

Abstract
Plasma membrane Na+/Ca2+ exchanger-1 (NCX1) helps regulate the cytosolic Ca2+ concentration ([Ca2+]CYT) in arterial myocytes. NCX1 mediates both Ca2+ entry and exit and tends to promote net Ca2+ entry in partially constricted arteries. Mean blood pressure (telemetry) is elevated by ≈10 mmHg in transgenic (TG) mice that overexpress NCX1 specifically in smooth muscle. We tested the hypothesis that NCX1 overexpression mediates Ca2+ gain and elevated [Ca2+]CYT in exposed femoral arteries that also express the Ca2+ biosensor exogenous myosin light chain kinase. [Ca2+]CYT and the NCX1-dependent (SEA0400-sensitive) component, ≈15% of total basal constriction in controls, were increased in TG arteries, but constrictions to phenylephrine and ANG II were comparable in TG and control arteries. Normalized phenylephrine dose-response curves and constriction to 30 and 300 ng/kg iv ANG II were virtually identical in control and TG arteries. ANG II-evoked constrictions, superimposed on elevated basal tone, accounted for the larger blood pressure responses to ANG II in TG arteries. TG and control mouse arteries fit the same pCa-constriction relationship over a wide range of pCa (≈125–500 nM). Vasodilation to acetylcholine, normalized to passive diameter, was also comparable in TG and control arteries, implying normal endothelial function. TG artery Na+ nitroprusside (nitric oxide donor)-induced dilations were, however, shifted to lower Na+ nitroprusside concentrations, indicating that TG myocyte vasodilator mechanisms were augmented. Maximum arterial dilation was comparable in TG and control mice, although passive diameter was ≈6–7% smaller in TG mice. The changes in TG arteries were apparently largely functional rather than structural, despite the congenital hypertension.
NEW & NOTEWORTHY Smooth muscle Na+/Ca2+ exchanger-1 transgene overexpression (TG mice) increases femoral artery basal cytosolic Ca2+ concentration ([Ca2+]CYT) and tone in vivo and raises blood pressure. Arterial constriction to phenylephrine and angiotensin II are normal but superimposed on the augmented basal [Ca2+]CYT and tone (constriction) in TG mouse arteries. Similar effects in resistance arteries would explain the elevated blood pressure. Acetylcholine-induced vasodilation is unimpaired, implying a normal endothelium, but TG arteries are hypersensitive to sodium nitroprusside.
Keywords: arterial tone; SEA0400, sodium nitroprusside; vasodilation
INTRODUCTION
Ca2+ homeostasis plays a fundamental role in vasoconstriction and hemodynamics because Ca2+ triggers contraction in vascular smooth muscle (47). The plasma membrane (PM) Na+/Ca2+ exchanger (NCX) is unique among the many ion channels and transporters that normally regulate the cytosolic Ca2+ concentration ([Ca2+]CYT) and signaling in arterial smooth muscle cells (ASMCs). NCX can transport Ca2+ either into or out of ASMCs, depending on the Na+ and Ca2+ concentration gradients and membrane potential (Vm). The net NCX-mediated Ca2+ flux direction, inward or outward, is determined by the net driving force on the exchanger, Vm – ENa/Ca, where ENa/Ca = 3ENa – 2ECa and ENa and ECa are, respectively, the Na+ and Ca2+ equilibrium potentials (9). In cardiomyocytes during diastole, and in quiescent neurons, Vm is on the order of −70 mV and is more negative than ENa/Ca; thus, NCX mediates Ca2+ efflux, primarily, in these cells. Most ASMCs are tonically contracted in vivo to maintain arterial tone, and Vm is generally about −30 to −45 mV (29, 32) and is more positive than calculated ENa/Ca.1 NCX should, therefore, mediate Ca2+ influx, primarily in ASMCs (23, 44, 61, 62). In this case, overexpression of NCX would be expected to enhance Ca2+ influx, elevate [Ca2+]CYT, augment Ca2+ signaling and arterial tone, and increase blood pressure (BP). In fact, transgenic (TG) mice that overexpress NCX 1 specifically in smooth muscle (SM-NCX1-TG mice) have elevated BP and enhanced salt sensitivity (23).
The discovery of NCX in arterial smooth muscle fostered the view that the cooperative action of PM Na+ pumps and NCX contributes to the regulation of ASMC [Ca2+]CYT and BP (6, 46). Indeed, NCX1 and ouabain-sensitive α2-Na+ pumps as well as certain receptor- and store-operated (transient receptor potential) channels that are permeable to Na+ and Ca2+ colocalize in PM microdomains at ASMC PM-sarcoplasmic reticulum (SR) junctions [“PLasmERosomes” (3, 8, 27, 65)]. Together, these transporters all help regulate local cytosolic Na+ concentration ([Na+]CYT) and [Ca2+]CYT and SR Ca2+ stores in human and rodent ASMCs (34, 39, 54).
Expression of NCX1 and of the SR Ca2+ pump (SERCA) as well as several Ca2+-selective channels is increased in ASMCs in many animal models of hypertension (10, 42, 65). This Ca2+ transporter “protein remodeling” appears to be due to a direct action of ouabain on α2-Na+ pumps, because incubation of primary cultured rodent or human ASMCs with nanomolar ouabain also upregulates these proteins (34, 43). Moreover, mice with mutant ouabain-resistant α2-Na+ pumps do not develop ouabain-induced or adrenocorticotropic hormone-induced hypertension, and the NCX blocker KBR7943 prevents the development of adrenocorticotropic hormone-induced hypertension (14, 15, 36). These studies have indicated that the α2-Na+ pump ouabain-binding site and NCX1 are intimately involved in the control of Ca2+ homeostasis and BP and contribute to the generation of many forms of hypertension.
Given this key role of NCX1 in arteries, the present report describes experiments designed to test the hypothesis that arterial NCX1-mediated Ca2+ entry is enhanced in SM-NCX1-TG mice, thereby increasing basal [Ca2+]CYT and augmenting Ca2+ signaling and constriction in vivo. The experiments were performed on exposed femoral arteries of live, anesthetized mice because of the following: 1) simultaneous [Ca2+]CYT and diameter measurements have been validated in this preparation (60); 2) the effects of local and systemic application of various pharmacological agents can be assessed in vivo, e.g., femoral artery [Ca2+]CYT is increased and diameter is decreased in mice with angiotensin (ANG) II-induced hypertension (58); and 3) the effects of genetic engineering (e.g., arterial NCX1 overexpression) on arterial [Ca2+]CYT and diameter also can be measured directly. Since NCX1-TG mice are modestly hypertensive (23) and endothelium-dependent vasodilation is often attenuated in hypertension (55), acetylcholine (ACh)-induced vasodilation was also examined.
BP is determined largely by the “tone” [constriction as a percentage of passive diameter (PD)] in small “resistance arteries,” with only a small contribution from constriction of large muscular arteries such as the femoral artery (4, 17). Nevertheless, most of the same vasoconstrictor and vasodilator receptors as well as Ca2+ transporter proteins, including NCX1, are expressed in resistance and muscular arteries (39, 41, 57, 61, 62), even though quantitative differences in Ca2+ handling surely contribute to functional dissimilarities. We reasoned that the effects of NCX1 upregulation and increased Ca2+ delivery on femoral artery Ca2+ signaling and diameter in vivo might also provide clues to the effects of NCX1 upregulation in resistance arteries. Indeed, the enhanced myogenic tone in isolated small resistance arteries of TG mice (unpublished observations) seems consistent with the enhanced sympathetic drive-dependent tone in the femoral arteries of TG mice (present study). Thus, despite the likely compensatory action of other transporters, which may be differently regulated in different vascular beds, enhanced Ca2+ delivery mediated by upregulated NCX1 and its effects on arterial constriction appeared to dominate.
Notwithstanding the elevated basal arterial [Ca2+]CYT and BP in TG mice, vasoconstrictor-evoked Ca2+ signals and ACh-induced vasodilation were comparable in control and TG mouse femoral arteries. These results, which are contrary to expectation, may provide novel insights into some of the peripheral mechanisms that contribute to the pathogenesis of hypertension.
MATERIALS AND METHODS
Experimental Mice and Ethical Approval
All husbandry and experimental procedures involving mice complied with the standards stated in the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of Maryland-Baltimore. Mice were housed in the University of Maryland-Baltimore Veterinary Resources facility and were fed standard laboratory chow.
SM-NCX1-TG mice (designated N1.3Tg/Tg-10) were originally generated by Prof. Takahiro Iwamoto (Fukuoka University, Fukuoka, Japan) (23). The transgene was randomly inserted into the genome so that mice would have several copies of the gene and greatly overexpress the protein (Fig. 1 and see Supplementary Fig. 3 in Ref. 23), but the copy number in each mouse was not determined. Mice were generously provided to us by Prof. Iwamoto, and the mouse line was rederived on a C57Bl/6 background. It was then crossed with a line that expresses a modified exogenous myosin light chain kinase transgene (exMLCK) in smooth muscles (60); exMLCK is a green fluorescent protein-based cyan/yellow fluorescent protein (CFP/YFP; eGFP) Förster resonance energy transfer (FRET) Ca2+ biosensor (18). The offspring of exMLCK/SM-NCX1-TG mice (on a C57Bl/6 background) were then inbred to generate mice homozygous for both transgenes. For convenience, inbred mice are designated as “NCX1-TG” or “TG” mice. Mice that expressed exMLCK, but not the NCX1 transgene, were used as control (Ctrl) mice. The presence of the exMLCK gene has no detectable effect on the contractile and hemodynamic properties of the arteries (60).
Fig. 1.
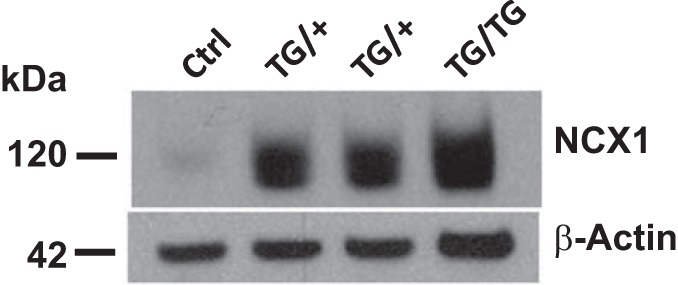
Na+/Ca2+ exchanger-1 (NCX1) is overexpressed in smooth muscle-specific NCX1-transgenic (TG) mice. Immunoblots show deendothelialized aortic membrane proteins from control (Ctrl) and heterozygous and homozygous TG mice that overexpress NCX1 specifically in smooth muscle (SM-NCX1-TG mice) (TG/+ and TG/TG, respectively). TG/TG mice were used for all physiological experiments in this study. The NCX1 transgene was randomly inserted into the genome, and the protein was, therefore, greatly overexpressed, as illustrated. Protein samples had to be markedly diluted so that both Ctrl and TG NCX1 bands could be visualized on the same gel (see also Supplemental Fig. S3 in Ref. 23). β-Actin was used as the loading control.
Fig. 3.

Effects of pretreatment (15 min) with 0.3 μM SEA0400 on basal cytosolic Ca2+ concentration ([Ca2+]CYT; A) and basal tone (C) in control (Ctrl) and transgenic (TG) femoral arteries in vivo. The SEA0400-induced decrease in [Ca2+]CYT (Δ[Ca2+]CYT; B) and vasodilation [diameter increase as %passive diameter (PD; D) are also shown. Effects of SEA0400 were augmented in TG mouse arteries. Values are means ± SE. Statistics were by two-way ANOVA. *P < 0.05 vs. the respective Ctrl group; #P < 0.05 and ##P < 0.01 vs. the basal TG group; ΔP < 0.05 vs. the basal TG group. Numbers of mice are in parentheses.
In a few experiments (see results), mice in which NCX1 was conditionally knocked out in smooth muscle (SM-NCX1-KO mice, abbreviated as “KO”) (57) were tested for comparison with Ctrl and TG mice.
Primer set sequences used for genotyping the mice with the two transgenes were as follows: 1) exMLCK, forward 5′-GCACGACTTCTTCAAGTCCGCCATGCC-3′ and reverse 5′-GCGGATCTTGAAGTTCACCTTGATGCC-3′; and NCX1, forward 5′-AGGTGAAGGTATTGCGAACATCTGGA-3′ and reverse 5′-GGCTCCAATGAAAGCTGTCAGTAT-3′.
BP Measurements
Telelemtry.
Telemetric BP transmitters (DSI TA11PA-C10, Data Sciences, Minneapolis, MN) were surgically implanted in 15 to 18 wk-old mice in the University of Maryland-Baltimore Institutional Animal Care and Use Committee-approved Physiology Phenotyping Core. The transmitter catheter was inserted into the right common carotid artery, and the tip was threaded into the ascending aorta. Details have been previously published (62). After 7–10 days of recovery from surgery, 24-h BPs were recorded with Data Sciences receivers and software. After stable baseline BPs were recorded, under isoflurane anesthesia, Alzet osmotic minipumps (Durect, Cupertino, CA) were implanted in the right abdominal wall for subcutaneous infusion of ANG II or vehicle. The 24-h BP data collection was performed “double blind”: the individual measuring BP did not know the genotype. Animal code numbers were matched with genotype and BP after the data had been collected.
Acute aortic BP measurements.
Mice (~15–18 wk old) were anesthetized with 2–3% isoflurane supplemented with 100% O2; core temperature was maintained at 37.5–38°C. An anterior midline incision was made in the neck, and the right carotid artery and jugular vein were isolated and the trunk of the carotid was ligated with a suture. A 1.1-Fr Mikro-tip pressure catheter (Millar Instruments, Houston, TX) was inserted into the artery proximal to the ligature and threaded forward to the ascending aorta. The jugular vein was then ligated, and a PE-10 catheter (Braintree Scientific, Braintree, MA) for intravenous infusions was inserted caudally and threaded down to the superior vena cava. The incision was then closed with 5-mm EZ clips (Braintree Scientific). Arterial BP was recorded under 1.5% isoflurane anesthesia (25) at a sampling rate of 1 kHz using a PowerLab data acquisition system (AD Instruments, Colorado Springs, CO). Data were later calculated offline (BioPac System, Santa Barbara, CA). After the experiment, the animal was deeply anesthetized with isoflurane and euthanized by cervical dislocation. Further details have been previously described in Ref. 60.
Femoral Artery In Vivo Imaging
The in vivo imaging methods and validation have been previously described in detail (58, 60). In brief, under microscope observation, a femoral artery of an isoflurane-anesthetized mouse was exposed through a cutaneous incision in the midthigh. The connective tissue was lightly dissected over a ~2-mm-long region of the distal part of the femoral artery, taking care to avoid severing nerves. The mouse was then moved to the stage of the fluorescence microscope, and superfusion of the exposed artery and surrounding muscle was initiated. The arrangement of the inlet and outlet for the superfusion fluid was made to achieve a laminar flow over the artery of ~9 ml/min.
The use of a volatile anesthetic (isoflurane) was desirable to maintain the animal in an optimal physiological state. Such agents have, however, been reported to influence sympathetic nerve activity (SNA) (49), alter the activity of the renin-angiotensin system (53), and activate K+ channels (30). Nevertheless, femoral arteries under the conditions of these experiments maintained a high degree of vasoconstriction, as evidenced by the large increase in diameter after total autonomic ganglion blockade (see results).
Fluorescence and Diameter Recordings in Femoral Arteries
A xenon arc lamp (Lambda LS, Sutter Instrument, Novato, CA) provided intensity-adjusted, gated fluorescent illumination at 436 ± 10 nm (excites CFP) that was controlled by a programmable shutter (Smart Shutter, Sutter Instrument). A long working distance, high numerical aperture Olympus MVX10 MacroView fluorescence microscope (Olympus America, Center Valley, PA) and a sensitive charge-coupled device ORCA ER camera (Hammamatsu, Bridgewater, NJ) fitted with a DualView image splitter (Photometrics, Tucson, AZ) were used. This provided “CFP” and “YFP” images (470 and 535 nm, respectively). The camera was controlled and images were acquired (1 frame/s) using HCImage (Hamamatsu). Image processing was performed with custom software written using IDL 6.4 (IIT Visual Information Solutions, Boulder, CO). Elevation of [Ca2+]CYT promotes the binding of Ca2+ to calmodulin and exMLCK [a calmodulin-based biosensor (22)]. In the latter case, CFP and YFP moieties then move apart, thereby reducing FRET from CFP to YFP and thus increasing the CFP-to-YFP (FRET) ratio. The FRET ratio was calibrated to determine [Ca2+]CYT (Ref. 60 provides details).
Arterial “tone” is the difference between the PD in Ca2+-free solution and the observed diameter under basal or other conditions tested (DObs) as a percentage of PD as follows:
Changes in DObs (ΔDObs) due to agent applications are presented as a percentage of PD [(ΔDObs/PD) × 100]. Vasodilation was graphed as upward bars or curves; vasoconstriction was graphed as downward bars or curves. Actual diameters were also graphed.
Immunoblot analysis
Previously published methods were used for immunoblot analysis of mouse aorta membrane proteins (34, 61, 62). NCX1 expression was identified by chemiluminescence with R3F1 anti-NCX1 monoclonal antibodies (R3F1; generously provided by Prof. Kenneth D. Philipson, University of California, Los Angeles, CA). β-Actin was used as a loading control and was identified with clone AC-74 anti-β-actin monoclonal antibodies (Sigma-Aldrich, St. Louis, MO).
Reagents and Solutions
Artery superfusion solution composition was as follows (in mM): 112 NaCl, 25.7 NaHCO3, 4.9 KCl, 2.5 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 11.5 glucose, and 10 HEPES (pH 7.4, equilibrated with gas of 5% O2-5% CO2-90% N2 at 37°C). Ca2+-free solution was made by omitting Ca2+ and adding 0.5 mM EGTA.
Reagents and sources were as follows. ACh, ANG II, hexamethonium chloride, sodium nitroprusside (SNP), ouabain, phenylephrine (PE), and prazosin HCl were from Sigma-Aldrich. Losartan was from Merck (Kenilworth, NJ). SEA0400 (2-{4-[(2,5-difluorophenyl) methoxy]phenoxy}-5-ethoxyaniline) was a gift from Prof. Iwamoto. All reagents were reagent grade or the highest grade available. SEA0400 was dissolved in DMSO; other agents were dissolved in double-distilled deionized water as stock solutions. The final bath concentration of DMSO did not exceed 0.01% (vol/vol) and did not affect vascular function.
Statistics
Data in Figs. 2–12 are shown as means ± SE. Shapiro-Wilk and Levene tests were performed for data normality and homogeneity, respectively; all data used for ANOVA passed both tests. Paired or unpaired t-tests, two-way ANOVA with post hoc correction using the Bonferroni approach, or linear regression (SigmaPlot 11.0, Systat Software, San Jose, CA) were used to test for significant differences (P < 0.05), as stated in the figures. SigmaPlot was used for post hoc calculation of statistical power.
Fig. 2.

Effects of smooth muscle-specific Na+/Ca2+ exchanger-1 (NCX1) overexpression on femoral artery cytosolic Ca2+ concentration ([Ca2+]CYT), diameter, and tone. Baseline diameter and passive diameter (PD; A), basal tone (PD − baseline diameter as %PD; B), and basal [Ca2+]CYT (C) in the exposed femoral arteries of control (Ctrl) and transgenic (TG) mice in vivo under light anesthesia are shown. Baseline diameter was reduced, and basal tone and [Ca2+]CYT were increased, in TG mouse arteries. *P < 0.05 vs. Ctrl mice (Student’s unpaired t-test); ***P < 0.001 vs. Ctrl mice; ###P < 0.001 vs. the respective basal diameter (two-way ANOVA with pairwise comparisons using the Holm-Sidak method). Numbers of Ctrl and TG mice are shown in parentheses at the top. The difference between mean Ctrl PD and TG PD was not significant (P = 0.085 by two-way ANOVA). When mean PD values from six independent experiments (with 51 Ctrl arteries and 51 TG arteries) were analyzed, however, the respective means of the means from each experiment (Ctrl PD: 397.2 ± 3.9 μm and TG PD: 379.5 ± 5.3 μm) were significantly different (P = 0.023 by a Student’s unpaired t-test). The 95% confidence interval for the difference of the means was 3.043–32.291; the power of the t-test was 0.617.
Fig. 12.

Sodium nitroprusside (SNP) cumulative dose-response curves for exposed femoral arteries in control (Ctrl) and transgenic (TG) mice in vivo; SNP was applied by superfusion. A: SNP dose versus artery diameter. Ctrl passive diameter (PD) = 407 ± 23 μm (n = 6); TG PD = 387 ± 13 μm (n = 6). B: SNP-induced decrease (Δ) in cytosolic Ca2+ concentration ([Ca2+]CYT). Basal [Ca2+]CYT was 219 ± 31 nM in Ctrl mouse arteries and 309 ± 27 nM in TG mouse arteries. C: SNP-induced vasodilation (as %PD). TG curves were all significantly shifted to the left (indicating higher affinity and lower SNP EC50 values) relative to the respective Ctrl curves (*P < 0.05). Values are means ± SE. Statistics were by two-way ANOVA. Numbers of mice are in parentheses.
RESULTS
NCX1 Expression Detected by Immunoblot Analysis and Function
Relative NCX1 expression was determined by immunoblot analysis. NCX1 expression was markedly upregulated in aortas from NCX1-TG heterozygous mice and even more so in homozygous TG mice (Fig. 1) because each mouse may have several copies of the transgene. These results resemble those obtained with the original TG line (see Supplemental Fig. 3a in Ref. 23). Homozygous overexpressors were used for all experiments described below.
Functional evidence of NCX1 overexpression was obtained in femoral arteries of anesthetized mice in vivo. First, arterial diameter and [Ca2+]CYT were measured in Ctrl and TG mouse arteries. Under basal conditions, the average diameter of TG arteries was ~20% narrower than that of Ctrl arteries, whereas the PDs of TG arteries (i.e., in Ca2+-free solution) were only ~5% narrower than the PDs of Ctrl arteries (Fig. 2A). Basal “tone” was, therefore, significantly greater in TG femoral arteries (Fig. 2B). This increased tone was associated with, and is likely due to, the significantly elevated basal [Ca2+]CYT in TG arteries (Fig. 2C). The modestly smaller PD of the TG arteries in age-matched mice raises the possibility that the congenital increase in arterial tone and BP (Ref. 23 and present study) may have induced some structural changes in TG arteries.
Second, to test for NCX1 function directly, the effects of the NCX1 inhibitor SEA0400 on [Ca2+]CYT and femoral artery diameter were determined. Superfusion with 0.3 μM SEA0400 reduced [Ca2+]CYT by ~50 nM in Ctrl arteries and by ~85 nM in TG arteries (Fig. 3, A and B). This was associated with a significantly greater decrease in tone in TG arteries than in Ctrl arteries (Fig. 3, C and D), as expected if the arterial constriction and tone depend, in part, on NCX1-mediated, SEA0400-sensitive Ca2+ entry. The SEA0400-induced 30% decline in basal tone in TG mouse arteries was statistically significant, but the 10% decline in Ctrl arteries was not, even though all six arteries dilated by a small amount.
Furthermore, when arteries were constricted by prolonged (15 min) exposure to 10 μM ouabain, which should elevate [Na+]CYT and promote Ca2+ entry via NCX1 (Fig. 4A), TG arteries constricted more than did Ctrl arteries (Fig. 4B). SEA0400 then reduced the constriction in TG arteries to a significantly greater extent than in Ctrl arteries (Fig. 4B). The elevated basal [Ca2+]CYT in arteries from TG mice and the difference in SEA0400-triggered decrease in tone between Ctrl and TG mice fit the view that ASMC NCX1 mediates Ca2+ entry, primarily in those arteries with tone. The data shown in Fig. 2, Fig. 3, and Fig. 4 are all consistent with the overexpression of NCX1 protein in TG mouse arteries (Fig. 1).
Fig. 4.

Effects of ouabain and SEA0400 on femoral artery cytosolic Ca2+ concentration ([Ca2+]CYT) and diameter. [Ca2+]CYT increase (Δ[Ca2+]CYT; A) and vasoconstriction [diameter decrease, as %passive diameter (PD); B] induced by a 15-min superfusion with 10 μM ouabain as well as the effects of 0.3 μM SEA0400 (15-min pretreatment before ouabain) on these changes. Ouabain-induced, SEA0400-sensitive effects were amplified in transgenic (TG) mouse arteries. Values are means ± SE. Statistics were by a paired t-test. #P < 0.05 vs. ouabain alone. Numbers of mice are in parentheses.
Basal BP and BP Responses to Chronic and Acute ANG II Infusion Are Augmented in TG Mice
The BP data shown in Fig. 5A support previous reports (23, 57, 62) showing that SM-NCX1-KO reduces basal BP and SM-NCX1 overexpression elevates BP relative to BP in Ctrl mice. Furthermore, the BP increase induced by chronic low-dose subcutaneous ANG II infusion (400 ng·kg−1·min−1 for 3 wk) was much greater in TG mice, and less in KO mice, than in Ctrl mice. Also, acute intravenous infusion of 30 and 300 ng/kg ANG II induced a significantly larger increment in BP in TG anesthetized mice than in Ctrl anesthetized mice (Fig. 5B). This raises the possibility that arteries from TG mice may be more sensitive to ANG II than arteries from Ctrl mice and that the ANG II dose-response curve may, therefore, be shifted to the left.
Fig. 5.

The acute and chronic angiotensin II (ANG II)-induced blood pressure elevation is augmented in transgenic (TG) mouse arteries. A: mean blood pressure (24-h MBP) in awake, mobile smooth muscle-specific NCX1 knockout mice (SM-NCX1-KO; KO), control (Ctrl) mice, and TG mice that overexpress NCX1 specifically in smooth muscle (SM-NCX1-TG; TG) mice infused with saline or ANG II (400 ng·kg−1·min−1 sc) for 3 wk. B: increases in aortic MBP (ΔMBP) induced by acute intravenous infusion of 30 or 300 ng/kg ANG II in lightly anesthetized KO, Ctrl, and TG mice. Values are means ± SE. Statistics were by two-way ANOVA. *P < 0.05 and ***P < 0.001 for the pairs indicated. NS, nonsignificant. Numbers of mice are in parentheses.
Femoral Artery Tone Is Largely Dependent on Sympathetic Drive
Femoral arteries in vivo constricted to superfused PE (an α1-adrenoceptor agonist) in a dose-dependent manner (Fig. 6). Basal [Ca2+]CYT was elevated in TG arteries (Figs. 2C and 3A), and they were modestly, but significantly, more constricted than Ctrl arteries at baseline (Figs. 2A and 6A; see Figs. 7–11). Furthermore, the TG artery diameter-PE dose-response curve was shifted downward relative to the curve for Ctrl arteries (Fig. 6A). When the data were normalized to PD and expressed as PE-induced vasoconstriction and Δ[Ca2+]CYT versus PE concentration curves, however, the Ctrl and TG artery curves were almost superimposable (Figs. 6, B and C). Thus, the sensitivity of TG arteries to PE appeared comparable to that of Ctrl arteries.
Fig. 6.

Phenylephrine (PE) cumulative dose-response curves for exposed femoral arteries in control (Ctrl) and transgenic (TG) mice in vivo; PE was applied by superfusion. A: PE dose versus artery diameter. Ctrl passive diameter (PD) = 404 ± 21 μm (n = 9); TG PD = 374 ± 5 μm (n = 11). B: PE-induced vasoconstriction (as %PD). C: PE-induced increase (Δ) in cytosolic Ca2+ concentration ([Ca2+]CYT). Basal [Ca2+]CYT was 221 ± 29 nM in Ctrl mouse arteries and 319 ± 36 nM in TG mouse arteries, but the normalized vasoconstriction curves (B) and Δ[Ca2+]CYT curves (C) from Ctrl and TG mice were virtually superimposable (two-way ANOVA). Values are means ± SE.
Fig. 7.

Effects of α1-adrenergic blockade [prazosin (Pra)] on femoral artery cytosolic Ca2+ concentration ([Ca2+]CYT), diameter, and tone in femoral arteries of control (Ctrl) and smooth muscle-specific Na+/Ca2+ exchanger-1 (NCX1) overexpression [transgenic (TG)] mice. A: diameters of exposed femoral arteries of Ctrl and TG mice in vivo as well as the effects of superfusion with 300 nM Pra. Passive diameter (PD) (arteries superfused with Ca2+-free medium) is also shown. Ctrl PD = 403 ± 13 μm (n = 6); TG PD = 397 ± 12 μm (n = 6). B and C: [Ca2+]CYT and arterial tone (as %PD), respectively, in Ctrl and TG mouse arteries under basal conditions and during superfusion with 300 nM Pra. Pra reduced [Ca2+]CYT and tone to a greater extent in TG mice than in Ctrl mice. [Ca2+]i, intracellular Ca2+ concentration. Values are means ± SE. Statistics were by two-way ANOVA. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. basal Ctrl mice; ##P < 0.01 and ###P < 0.001 vs. basal TG mice. Numbers of mice are in parentheses.
Fig. 11.

Acetylcholine (ACh) cumulative dose-response curves for exposed femoral arteries in control (Ctrl) and transgenic (TG) mice in vivo; ACh was applied by superfusion. A: ACh dose versus artery diameter. Ctrl passive diameter (PD) = 390 ± 19 μm (n = 6); TG PD = 361 ± 16 μm (n = 10). B: phenylephrine-induced vasodilation (as %PD). C: ACh-induced decrease (Δ) in cytosolic Ca2+ concentration ([Ca2+]CYT). Basal [Ca2+]CYT was 230 ± 23 nM in Ctrl mouse arteries and 335 ± 10 nM in TG mouse arteries, but the normalized vasodilation curves (B) and [Ca2+]CYT curves (C) from Ctrl and TG mice were virtually superimposable. Values are means ± SE.
Superfusion of femoral arteries with 300 nM prazosin (an α1-adrenoceptor antagonist) reduced arterial [Ca2+]CYT to ≈80–85 nM in both TG and Ctrl anesthetized mice and dilated the arteries and decreased basal tone by ~55% in Ctrl arteries and 70% in TG arteries (Fig. 7). The decrease in tone in wild-type (WT) arteries was only modestly greater than the 46% decline observed with 100 nM prazosin (59). Of note, prazosin reduced tone to about the same level (≈12–14% of PD) in TG and Ctrl arteries. This implies that the TG-induced increase in tone apparently depended on both the NCX1-mediated increase in [Ca2+]CYT (Fig. 3) and SNA.
Blockade of the autonomic ganglia by intravenous bolus infusion of 3 mg/kg hexamethonium reduced [Ca2+]CYT to barely measurable levels and dilated the arteries almost to PD, thereby nearly abolishing tone in both TG and Ctrl mice (Fig. 8). These effects of prazosin and hexamethonium indicate that the higher [Ca2+]CYT and greater tone in TG mouse femoral arteries both depend largely on SNA. The hexamethonium data signify that essentially all of the basal tone in TG arteries, as in Ctrl arteries (see Ref. 59), was due to sympathetic drive. Also, mean PDs of Ctrl and TG arteries were nearly identical (see Fig. 2), even though TG arteries had significantly more basal tone (Fig. 2B, Fig. 3C, and Fig. 7A).
Fig. 8.

Effects of intravenous injection of 3 mg/kg hexamethonium on arterial diameter (representative original recording; A) and mean arterial tone (B) and on cytosolic Ca2+ concentration ([Ca2+]CYT) representative original data (C) and mean data (D) in exposed femoral arteries of control (Ctrl) and smooth muscle-specific Na+/Ca2+ exchanger-1 transgenic (TG) mice in vivo. [Ca2+]i, intracellular Ca2+ concentration. Hexamethonium redued [Ca2+]CYT to ≪50 nM and virtually abolished tone in both Ctrl and TG arteries. Ctrl passive diameter (PD) = 382 ± 14 μm (n = 8); TG PD = 386 ± 12 μm (n = 8). Values are means ± SE. Statistics were by two-way ANOVA. *P < 0.05 and ***P < 0.001 vs. basal Ctrl mice; ###P < 0.001 vs. basal TG mice. Numbers of mice are in parentheses.
In both Ctrl and TG femoral arteries, 100 nM ANG II superfusion elevated [Ca2+]CYT, induced vasoconstriction, and increased tone, and the magnitudes of these changes were comparable in TG and Ctrl arteries (Fig. 9). Furthermore, 100 nM losartan [an ANG II type 1 receptor (AT1R) antagonist] abolished all of these ANG II-induced changes (Fig. 9). Both the PE and ANG II data (Figs. 6 and 9) indicate that arterial contractility was comparable in Ctrl and TG mice. In comparison with the sympatholytic agents (Figs. 7 and 8), however, losartan had a negligible effect on [Ca2+]CYT, basal diameter, and tone in either Ctrl or TG arteries (Fig. 10). Thus, ANG II activation apparently contributes very little to basal tone, in contrast to the dominant role of the sympathetic nervous system and norepinephrine (Figs. 6 and 7) (see Ref. 59).
Fig. 9.

Effects of superfusion of 100 nM angiotensin II (ANG II) with or without 100 nM losartan on the diameter (A), cytosolic Ca2+ concentration ([Ca2+]CYT; B), and tone [as %passive diameter (PD); C] of exposed femoral arteries in control (Ctrl) and transgenic (TG) mice in vivo. Losartan abolished nearly all of the ANG II-induced increases in [Ca2+]CYT and tone in both Ctrl and TG arteries. Values are means ± SE. Statistics were by two-way ANOVA. ***P < 0.001 vs. the respective basal Ctrl or TG mice; ##P < 0.01 vs. ANG II alone; ¶P < 0.05 vs. basal Ctrl mice. Numbers of mice are in parentheses.
Fig. 10.

Effects of superfusion with 100 nM losartan on the diameter (A), cytosolic Ca2+ concentration ([Ca2+]CYT; B), and tone [as %passive diameter (PD); C] on exposed femoral arteries in control (Ctrl) and transgenic (TG) mice in vivo. Losartan had a negligible effect on [Ca2+]CYT and tone in both Ctrl and TG arteries. Values are means ± SE. Statistics were by two-way ANOVA. *P < 0.05 vs. basal Ctrl mice. Numbers of mice are in parentheses.
Vasodilation Is Not Attenuated in TG Mouse Femoral Arteries
ACh-induced vasodilation is impaired in human hypertension and in many animal hypertension models (for a review, see Ref. 55). Despite the modestly elevated basal BP in TG mice (Fig. 5A and Ref. 23), however, ACh-induced vasodilation of femoral arteries was comparable in TG and Ctrl mouse femoral arteries (Fig. 11): the ACh-induced decline in arterial [Ca2+]CYT and vasodilation, normalized to PD, was virtually identical in Ctrl and TG mice (Fig. 11, B and C). This implies that endothelium-mediated vasodilator mechanisms, including the release of nitric oxide (NO), were not compromised in TG mice, despite the chronic BP elevation. Although the PDs of TG mouse arteries were only slightly smaller than those of Ctrl mouse arteries (Fig. 11), a similar difference was seen when all arteries were averaged (Ctrl PD: 397 ± 5 μM, n = 51; TG PD: 380 ± 5, n = 51; P < 0.03; data from experiments shown in Figs. 2, 6–8, 11, and 12).
Surprisingly, comparison of the vasodilation activated by the NO donor SNP in the two phenotypes revealed that TG mouse femoral arteries were significantly more sensitive to SNP than Ctrl mouse femoral arteries (Fig. 12). TG artery diameter, normalized vasodilation, and Δ[Ca2+]CYT SNP dose-response curves were all shifted leftward (toward lower SNP doses) compared with those from Ctrl arteries (Fig. 11, A–C, respectively). Thus, although the endothelium-mediated vasodilator mechanisms apparently were normal (Fig. 11), either the myocyte PKG response to NO was enhanced or some downstream vasodilator mechanism in ASMCs was upregulated in TG mice.
DISCUSSION
ASMC NCX1 Preferentially Mediates Ca2+ Entry Under Basal Conditions In Vivo
NCX1, along with the PM Ca2+ pump (50) and various Ca2+-permeable channels (16, 24), helps to maintain Ca2+ homeostasis in ASMCs. NCX1 can mediate Ca2+ influx as well as efflux and is important not only for clearing Ca2+ after Ca2+ transients (39, 41) but also for mediating net Ca2+ entry and maintaining elevated [Ca2+]CYT during the development and maintenance of arterial tone (23, 44, 45, 61). The latter is exemplified by the observations that local inhibition of NCX1 modestly lowers basal arterial [Ca2+]CYT and dilates tonically constricted Ctrl arteries (Fig. 3, B and C). Therefore, it is not surprising that increasing NCX1 capacity by TG overexpression of NCX1 in smooth muscle (Fig. 1) increases basal [Ca2+]CYT and arterial tone and elevates BP (Fig. 2) (23). This demonstrates that NCX1 mediates Ca2+ entry in vivo under basal conditions.
To better understand these effects, the influence of vasoconstrictors and vasodilators on BP, and on femoral arteries in vivo (under anesthesia), was studied in Ctrl and smooth muscle-specific NCX1-TG mice. Homozygous TG/TG mice markedly overexpress NCX1 in arterial myocytes (Fig. 1) because of random insertion of the transgene into the genome. The overexpression was functionally confirmed by the augmented SEA0400-induced vasodilation and decline in Δ[Ca2+]CYT (Fig. 3) and the increase in ouabain-induced, SEA0400-sensitive vasoconstriction in TG mouse arteries (Fig. 4).
Role of Arterial NCX1 Activity in the Control of Basal [Ca2+]CYT and Arterial Tone
If NCX1 helps regulate [Ca2+]CYT in ASMCs primarily by promoting Ca2+ entry (23, 62), and if arterial constriction and tone depend on Ca2+ signals (13, 29, 60), it follows that knockout of myocyte NCX1 would be expected to lower [Ca2+]CYT and reduce arterial tone and BP, as observed (44, 57, 62). Conversely, overexpression of NCX1 in ASMCs may be expected to raise [Ca2+]CYT and augment arterial tone (Figs. 2 and 3). Furthermore, the elevated BP in TG mice (Fig. 5A) (23) fits with the inference that the role of NCX1 observed in muscular femoral arteries also applies to small resistance arteries with myogenic tone (57, 62).
The contribution of NCX1 to basal [Ca2+]CYT and arterial tone is indicated by the effects of genetically altered NCX1 expression and NCX1 blockade by SEA0400. The amplification of the effects of blockade in TG arteries (Fig. 3) and, especially, the absence of effects on arterial tone and BP in NCX1 KO mice (57, 62), indicates the specificity of low-dose (0.3 μM) SEA0400 action. While the SEA0400-induced fall in [Ca2+]CYT was significant, the 17% decline in Ctrl muscular artery basal tone did not reach statistical significance (Fig. 3). (Power analysis indicated that a far larger number of arteries would be required, given the small magnitude of the decrease and the variance of the measurements.) Nevertheless, the trend conforms with the similar, but statistically significant, ≈15% decrease in resistance artery myogenic tone previously observed in 0.3 μM SEA0400-treated isolated Ctrl arteries (23, 44). The SEA0400-induced changes indicate that NCX1 accounted for ~27% of basal [Ca2+]CYT and ~19% of basal tone in TG mouse femoral arteries.
In a post hoc analysis after completion of all experiments, the relationship between femoral artery [Ca2+]CYT and tone was examined for the myriad conditions tested. The data [shown as an in vivo (under light anesthesia) pCa-vs.-arterial constriction plot in Fig. 13] revealed that constriction was nearly linearly related to log[Ca2+]CYT between 100 and 550 nM [Ca2+]CYT. The exMLCK FRET calibration data (58) are included in Fig. 13. For comparison, a plot of pCa versus myogenic constriction in isolated, pressurized rat small cerebral arteries (29) is also shown (Fig. 13), because myogenic tone in small arteries is directly related to intracellular Ca2+ concentration measured with fura-2 (13, 29). The inflection point (~120 nM [Ca2+]CYT, pCa = 6.92) is similar for femoral and small cerebral arteries, presumably because this is close to the contraction threshold. However, the normalized small-artery constriction curve is steeper than that for femoral arteries and reaches maximal constriction (diameter ≈40 μm) at [Ca2+]CYT ~ 325 nM (pCa = 6.5). The larger arteries have a larger range of constriction (from ≈400 to ≈150 μm vs. ≈200 to ≈40 μm for small arteries) and are not yet maximally constricted at [Ca2+]CYT = 550 nM (pCa = 6.26). This relationship implies that small NCX1-dependent changes in [Ca2+]CYT may have a large effect on arterial constriction and BP. The evidence that Ctrl and TG artery data fit the same curve implies that the sensitivity of the contractile apparatus to Ca2+ did not vary under the conditions examined here, consistent with the data shown in Figs. 6 and 8.
Fig. 13.
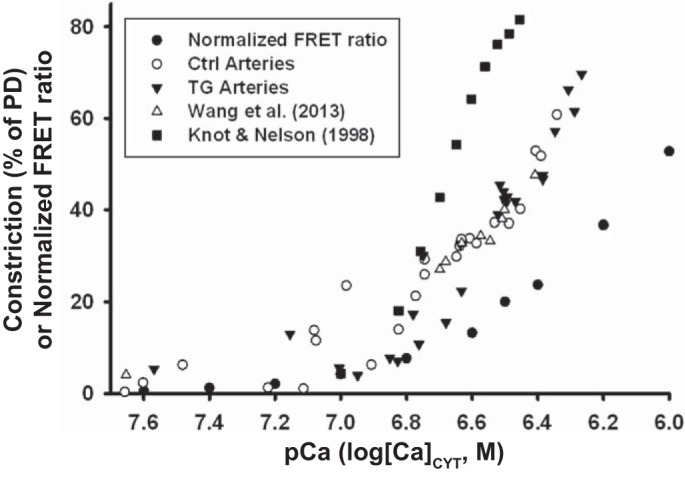
Femoral artery constriction [%passive diameter (PD)] in vivo graphed as a function of pCa (−log[Ca2+]CYT; cytosolic Ca2+ concentration). Data were taken from Figs. 2–12 [○, control (Ctrl); ▼, transgenic (TG)] and from Wang et al. (58) (Δ). The regression lines for the three sets of data for [Ca2+]CYT = 100–500 nM (R = 0.91, 0.69 and, 0.97, respectively) were indistinguishable and correspond to a constriction of ~10% of PD per 100 nM increase in [Ca2+]CYT. For comparison, the myogenic constriction versus pCa data in isolated, pressurized (60 mmHg) rat small posterior cerebral arteries taken from Fig. 10 of Knot and Nelson (29) are also shown (■); the constriction, as %PD, was calculated based on a PD of 210 μm. The graph also contains the normalized exogenous myosin light chain kinase transgene Förster resonance energy transfer (FRET) ratio curve (●; where 0 = “0” [Ca2+]CYT; 100 = saturating [Ca2+]CYT) from Fig. 1B of Wang et al. (58).
Dominant Role of Sympathetic Drive in Maintaining Basal Femoral Artery Tone
In contrast to the modest effects of SEA0400 in Ctrl mice, blockade of α1-adrenoceptors with prazosin reduced femoral artery [Ca2+]CYT by ≈65% and basal tone by ≈55% (Fig. 7). Blockade of sympathetic ganglia with hexamethonium reduced [Ca2+]CYT and basal tone by ≥90% in Ctrl mice and by even more in TG mice (Fig. 8). Nevertheless, the data shown in Fig. 3 suggest that a modest fraction of the almost entirely SNA-driven tone (59), perhaps ≈15% in normal femoral arteries, also depends on NCX1-mediated Ca2+ entry, but the majority apparently depends on other Ca2+ transporters and channels. Basal [Ca2+]CYT and tone in TG as well as Ctrl mouse arteries were, however, negligibly affected by the AT1R inhibitor losartan (Fig. 10), even though it effectively blocked ANG II-evoked [Ca2+]CYT elevation and vasoconstriction (Fig. 9). These findings corroborate earlier reports showing that in vivo basal arterial tone in mouse femoral arteries is largely determined by sympathetic drive (58, 59).
Why Is ANG II-Induced BP Elevation Greater in SM-NCX1-TG Mice Than in Ctrl Mice?
ANG II, whether infused acutely (intravenously) or chronically (subcutaneously), is widely known to increase BP (37, 48, 58). Interestingly, not only is basal BP modestly elevated in SM-NCX1-TG mice but also both acute and chronic ANG II induced a larger BP increment in TG mice than in Ctrl mice (Fig. 5). Conversely, SM-NCX1-KO mice are modestly hypotensive (57, 62), and the chronic ANG II-induced BP elevation tends to be smaller than in either Ctrl or TG mice (Fig. 5A). Thus, the ANG II-induced BP elevation appears to be arterial NCX1 “dose dependent”.
The results summarized above might suggest that TG mouse arteries are more sensitive to vasoconstrictors than are Ctrl mouse arteries. For example, if basal [Ca2+]CYT is elevated, SR Ca2+ stores and evoked Ca2+ signals might be expected to increase, but this apparently is not the case. Femoral arteries in anesthetized TG and Ctrl mice exhibited virtually identical vasoconstrictor responses (measured as “Δdiameter”) to locally superfused PE (Fig. 6) and ANG II (Fig. 9A). The downward shift of the PE dose versus TG artery diameter curve (i.e., the dose-response curve), relative to the Ctrl artery curve (Fig. 6A), implies that the PE constriction of TG arteries is superimposed on the augmented basal constriction. The normalized curves (relative to PD; Fig. 6B) are nearly identical, however, indicating that there is no difference in PE sensitivity (i.e., half-maximal activation or K0.5) between Ctrl and TG mice. It is noteworthy that a very similar result was obtained when PE-induced femoral artery vasoconstriction was compared in Ctrl mice and in mice infused chronically with ANG II to induce hypertension (58).
Complementary effects were observed in SM-NCX1-KO mice, which have low BP. Basal myogenic tone was lower in KO resistance arteries than in Ctrl resistance arteries, and the PE dose-response curve was simply displaced downward in the KOs (62). In vivo infused ANG II dose-BP response data also indicate that the curve for KO mouse arteries paralleled that of Ctrl mouse arteries but was displaced downward (63). Thus basal tone and BP are directly related to the level of NCX1 expression in Ctrl, TG, and KO arteries.
TG and Ctrl arteries also constricted to the same extent when tested with 100 nM ANG II (Fig. 9A). Furthermore, the amplitude of the ANG II-induced rise in [Ca2+]CYT was also virtually identical in Ctrl and TG arteries (i.e., the signal, itself, was not amplified), but it was superimposed on a higher basal [Ca2+]CYT in TG arteries (Fig. 9B). A possible explanation for the lack of signal amplification is that compensatory mechanisms (e.g., reduced SERCA expression) might be triggered to minimize the effects of the enhanced Ca2+ entry in TG mouse ASMCs; this needs verification. Importantly, our measurements of [Ca2+]CYT are spatial averages of cytoplasmic Ca2+. We cannot eliminate the possibility that overexpression of NCX1 changed Ca2+ concentration in other cellular compartments in which Ca2+ signaling occurs, such as the endoplasmic reticulum or nuclear envelope. Thus, differences between Ctrl and SM-NCX1-TG mice could, in ways yet unknown, be due also to changes in Ca2+ concentration in these compartments.
In contrast, femoral arteries of mice with ANG II-induced hypertension give attenuated [Ca2+]CYT and vasoconstrictor responses to superfused ANG II relative to Ctrl mice (58). In that case, the sustained elevation of plasma ANG II likely caused downregulation of AT1Rs (56) to compensate (partially) for the hyperstimulation. There is no reason to expect hyperstimulation of either α1-adrenoceptors or AT1Rs in NCX1-TG hypertension; consequently, agonist-induced downregulation of the cognate receptors seems unlikely.
If TG arteries do not have enhanced sensitivity to vasoconstrictors, what accounts for the augmented BP elevation in TG mice, versus Ctrl mice, when ANG II is infused (Fig. 5)? A likely explanation is based on the evidence that basal arterial diameter is smaller and arterial tone is greater not only in TG mouse muscular arteries than in Ctrl mouse muscular arteries (Fig. 2), but, importantly, also in resistance arteries (unpublished observations), which account for most of the total peripheral resistance (TPR). Furthermore, artery resistance (R) is proportional to 1/r4, where r is artery radius (4). Under these circumstances, the same vasoconstrictor-induced artery diameter decrease in Ctrl and TG mice should cause a greater increase in TPR and BP in TG mice [assuming constant cardiac output, since BP = cardiac output × TPR (4)]. In other words, the vasoconstrictor-induced increase in TPR, and thus in BP, is amplified in TG mice simply because the increase is superimposed on a greater basal tone due to the increased basal [Ca2+]CYT.
SNP- But Not ACh-Induced Vasodilation Is Enhanced in TG Mouse Arteries
Endothelium-dependent vasodilation, as measured with ACh stimulation (38), is often impaired in arteries of hypertensive humans and animal models (55). One might anticipate, therefore, that ACh-induced vasodilation would be attenuated in TG mice with chronically elevated BP. In contrast, the data shown in Fig. 11 reveal that the response of TG mouse arteries to ACh is normal. This implies that the endothelial impairment in hypertension may not simply be the result of the elevated intravascular pressure (19). When vasodilation was induced with SNP, however, the dose-response curve was shifted to the left in TG arteries; i.e., sensitivity to the NO donor was increased: the apparent EC50 was ~100 nM in Ctrl arteries but only ~20 nM in TG arteries (Fig. 11). This is not totally unexpected because there are reports showing that the sensitivity to SNP, largely an endothelium-independent vasodilator (for evidence of endothelium-dependent effects, see Ref. 28), is increased in some forms of hypertension (12, 52). In arterial myocytes, NO stimulates PKG to increase the cGMP level; this activates myosin light chain phosphatase, which promotes vasodilation by reducing myocyte sensitivity to Ca2+ (11), although other mechanisms may also contribute (33, 64). It seems possible that one or more of these vasodilator pathways were augmented to help compensate for the tendency of NCX1 overexpression to drive BP up. Indeed, others have suggested that Ca2+ sensitivity may be reduced in hypertension (1).
Conclusions
The aim of the present in vivo study was to test the seemingly intuitive hypothesis that elevated arterial myocyte [Ca2+]CYT and enhanced myocyte Ca2+ signaling can explain the mild hypertension in NCX1-TG mice. While TG mice have high basal [Ca2+]CYT and enhanced arterial tone, the data indicate that arterial myocytes are not hypersensitive to vasoconstrictors, as also has been reported for human small arteries (1, 51). Aalkjaer and colleagues (1) attributed the enhanced agonist-induced pressor response without a change in agonist sensitivity to “altered vascular structure” but “unchanged or possibly depressed” myocyte function. In contrast, however, the present results indicate that the increased pressor response in TG hypertensive mice is largely due to the superimposition of normal agonist-induced contraction on an artery with already enhanced basal tone. Despite the (presumably) congenitally high BP, the arterial functional responses appeared normal: arteries constricted normally to PE and ANG II and dilated to a PD that was only ~6–7% narrower than that of Ctrl arteries in response to ACh and SNP. This suggests that there is relatively little structural remodeling (21, 31) of TG arteries, despite longstanding hypertension. Thus, at least in NCX1-TG hypertension, the primary defect appears to be the NCX1-mediated elevation of [Ca2+]CYT and its functional consequences, most notably, enhanced basal tone, that lead directly to increased TPR. Because arterial NCX1 is upregulated in many forms of hypertension (10, 42, 43, 65), it seems reasonable to anticipate that similar functional (active contraction) changes likely also apply at least during the early stages of hypertension. We also made the unexpected observation that endothelial function is not attenuated in femoral arteries of modestly hypertensive SM-NCX1-TG mice (Fig. 11) and that vasodilator mechanisms are even augmented (Fig. 12). These observations seem inconsistent with the popular view that BP elevation, per se, impairs endothelium-dependent vasodilation (35).
The key observation is that, while sympathetic drive is the dominant mechanism responsible for sustaining arterial tone, Ca2+ entry via NCX1 plays a small but crucial role in helping to maintain [Ca2+]CYT above the contraction threshold. The critical relationship between [Ca2+]CYT and arterial tone (Fig. 13) illustrates how enhanced net NCX1-mediated Ca2+ entry can increase tone. Net Ca2+ transport by NCX1 is governed by the PM Na+ electrochemical gradient (see the introduction and Ref. 9) that is controlled by adjacent α2-Na+ pumps in myocyte PLasmERosomes (see the introduction and Refs. 8, 27, and 39). Apparently, α2-Na+ pump activity is ultimately responsible for modulating arterial tone, TPR, and BP through NCX1 (7). The steep relationship between [Ca2+]CYT and arterial tone (Fig. 13) suggests that very modest increases in SNA (40) and plasma endogenous ouabain (5, 20), which increases arterial NCX1 expression (43, 66), may act in concert to elevate BP in many forms of hypertension.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants R01-HL-107654 (to W. G. Wier) and R01-HL-45215 and R01-HL-107555 (to M. P. Blaustein), American Heart Association Grant 15GRNT24940022 (to J. Zhang and then to M. P. Blaustein), National Natural Science Foundation of China Grants 81100174 and 81570449 (to Y. Wang), Fundamental Research Funds for the Central Universities Grant GK201803097 (to Y. Wang), College Students’ Innovative and Training Project of Shaanxi Normal University Grant cx2018197 (to Y. Wang), and the University of Maryland Baltimore Foundation (Center for Heart Hypertension and Kidney Diseases Fund). This work was also supported by the University of Maryland School of Medicine Physiological Phenotyping Core (L. Chen, Core Manager).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.Z., Y.W., and L.C. performed experiments; J.Z., Y.W., L.C., and M.P.B. analyzed data; J.Z., Y.W., W.G.W., and M.P.B. interpreted results of experiments; J.Z. and M.P.B. drafted manuscript; J.Z., Y.W., L.C., W.G.W., and M.P.B. approved final version of manuscript; Y.W. and M.P.B. prepared figures; Y.W., L.C., W.G.W., and M.P.B. edited and revised manuscript.
ACKNOWLEDGMENTS
We thank Meng Li for maintaining the mouse colony and for laboratory support and Prof. Soren Bentzen for advice and assistance with statistical analyses.
Present address of Y. Wang: Dept. of Physical Education, Shaanxi Normal University, Xi’an, Shaanxi 710119, China.
Present address of W. G. Wier: Dept. of Physiology, University of Maryland School of Medicine, Baltimore, MD 21201.
Footnotes
ENa/Ca should be determined from the local cytosolic Na+ concentration ([Na+]CYT) in the vicinity of NCX, assuming that the extracellular Na+ and Ca2+ concentrations and [Ca2+]CYT are known. In ASMCs, the calculation is based on an estimated global [Na+]CYT (2, 26). In tonically constricted arteries, however, local [Na+]CYT (and [Ca2+]CYT) in the vicinity of NCX might be elevated due to Na+ entry through various transient receptor potential channels, which admit Na+ and Ca2+ (16). Furthermore, Vm, [Na+]CYT, and [Ca2+]CYT, and thus ENa/Ca and Vm – ENa/Ca, all vary dynamically.
REFERENCES
- 1.Aalkjaer C, Heagerty AM, Petersen KK, Swales JD, Mulvany MJ. Evidence for increased media thickness, increased neuronal amine uptake, and depressed excitation−contraction coupling in isolated resistance vessels from essential hypertensives. Circ Res 61: 181–186, 1987. doi: 10.1161/01.RES.61.2.181. [DOI] [PubMed] [Google Scholar]
- 2.Aalkjaer C, Mulvany MJ. Sodium metabolism in rat resistance vessels. J Physiol 343: 105–116, 1983. doi: 10.1113/jphysiol.1983.sp014883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baryshnikov SG, Pulina MV, Zulian A, Linde CI, Golovina VA. Orai1, a critical component of store-operated Ca2+ entry, is functionally associated with Na+/Ca2+ exchanger and plasma membrane Ca2+ pump in proliferating human arterial myocytes. Am J Physiol Cell Physiol 297: C1103–C1112, 2009. doi: 10.1152/ajpcell.00283.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berne RM, Levy MN. Cardiovascular Physiology. St. Louis, MO: Mosby, 2001, p. 115–153. [Google Scholar]
- 5.Blaustein MP. The pump, the exchanger, and the holy spirit: origins and 40-year evolution of ideas about the ouabain-Na+ pump endocrine system. Am J Physiol Cell Physiol 314: C3–C26, 2018. doi: 10.1152/ajpcell.00196.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blaustein MP. Sodium ions, calcium ions, blood pressure regulation, and hypertension: a reassessment and a hypothesis. Am J Physiol Cell Physiol 232: C165–C173, 1977. doi: 10.1152/ajpcell.1977.232.5.C165. [DOI] [PubMed] [Google Scholar]
- 7.Blaustein MP, Chen L, Hamlyn JM, Leenen FH, Lingrel JB, Wier WG, Zhang J. Pivotal role of α2 Na+ pumps and their high affinity ouabain binding site in cardiovascular health and disease. J Physiol 594: 6079–6103, 2016. doi: 10.1113/JP272419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blaustein MP, Juhaszova M, Golovina VA. The cellular mechanism of action of cardiotonic steroids: a new hypothesis. Clin Exp Hypertens 20: 691–703, 1998. doi: 10.3109/10641969809053247. [DOI] [PubMed] [Google Scholar]
- 9.Blaustein MP, Lederer WJ. Sodium/calcium exchange: its physiological implications. Physiol Rev 79: 763–854, 1999. doi: 10.1152/physrev.1999.79.3.763. [DOI] [PubMed] [Google Scholar]
- 10.Blaustein MP, Leenen FH, Chen L, Golovina VA, Hamlyn JM, Pallone TL, Van Huysse JW, Zhang J, Wier WG. How NaCl raises blood pressure: a new paradigm for the pathogenesis of salt-dependent hypertension. Am J Physiol Heart Circ Physiol 302: H1031–H1049, 2012. doi: 10.1152/ajpheart.00899.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bolz SS, Vogel L, Sollinger D, Derwand R, de Wit C, Loirand G, Pohl U. Nitric oxide-induced decrease in calcium sensitivity of resistance arteries is attributable to activation of the myosin light chain phosphatase and antagonized by the RhoA/Rho kinase pathway. Circulation 107: 3081–3087, 2003. doi: 10.1161/01.CIR.0000074202.19612.8C. [DOI] [PubMed] [Google Scholar]
- 12.Cohen DM, Webb RC, Bohr DF. Nitroprusside-induced vascular relaxation in DOCA hypertensive rats. Hypertension 4: 13–19, 1982. doi: 10.1161/01.HYP.4.1.13. [DOI] [PubMed] [Google Scholar]
- 13.D’Angelo G, Davis MJ, Meininger GA. Calcium and mechanotransduction of the myogenic response. Am J Physiol Heart Circ Physiol 273: H175–H182, 1997 10.1152/ajpheart.1997.273.1.H175. [DOI] [PubMed] [Google Scholar]
- 14.Dostanic-Larson I, Van Huysse JW, Lorenz JN, Lingrel JB. The highly conserved cardiac glycoside binding site of Na,K-ATPase plays a role in blood pressure regulation. Proc Natl Acad Sci USA 102: 15845–15850, 2005. doi: 10.1073/pnas.0507358102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dostanic I, Paul RJ, Lorenz JN, Theriault S, Van Huysse JW, Lingrel JB. The α2-isoform of Na-K-ATPase mediates ouabain-induced hypertension in mice and increased vascular contractility in vitro. Am J Physiol Heart Circ Physiol 288: H477–H485, 2005. doi: 10.1152/ajpheart.00083.2004. [DOI] [PubMed] [Google Scholar]
- 16.Earley S, Brayden JE. Transient receptor potential channels in the vasculature. Physiol Rev 95: 645–690, 2015. doi: 10.1152/physrev.00026.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Folkow B, Neil E. Circulation. New York: Oxford University Press, 1971, p. 593. [Google Scholar]
- 18.Geguchadze R, Zhi G, Lau KS, Isotani E, Persechini A, Kamm KE, Stull JT. Quantitative measurements of Ca2+/calmodulin binding and activation of myosin light chain kinase in cells. FEBS Lett 557: 121–124, 2004. doi: 10.1016/S0014-5793(03)01456-X. [DOI] [PubMed] [Google Scholar]
- 19.Gündüz F, Baskurt OK, Meiselman HJ. Vascular dilation responses of rat small mesenteric arteries at high intravascular pressure in spontaneously hypertensive rats. Circ J 73: 2091–2097, 2009. doi: 10.1253/circj.CJ-09-0392. [DOI] [PubMed] [Google Scholar]
- 20.Hamlyn JM, Blaustein MP. Endogenous ouabain: recent advances and controversies. Hypertension 68: 526–532, 2016. doi: 10.1161/HYPERTENSIONAHA.116.06599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heagerty AM, Aalkjaer C, Bund SJ, Korsgaard N, Mulvany MJ. Small artery structure in hypertension. Dual processes of remodeling and growth. Hypertension 21: 391–397, 1993. doi: 10.1161/01.HYP.21.4.391. [DOI] [PubMed] [Google Scholar]
- 22.Isotani E, Zhi G, Lau KS, Huang J, Mizuno Y, Persechini A, Geguchadze R, Kamm KE, Stull JT. Real-time evaluation of myosin light chain kinase activation in smooth muscle tissues from a transgenic calmodulin-biosensor mouse. Proc Natl Acad Sci USA 101: 6279–6284, 2004. doi: 10.1073/pnas.0308742101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iwamoto T, Kita S, Zhang J, Blaustein MP, Arai Y, Yoshida S, Wakimoto K, Komuro I, Katsuragi T. Salt-sensitive hypertension is triggered by Ca2+ entry via Na+/Ca2+ exchanger type-1 in vascular smooth muscle. Nat Med 10: 1193–1199, 2004. doi: 10.1038/nm1118. [DOI] [PubMed] [Google Scholar]
- 24.Jaggar JH, Wellman GC, Heppner TJ, Porter VA, Perez GJ, Gollasch M, Kleppisch T, Rubart M, Stevenson AS, Lederer WJ, Knot HJ, Bonev AD, Nelson MT. Ca2+ channels, ryanodine receptors and Ca(2+)-activated K+ channels: a functional unit for regulating arterial tone. Acta Physiol Scand 164: 577–587, 1998. doi: 10.1046/j.1365-201X.1998.00462.x. [DOI] [PubMed] [Google Scholar]
- 25.Janssen BJ, De Celle T, Debets JJ, Brouns AE, Callahan MF, Smith TL. Effects of anesthetics on systemic hemodynamics in mice. Am J Physiol Heart Circ Physiol 287: H1618–H1624, 2004. doi: 10.1152/ajpheart.01192.2003. [DOI] [PubMed] [Google Scholar]
- 26.Jones AW. Electrolyte metabolism of the arterial wall. : Vascular Smooth Muscle: Metabolic, Ionic and Contractile Mechanisms, edited by Crass MF III, Barnes CD. New York: Academic, 1982, p. 37–70. [Google Scholar]
- 27.Juhaszova M, Blaustein MP. Distinct distribution of different Na+ pump alpha subunit isoforms in plasmalemma. Physiological implications. Ann N Y Acad Sci 834: 524–536, 1997. doi: 10.1111/j.1749-6632.1997.tb52310.x. [DOI] [PubMed] [Google Scholar]
- 28.Kangussu LM, Olivon VC, Arifa RD, Araújo N, Reis D, Assis MT, Soriani FM, de Souza DG, Bendhack LM, Bonaventura D. Enhancement on reactive oxygen species and COX-1 mRNA levels modulate the vascular relaxation induced by sodium nitroprusside in denuded mice aorta. Fundam Clin Pharmacol 29: 150–163, 2015. doi: 10.1111/fcp.12103. [DOI] [PubMed] [Google Scholar]
- 29.Knot HJ, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol 508: 199–209, 1998. doi: 10.1111/j.1469-7793.1998.199br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kokita N, Stekiel TA, Yamazaki M, Bosnjak ZJ, Kampine JP, Stekiel WJ. Potassium channel-mediated hyperpolarization of mesenteric vascular smooth muscle by isoflurane. Anesthesiology 90: 779–788, 1999. doi: 10.1097/00000542-199903000-00021. [DOI] [PubMed] [Google Scholar]
- 31.Korsgaard N, Aalkjaer C, Heagerty AM, Izzard AS, Mulvany MJ. Histology of subcutaneous small arteries from patients with essential hypertension. Hypertension 22: 523–526, 1993. doi: 10.1161/01.HYP.22.4.523. [DOI] [PubMed] [Google Scholar]
- 32.Kotecha N, Hill MA. Myogenic contraction in rat skeletal muscle arterioles: smooth muscle membrane potential and Ca2+ signaling. Am J Physiol Heart Circ Physiol 289: H1326–H1334, 2005. doi: 10.1152/ajpheart.00323.2005. [DOI] [PubMed] [Google Scholar]
- 33.Kyle BD, Mishra RC, Braun AP. The augmentation of BK channel activity by nitric oxide signaling in rat cerebral arteries involves co-localized regulatory elements. J Cereb Blood Flow Metab 37: 3759–3773, 2017. doi: 10.1177/0271678X17691291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Linde CI, Antos LK, Golovina VA, Blaustein MP. Nanomolar ouabain increases NCX1 expression and enhances Ca2+ signaling in human arterial myocytes: a mechanism that links salt to increased vascular resistance? Am J Physiol Heart Circ Physiol 303: H784–H794, 2012. doi: 10.1152/ajpheart.00399.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lockette W, Otsuka Y, Carretero O. The loss of endothelium-dependent vascular relaxation in hypertension. Hypertension 8: II61–II66, 1986. doi: 10.1161/01.HYP.8.6_Pt_2.II61. [DOI] [PubMed] [Google Scholar]
- 36.Lorenz JN, Loreaux EL, Dostanic-Larson I, Lasko V, Schnetzer JR, Paul RJ, Lingrel JB. ACTH-induced hypertension is dependent on the ouabain-binding site of the α2-Na+-K+-ATPase subunit. Am J Physiol Heart Circ Physiol 295: H273–H280, 2008. doi: 10.1152/ajpheart.00183.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luft FC, Wilcox CS, Unger T, Kühn R, Demmert G, Rohmeiss P, Ganten D, Sterzel RB. Angiotensin-induced hypertension in the rat. Sympathetic nerve activity and prostaglandins. Hypertension 14: 396–403, 1989. doi: 10.1161/01.HYP.14.4.396. [DOI] [PubMed] [Google Scholar]
- 38.Lüscher TF, Raij L, Vanhoutte PM. Endothelium-dependent vascular responses in normotensive and hypertensive Dahl rats. Hypertension 9: 157–163, 1987. doi: 10.1161/01.HYP.9.2.157. [DOI] [PubMed] [Google Scholar]
- 39.Lynch RM, Weber CS, Nullmeyer KD, Moore ED, Paul RJ. Clearance of store-released Ca2+ by the Na+-Ca2+ exchanger is diminished in aortic smooth muscle from Na+-K+-ATPase α2-isoform gene-ablated mice. Am J Physiol Heart Circ Physiol 294: H1407–H1416, 2008. doi: 10.1152/ajpheart.00855.2007. [DOI] [PubMed] [Google Scholar]
- 40.Mark AL. Regulation of sympathetic nerve activity in mild human hypertension. J Hypertens Suppl 8: S67–S75, 1990. [PubMed] [Google Scholar]
- 41.Pritchard TJ, Bowman PS, Jefferson A, Tosun M, Lynch RM, Paul RJ. Na+-K+-ATPase and Ca2+ clearance proteins in smooth muscle: a functional unit. Am J Physiol Heart Circ Physiol 299: H548–H556, 2010. doi: 10.1152/ajpheart.00527.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pulina MV, Zulian A, Baryshnikov SG, Linde CI, Karashima E, Hamlyn JM, Ferrari P, Blaustein MP, Golovina VA. Cross talk between plasma membrane Na+/Ca2+ exchanger-1 and TRPC/Orai-containing channels: key players in arterial hypertension. Adv Exp Med Biol 961: 365–374, 2013. doi: 10.1007/978-1-4614-4756-6_31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pulina MV, Zulian A, Berra-Romani R, Beskina O, Mazzocco-Spezzia A, Baryshnikov SG, Papparella I, Hamlyn JM, Blaustein MP, Golovina VA. Upregulation of Na+ and Ca2+ transporters in arterial smooth muscle from ouabain-induced hypertensive rats. Am J Physiol Heart Circ Physiol 298: H263–H274, 2010. doi: 10.1152/ajpheart.00784.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raina H, Ella SR, Hill MA. Decreased activity of the smooth muscle Na+/Ca2+ exchanger impairs arteriolar myogenic reactivity. J Physiol 586: 1669–1681, 2008. doi: 10.1113/jphysiol.2007.150268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rebolledo A, Speroni F, Raingo J, Salemme SV, Tanzi F, Munin V, Añón MC, Milesi V. The Na+/Ca2+ exchanger is active and working in the reverse mode in human umbilical artery smooth muscle cells. Biochem Biophys Res Commun 339: 840–845, 2006. doi: 10.1016/j.bbrc.2005.11.084. [DOI] [PubMed] [Google Scholar]
- 46.Reuter H, Blaustein MP, Haeusler G. Na-Ca exchange and tension development in arterial smooth muscle. Philos Trans R Soc Lond B Biol Sci 265: 87–94, 1973. doi: 10.1098/rstb.1973.0011. [DOI] [PubMed] [Google Scholar]
- 47.Rüegg JC. Calcium in Muscle Contraction: Cellular and Molecular Physiology. Berlin: Springer-Verlag, 1992, p. 206–212. doi: 10.1007/978-3-642-77560-4. [DOI] [Google Scholar]
- 48.Saxena PR. Interaction between the renin-angiotensin-aldosterone and sympathetic nervous systems. J Cardiovasc Pharmacol 19, Suppl 6: S80–S88, 1992. doi: 10.1097/00005344-199219006-00013. [DOI] [PubMed] [Google Scholar]
- 49.Seagard JL, Hopp FA, Bosnjak ZJ, Osborn JL, Kampine JP. Sympathetic efferent nerve activity in conscious and isoflurane-anesthetized dogs. Anesthesiology 61: 266–270, 1984. doi: 10.1097/00000542-198409000-00006. [DOI] [PubMed] [Google Scholar]
- 50.Shimizu H, Borin ML, Blaustein MP. Use of La3+ to distinguish activity of the plasmalemmal Ca2+ pump from Na+/Ca2+ exchange in arterial myocytes. Cell Calcium 21: 31–41, 1997. doi: 10.1016/S0143-4160(97)90094-4. [DOI] [PubMed] [Google Scholar]
- 51.Sivertsson R. The hemodynamic importance of structural vascular changes in essential hypertension. Acta Physiol Scand Suppl 343: 1–56, 1970. [PubMed] [Google Scholar]
- 52.Tesfamariam B, Halpern W. Endothelium-dependent and endothelium-independent vasodilation in resistance arteries from hypertensive rats. Hypertension 11: 440–444, 1988. doi: 10.1161/01.HYP.11.5.440. [DOI] [PubMed] [Google Scholar]
- 53.Ullman J, Härgestam R, Lindahl S, Chan SH, Eriksson S, Rundgren M. Circulatory effects of angiotensin II during anaesthesia, evaluated by real-time spectral analysis. Acta Anaesthesiol Scand 47: 532–540, 2003. doi: 10.1034/j.1399-6576.2003.00114.x. [DOI] [PubMed] [Google Scholar]
- 54.van Breemen C, Laher I, Chen Q. Superficial buffer barrier function of smooth muscle sarcoplasmic reticulum. Trends Pharmacol Sci 16: 98–105, 1995. doi: 10.1016/S0165-6147(00)88990-7. [DOI] [PubMed] [Google Scholar]
- 55.Vanhoutte PM, Shimokawa H, Feletou M, Tang EH. Endothelial dysfunction and vascular disease−a 30th anniversary update. Acta Physiol (Oxf) 219: 22–96, 2017. doi: 10.1111/apha.12646. [DOI] [PubMed] [Google Scholar]
- 56.Wang DH, Du Y, Yao A. Regulation of the gene-encoding angiotensin II receptor in vascular tissue. Microcirculation 3: 237–239, 1996. doi: 10.3109/10739689609148295. [DOI] [PubMed] [Google Scholar]
- 57.Wang Y, Chen L, Li M, Cha H, Iwamoto T, Zhang J. Conditional knockout of smooth muscle sodium calcium exchanger type-1 lowers blood pressure and attenuates angiotensin II-salt hypertension. Physiol Rep 3: e12273, 2015. doi: 10.14814/phy2.12273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Y, Chen L, Wier WG, Zhang J. Intravital Förster resonance energy transfer imaging reveals elevated [Ca2+]i and enhanced sympathetic tone in femoral arteries of angiotensin II-infused hypertensive biosensor mice. J Physiol 591: 5321–5336, 2013. doi: 10.1113/jphysiol.2013.257808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zacharia J, Mauban JR, Raina H, Fisher SA, Wier WG. High vascular tone of mouse femoral arteries in vivo is determined by sympathetic nerve activity via α1A- and α1D-adrenoceptor subtypes. PLoS One 8: e65969, 2013. doi: 10.1371/journal.pone.0065969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang J, Chen L, Raina H, Blaustein MP, Wier WG. In vivo assessment of artery smooth muscle [Ca2+]i and MLCK activation in FRET-based biosensor mice. Am J Physiol Heart Circ Physiol 299: H946–H956, 2010. doi: 10.1152/ajpheart.00359.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang J, Lee MY, Cavalli M, Chen L, Berra-Romani R, Balke CW, Bianchi G, Ferrari P, Hamlyn JM, Iwamoto T, Lingrel JB, Matteson DR, Wier WG, Blaustein MP. Sodium pump α2 subunits control myogenic tone and blood pressure in mice. J Physiol 569: 243–256, 2005. doi: 10.1113/jphysiol.2005.091801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang J, Ren C, Chen L, Navedo MF, Antos LK, Kinsey SP, Iwamoto T, Philipson KD, Kotlikoff MI, Santana LF, Wier WG, Matteson DR, Blaustein MP. Knockout of Na+/Ca2+ exchanger in smooth muscle attenuates vasoconstriction and L-type Ca2+ channel current and lowers blood pressure. Am J Physiol Heart Circ Physiol 298: H1472–H1483, 2010. doi: 10.1152/ajpheart.00964.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhao D, Zhang J, Blaustein MP, Navar LG. Attenuated renal vascular responses to acute angiotensin II infusion in smooth muscle-specific Na+/Ca2+ exchanger knockout mice. Am J Physiol Renal Physiol 301: F574–F579, 2011. doi: 10.1152/ajprenal.00065.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhao Y, Vanhoutte PM, Leung SW. Vascular nitric oxide: beyond eNOS. J Pharmacol Sci 129: 83–94, 2015. doi: 10.1016/j.jphs.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 65.Zulian A, Baryshnikov SG, Linde CI, Hamlyn JM, Ferrari P, Golovina VA. Upregulation of Na+/Ca2+ exchanger and TRPC6 contributes to abnormal Ca2+ homeostasis in arterial smooth muscle cells from Milan hypertensive rats. Am J Physiol Heart Circ Physiol 299: H624–H633, 2010. doi: 10.1152/ajpheart.00356.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zulian A, Linde CI, Pulina MV, Baryshnikov SG, Papparella I, Hamlyn JM, Golovina VA. Activation of c-SRC underlies the differential effects of ouabain and digoxin on Ca2+ signaling in arterial smooth muscle cells. Am J Physiol Cell Physiol 304: C324–C333, 2013. doi: 10.1152/ajpcell.00337.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]