary antibodies overnight at 4°C. Attached primary antibodies were identified using Alexa Fluor 680-linked anti-rabbit IgG (Molecular Probes, Eugene, OR) and visualized using infrared detection with the LICOR Odyssey protein analysis system (Lincoln, NE). Antibodies to AQP2, AQP2 phosphorylated at serine 256 or 261, AMPK, and pThr172-AMPK were purchased from Cell Signaling Technology (Danvers, MA).
Statistics.
All data are presented as means ± SE. Data from tubule perfusion studies were analyzed using a paired Student’s t-test or repeated measures ANOVA, depending on whether two conditions or more than two conditions were assessed in each tubule (31). Each perfused tubule serves as its own control, allowing a paired analysis of the data. Data from protein analysis studies were analyzed using a nonpaired Student’s t-test or ANOVA. The criterion for statistical significance is P < 0.05.
RESULTS
Hypertonicity increases water transport.
To determine whether hypertonicity increases Pf, the terminal IMCDs were first perfused under isotonic conditions and then perfused under hypertonic conditions. The Pf was close to zero when the osmotic gradient was created by hypotonic perfusate and isotonic bath solution under isotonic conditions. Increasing the osmolality of the perfusate and bath solutions from 150 to 290 mosmol/kgH2O and from 290 to 430 mosmol/kgH2O, respectively, resulted in a significant increase in Pf from 1.4 ± 0.9 to 189 ± 17 µm/s (n = 4, P < 0.01; Fig. 1). The increased Pf was significantly reduced to 133 ± 11 µm/s by addition of the PKC inhibitor chelerythrine (10 µM) to the hypertonic bath solution (n = 4, P < 0.05; Fig. 1). To further confirm the role of PKC in the regulation of water permeability, 50 µM rottlerin (a second chemical inhibitor of PKC that inhibits a wide spectrum of PKCs at 50 µM) was added to the hypertonic bath. As before, hypertonicity increased osmotic water permeability from 1.2 ± 0.5 to 149 ± 23 µm/s (n = 4, P < 0.01; Fig. 2). The addition of rottlerin significantly decreased osmotic water permeability to 91 ± 12 µm/s (n = 4, P < 0.05; Fig. 2).
Fig. 1.
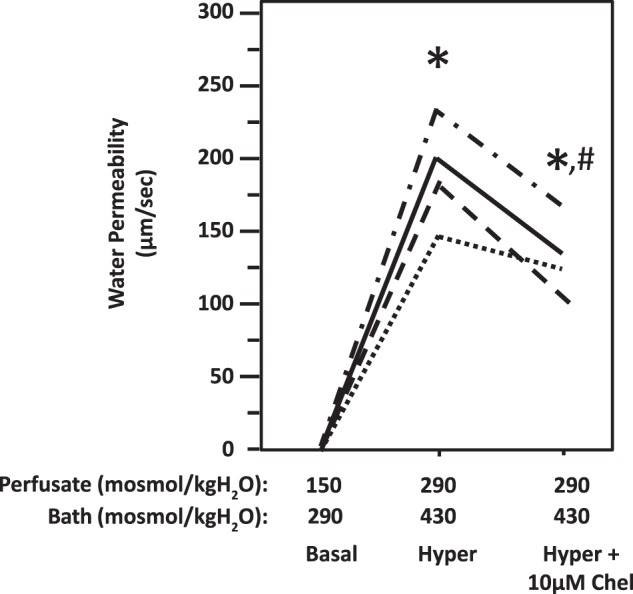
Hypertonicity-induced osmotic water permeability in terminal inner medullary collecting ducts (IMCDs) was reversed by inhibition of protein kinase C with chelerythrine (Chel). Terminal IMCDs were perfused with isotonic conditions (perfusate: 150 mosmol/kgH2O; bath: 290 mosmol/kgH2O), switched to hypertonic (Hyper) condition (perfusate: 290 mosmol/kgH2O; bath: 430 mosmol/kgH2O), and then supplemented with 10 µM chelerythrine. Each line represents a separate IMCD from a different rat. *P < 0.01 vs. 150 mosmol/kgH2O perfusate and 290 mosmol/kgH2O bath. #P < 0.05 vs. 290 mosmol/kgH2O perfusate and 430 mosmol/kgH2O bath without inhibitor; n = 4 rats/condition.
Fig. 2.
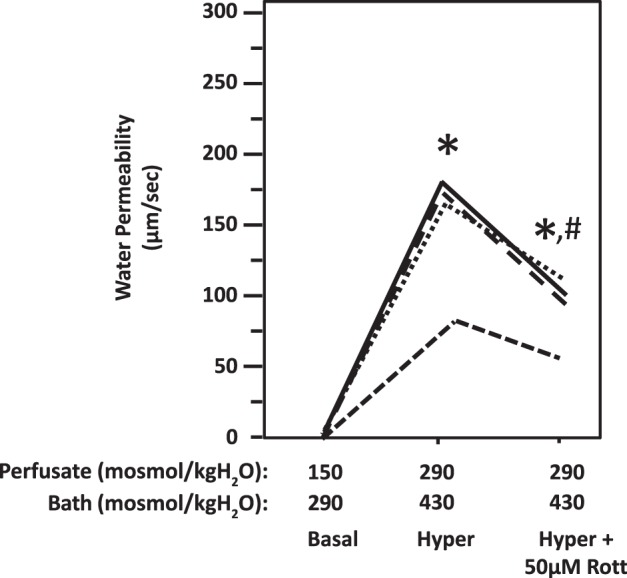
Hypertonicity-induced osmotic water permeability in terminal inner medullary collecting ducts (IMCDs) was reversed by inhibition of protein kinase C with rottlerin (Rott). Terminal IMCDs were perfused with isotonic conditions (perfusate: 150 mosmol/kgH2O; bath: 290 mosmol/kgH2O), switched to hypertonic (Hyper) condition (perfusate: 290 mosmol/kgH2O; bath: 430 mosmol/kgH2O), and then supplemented with 50 µM rottlerin. Each line represents a separate IMCD from a different rat. *P < 0.01 vs. 150 mosmol/kgH2O perfusate and 290 mosmol/kgH2O bath. #P < 0.05 vs 290 mosmol/kgH2O perfusate and 430 mosmol/kgH2O bath without inhibitor. n = 4 rats/condition.
To determine the role of AMPK in the regulation of hypertonicity-stimulated water permeability, 10 µM Compound C (an AMPK inhibitor) were added to the hypertonic bath. As before, hypertonicity increased the Pf from 1 ± 0.6 to 208 ± 18 µm/s (n = 4, P < 0.01; Fig. 3). The addition of Compound C significantly decreased the Pf to 166 ± 14 µm/s (n = 4, P < 0.05; Fig. 3).
Fig. 3.
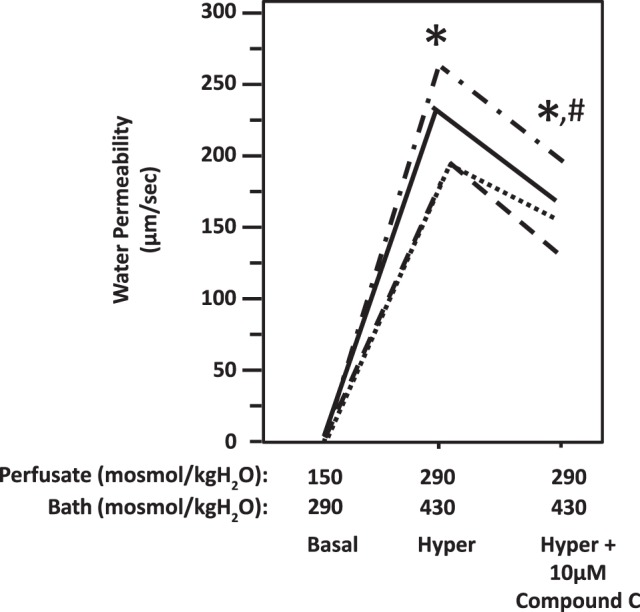
Hypertonicity-induced osmotic water permeability in terminal inner medullary collecting ducts (IMCDs) was reversed by inhibition of adenosine monophosphate kinase with Compound C (Comp C). Terminal IMCDs were perfused with isotonic conditions (perfusate: 150 mosmol/kgH2O; bath: 290 mosmol/kgH2O), switched to hypertonic (Hyper) condition (perfusate: 290 mosmol/kgH2O; bath: 430 mosmol/kgH2O), and then supplemented with 10 µM Compound C. Each line represents a separate IMCD from a different rat. *P < 0.01 vs. 150 mosmol/kgH2O perfusate and 290 mosmol/kgH2O bath. #P < 0.05 vs. 290 mosmol/kgH2O perfusate and 430 mosmol/kgH2O bath without inhibitor. n = 4 rats/condition.
Hypertonicity stimulates osmotic water permeability in the presence of urea.
Previous studies by us and others did not add urea to the perfusate or bath when measuring osmotic water permeability. To determine if urea affects hypertonicity-stimulated osmotic water permeability, we perfused IMCDs in a medium with urea. As in the absence of urea, hypertonicity increased the Pf from 3 ± 0.8 to 146 ± 15 µm/s (n = 3, P < 0.01) in the presence of urea. Since there was no significant difference in hypertonicity-stimulated osmotic water permeability with and without urea, we did not perfuse and bathe the IMCDs in a medium with urea in the tubule perfusion experiments in this study.
Hypertonicity phosphorylates AQP2.
To test the role of hypertonicity alone in the phosphorylation of AQP2, IM tissues were incubated in isotonic medium for 15 min and then stimulated with hypertonic medium for 15 min. Control tissues maintained the isotonic condition throughout the entire experiment. Hypertonicity significantly increased the phosphorylation of AQP2 at serine 256 [band densities (arbitrary units) control: 1.16 ± 0.11, hypertonicity-stimulated: 1.5 ± 0.04; n = 4, P < 0.05, Fig. 4]. Hypertonicity also significantly decreased the phosphorylation of AQP2 at serine 261 (control: 2.96 ± 0.18, hypertonicity-stimulated: 2.69 ± 0.15; n = 7, P < 0.05, Fig. 5).
Fig. 4.

Hypertonicity increased phosphorylation of aquaporin-2 (AQP2) at serine 256 in rat inner medulla (IM). A: representative Western blot showing AQP2 abundance and phosphorylated AQP2 in rat IM with hypertonic (Hyper) stimulation (690 mosmol/kgH2O) for 15 min. Brackets indicate AQP2 glycosylated protein between 35 and 45 kDa and arrows indicate unglycosylated protein at 29 kDa. Samples from a different rat were loaded into each lane. B: bar graph showing the ratio of the band densities of phosphorylated AQP2 to total AQP2 abundance. Bars = means ± SE. *P < 0.05 vs. isotonic (Iso) treatment (290 mosmol/kgH2O); n = 4 rats/condition.
Fig. 5.

Hypertonicity decreased phosphorylation of aquaporin-2 (AQP2) at serine 261 in rat inner medulla (IM). A: representative Western blot showing AQP2 abundance and phosphorylated AQP2 in rat IM with hypertonic (Hyper) stimulation (690 mosmol/kgH2O) for 15 min. Brackets indicate AQP2 glycosylated protein between 35 and 45 kDa and arrows indicate unglycosylated protein at 29 kDa. Samples from a different rat were loaded into each lane. B: bar graph showing the ratio of the band densities of phosphorylated AQP2 to total AQP2 abundance. Bars = means ± SE. *P < 0.05 vs. isotonic (Iso) treatment (290 mosmol/kgH2O); n = 7 rats/condition.
To test the role of chelerythrine (PKC inhibitor) or Compound C (AMPK inhibitor) in the hypertonicity-stimulated phosphorylation of AQP2, IM tissues were incubated in isotonic medium for 15 min and then treated with chelerythrine or Compound C in hypertonic medium for 15 min. Control tissues were subjected to hypertonic medium without chelerythrine or Compound C added. Chelerythrine did not significantly change the phosphorylation of AQP2 at serine 256 (control: 0.51 ± 0.08, chelerythrine-treated: 0.53 ± 0.06; n = 4, Fig. 6). However, chelerythrine dramatically increased the phosphorylation of AQP2 at serine 261 (control: 1.80 ± 0.07, chelerythrine-treated: 2.34 ± 0.03; n = 4, P < 0.01, Fig. 7). Compound C did not significantly change the phosphorylation of AQP2 either at serine 256 (control: 0.42 ± 0.03, Compound C-treated: 0.41 ± 0.04; n = 4, Fig. 8) or at serine 261 (control: 1.20 ± 0.04, Compound C-treated: 1.14 ± 0.03; n = 4, Fig. 9).
Fig. 6.

Inhibition of protein kinase C (PKC) with chelerythrine (Chel) did not change phosphorylation of aquaporin-2 (AQP2) at serine 256 in rat inner medulla (IM). A: representative Western blot showing AQP2 abundance and phosphorylated AQP2 in rat IM with hypertonic (Hyper) stimulation (690 mosmol/kgH2O) and treatment of 10 µM chelerythrine (PKC inhibitor) for 15 min. Brackets indicate AQP2 glycosylated protein between 35 and 45 kDa and arrows indicate unglycosylated protein at 29 kDa. Samples from a different rat were loaded into each lane. B: bar graph showing the ratio of the band densities of phosphorylated AQP2 to total AQP2 abundance. Bars = means ± SE; results were not significantly different between hypertonic treatment alone and combination of hypertonicity and chelerythrine; n = 4 rats/condition.
Fig. 7.

Inhibition of protein kinase C with chelerythrine (Chel) increased phosphorylation of aquaporin-2 (AQP2) at serine 261 in rat inner medulla (IM). A: representative Western blot showing AQP2 abundance and phosphorylated AQP2 in rat IM with hypertonic (Hyper) stimulation (690 mosmol/kgH2O) and treatment of 10 µM chelerythrine for 15 min. Brackets indicate AQP2 glycosylated protein between 35 and 45 kDa and arrows indicate unglycosylated protein at 29 kDa. Samples from a different rat were loaded into each lane. B: bar graph showing the ratio of the band densities of phosphorylated AQP2 to total AQP2 abundance. Bars = means ± SE. *P < 0.01 vs. hypertonic treatment (690 mosmol/kgH2O) only; n = 4 rats/condition.
Fig. 8.

Inhibition of adenosine monophosphate kinase (AMPK) with Compound C (Comp C) did not change phosphorylation of aquaporin-2 (AQP2) at serine 256 in rat inner medulla (IM). A: representative Western blot showing AQP2 abundance and phosphorylated AQP2 in rat IM with hypertonic (Hyper) stimulation (690 mosmol/kgH2O) and treatment of 10 µM Compound C (AMPK inhibitor) for 15 min. Brackets indicate AQP2 glycosylated protein between 35 and 45 kDa and arrows indicate unglycosylated protein at 29 kDa. Samples from a different rat were loaded into each lane. B: bar graph showing the ratio of the band densities of phosphorylated AQP2 to total AQP2 abundance. Bars = means ± SE; results were not significantly different between hypertonic treatment alone and combination of hypertonicity and compound C; n = 4 rats/condition.
Fig. 9.

Inhibition of adenosine monophosphate kinase (AMPK) with Compound C (Comp C) did not change phosphorylation of aquaporin-2 (AQP2) at serine 261 in rat inner medulla (IM). A: representative Western blot showing AQP2 abundance and phosphorylated AQP2 in rat IM with hypertonic (Hyper) stimulation (690 mosmol/kgH2O) and treatment of 10 µM Compound C (AMPK inhibitor) for 15 min. Brackets indicate AQP2 glycosylated protein between 35 and 45 kDa and arrows indicate unglycosylated protein at 29 kDa. Samples from a different rat were loaded into each lane. B: bar graph showing the ratio of the band densities of phosphorylated AQP2 to total AQP2 abundance. Bars = means ± SE; results were not significantly different between hypertonic treatment alone and combination of hypertonicity and Compound C; n = 4 rats/condition.
To verify that Compound C inhibits AMPK, we treated rat IMCDs with 10 µM Compound C and observed the level of phospho-AMPK by Western blotting. Compound C inhibited phosphorylation of AMPK at threonine 172 (data not shown).
AICAR stimulates osmotic water permeability.
AICAR is a commonly used AMPK activator. To examine whether AMPK regulates water transport under isotonic conditions, the tubule was first perfused with a hypotonic perfusate (150 mosmol/kgH2O) and an isotonic bath (290 mosmol/kgH2O) fluid. As stated above, the Pf was close to zero when the osmotic gradient was created by hypotonic perfusate and isotonic bath solution. The addition of 1 mM AICAR to the bath caused a rapid increase in osmotic water permeability from 0.4 ± 0.4 to 60 ± 17 µm/s (n = 4, P < 0.05; Fig. 10) in 10 min. However, the Pf returned to zero 30 min after stimulation with AICAR.
Fig. 10.
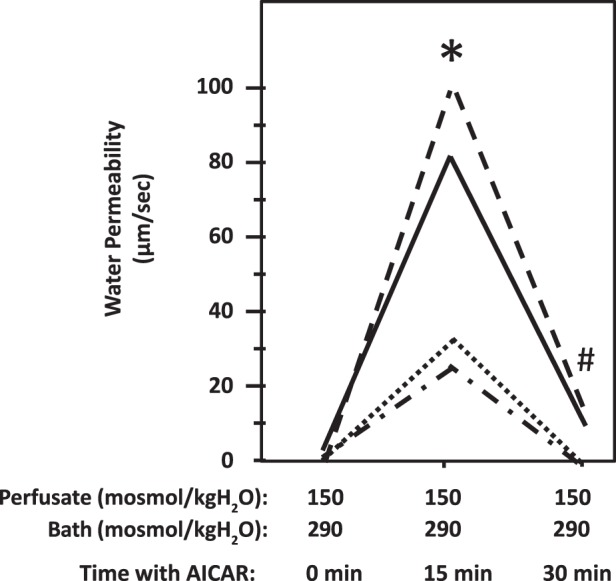
Stimulation of adenosine monophosphate kinase (AMPK) with AICAR increased osmotic water permeability in terminal inner medulla collecting ducts (IMCDs). A. terminal IMCDs were perfused with isotonic conditions (perfusate: 150 mosmol/kgH2O; bath: 290 mosmol/kgH2O) and then supplemented with 1 mM AICAR (AMPK stimulator). Each line represents a separate IMCD from a different rat. *P < 0.05 vs. isotonic control groups without AICAR; n = 4 rats/condition. #P < 0.05 vs. isotonic groups treated with AICAR for 15 min; n = 4 rats/condition.
AICAR phosphorylates AQP2.
To test the role of AICAR in the phosphorylation of AQP2, IM tissue was incubated in isotonic medium for 15 min and then stimulated with 1 mM AICAR for 15 min. AICAR significantly increased phosphorylation of AQP2 at serine 256 (control: 0.21 ± 0.13, AICAR: 0.56 ± 0.09; n = 4, P < 0.05, Fig. 11) but did not change phosphorylation of AQP2 at serine 261 (control: 4.99 ± 2.36, AICAR: 7.16 ± 1.84; n = 4, Fig. 12).
Fig. 11.

Stimulation of adenosine monophosphate kinase with AICAR increased phosphorylation of aquaporin-2 (AQP2) at serine 256 in rat inner medulla (IM). A: representative Western blot showing AQP2 abundance and phosphorylated AQP2 in rat IM with AICAR stimulation for 15 min. Brackets indicate AQP2 glycosylated protein between 35 and 45 kDa and arrows indicate unglycosylated protein at 29 kDa. Samples from a different rat were loaded into each lane. B: bar graph showing the ratio of the band densities of phosphorylated AQP2 to total AQP2 abundance. Bars = means ± SE. *P < 0.05 vs. control tissues (290 mosmol/kgH2O, without AICAR treatment); n = 4 rats/condition.
Fig. 12.

Stimulation of adenosine monophosphate kinase with AICAR did not change phosphorylation of aquaporin-2 (AQP2) at serine 261 in rat inner medulla (IM). A: representative Western blot showing AQP2 abundance and phosphorylated AQP2 in rat IM with AICAR stimulation for 15 min. Brackets indicate AQP2 glycosylated protein between 35 and 45 kDa and arrows indicate unglycosylated protein at 29 kDa. Samples from a different rat were loaded into each lane. B: bar graph showing the ratio of the band densities of phosphorylated AQP2 to total AQP2 abundance. Bars = means ± SE; results were not significantly different between control and AICAR-treated tissues; n = 4 rats/condition.
Hypertonicity increases the phosphorylation of AMPK.
IM tissues were incubated in isotonic medium for 15 min and then stimulated with hypertonic medium for 45 min. Control tissues maintained the isotonic condition throughout the entire experiment. Hypertonicity significantly increased the phosphorylation of AMPK at threonine 172 (control: 0.74 ± 0.09, hypertonicity-stimulated: 1.67 ± 0.09; n = 4, P < 0.01; Fig. 13).
Fig. 13.

Hypertonicity increased phosphorylation of adenosine monophosphate kinase (AMPK) at threonine 172 in rat inner medulla (IM). A: representative Western blot showing total AMPK abundance (bottom) and phosphorylated AMPK (top) in rat IM with hypertonic (Hyper) stimulation (690 mosmol/kgH2O) for 15 min. Arrows show the AMPK glycosylated protein band at 63–64 kDa. Samples from a different rat were loaded into each lane. B: bar graph showing the ratio of the band densities of phosphorylated AMPK to total AMPK abundance. Bars = means ± SE. *P < 0.01 vs. isotonic (Iso) treatment (290 mosmol/kgH2O) only; n = 4 rats/condition.
PKC and AMPK do not provide an additive effect on water reabsorption.
To determine if the two signaling pathways regulate water transport independently, IM tissues were treated with PDBu, metformin, and PDBu and metformin together. Neither reagent individually nor the combination of PDBu with metformin significantly altered the phosphorylation of AQP2 at serine 256 (control: 2.06 ± 0.39, PDBu: 3.45 ± 0.76, metformin: 3.56 ± 0.62, PDBu+metformin: 3.55 ± 0.43; n = 6, Fig. 14). Treatment with PDBu or metformin separately significantly reduced the phosphorylation of AQP2 at serine 261 (control: 10.68 ± 0.99, PDBu: 5.75 ± 0.81, n = 6, P < 0.01; metformin: 5.45 ± 0.59, n = 4, P < 0.01, Fig. 15), whereas the addition of PDBu with metformin did not cause an additive decrease in the phosphorylation of AQP2 at serine 261 (PDBu+metformin: 4.96 ± 0.61, n = 4, Fig. 15).
Fig. 14.

Protein kinase C (PKC) and adenosine monophosphate kinase (AMPK) did not change phosphorylation of AQP2 at serine 256 in rat inner medulla (IM). A: representative Western blot showing AQP2 abundance and phosphorylated AQP2 in rat IM with phorbol dibutyrate (PDBu) (PKC activator), metformin (Met; AMPK stimulator), or both for 30 min. Brackets indicate AQP2 glycosylated protein between 35 and 45 kDa and arrows indicate unglycosylated protein at 29 kDa. Samples from a different rat were loaded into each lane. B: bar graph showing the ratio of the band densities of phosphorylated AQP2 to total AQP2 abundance. Bars = means ± SE; results were not significantly different from the isotonic control group without PDBu and metformin. n = 6 rats/condition. Ctrl, control.
Fig. 15.

Protein kinase C and adenosine monophosphate kinase decreased phosphorylation of aquaporin-2 (AQP2) at serine 261, but combined treatment did not provide an additive effect in rat inner medulla (IM). A: representative Western blot showing AQP2 abundance and phosphorylated AQP2 in rat IM with phorbol dibutyrate (PDBu), metformin (Met), or both for 30 min. Brackets indicate AQP2 glycosylated protein between 35 and 45 kDa and arrows indicate unglycosylated protein at 29 kDa. Samples from a different rat were loaded into each lane. B: bar graph showing the ratio of the band densities of phosphorylated AQP2 to total AQP2 abundance. Bars = means ± SE; *P < 0.01 vs. isotonic control groups without PDBu and metformin; n = 6 rats/condition. Ctrl, control.
DISCUSSION
It is well established that water transport is regulated by vasopressin via cAMP-dependent signaling pathways. Vasopressin regulates several key transport proteins in the renal medulla, such as the Na-K-2Cl cotransporter, NKCC2 (11, 21), in the outer medulla, and AQP2 (26, 33) and the urea transporter, UT-A1 (3, 4, 34), in the inner medulla. These transporters play an important role in the concentration of urine. This study provides evidence that hypertonicity can regulate AQP2 activity and water permeability through a vasopressin-independent mechanism involving PKC. AMPK may also be a potential mechanism for hypertonicity-stimulated osmotic water permeability, but further investigation is necessary to confirm the role of AMPK in osmotic water permeability.
Previous studies show that hypertonicity increases ADH-stimulated water permeability (25) and plasma membrane accumulation of AQP2 in rat inner medulla (13). The response to hypertonicity occurs through increases in intracellular calcium, and the activation of PKC plays a role in urea transport (32). To determine the role of PKC in hypertonicity-stimulated osmotic water permeability, hypertonicity was used to stimulate PKC in the present study. Our data show that hypertonicity significantly increases osmotic water permeability (Figs. 1 and 2) and phosphorylation of AQP2 at serine 256 (Fig. 4) independent of vasopressin, whereas it decreases phosphorylation of AQP2 at serine 261 (Fig. 5). Phosphorylation of AQP2 at serine 256 promotes AQP2 trafficking to the apical plasma membrane (6), and phosphorylation of AQP2 at serine 261 is linked to ubiquitination (15). This suggests that hypertonicity increases plasma membrane accumulation of AQP2 through phosphorylation at serine 256 and decreases ubiquitination of AQP2 associated with phosphorylation at serine 261. To verify that the increase in osmotic water permeability is in response to the activation of PKC, two PKC inhibitors, chelerythrine or rottlerin, were used to decrease osmotic water permeability. The data in the present study show that both chelerythrine and rottlerin significantly reversed the increase in osmotic water permeability by hypertonicity (Fig. 1 and 2), which supports the concept that hypertonicity increases water permeability by stimulating PKC. Chelerythrine did not change phosphorylation of AQP2 at serine 256 (Fig. 6) but significantly increased phosphorylation at serine 261 (Fig. 7), which suggests that the decrease in hypertonicity-stimulated water permeability by PKC inhibition may be related to the increase in endocytosis of AQP2 instead of the decrease in AQP2 trafficking to the plasma membrane.
An osmotic gradient alone may not cause water transport across the plasma membrane. If an osmotic gradient of 140 mosmol/kgH2O is created by a hypotonic perfusate (150 mosmol/kgH2O) and an isotonic bath (290 mosmol/kgH2O), water permeability is not detected in the tubule (Figs. 1 and 2). By maintaining a constant osmotic gradient, we confirm that hypertonicity alone stimulates osmotic water permeability.
Another response to hypertonicity is activation of AMPK. AMPK is a heterotrimeric kinase with one catalytic (α) and two regulatory subunits (β and γ) (24). There are two AMPKα subunits: α1 and α2. Both α isoforms are expressed in the inner medulla (18). However, reports of the AMPK response to hypertonicity are not consistent. Hypertonicity increases phosphorylation of AMPK in cultured inner medullary collecting duct cells but decreases phosphorylation of AMPK in cultured renal medullary interstitial cells (12). To verify the response of AMPK to hypertonicity in the inner medulla, we examined AMPK phosphorylation in hypertonic rat IM tissues. Our data show that hypertonicity significantly increases AMPK phosphorylation in rat IM tissues (Fig. 13). Previous studies show that AMPK upregulates NKCC2 (8, 9, 14), and metformin, an AMPK stimulator, stimulates water transport independent of vasopressin (18). Combining the information, we propose that AMPK activity may be involved in the regulation of hypertonicity-stimulated water permeability. To verify that the increase in osmotic water permeability is in response to the activation of AMPK, the AMPK inhibitor, Compound C, was used to decrease osmotic water permeability. Data from isolated perfused tubules show that Compound C significantly decreases hypertonicity-stimulated osmotic water permeability (Fig. 3), which indicates that hypertonicity regulates water transport through AMPK. However, the phosphorylation of AQP2 is unchanged by blocking AMPK with Compound C (Figs. 8 and 9). Therefore, AMPK does not regulate water transport through direct phosphorylation of AQP2 at serines 256 and 261 under hypertonic conditions. We also verified that Compound C inhibits AMPK phosphorylation at threonine 172 in rat inner medulla (data not shown).
Dr. Hallows’ group reported that the AICAR-stimulated AMPK signaling pathway inhibits AQP2 function in cortical collecting duct cells (1). We previously found that the metformin-stimulated AMPK pathway stimulates water permeability in inner medullary collecting ducts. The discrepancy in the proposed roles for activated AMPK may be due to the different ways that AMPK was stimulated. AMPK is an energy-sensitive kinase and is stimulated by a high AMP or low ATP level. AICAR is an adenosine analog taken up into cells by adenosine transporters and phosphorylated by adenosine kinase, thus generating the AMP-mimetic and stimulating AMPK. However, AICAR does not change the intracellular ATP level as do many other AMPK activators, such as metformin (17). Metformin inhibits complex I of the mitochondrial respiratory chain, leading to an increased AMP-ATP ratio. Therefore, the discrepancy in the role of these two AMPK activators may be due to the different reagents used to stimulate AMPK or might reflect different responses to the different cell types in cortical versus inner medullary collecting ducts.
To further examine the discrepancy, we used AICAR to stimulate tubules for water permeability and compared the results to the action of metformin on water permeability reported in our previous publication (18). We also directly compared AQP2 phosphorylation in inner medullary tissue treated with metformin and AICAR. We found that AICAR is able to stimulate osmotic water permeability in 15 min, but beyond 15 min, AICAR-stimulated water permeability decreased (Fig. 10). Given that AMPK is an energy-sensitive kinase, we speculate that the transient stimulation by AICAR under the isotonic condition may be due to the recovery of energy in the cells. AICAR significantly increased phosphorylation of AQP2 at serine 256 (Fig. 11) but showed no change in the phosphorylation level of serine 261-AQP2 (Fig. 12). Metformin increased phosphorylation of AQP2 at serine 256, but this did not reach statistical significance (Fig. 14). Metformin treatment resulted in significantly decreased phosphorylation of AQP2 at serine 261 (Fig. 15). The explanation for the discrepancy in the phosphorylation of AQP2 is unclear; however, we speculate that the stimulation of AMPK by metformin was either of longer duration than that of AICAR (suggested by the tubule perfusion response time course), or a secondary dephosphorylation pathway was stimulated by metformin either directly as an off-target metformin effect, or by AMPK. In light of this data, we acknowledged our inability to know what mechanism may be responsible for these discrepancies.
Our results suggest that PKC is able to regulate water permeability, and AMPK is a potential regulator of water permeability. To investigate whether the two signals may regulate water reabsorption independently or synergistically, we tested phosphorylation of AQP2 by PDBu (PKC stimulator), metformin (AMPK stimulator), and PDBu plus metformin. PDBu did not alter the phosphorylation of AQP2 at serine 256 (Fig. 14) but decreased the phosphorylation at serine 261 (Fig. 15), which further supports the concept that PKC increases water permeability by dephosphorylation of serine 261 as suggested in Fig. 7. Like PDBu, metformin did not change the phosphorylation at serine 256 (Fig. 14) but decreased the phosphorylation at serine 261 (Fig. 15), suggesting that AMPK increases water permeability by dephosphorylation of AQP2 at serine 261. However, our previous data from immunoprecipitation of radiolabeled AQP2 show that metformin significantly increases phosphorylation of AQP2 (18), which suggests that AMPK may phosphorylate other sites of AQP2 instead of serine 256. When PDBu and metformin were added simultaneously in IM tissues, they did not provide an additive effect on AQP2 phosphorylation (Figs. 14 and 15), which suggests that PKC and AMPK may not act synergistically to regulate water reabsorption.
In conclusion, the present study shows that hypertonicity is an independent regulator of water permeability. Hypertonicity stimulates water transport by activating PKC. PKC mediates the hypertonicity-stimulated increase in water permeability. The increase in water permeability appears to be a result of a decrease in AQP2 phosphorylation at serine 261, which may promote AQP2 retention at the plasma membrane. Hypertonicity also increases AMPK phosphorylation, suggesting that AMPK may mediate the hypertonicity-stimulated increase in water permeability, but the mechanism for AMPK to regulate water permeability remains to be determined.
GRANTS
This work was supported by NIH National Institute of Diabetes and Digestive and Kidney Diseases Grant R01-DK-41707, Emory University Research Committee Grant 00067457, and American Heart Association Career Development Grant 18CDA34060053.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Y.W. conceived and designed research; J.K.L., J.A.R., L.M.L., F.R., F.M., and Y.W. performed experiments; J.K.L., J.A.R., and Y.W. analyzed data; J.A.R. and Y.W. interpreted results of experiments; Y.W. prepared figures; Y.W. drafted manuscript; Y.W. edited and revised manuscript; Y.W. approved final version of manuscript.
REFERENCES
- 1.Al-Bataineh MM, Li H, Ohmi K, Gong F, Marciszyn AL, Naveed S, Zhu X, Neumann D, Wu Q, Cheng L, Fenton RA, Pastor-Soler NM, Hallows KR. Activation of the metabolic sensor AMP-activated protein kinase inhibits aquaporin-2 function in kidney principal cells. Am J Physiol Renal Physiol 311: F890–F900, 2016. doi: 10.1152/ajprenal.00308.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bichet DG. Nephrogenic diabetes insipidus. Adv Chronic Kidney Dis 13: 96–104, 2006. doi: 10.1053/j.ackd.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 3.Blount MA, Klein JD, Martin CF, Tchapyjnikov D, Sands JM. Forskolin stimulates phosphorylation and membrane accumulation of UT-A3. Am J Physiol Renal Physiol 293: F1308–F1313, 2007. doi: 10.1152/ajprenal.00197.2007. [DOI] [PubMed] [Google Scholar]
- 4.Blount MA, Mistry AC, Fröhlich O, Price SR, Chen G, Sands JM, Klein JD. Phosphorylation of UT-A1 urea transporter at serines 486 and 499 is important for vasopressin-regulated activity and membrane accumulation. Am J Physiol Renal Physiol 295: F295–F299, 2008. doi: 10.1152/ajprenal.00102.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown D, Fenton RA. The cell biology of vasopressin action. In: Brenner and Rector’s The Kidney, edited by Taal MW, Chertow GM, Marsden PA, Skorecki K, Yu ASL, Brenner BM. Philadelphia, PA: Elsevier, 2011, p. 353–383. [Google Scholar]
- 6.Brown D, Hasler U, Nunes P, Bouley R, Lu HA. Phosphorylation events and the modulation of aquaporin 2 cell surface expression. Curr Opin Nephrol Hypertens 17: 491–498, 2008. doi: 10.1097/MNH.0b013e3283094eb1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chou CL, Yip KP, Michea L, Kador K, Ferraris JD, Wade JB, Knepper MA. Regulation of aquaporin-2 trafficking by vasopressin in the renal collecting duct. Roles of ryanodine-sensitive Ca2+ stores and calmodulin. J Biol Chem 275: 36839–36846, 2000. doi: 10.1074/jbc.M005552200. [DOI] [PubMed] [Google Scholar]
- 8.Fraser S, Mount P, Hill R, Levidiotis V, Katsis F, Stapleton D, Kemp BE, Power DA. Regulation of the energy sensor AMP-activated protein kinase in the kidney by dietary salt intake and osmolality. Am J Physiol Renal Physiol 288: F578–F586, 2005. doi: 10.1152/ajprenal.00190.2004. [DOI] [PubMed] [Google Scholar]
- 9.Fraser SA, Gimenez I, Cook N, Jennings I, Katerelos M, Katsis F, Levidiotis V, Kemp BE, Power DA. Regulation of the renal-specific Na+-K+-2Cl− co-transporter NKCC2 by AMP-activated protein kinase (AMPK). Biochem J 405: 85–93, 2007. doi: 10.1042/BJ20061850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gillin AG, Star RA, Sands JM. Osmolarity-stimulated urea transport in rat terminal IMCD: role of intracellular calcium. Am J Physiol Renal Physiol 265: F272–F277, 1993. doi: 10.1152/ajprenal.1993.265.2.F272. [DOI] [PubMed] [Google Scholar]
- 11.Giménez I, Forbush B. Short-term stimulation of the renal Na-K-Cl cotransporter (NKCC2) by vasopressin involves phosphorylation and membrane translocation of the protein. J Biol Chem 278: 26946–26951, 2003. doi: 10.1074/jbc.M303435200. [DOI] [PubMed] [Google Scholar]
- 12.Han Q, Zhang X, Xue R, Yang H, Zhou Y, Kong X, Zhao P, Li J, Yang J, Zhu Y, Guan Y. AMPK potentiates hypertonicity-induced apoptosis by suppressing NFκB/COX-2 in medullary interstitial cells. J Am Soc Nephrol 22: 1897–1911, 2011. doi: 10.1681/ASN.2010080822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hasler U, Nunes P, Bouley R, Lu HA, Matsuzaki T, Brown D. Acute hypertonicity alters aquaporin-2 trafficking and induces a MAPK-dependent accumulation at the plasma membrane of renal epithelial cells. J Biol Chem 283: 26643–26661, 2008. doi: 10.1074/jbc.M801071200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hayashi T, Hirshman MF, Fujii N, Habinowski SA, Witters LA, Goodyear LJ. Metabolic stress and altered glucose transport: activation of AMP-activated protein kinase as a unifying coupling mechanism. Diabetes 49: 527–531, 2000. doi: 10.2337/diabetes.49.4.527. [DOI] [PubMed] [Google Scholar]
- 15.Hoffert JD, Nielsen J, Yu MJ, Pisitkun T, Schleicher SM, Nielsen S, Knepper MA. Dynamics of aquaporin-2 serine-261 phosphorylation in response to short-term vasopressin treatment in collecting duct. Am J Physiol Renal Physiol 292: F691–F700, 2007. doi: 10.1152/ajprenal.00284.2006. [DOI] [PubMed] [Google Scholar]
- 16.Hoffert JD, Pisitkun T, Wang G, Shen RF, Knepper MA. Quantitative phosphoproteomics of vasopressin-sensitive renal cells: regulation of aquaporin-2 phosphorylation at two sites. Proc Natl Acad Sci USA 103: 7159–7164, 2006. doi: 10.1073/pnas.0600895103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim J, Yang G, Kim Y, Kim J, Ha J. AMPK activators: mechanisms of action and physiological activities. Exp Mol Med 48: e224, 2016. doi: 10.1038/emm.2016.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klein JD, Wang Y, Blount MA, Molina PA, LaRocque LM, Ruiz JA, Sands JM. Metformin, an AMPK activator, stimulates the phosphorylation of aquaporin 2 and urea transporter A1 in inner medullary collecting ducts. Am J Physiol Renal Physiol 310: F1008–F1012, 2016. doi: 10.1152/ajprenal.00102.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knepper MA, Good DW, Burg MB. Ammonia and bicarbonate transport by rat cortical collecting ducts perfused in vitro. Am J Physiol Renal Physiol 249: F870–F877, 1985. doi: 10.1152/ajprenal.1985.249.6.F870. [DOI] [PubMed] [Google Scholar]
- 20.Knepper MA, Inoue T. Regulation of aquaporin-2 water channel trafficking by vasopressin. Curr Opin Cell Biol 9: 560–564, 1997. doi: 10.1016/S0955-0674(97)80034-8. [DOI] [PubMed] [Google Scholar]
- 21.Knepper MA, Kim GH, Fernández-Llama P, Ecelbarger CA. Regulation of thick ascending limb transport by vasopressin. J Am Soc Nephrol 10: 628–634, 1999. [DOI] [PubMed] [Google Scholar]
- 22.Kudo LH, César KR, Ping WC, Rocha AS. Effect of peritubular hypertonicity on water and urea transport of inner medullary collecting duct. Am J Physiol Renal Fluid Electrolyte Physiol 262: F338–F347, 1992. doi: 10.1152/ajprenal.1992.262.3.F338. [DOI] [PubMed] [Google Scholar]
- 23.Mamenko M, Dhande I, Tomilin V, Zaika O, Boukelmoune N, Zhu Y, Gonzalez-Garay ML, Pochynyuk O, Doris PA. Defective store-operated calcium entry causes partial nephrogenic diabetes insipidus. J Am Soc Nephrol 27: 2035–2048, 2016. doi: 10.1681/ASN.2014121200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moffat C, Harper ME. Metabolic functions of AMPK: aspects of structure and of natural mutations in the regulatory gamma subunits. IUBMB Life 62: 739–745, 2010. doi: 10.1002/iub.387. [DOI] [PubMed] [Google Scholar]
- 25.Nadler SP. Effects of hypertonicity on ADH-stimulated water permeability in rat inner medullary collecting duct. Am J Physiol Renal Fluid Electrolyte Physiol 258: F266–F272, 1990. doi: 10.1152/ajprenal.1990.258.2.F266. [DOI] [PubMed] [Google Scholar]
- 26.Nielsen S, Kwon TH, Christensen BM, Promeneur D, Frøkiaer J, Marples D. Physiology and pathophysiology of renal aquaporins. J Am Soc Nephrol 10: 647–663, 1999. [DOI] [PubMed] [Google Scholar]
- 27.Pastor-Soler NM, Hallows KR. AMP-activated protein kinase regulation of kidney tubular transport. Curr Opin Nephrol Hypertens 21: 523–533, 2012. doi: 10.1097/MNH.0b013e3283562390. [DOI] [PubMed] [Google Scholar]
- 28.Sands JM, Bichet DG; American College of Physicians; American Physiological Society . Nephrogenic diabetes insipidus. Ann Intern Med 144: 186–194, 2006. doi: 10.7326/0003-4819-144-3-200602070-00007. [DOI] [PubMed] [Google Scholar]
- 29.Sands JM, Knepper MA. Urea permeability of mammalian inner medullary collecting duct system and papillary surface epithelium. J Clin Invest 79: 138–147, 1987. doi: 10.1172/JCI112774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sands JM, Nonoguchi H, Knepper MA. Vasopressin effects on urea and H2O transport in inner medullary collecting duct subsegments. Am J Physiol Renal Fluid Electrolyte Physiol 253: F823–F832, 1987. doi: 10.1152/ajprenal.1987.253.5.F823. [DOI] [PubMed] [Google Scholar]
- 31.Snedecor GW, Cochran WG. Statistical Methods. Ames, IA: Iowa State Univ.Press, 1980. [Google Scholar]
- 32.Wang Y, Klein JD, Liedtke CM, Sands JM. Protein kinase C regulates urea permeability in the rat inner medullary collecting duct. Am J Physiol Renal Physiol 299: F1401–F1406, 2010. doi: 10.1152/ajprenal.00322.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilson JL, Miranda CA, Knepper MA. Vasopressin and the regulation of aquaporin-2. Clin Exp Nephrol 17: 751–764, 2013. doi: 10.1007/s10157-013-0789-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang C, Sands JM, Klein JD. Vasopressin rapidly increases phosphorylation of UT-A1 urea transporter in rat IMCDs through PKA. Am J Physiol Renal Physiol 282: F85–F90, 2002. doi: 10.1152/ajprenal.0054.2001. [DOI] [PubMed] [Google Scholar]