

Abstract
Hypokalemia contributes to the progression of chronic kidney disease, although a definitive pathophysiological theory to explain this remains to be established. K+ deficiency results in profound alterations in renal epithelial transport. These include an increase in salt reabsorption via the Na+-Cl− cotransporter (NCC) of the distal convoluted tubule (DCT), which minimizes electroneutral K+ loss in downstream nephron segments. In experimental conditions of dietary K+ depletion, punctate structures in the DCT containing crucial NCC-regulating kinases have been discovered in the murine DCT and termed “WNK bodies,” referring to their component, with no K (lysine) kinases (WNKs). We hypothesized that in humans, WNK bodies occur in hypokalemia as well. Renal needle biopsies of patients with chronic hypokalemic nephropathy and appropriate controls were examined by histological stains and immunofluorescence. Segment- and organelle-specific marker proteins were used to characterize the intrarenal and subcellular distribution of established WNK body constituents, namely, WNKs and Ste20-related proline-alanine-rich kinase (SPAK). In both patients with hypokalemia, WNKs and SPAK concentrated in non-membrane-bound cytoplasmic regions in the DCT, consistent with prior descriptions of WNK bodies. The putative WNK bodies were located in the perinuclear region close to, but not within, the endoplasmic reticulum. They were closely adjacent to microtubules but not clustered in aggresomes. Notably, we provide the first report of WNK bodies, which are functionally challenging structures associated with K+ deficiency, in human patients.
Keywords: hypokalemic nephropathy, microtubules, NCC, SPAK, trafficking
INTRODUCTION
Hypokalemia contributes to renal disease (10, 11). It is associated with the distinct histopathological entity of hypokalemic nephropathy, including tubular vacuolization and dilation, interstitial fibrosis, and lymphocyte and macrophage infiltrates (17, 18). The condition has often been described in patients with anorexia, particularly of the purging type involving diuretic or laxative abuse. Renovascular dysregulation (2, 17, 21) and complement system activation induced by increased ammoniagenesis (7, 15, 25) occur in hypokalemic nephropathy and have been proposed as crucial factors of its pathogenesis. To date, the distal convoluted tubule (DCT) has received little attention in the context of hypokalemic nephropathy. However, the DCT is an indispensable segment for systemic K+ homeostasis by its action on two downstream segments, the connecting tubule and collecting duct. Dietary K+ deficiency stimulates salt reabsorption via the Na+-Cl− cotransporter (NCC) of the DCT (23). The resulting Na+ depletion of urine before its passage through the connecting tubule and collecting duct, where electrogenic Na+ uptake is balanced by K+ secretion into the urine, prevents further K+ wasting (26). NCC is activated by Ste20-related proline-alanine-rich kinase (SPAK) and its homolog, oxidative stress-responsive kinase-1 (OSR1), after their phosphorylation by with no lysine kinases (WNKs). In murine models of dietary K+ deficiency, components of this pathway have been found in discrete, punctate structures in the DCT by Boyd-Shiwarski et al. (4), who termed them “WNK bodies.” Similar phenomena were observed in transgenic SPAK knockout (KO) mice, which display deficient NCC phosphorylation and hypokalemia (13). It has been discussed whether WNK bodies represent pathological aggregates arising in conditions of inadequate protein quality control (5) or serve to concentrate signaling processes involved in the distal tubular response to K+ stress (4).
Whether WNK bodies occur in patients with hypokalemia is unknown. To investigate this, we selected renal needle biopsies of two patients with medical histories of both chronic hypokalemia and nephropathy (estimated glomerular filtration rate <15) and compared them with appropriate controls. One case, previously described by Elitok et al. (6), involved a patient who developed cachexia over the course of 2 yr because of an eating disorder. She ingested laxatives (bisacodyl) and nonsteroidal anti-inflammatory drugs (NSAIDs) regularly. The second patient suffered from Munchausen syndrome and self-administered calcineurin inhibitors (CnI). She had a 22-yr history of hypokalemia and hypertension of unclear causes. For an overview of diagnostic data, see Supplement S1 (Supplemental Material for this article is available online at the American Journal of Physiology Renal Physiology website).
MATERIALS AND METHODS
Biopsy specimens.
This study was communicated to the Institutional Review Board (Ethics Commission) of Charité-Universitätsmedizin Berlin. It was classified as part of an ongoing pathological investigation (Department of Pathology of the Charité) with no requirement of informed consent and was undertaken in accordance with the Declaration of Helsinki. Renal needle biopsies of the two patients with chronic hypokalemia were obtained percutaneously. In the case of the patient with an eating disorder, two tissue cylinders with lengths of 11 and 10 mm were fixed in formalin and embedded in paraffin wax. Two cylinders of 15- and 16-mm length were taken from the kidney of the patient with Munchausen syndrome; 2 mm of each were fixed in glutaraldehyde and embedded in Epon for examination by electron microscopy. The remainder was formalin-fixed and paraffin-embedded. In both the patient with an eating disorder and the patient with Munchausen syndrome, renal histology was found to be consistent with chronic hypokalemic nephropathy by the Department of Pathology. Two renal biopsies were selected to serve as controls. One was obtained by percutaneous needle biopsy to determine the cause of a patient’s deteriorating renal function after 3 days of cefuroxime and ibuprofen therapy. Two tissue cylinders (13- and 12-mm length) were formalin-fixed and paraffin-embedded. Pathology reports on the control needle biopsy noted sclerosis of 4 of 28 glomeruli, with microangiopathy occurring in 2 of the remaining 24 nonsclerotized glomeruli, but no significant tubulointerstitial nephritis. Nontumorous and otherwise morphologically normal sections from a formalin-fixed, paraffin-embedded nephrectomy specimen of a patient with renal carcinoma served as a second control.
Antibodies.
The pS-WNK antibody (obtained from MRC-PPU Reagents, University of Dundee) was raised in sheep against residues 375–389 of human WNK1 phosphorylated on Ser382. It was first used by Thastrup et al., who performed validation studies by Western blot analysis using HEK cell lysates transfected with wild-type or T-loop-mutated human WNK1 and WNK4 isoforms (24). The pS373-SPAK antibody (MRC-PPU Reagents, University of Dundee) was raised in sheep against residues 367–379 of human SPAK phosphorylated on Ser373. Its use was first reported by Zagórska et al., who examined the phosphospecificity of the antibody by Western blot analysis using HeLa cell lysates transfected with human WNK1 siRNA or an S-motif-mutated OSR1 mutant (28). The rabbit NCC (3) and guinea pig type 2 Na+-K+-2Cl− cotransporter (NKCC2; 19) antibodies, generated by David Ellison and Kerim Mutig, respectively, are well established in our groups and have previously been used to stain human kidney sections (14). The WNK4 antibody was generated by David H. Ellison and has been validated in transgenic WNK4 KO mice (22). The remaining primary antibodies used in this study and validation methods employed by their respective commercial suppliers are listed in Supplement S2.
Immunofluorescence.
Formalin-fixed, paraffin-embedded, 3-µm-thick human kidney needle biopsy and nephrectomy sections were dewaxed in xylene and rehydrated via graded ethanol. Heat-induced epitope retrieval was performed for 6 min with a pressure cooker in citrate buffer, pH 6. Sections were blocked with 10% normal donkey serum (Abcam) and 1% bovine serum albumin (BSA; Serva) in TBS for 30 min at room temperature (RT). Five percent BSA in TBS was substituted as a blocking buffer for triple immunolabeling of p62, pS-WNK, and SPAK. The samples were incubated overnight at 4°C with primary antibodies (Supplement S2). Secondary antibodies (Supplement S3) were applied for 1 h at RT. Primary and secondary antibodies were dissolved in 1% normal donkey serum and 1% BSA in TBS. Polyclonal antibodies against phosphoproteins (pS-WNK and pS373-SPAK) were incubated with 10 µg/ml of their nonphosphorylated epitopes for 30 min at RT before application to block binding of nonphosphorylated WNKs and SPAK/OSR1. DAPI (1 µg/ml; Sigma-Aldrich) was added to all secondary antibody mixtures without Alexa Fluor 405. Sections were mounted in 1:9 PBS-glycerol and examined under a confocal microscope. Elution buffer for sequential immunostaining was prepared by mixing 20 ml 10% SDS and 17.5 ml 150 mM Tris·HCl, pH 6.8, with 62.5 ml distilled water (dH2O) and then adding 0.8 ml of 2-mercaptoethanol, as described by Gendusa et al. (8). Sections were transferred to elution buffer preheated to 50°C, maintained at 50°C with constant agitation for 60 min, and cooled to RT. Sections were mounted in 1:9 PBS-glycerol. Successful elution of the first round of staining was confirmed under a confocal microscope. Sections were restained with rabbit anti-WNK4 primary and donkey anti-rabbit Alexa Fluor 647-conjugated secondary antibodies.
Confocal microscopy and image processing.
Images and z-stacks were acquired using an LSM 5 Exciter confocal microscope (Zeiss) equipped with an Imager.M1 stand and a NeoFluar objective lens [×63/numerical aperture (NA) 1.40]. Laser lines of 405, 488, 543, and 633 nm were used. Differential interference contrast optics were used in some images. The system was operated with Zen 2008 software (Zeiss). All image processing was performed in Fiji v2.0. The z-stacks were converted to maximum-intensity projections, background was subtracted by rolling-ball function (20), and brightness and contrast were adjusted.
Immunoperoxidase staining.
Immunoperoxidase staining followed the immunofluorescence protocol until incubation of primary antibodies. Samples were bleached with 0.3% H2O2 for 15 min and incubated with a horseradish peroxidase-conjugated secondary antibody in 1% normal donkey serum and 1% BSA in TBS for 1 h at RT. Diaminobenzidine (Sigma) in dH2O with 1% H2O2 was applied as a chromogen. Staining was monitored under a microscope; the reaction was stopped by extensive washing with TBS. Samples were dehydrated with graded ethanol, cleared with xylene, and mounted in Eukitt quick-hardening mounting medium (Sigma-Aldrich).
Periodic acid-Schiff staining.
A periodic acid-Schiff (PAS) stain was applied to renal biopsy sections previously used for immunofluorescence. Sections were postfixed in Bouin’s fluid (1 part glacial acetic acid, 5 parts 37% formaldehyde solution, and 15 parts saturated aqueous picric acid) at 37°C for 4 h. After washing out excess picric acid with dH2O, they were placed in 1% periodic acid for 10 min and in Schiff reagent (Merck) for 25 min. Sections were incubated in running warm tap water for 10 min and then transferred to Mayer’s hemalum (Roth) for 10 min to counterstain nuclei. After bluing under running tap water for 10 min, sections were dehydrated with graded ethanol, cleared with xylene, and mounted in Eukitt.
Semithin section preparation and toluidine blue O staining.
Of the glutaraldehyde-fixed, Epon-embedded tissue cylinders belonging to the patient with Munchausen syndrome, sections of 300-nm thickness were cut using an ultramicrotome and stained with toluidine blue O.
Bright-field microscopy.
Immunoperoxidase-, PAS-, and toluidine blue O-stained sections were evaluated by bright-field microscopy under a Leica DMRB microscope equipped with PL Fluotar ×20/NA 0.50, ×40/NA 1.00, ×63/NA 1.40, and ×100/NA 1.30 objectives. An AxioCam MRc (Zeiss) camera and AxioVision SE64 software v4.8.3.0 (Zeiss) were used for image acquisition. Brightness and contrast were adjusted in Fiji v2.0.
Electron microscopy.
From Epon-embedded tissue of the patient with Munchausen syndrome, ultrathin sections of 80-nm thickness were transferred to Formvar-coated grids and stained with uranyl acetate and lead citrate. They were examined using a Leo EM 906 transmission electron microscope (Zeiss). Images were acquired at ×1,293–16,700 magnification. The tissue obtained from the patient with an eating disorder and controls had not been processed for electron microscopy.
RESULTS
Renal biopsies of patients with chronic hypokalemia display punctate accumulations of NCC-activating kinases specific to the DCT.
We used formalin-fixed, paraffin-embedded renal biopsy tissue from the two patients with hypokalemia and controls for immunofluorescence studies. Antibodies directed against SPAK and pS-WNK detected punctate accumulations of the kinases in the DCT cytoplasm of both patients with hypokalemia but not controls (Fig. 1A). SPAK and pS-WNK signals were colocalized within the puncta, which we recognized as the WNK bodies previously described in rodents and cultured cells (4, 5, 13). The pS-WNK antibody detects WNK isoforms phosphorylated within the activation loop at a site conserved between all four human WNK isoforms. Phosphorylation of this site is a prerequisite for their catalytic activity (24). pS373-SPAK was also detected in cytoplasmic puncta in the DCT, as well as its luminal membrane (identified by NCC expression; Fig. 1B). Besides NCC, the WNK-SPAK signaling pathway is involved in the regulation of multiple other cation-chloride cotransporters, including NKCC2 of the thick ascending limb (TAL; 16). Whereas patients with hypokalemia expressed pS373-SPAK in the luminal membrane of the TAL (identified by NKCC2 expression), no cytoplasmic puncta were found in the TAL (Fig. 1B).
Fig. 1.

Punctate inclusions of Na+-Cl− cotransporter (NCC)-activating kinases in the cytoplasm of the distal convoluted tubule (DCT) of patients with hypokalemia. A: the DCT is enriched with Ste20-related proline-alanine-rich kinase (SPAK) accumulations in both patients with hypokalemia but not in a morphologically normal control. The SPAK-positive structures in the patients with hypokalemia are also labeled by antibody against residues 375–389 of human with no lysine kinase-1 phosphorylated on Ser382 (pS-WNK). B: antibody against residues 367–379 of human SPAK phosphorylated on Ser373 (pS373-SPAK) is found in the luminal membranes of both the thick ascending limb [where it is colocalized with the type 2 Na+-K+-2Cl− cotransporter (NKCC2)] and the DCT (where it is colocalized with NCC). Its punctate cytoplasmic signal is limited to the DCT. Representative image of a patient with hypokalemia. Blue channels show DAPI nuclear counterstain. CnI, calcineurin inhibitors.
SPAK locates to the vicinity of the endoplasmic reticulum and microtubules in chronic hypokalemia.
To assess the subcellular location at which SPAK/OSR1 and WNKs accumulate, we conducted further double-immunofluorescence studies. Concomitant staining of pS373-SPAK and calreticulin, an endoplasmic reticulum (ER) lumen-resident protein, detected the presence of calreticulin in the close vicinity of pS373-SPAK puncta (Fig. 2A). Both calreticulin and pS373-SPAK puncta concentrated in the perinuclear compartment. When microtubule component α-tubulin and pS373-SPAK were double stained, the kinase accumulations were shown to be arranged along the α-tubulin filaments (Fig. 2B).
Fig. 2.

Inclusions of Na+-Cl− cotransporter (NCC)-activating kinases are found adjacent to the endoplasmic reticulum and microtubules. All images in this figure are from biopsies of patients with chronic hypokalemia. A: the endoplasmic reticulum lumen protein calreticulin is consistently localized in the vicinity of puncta labeled by antibody against residues 367–379 of human Ste20-related proline-alanine-rich kinase (SPAK) phosphorylated on Ser373 (pS373-SPAK) puncta in the perinuclear compartment. B: pS373-SPAK-positive puncta are arranged alongside filamentous α-tubulin (microtubular) immunofluorescence. C: pS373-SPAK does not accumulate around microtubule organizing centers (MTOCs). MTOCs are visible as sites of concentrated γ-tubulin expression, appearing once per cell. D: Histone deacetylase 6 (HDAC6) is expressed in all renal segments and compartments. In the distal convoluted tubule (DCT), it displays an intense, punctated signal colocalized with pS373-SPAK. E: p62 shows a speckled cytoplasmic pattern in all renal segments and compartments, with no apparent spatial relationship to the pS373-SPAK signal in the DCT. F: immunoperoxidase staining of vimentin is limited to interstitial and vascular compartments, whereas tubular epithelia are vimentin-negative (DT, distal tubule; IA, interlobular artery; PT, proximal tubule). G: the hypokalemic DCT contains punctate with no lysine kinase-4 (WNK4) accumulations. WNK4 immunostaining was performed after eluting several other primary antibodies. In the first round of staining, pS373-SPAK was detected in equally located cytoplasmic puncta. Blue channels show DAPI nuclear counterstain.
WNK bodies lack several defining properties of aggresomes.
Cells may react to pathological protein aggregation by microtubular transport of polyubiquitinated, misfolded polypeptides to the aggresome, a perinuclear organelle that forms around microtubule organizing centers (MTOCs) to facilitate autophagolysosomal degradation at a central processing site (8a). Transport of cargo along the microtubule to the aggresome requires the ubiquitin-binding domain of the microtubule-associated deacetylase histone deacetylase 6 (HDAC6), which links polyubiquitinated polypeptides to dynein motors (9a). Characteristics of aggresomes include p62/sequestosome-1 content and a surrounding “cage” of the intermediate filament vimentin (6a).
The WNK body-microtubule association prompted us to investigate whether some of the WNK bodies were located around MTOCs. Immunostaining of γ-tubulin, a protein enriched in MTOCs, displayed a single site of especially strong expression per epithelial cell. We presumed this to represent the basal body/kinetosome of cilia. WNK bodies did not form around these putative MTOCs (Fig. 2C). HDAC6 immunofluorescence was granular and concentrated in the apical region of renal tubules, consistent with the observed apical polarization of α-tubulin immunofluorescence. In the DCT of the patients with hypokalemia, we found large cytoplasmic HDAC6 foci that colocalized with WNK bodies in double stainings (Fig. 2D). The p62 immunostaining detected non-segment-specific cytoplasmic expression in renal epithelia with a speckled appearance. There was no p62 found in the WNK bodies of the DCT of the patients with hypokalemia (Fig. 2E). Vimentin expression was observed in the glomerular, interstitial, and vascular compartments, whereas all tubular epithelia, including the DCT of both patients with hypokalemia, were vimentin-negative (Fig. 2F).
The WNK4 isoform is contained in SPAK/WNK puncta.
The pS-WNK antibody we used (Fig. 1A) could not differentiate between T-loop-phosphorylated WNK isoforms. To identify a specific isoform within WNK bodies, we restained sections with a WNK4 antibody after elution of previously applied antibodies. WNK4 was detected in cytoplasmic puncta in the hypokalemic biopsies. The pS373-SPAK antibody had shown the same pattern in the first round of staining (Fig. 2G).
Ultrastructure of WNK bodies.
We examined the biopsy of the patient with Munchausen syndrome by transmission electron microscopy to verify the presence of WNK bodies. In the DCT (Fig. 3A), spherical, hypodense regions in the cytosol were common (Fig. 3B). Their appearance mirrored the WNK-positive structures previously characterized by immunoelectron microscopy in murine and HEK-293 models of WNK body formation (4, 5). Rough ER was concentrated at the border of these putative WNK bodies (Fig. 3B), consistent with our finding of intensified calreticulin staining around immunoreactive SPAK/WNK puncta (Fig. 2A).
Fig. 3.
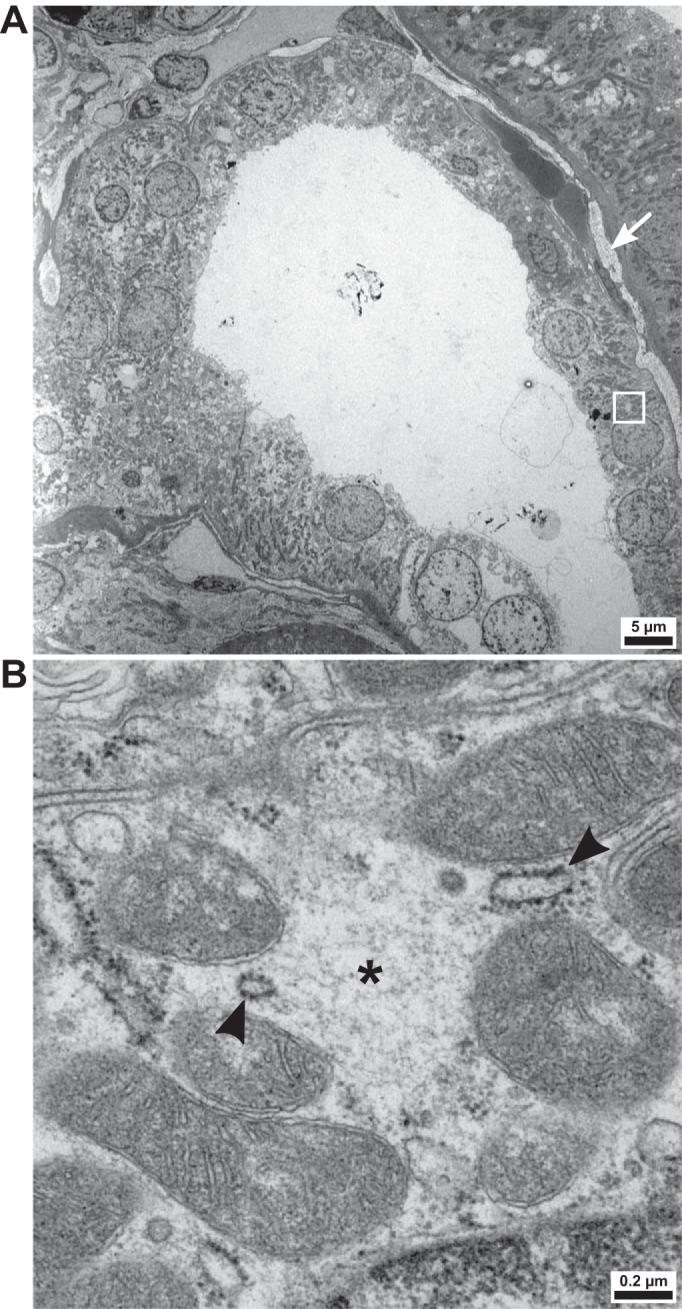
Ultrastructure of the distal convoluted tubule (DCT) of a patient with hypokalemia. A: overview of a cross section of the DCT in the patient with Munchausen syndrome. The region of interest (see B) is indicated by a white rectangle. To the right of the tubule, interstitial collagen deposits are visible (arrow). B: the DCT is enriched with hypodense, spherical cytoplasmic regions not delineated by membranes (*). Their ultrastructure is compatible with prior descriptions of with no lysine kinase (WNK) bodies. Rough endoplasmic reticulum (arrowheads) is found at their borders.
Comparative histopathology of patients with hypokalemia.
We used differential interference contrast and PAS staining for the light microscopic examination of gross DCT morphology. Kinase accumulation was not specific to the damaged tubules, demonstrated by the comparable appearance of tubules containing WNK bodies in patients with hypokalemia to the WNK body-free DCT of a control needle biopsy (Fig. 4A). Morphologically intact DCTs remained frequent in both patients (Fig. 4, B and C; normal control in Fig. 4D) despite their pathology. Segmentally, tubular vacuolization had occurred in both patients with hypokalemia (Fig. 5, A, B, and D; normal control in Fig. 5C). Severe damage to tubules by larger and less numerous vacuoles was found in the patient with an eating disorder; the biopsy of the patient with Munchausen syndrome displayed finer and isometric vacuolization of the proximal tubules. Hyaline casts in distal tubules were frequent in both cases. Dilation of the distal tubules, often accompanied by cellular detritus inside the lumen, was specific to the patient with an eating disorder, as were lymphocytic infiltrates. The patient with Munchausen syndrome showed a lesser degree of interstitial fibrosis (Fig. 3A). Furthermore, fibrosis was detected in both patients by picrosirius red staining (not shown).
Fig. 4.

With no lysine kinase (WNK) bodies appear in morphologically normal tubules. A: antibody against residues 367–379 of human Ste20-related proline-alanine-rich kinase (SPAK) phosphorylated on Ser373 (pS373-SPAK) displays a punctate signal in hypokalemic patients, shown here in a distal convoluted tubule (DCT) with normal morphology as visualized by differential interference contrast optics. Na+-Cl− cotransporter (NCC) locates to the luminal DCT membrane in the biopsies of patients with hypokalemia and control biopsies. B–D: renal cortex of both patients with hypokalemia (B and C) and a control needle biopsy (D). Periodic acid-Schiff stain, ×400 magnification with regions of interest containing DCTs at ×1,000 magnification. In both patients with hypokalemia, the DCTs that have not been affected by vacuolization display morphology comparable to that of the control. CnI, calcineurin inhibitors.
Fig. 5.

Histopathology of two patients with hypokalemia with nephropathy. A: renal needle biopsy of the patient with an eating disorder. Distal tubules are frequently dilated and contain hyaline casts (*). There is hyperplasia of collecting duct cells, accompanied by massive dilation of the lumen, which is obstructed by cellular debris (arrow). Tubular vacuolization (arrowheads) and atrophy are seen alongside interstitial fibrosis with cellular infiltrates, whereas glomeruli are normal. B: renal biopsy of the patient with Munchausen syndrome. Pathological changes are discrete relative to the patient with an eating disorder. Tubular vacuolization (arrowheads) has occurred in a microvesicular fashion and principally in proximal tubules. Interstitial changes are mild, and glomeruli show normal morphology. C: control needle biopsy, obtained after the patient’s renal function declined upon cefuroxime and ibuprofen administration. Two normal glomeruli and intact proximal and distal tubules are visible. D: microvesicular alterations (arrowheads) of the proximal tubular cytoplasm in the patient with Munchausen syndrome. A–C show periodic acid-Schiff-stained, paraffin-embedded sections, and D shows a toluidine blue O-stained, semithin, Epon-embedded section. CnI, calcineurin inhibitors.
DISCUSSION
Equivalent structures to the WNK bodies reported here in patients have been documented in the DCT in several mouse models, including dietary K+ depletion (4) and aldosterone infusion (5). There, WNK bodies developed after a few days of a K+-deficient diet or aldosterone injection. WNK body formation thus cannot be exclusive to the later stages of hypokalemic kidney injury; that is, it is secondary to hypokalemia rather than hypokalemic nephropathy. Our findings appear to corroborate this: despite extensive tubular vacuolization, both biopsies did contain numerous DCTs with normal morphology (Fig. 4, B and C; normal control in Fig. 4D) that had already formed WNK bodies (Fig. 4A). The possibility that WNK bodies may advance nephropathy in hypokalemic conditions remains. This has been previously addressed in the context of aldosterone administration in mice (5), where vimentin foci and deficient E-cadherin expression in DCT cells were observed. The authors proposed that proteotoxicity induced by aldosterone might have provoked these signs of cellular stress.
A common form of proteotoxicity is the contact of cell content with hydrophobic sequences exposed by protein misfolding. Misfolded proteins are prone to aggregation, and WNK body formation requires a hydrophobic motif of kidney-specific WNK1 (4), suggesting that hydrophobic forces akin to those in misfolded protein aggregation could be critical to WNK body formation. However, WNK bodies do not display several properties typical of protein aggregates. By live-cell imaging, Boyd-Shiwarski et al. (4) documented a considerable motility of WNK bodies in HEK-293 cells expressing enhanced green fluorescent protein-tagged kidney-specific WNK1, thus demonstrating their dynamic nature in vitro. Furthermore, the kinases are not concentrated in aggresomes. They form at, or are transported to, locations separate from the DCT cells’ MTOCs. Of aggresomal proteins, neither p62 nor vimentin are components of WNK bodies, though we detected HDAC6 within them. HDAC6 could facilitate microtubular transport of WNK bodies to the perinuclear region, where they are found adjacently to the ER. Although the constellation of microtubular adjacency and the documented mobility of WNK bodies suggests their participation in cellular trafficking, they do not appear to be of a vesicular nature, as they lacked a delineating membrane in our ultrastructural studies.
Comparative assessment of the renal biopsies of the patients with hypokalemia revealed marked differences (Fig. 5, A, B, and D; normal control in Fig. 5C). We observed the hallmarks of chronic hypokalemic nephropathy in the patient with an eating disorder, including large cytoplasmic vacuoles in proximal and distal tubules, pronounced hyperplasia and dilation of collecting ducts (9, 21), and interstitial fibrosis. The histopathology of the Munchausen-associated case [fine isometric vacuolization principally affecting the proximal tubules, some interstitial fibrosis (Fig. 3A), with clinical pathology reports noting mild arterial hyalinosis] may reflect acute CnI nephrotoxicity (12). Factors common to hypokalemia- and CnI-induced renal injury may have acted synergistically in the patient, as both pathologies are linked to ischemia and renin-angiotensin-aldosterone system (RAAS) activation. Cyclooxygenase suppression from NSAID intake could similarly have exacerbated hypokalemic vascular dysregulation in the patient with an eating disorder. An overactive RAAS was documented in the patient with Munchausen syndrome. Although renin and aldosterone were not measured in the patient with an eating disorder, laxative abuse and anorexia are strong stimulants of aldosterone secretion. Furthermore, the RAAS is locally augmented in the kidney during hypokalemia despite systemic downregulation (2). Walsh et al. argue that secondary hyperaldosteronism is the key mediator of the nephropathy observed in cases of chronic hypokalemia, as patients with Bartter syndrome are prone to nephropathy and patients with Gitelman syndrome are not (27). Whereas both patient groups develop hypokalemia, patients with Bartter syndrome characteristically display higher plasma aldosterone levels and salt and water losses. WNK bodies appear when the salt transport load of the DCT is increased under hypokalemic conditions, which includes aldosterone action (5), but they occur in the aldosterone-insensitive DCT1 and the aldosterone-sensitive DCT2. Thus, the prominent involvement of WNK bodies in an aldosterone-based theory of chronic hypokalemic nephropathy appears unlikely. It is also notable that SPAK KO mice, which display a Gitelman-like phenotype, form WNK bodies with WNK and OSR1 content (13).
An unexplored avenue of investigation concerns the widespread nature of high-Na+, low-K+ “Western diets” as they relate to WNK body formation. In the United States, average daily K+ intake among adults is 2,795 mg, falling short of the recommended 4,700 mg (1). Nonetheless, normokalemia is typically maintained on a Western diet, whereas mouse models of WNK body formation involving dietary K+ deficiency are based on essentially K+-free diets. The latter trigger responses aimed at maintaining K+ homeostasis, including phosphorylation of NCC (23), but that are insufficient to maintain adequate K+ blood levels (4). Plasma K+ is not maintained within its physiological range in any other animal models in which WNK bodies occur. SPAK KO mice show a trend toward hypokalemia at baseline and are predisposed to hypokalemia upon K+ deprivation (13). Aldosterone-treated mice are expected to display hypokalemia, though Cheema et al. did not measure plasma K+ in their study involving 7 days of aldosterone infusion (5). Although we detected WNK bodies only in the two biopsies obtained from patients with hypokalemia, the possibility that WNK bodies are common in individuals consuming Western diets, even in normokalemia, cannot currently be excluded. Experiments concerning WNK body formation in animals maintained on a diet mimicking the typical Western diet, as opposed to settings of decompensated K+ depletion, have yet to be performed.
In sum, we confirm the occurrence of WNK bodies, described in several in vitro and rodent models, in a human patient with hypokalemia. WNK bodies were concentrated alongside microtubules but lacked several characteristics of aggresomes. They preferentially located to perinuclear sites adjacent to the ER. Whether WNK bodies contribute to the progression of renal and electrolyte disorders or represent a homeostatic phenomenon without adverse effects is a question that exceeds the scope of the present study.
GRANTS
This study was financially supported by the Deutsche Forschungsgemeinschaft (Grants MU 2924/2-1,2 and BA 700/22-1,2; INST 335/596-1 FUGG to S. Bachmann). Furthermore, D. H. Ellison was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants R01-DK-051496 and R01-DK-054983, Department of Veterans Affairs Grant I01 BX002228, and a Transatlantic Network of Excellence award from Fondation LeDucq.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.N.T., W.S., and S.B. conceived and designed research; M.N.T. performed experiments; M.N.T. and W.S. analyzed data; M.N.T. interpreted results of experiments; M.N.T. prepared figures; M.N.T. and S.B. drafted manuscript; K.M., D.H.E., and R.K. edited and revised manuscript; M.N.T., W.S. , K.M., D.H.E., R.K., and S.B. approved final version of manuscript.
Supplemental Data
Diagnostic data of two chronically hypokalemic patients - .docx (17 KB)
Primary antibodies used in this study - .docx (15 KB)
Secondary antibodies used in this study - .docx (13 KB)
ACKNOWLEDGMENTS
We thank John Horn and Kerstin Riskowsky for expert technical assistance. We are grateful to Friedrich C. Luft for advice in the editing of the manuscript.
REFERENCES
- 1.Bailey RL, Parker EA, Rhodes DG, Goldman JD, Clemens JC, Moshfegh AJ, Thuppal SV, Weaver CM. Estimating sodium and potassium intakes and their ratio in the American diet: data from the 2011-2012 NHANES. J Nutr 146: 745–750, 2016. doi: 10.3945/jn.115.221184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beck N, Shaw JO. Thromboxane B2 and prostaglandin E2 in the K+-depleted rat kidney. Am J Physiol Renal Physiol 240: F151–F157, 1981. doi: 10.1152/ajprenal.1981.240.2.F151. [DOI] [PubMed] [Google Scholar]
- 3.Bostanjoglo M, Reeves WB, Reilly RF, Velázquez H, Robertson N, Litwack G, Morsing P, Dørup J, Bachmann S, Ellison DH. 11Beta-hydroxysteroid dehydrogenase, mineralocorticoid receptor, and thiazide-sensitive Na-Cl cotransporter expression by distal tubules. J Am Soc Nephrol 9: 1347–1358, 1998. [DOI] [PubMed] [Google Scholar]
- 4.Boyd-Shiwarski CR, Shiwarski DJ, Roy A, Namboodiri HN, Nkashama LJ, Xie J, McClain KL, Marciszyn A, Kleyman TR, Tan RJ, Stolz DB, Puthenveedu MA, Huang CL, Subramanya AR. Potassium-regulated distal tubule WNK bodies are kidney-specific WNK1 dependent. Mol Biol Cell 29: 499–509, 2018. doi: 10.1091/mbc.E17-08-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheema MU, Damkier HH, Nielsen J, Poulsen ET, Enghild JJ, Fenton RA, Praetorius J. Distal renal tubules are deficient in aggresome formation and autophagy upon aldosterone administration. PLoS One 9: e101258, 2014. doi: 10.1371/journal.pone.0101258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elitok S, Bieringer M, Schneider W, Luft FC. Kaliopenic nephropathy revisited. Clin Kidney J 9: 543–546, 2016. doi: 10.1093/ckj/sfv154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6a.Garcia-Mata R, Gao YS, Sztul E. Hassles with taking out the garbage: aggravating aggresomes. Traffic 3: 388–396, 2002. [DOI] [PubMed] [Google Scholar]
- 7.Fervenza FC, Rabkin R. The role of growth factors and ammonia in the genesis of hypokalemic nephropathy. J Ren Nutr 12: 151–159, 2002. doi: 10.1053/jren.2002.33511. [DOI] [PubMed] [Google Scholar]
- 8.Gendusa R, Scalia CR, Buscone S, Cattoretti G. Elution of high-affinity (>10−9 KD) antibodies from tissue sections: clues to the molecular mechanism and use in sequential immunostaining. J Histochem Cytochem 62: 519–531, 2014. doi: 10.1369/0022155414536732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8a.Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol 143: 1883–1898, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kimura T, Nishino T, Maruyama N, Hamano K, Kubo A, Iwano M, Shiiki H. Expression of Bcl-2 and Bax in hypokalemic nephropathy in rats. Pathobiology 69: 237–248, 2001. doi: 10.1159/000064334. [DOI] [PubMed] [Google Scholar]
- 9a.Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115: 727–738, 2003. [DOI] [PubMed] [Google Scholar]
- 10.Korgaonkar S, Tilea A, Gillespie BW, Kiser M, Eisele G, Finkelstein F, Kotanko P, Pitt B, Saran R. Serum potassium and outcomes in CKD: insights from the RRI-CKD cohort study. Clin J Am Soc Nephrol 5: 762–769, 2010. doi: 10.2215/CJN.05850809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liang CC, Yeh HC. Hypokalemic nephropathy in anorexia nervosa. CMAJ 183: E761, 2011. doi: 10.1503/cmaj.101790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liptak P, Ivanyi B. Primer: histopathology of calcineurin-inhibitor toxicity in renal allografts. Nat Clin Pract Nephrol 2: 398–404, 2006. doi: 10.1038/ncpneph0225. [DOI] [PubMed] [Google Scholar]
- 13.McCormick JA, Mutig K, Nelson JH, Saritas T, Hoorn EJ, Yang CL, Rogers S, Curry J, Delpire E, Bachmann S, Ellison DH. A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab 14: 352–364, 2011. doi: 10.1016/j.cmet.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mutig K, Paliege A, Kahl T, Jöns T, Müller-Esterl W, Bachmann S. Vasopressin V2 receptor expression along rat, mouse, and human renal epithelia with focus on TAL. Am J Physiol Renal Physiol 293: F1166–F1177, 2007. doi: 10.1152/ajprenal.00196.2007. [DOI] [PubMed] [Google Scholar]
- 15.Nath KA, Hostetter MK, Hostetter TH. Increased ammoniagenesis as a determinant of progressive renal injury. Am J Kidney Dis 17: 654–657, 1991. doi: 10.1016/S0272-6386(12)80344-1. [DOI] [PubMed] [Google Scholar]
- 16.Ponce-Coria J, San-Cristobal P, Kahle KT, Vazquez N, Pacheco-Alvarez D, de Los Heros P, Juárez P, Muñoz E, Michel G, Bobadilla NA, Gimenez I, Lifton RP, Hebert SC, Gamba G. Regulation of NKCC2 by a chloride-sensing mechanism involving the WNK3 and SPAK kinases. Proc Natl Acad Sci USA 105: 8458–8463, 2008. doi: 10.1073/pnas.0802966105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reungjui S, Roncal CA, Sato W, Glushakova OY, Croker BP, Suga S, Ouyang X, Tungsanga K, Nakagawa T, Johnson RJ, Mu W. Hypokalemic nephropathy is associated with impaired angiogenesis. J Am Soc Nephrol 19: 125–134, 2008. doi: 10.1681/ASN.2007030261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Riemenschneider T, Bohle A. Morphologic aspects of low-potassium and low-sodium nephropathy. Clin Nephrol 19: 271–279, 1983. [PubMed] [Google Scholar]
- 19.Schmitt R, Klussmann E, Kahl T, Ellison DH, Bachmann S. Renal expression of sodium transporters and aquaporin-2 in hypothyroid rats. Am J Physiol Renal Physiol 284: F1097–F1104, 2003. doi: 10.1152/ajprenal.00368.2002. [DOI] [PubMed] [Google Scholar]
- 20.Sternberg SR. Biomedical image processing. Computer 16: 22–34, 1983. doi: 10.1109/MC.1983.1654163. [DOI] [Google Scholar]
- 21.Suga SI, Phillips MI, Ray PE, Raleigh JA, Vio CP, Kim YG, Mazzali M, Gordon KL, Hughes J, Johnson RJ. Hypokalemia induces renal injury and alterations in vasoactive mediators that favor salt sensitivity. Am J Physiol Renal Physiol 281: F620–F629, 2001. doi: 10.1152/ajprenal.2001.281.4.F620. [DOI] [PubMed] [Google Scholar]
- 22.Terker AS, Castañeda-Bueno M, Ferdaus MZ, Cornelius RJ, Erspamer KJ, Su XT, Miller LN, McCormick JA, Wang WH, Gamba G, Yang CL, Ellison DH. With no lysine kinase 4 modulates sodium potassium 2 chloride cotransporter activity in vivo. Am J Physiol Renal Physiol 315: F781–F790, 2018. doi: 10.1152/ajprenal.00485.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Terker AS, Zhang C, McCormick JA, Lazelle RA, Zhang C, Meermeier NP, Siler DA, Park HJ, Fu Y, Cohen DM, Weinstein AM, Wang WH, Yang CL, Ellison DH. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride. Cell Metab 21: 39–50, 2015. doi: 10.1016/j.cmet.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thastrup JO, Rafiqi FH, Vitari AC, Pozo-Guisado E, Deak M, Mehellou Y, Alessi DR. SPAK/OSR1 regulate NKCC1 and WNK activity: analysis of WNK isoform interactions and activation by T-loop trans-autophosphorylation. Biochem J 441: 325–337, 2012. doi: 10.1042/BJ20111879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tolins JP, Hostetter MK, Hostetter TH. Hypokalemic nephropathy in the rat. Role of ammonia in chronic tubular injury. J Clin Invest 79: 1447–1458, 1987. doi: 10.1172/JCI112973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wade JB, Liu J, Coleman R, Grimm PR, Delpire E, Welling PA. SPAK-mediated NCC regulation in response to low-K+ diet. Am J Physiol Renal Physiol 308: F923–F931, 2015. doi: 10.1152/ajprenal.00388.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walsh SB, Unwin E, Vargas-Poussou R, Houillier P, Unwin R. Does hypokalaemia cause nephropathy? An observational study of renal function in patients with Bartter or Gitelman syndrome. QJM 104: 939–944, 2011. doi: 10.1093/qjmed/hcr095. [DOI] [PubMed] [Google Scholar]
- 28.Zagórska A, Pozo-Guisado E, Boudeau J, Vitari AC, Rafiqi FH, Thastrup J, Deak M, Campbell DG, Morrice NA, Prescott AR, Alessi DR. Regulation of activity and localization of the WNK1 protein kinase by hyperosmotic stress. J Cell Biol 176: 89–100, 2007. doi: 10.1083/jcb.200605093. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Diagnostic data of two chronically hypokalemic patients - .docx (17 KB)
Primary antibodies used in this study - .docx (15 KB)
Secondary antibodies used in this study - .docx (13 KB)