

Abstract
The aim of this study was to investigate the acute effect of hydration status on glycemic regulation in healthy adults and explore underlying mechanisms. In this randomized crossover trial, 16 healthy adults (8 men, 8 women) underwent an oral glucose tolerance test (OGTT) when hypohydrated and rehydrated after 4 days of pretrial standardization. One day before OGTT, participants were dehydrated for 1 h in a heat tent with subsequent fluid restriction (HYPO) or replacement (RE). The following day, an OGTT was performed with metabolic rate measurements and pre- and post-OGTT muscle biopsies. Peripheral quantitative computer tomography thigh scans were taken before and after intervention to infer changes in cell volume. HYPO (but not RE) induced 1.9% (SD 1.2) body mass loss, 2.9% (SD 2.7) cell volume reduction, and increased urinary hydration markers, serum osmolality, and plasma copeptin concentration (all P ≤ 0.007). Fasted serum glucose [HYPO 5.10 mmol/l (SD 0.42), RE 5.02 mmol/l (SD 0.40); P = 0.327] and insulin [HYPO 27.1 pmol/l (SD 9.7), RE 27.6 pmol/l (SD 9.2); P = 0.809] concentrations were similar between HYPO and RE. Hydration status did not alter the serum glucose (P = 0.627) or insulin (P = 0.200) responses during the OGTT. Muscle water content was lower before OGTT after HYPO compared with RE [761 g/kg wet wt (SD 13) vs. 772 g/kg wet wt (SD 18) RE] but similar after OGTT [HYPO 779 g/kg wet wt (SD 15) vs. RE 780 g/kg wet wt (SD 20); time P = 0.011; trial × time P = 0.055]. Resting energy expenditure was similar between hydration states (stable between −1.21 and 5.94 kJ·kg−1·day−1; trial P = 0.904). Overall, despite acute mild hypohydration increasing plasma copeptin concentrations and decreasing fasted cell volume and muscle water, we found no effect on glycemic regulation.
NEW & NOTEWORTHY We demonstrated for the first time that an acute bout of hypohydration does not impact blood sugar control in healthy adults. Physiological responses to mild hypohydration (<2% body mass loss) caused an elevation in copeptin concentrations similar to that seen in those with diabetes as well as reducing cell volume by ~3%; both of these changes had been hypothesized to cause a higher blood sugar response.
Keywords: copeptin, glycemia, health, hydration, metabolism
INTRODUCTION
Although it is well established that several dietary factors are implicated in glycemic regulation, research into the effects of hydration status is lacking. Observationally, plain water intake is typically associated with better glycemic regulation (6, 7, 26), which suggests that this could represent an inexpensive and time-efficient health intervention. However, rather than demonstrating a causal relationship, such associations may reflect water intake being a marker of a generally healthy lifestyle encompassing higher fiber intake and physical activity (19).
Understanding how hydration status impacts glycemia has consequences for both clinical practice and research, whereby fasted glucose concentrations and oral glucose tolerance tests (OGTTs) are commonly used for diabetes diagnostics or to ascertain the efficacy of an intervention. Standardization of food and fluid intake before these measures is required to prevent confounding influences, yet hydration status is not uniformly controlled for, despite studies in adults with type 1 and 2 diabetes finding higher glycemic responses when participants were hypohydrated vs. euhydrated (5, 18). One study has also shown a similar deterioration in fasting glucose in healthy adults when manipulating extracellular osmolality, which mimics some of the physiological effects of hypohydration (20). Yet the effect of directly manipulating hydration status on glycemic control in healthy adults has, to our knowledge, never been investigated.
There are several causal mechanisms through which hydration status could influence glycemia (6). Briefly, hypohydration decreases cell volume, which has been hypothesized to influence glucoregulation (16, 20). Serum osmolality and arginine vasopressin (AVP) concentrations concurrently increase, potentially stimulating hepatic glucose output via V1aR binding in the liver (21, 29) and/or via adrenocorticotropic hormone (ACTH) and cortisol secretion. Accordingly, high plasma copeptin (a surrogate marker of AVP) concentrations (≥10.70 pmol/l in men and ≥6.47 pmol/l in women) have been associated with worse cardiometabolic health outcomes compared with those with low copeptin concentrations (≤4.59 pmol/l and ≤2.71 pmol/l, respectively) (10, 11).
Considering the implications for clinical practice, research, and public health, we conducted a pilot study (n = 5) in which ~12-h hypohydration (sauna plus fluid restriction) induced a higher glycemic response to an OGTT compared with sauna plus fluid replacement (8). Such findings warranted further exploration in a tightly controlled study. Therefore we aimed to investigate the role of hydration status in glycemic regulation as well as examine key mechanisms (change in cell volume and AVP secretion), hypothesizing that compared with rehydration hypohydration would cause a higher glycemic response to an OGTT.
METHODS
Participants
Sixteen healthy adults volunteered to participate (n = 8 men, 8 women), with mean age of 30 yr (SD 9), body mass of 71.7 kg (SD 9.6), and body mass index of 24.0 kg/m2 (SD 3.4). Participants were randomized with simple randomization (no strata) by H. A. Carroll upon consent with a random number generator (Excel 2013, Microsoft). Exclusion criteria were age < 18 yr or ≥ 60 yr, metabolic disease (no body mass restrictions, except self-reported weight loss > 5 kg in previous 6 mo), drug dependence, and pregnancy/breastfeeding; thus all participants were considered healthy and not taking medication or necessary supplements (except contraceptives). Women not taking continuous hormonal contraceptives were tested during their estimated follicular phase (3–10 days after onset of menses). Data were collected in South West England between June 2016 and January 2017, inclusive. Participants provided written informed consent.
The sample size estimate was based on our pilot project in five participants (8) showing the largest magnitude of effect at 45 min after glucose ingestion (d = 1.1 mmol/l). The standard deviation at this time point in the control (rehydration) group was also 1.1 mmol/l, resulting in an effect size (dz) of 1. To provide a 95% power (β) to detect this effect at an α (P) of ≤0.05 with a two-tailed paired t-test required 16 participants.
Experimental Design
This was a randomized crossover trial, with 5- to 35-day washout to account for the menstrual cycle where applicable. Each trial arm consisted of 3 days of between-trial physical activity and diet replication (“pretrial monitoring phase”), a within-participant standardized “intervention day,” and a full “laboratory testing day” (as detailed below and in Fig. 1; trial registration can be found at Clinicaltrials.gov: NCT02841449 and Open Science Framework: https://osf.io/ptq7m; deviations from the registered protocol are explained in full in the data set available at https://researchdata.bath.ac.uk/id/eprint/547). The research received ethical approval from National Health Service Health Research Authority Frenchay (ref: 16/SW/0057) and was conducted in accordance with the Declaration of Helsinki.
Fig. 1.

Protocol schematic. Resting metabolic rate (RMR) at 30 min taken in subsample (n = 9). BM, body mass; BW, body water; OGTT, oral glucose tolerance test; pQCT, peripheral quantitative computer tomography scan; Uosm, urine osmolality; USG, urine specific gravity. *Muscle biopsies were taken only in those who opted in.
Pretrial Monitoring Phase
Three days before trial, participants were asked to replicate their food and fluid intake (weighed food and fluid intake diaries; analyzed with Nutritics Nutrition Analysis Software, Nutritics, Dublin, Ireland) and physical activity (combined heart rate and accelerometry; Actiheart; CamNtech, Cambridge, UK), morning body mass (Inner scan body composition monitor model BC-543; Tanita) and urine specific gravity (Table 1). On the third monitoring day, participants were instructed to limit activity and to consume ≥40 ml/kg lean body mass (assessed via bioelectrical impedance) of nonalcoholic fluid to ensure euhydration before starting the intervention. This is in line with previous research aiming to achieve euhydration (9, 22). No restrictions were placed on caffeinated beverages during this phase, although it was emphasized to participants that they would need to replicate their fluid intake in the subsequent trial arm.
Table 1.
Lifestyle factors and markers of hydration status during 3-day pretrial monitoring phase
| Hypohydration | Rehydration | P Value* | |
|---|---|---|---|
| Energy intake, kJ/day | 9,777 (SD 3,765) | 10,091 (SD 3,513) | 0.541 |
| Carbohydrate, g/day | 274 (SD 125) | 271 (SD 125) | 0.829 |
| Fat, g/day | 84 (SD 51) | 93 (SD 53) | 0.258 |
| Protein, g/day | 105 (SD 52) | 103 (SD 41) | 0.729 |
| Sodium, mg/day | 2,487 (SD 1,774) | 2,497 (SD 1,156) | 0.970 |
| Potassium, mg/day | 3,486 (SD 3,417) | 2,778 (SD 1,347) | 0.128 |
| Water, l/day (food + fluid) | 3.3 (SD 1.5) | 3.2 (SD 1.4) | 0.315 |
| Physical activity energy expenditure, kJ/day | 4,462 (SD 2,276) | 4,314 (SD 1,764) | 0.381 |
| Body mass, kg | 71.7 (SD 9.6) | 71.8 (SD 9.8) | 0.340 |
| Body mass index, kg/m2 | 23.6 (SD 4.2) | 23.2 (SD 4.8) | 0.409 |
| USG | 1.018 (SD 0.005) | 1.018 (SD 0.005) | 0.932 |
Values are means [standard deviation (SD)]; n = 16. USG, urine specific gravity.
Statistical significance calculated with 2-tailed paired t-test.
Diet diaries were analyzed by the same coder within participant, and accuracy was verified by the lead author. Coding discrepancies were shared among all diet analysts to help ensure consistency between participants. Nutrient intakes for each day of diet recording were taken from the software and an average created for each participant. Physical activity energy expenditure was analyzed with Actiheart 4 software, which utilizes a two-branch equation to estimate physical activity energy expenditure based on heart rate and accelerometry. Each Actiheart was calibrated against measured resting metabolic rate for each participant and energy expenditure calculated with the “Group Cal JAP 2007” model. Physical activity energy expenditure for each day was then averaged for each participant.
Experimental Protocol
Intervention day.
After the pretrial monitoring phase, participants came to the laboratory between 0600 and 1000 in a euhydrated state, following overnight fluid and food abstention from 2200 the previous day. A peripheral quantitative computer tomography (pQCT; Stratec, Pforzheim, Germany) scan of a cross section of the midpoint of the right thigh was taken as a proxy for muscle cell volume, after which a 10-ml euhydrated baseline blood sample was taken from an antecubital vein. Blood analytes from this venipuncture further confirmed compliance with the pretrial monitoring phase, showing that participants were in a similar metabolic state before starting each trial arm (Table 2).
Table 2.
Overnight fasted concentrations of blood hormones and metabolites in euhydrated state at baseline before each trial arm
| Prehypohydration | Prerehydration | P Value* | |
|---|---|---|---|
| Plasma ACTH, pmol/l | 3.54 (SD 1.98) | 3.61 (SD 1.48) | 0.841 |
| Plasma copeptin, pmol/l | 4.45 (SD 2.01) | 4.25 (SD 1.83) | 0.462 |
| Plasma cortisol, nmol/l | 316 (SD 162) | 330 (SD 269) | 0.687 |
| Serum glucose, mmol/l | 5.04 (SD 0.30) | 4.98 (SD 0.46) | 0.493 |
| Serum insulin, pmol/l | 30.4 (SD 11.3) | 29.8 (SD 11.0) | 0.852 |
| Serum osmolality, mosmol/kg | 287 (SD 6) | 285 (SD 4) | 0.152 |
Values are means [standard deviation (SD)]; n = 16. ACTH, adrenocorticotropic hormone.
Statistical significance calculated with 2-tailed paired t-test.
Participants then sat in a heat tent [hypohydrated trial arm (HYPO) 45.2°C (SD 1.6), rehydrated trial arm (RE) 44.6°C (SD 1.3); P = 0.292] wearing a sweat suit (RDX EVA Nylon Sauna Sweat Suit) for 60 min. Participants were nude weighed three times (Seca 803; Seca, Birmingham, UK) immediately before and after the heat tent to determine body water losses. After the heat tent, participants were provided with a sandwich containing ≥1 g salt (Co-Operative Group) of their choosing [standardized within participant; sandwiches contained 1.5 g (SD 0.5), range 1.1–2.4 g], to maximize fluid retention and serum osmolality changes (17), and either 3 ml/kg body mass (HYPO) or 40 ml/kg lean body mass plus 150% sweat losses (RE) of plain water to replace losses and account for the increased drink-induced diuresis (12). All other fluids were prohibited, including those containing caffeine and alcohol. Participants were only allowed to eat from a list of low-water-content foods (e.g., pizza, biscuits, nuts; avoiding fruit, vegetables, soups, and other fluids). Physical activity energy expenditure and nutrient intake profiles were similar during the intervention day (all measured nutrients P ≥ 0.102), except water intake [HYPO 0.52 l/day (SD 0.11) vs. RE 3.7 l/day (SD 0.8); P < 0.001], confirming compliance with the protocol.
Laboratory testing day.
Participants arrived at the laboratory between 0700 and 0730 after overnight food and fluid abstention from at least 2200 the previous day, provided a urine sample, and had their body mass recorded (Inner scan; body composition monitor, model BC-543, Tanita). A second pQCT scan of the midpoint of the right thigh was taken, after which participants were asked to rest semisupine for 10–15 min. Their resting metabolic rate was recorded via indirect calorimetry from gaseous exchange (13), whereby 2 × 5-min measures were taken. Expired gas samples were collected in a Douglas bag (Hans Rudolph, Kansas City, KS) through Falconia tubing (Baxter, Woodhouse and Taylor, Macclesfield, UK). Inspired air was simultaneously measured to adjust for ambient O2 and CO2 concentrations (4). Inspired and expired O2 and CO2 concentrations were measured with paramagnetic and infrared analyzers (Mini HF 5200; Servomex Group, Crowborough, UK). For metabolic rate data n = 14, as two participants were excluded from all metabolic rate and respiratory exchange ratio analyses because of fasted values > 1, which appeared to be caused by hyperventilation at these time points. In a subgroup of participants (n = 9) an additional 5-min Douglas bag was taken at 25–30 min after glucose ingestion to establish whether the initial diet-induced thermogenesis trend was linear between 0 and 60 min.
Participants then placed their hand in a hotbox (Medical Engineering Unit, University of Nottingham) set to 55°C for 5 min before an indwelling cannula was fitted in an antecubital vein and a fasted blood sample was drawn. An opt-in muscle biopsy was taken (n = 9), followed by a second fasted blood sample to quantify any changes from the expected stress response of the biopsy. The muscle samples were acquired via an ~3-mm incision at the anterior aspect of the thigh under local anesthetic (lidocaine 1%, without epinephrine; Hameln Pharmacueticals, Brockworth, UK) from the vastus lateralis by percutaneous needle biopsy technique (3). Samples were immediately removed from the needle and snap frozen in liquid nitrogen before storage at −80°C.
A 75-g OGTT (Polycal; Nutricia) was subsequently conducted. Arterialized venous blood samples (10 ml) were drawn at 15-min intervals for 120 min. Expired gas samples (1 × 5-min Douglas bag as described above) were collected hourly. A second muscle biopsy was then taken (for those who had opted in; n = 7) after 120 min.
Muscle Water Content Analysis
Total muscle water content was determined by weighing the biopsy samples before and after freeze drying (23). Samples were weighed on a high-precision (resolution 0.01 mg) electronic balance (Mettler Toledo AE240) while frozen (wet weight). The time elapsed between sample removal from storage and weighing was fixed and recorded to allow for standardization of tissue water evaporation. Samples were then freeze dried with a LyoDry Compact (MechaTech Systems) freeze dryer for 24 h at −55°C before being weighed again (dry weight) with the same precision balance. Total muscle water content was calculated as grams of water per kilogram of wet muscle with the following equation: (wet weight − dry weight/wet weight) × 1,000.
Biochemical Analysis
Six milliliters of blood was decanted into two EDTA tubes and spun for 10 min at 2,500–3,446 g at 4°C. The remaining 4 ml of blood was decanted into a serum tube, left for at least 30 min at room temperature, and then spun as per the plasma. The plasma and serum were divided into aliquots in separate Eppendorf tubes and frozen at −20°C before being moved to a −80°C freezer for longer-term storage.
Metabolites and hormones were measured with commercially available ELISAs (plasma arginine8 vasopressin, Enzo Life Sciences; serum insulin, Mercodia), electrochemiluminescence immunoassays (plasma ACTH and cortisol, Roche), automated immune analyzers (plasma copeptin, ThermoFisher Kryptor Compact Plus), and spectrophotometric assays (serum glucose, RX Daytona, Randox Laboratories). Osmolality was measured with freezing-point depression (serum osmolality, Gonotec Osmomat auto; urine osmolality, Micro-Osmometer 3300), and urine specific gravity was measured with a handheld refractometer (SUR-NE Clinical Refractometer; Atago).
Statistical Analysis
The primary aim of the study was to investigate the glycemic response to an OGTT. Secondary outcomes were the insulin, vasopressin (copeptin), rested energy expenditure, and substrate utilization responses to the OGTT and the change in urine osmolality, urine specific gravity, cell volume, serum osmolality, and muscle water content from the intervention. We were additionally able to investigate ACTH and cortisol.
Data were analyzed with paired-samples t-test or two-way repeated measures (trial, time, trial × time) analysis of variance or appropriate nonparametric tests (SPSS, version 22; IBM). Normality was checked visually via P-P plots, histograms of standardized residuals, and scatterplots of the standardized predicted vs. residual values. Asphericity was determined with Greenhouse-Geisser epsilon; values < 0.75 were corrected for with Greenhouse-Geisser correction and values > 0.75 with Huynh-Feldt correction. Total area under the curve (AUC) and incremental AUC (iAUC) were calculated as per Wolever (30). Analyses were repeated excluding those who had muscle biopsies to explore whether their inclusion skewed the overall findings. All analyses were two tailed with an α level of ≤0.05.
RESULTS
Markers of hydration status (body mass, urine specific gravity, urine osmolality, and cross-sectional muscle area) suggested compliance with both the HYPO and RE protocols (Table 3).
Table 3.
Markers of hydration status between morning of intervention day (euhydrated state) and full laboratory testing day
| Hypohydration |
Rehydration |
Pdifference |
||||||
|---|---|---|---|---|---|---|---|---|
| BL [mean (SD)] | FT [mean (SD)] | Δ BL to FT HYPO [mean (95% CI)] | BL [mean (SD)] | FT [mean (SD)] | Δ BL to FT RE [mean (95% CI)] | BL HYPO vs. RE* | FT HYPO vs. RE* | |
| Body mass, kg | 71.6 (SD 9.8) | 70.3 (SD 9.7) | −1.3 (−1.8, −0.8) | 71.5 (SD 9.8) | 71.4 (SD 9.9) | −0.1 (−0.3, −0.1) | 0.675 | <0.001 |
| Body water, kg | 37.3 (SD 7.0) | 36.8 (SD 6.5) | −0.4 (−0.8, 0.0) | 37.6 (SD 5.8) | 37.8 (SD 5.3) | 0.1 (−0.3, 0.6) | 0.669 | 0.226 |
| CSMA, mm2 | 12,773 (SD 2,829) | 12,408 (SD 2,662) | −365 (−587, −138) | 12,682 (SD 2,684) | 12,688 (SD 2,739) | 6 (−137, 148) | 0.304 | 0.002 |
| USG | 1.017 (SD 0.005) | 1.027 (SD 0.003) | 0.010 (0.007, 0.014) | 1.013 (SD 0.005) | 1.016 (SD 0.003) | 0.003 (0.000, 0.005) | 0.017 | <0.001 |
| Urine osmolality, mosmol/kg | 554 (SD 185) | 965 (SD 84) | 442 (355, 529) | 419 (SD 147) | 532 (SD 126) | 93 (9, 178) | 0.023 | <0.001 |
Values are means [standard deviation (SD] or means [95% confidence interval (CI)]. BL, euhydrated baseline measurement from the morning of the intervention day; CSMA, cross-sectional muscle area; HYPO, hypohydrated trial arm; FT, full trial day; RE, rehydrated trial arm; USG, urine specific gravity.
Statistical significance calculated with paired t-test.
Serum Glucose Concentration
Fasting serum glucose concentrations were similar between HYPO [5.10 mmol/l (SD 0.42)] and RE [5.02 mmol/l (SD 0.40); P = 0.327]. There were no differences in the glycemic response between HYPO and RE during the OGTT (trial F = 0.246, P = 0.627; time F = 41.128, P < 0.001; trial × time F = 0.944, P = 0.430; Fig. 2A). No differences were found in the serum glucose iAUC [HYPO 303 mmol·120 min·l−1 (SD 121), RE 306 mmol·120 min·l−1 (SD 113); P = 0.866], AUC [HYPO 926 mmol·120 min·l−1 (SD 169), RE 934 mmol·120 min·l−1 (SD 120); P = 0.819], or time to peak [HYPO 46 min (SD 14), RE 48 min (SD 14) min; P = 0.609].
Fig. 2.
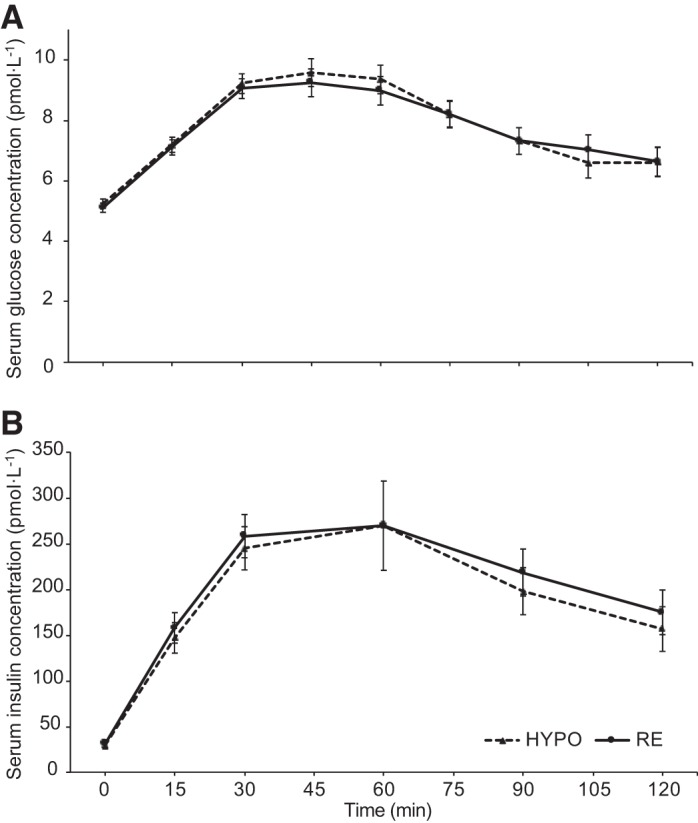
Serum glucose (A; trial F = 0.246, P = 0.627; time F = 41.128, P < 0.001; trial × time F = 0.944, P = 0.430) and insulin (B; trial F = 1.800, P = 0.200; time F = 29.597, P < 0.001; trial × time F = 0.232, P = 0.859) responses to a 75-g oral glucose tolerance test (n = 16). Data are means and normalized 95% confidence intervals. HYPO, hypohydrated trial arm; RE, rehydrated trial arm.
Serum Insulin Concentration
Serum insulin concentrations were similar in the fasted state [HYPO 27.09 pmol/l (SD 9.66), RE 27.62 pmol/l (SD 9.21); P = 0.809]. During the OGTT, there were no differences between HYPO and RE in the insulinemic response (trial F = 1.800, P = 0.200; time F = 29.597, P < 0.001; trial × time F = 0.232, P = 0.859; Fig. 2B). There were no differences in the iAUC [HYPO 20,860 pmol·120 min·l−1 (SD 8,311), RE 21,937 pmol·120 min·l−1 (SD 8,340); P = 0.369], AUC [HYPO 23,958 pmol·120 min·l−1 (SD 9,275), RE 25,326 mmol·120 min·l−1 (SD 8,679); P = 0.359], or time to peak [HYPO 54 min (SD 25), RE 58 min (SD 26); = P = 0.633] serum insulin concentrations.
Serum Osmolality
There was an increase from baseline in serum osmolality during HYPO of 9 mosmol/kg (SD 6), with relative stability from baseline during RE [Δ 1 mosmol/kg (SD 4); HYPO vs. RE P < 0.001; Table 3). Similar differences remained throughout the OGTT (trial F = 74.457, P < 0.001), reflected in a greater AUC during HYPO compared with RE [HYPO 35,355 mosmol·120 min·l−1 (SD 692), RE 34,232 mosmol·120 min·l−1 (SD 701); P < 0.001].
Plasma Copeptin Concentration
Because of the difficulties in measuring AVP (24), our data were unreliable but are available in the data set archived at https://researchdata.bath.ac.uk/id/eprint/547. Plasma copeptin was measured as a reliable marker of AVP secretion (24). Fasted (prebiopsy) plasma copeptin concentrations increased significantly from baseline after HYPO [Δ 14.32 pmol/l (SD 9.32); P < 0.001] but not after RE [Δ 0.46 pmol/l (SD 2.34); P = 0.457]. Plasma copeptin concentrations were consistently higher throughout the OGTT during HYPO compared with RE (trial F = 14.193, P = 0.002; time F = 1.285, P = 0.282; trial × time F = 1.396, P = 0.261; Fig. 3A), confirmed by a higher AUC [HYPO 2,704 pmol·120 min·l−1 (SD 2,398), RE 961 pmol·120 min·l−1 (SD 1,488); P = 0.001].
Fig. 3.

A: plasma copeptin response during an oral glucose tolerance test (n = 16; trial F = 14.193, P = 0.002; time F = 1.285, P = 0.282; trial × time F = 1.396, P = 0.261). B: plasma copeptin response during an oral glucose tolerance test, with those who had muscle biopsies on both trials (n = 7) separated from those who had no biopsies on either trial (n = 7). For those who had biopsies a fasted blood sample was taken before the biopsy (time point “Pre-biopsy”) and after the biopsy immediately before the glucose was ingested (time point “0”), and for those who did not have biopsies only 1 fasted sample was taken (time point “0”). *P < 0.050, **P < 0.010 comparing hypohydrated trial arm (HYPO) and rehydrated trial arm (RE) (trial effect) after Bonferroni adjustment for multiple comparisons. Error bars show normalized 95% confidence intervals.
Plasma ACTH Concentration
There were no differences in the ACTH response between HYPO and RE (trial F = 2.541, P = 0.132; time F = 6.120, P = 0.025; trial × time F = 1.343, P = 0.266) during the OGTT, nor were there differences in the AUC [HYPO 405 pmol·120 min·l−1 (SD 195), RE 468 pmol·120 min·l−1 (SD 255); P = 0.121].
Plasma Cortisol Concentration
Plasma cortisol concentrations were similar between HYPO and RE (trial F = 0.216, P = 0.649; time F = 19.416, P < 0.001; trial × time F = 0.275, P = 0.674), with no differences in the plasma cortisol AUC [HYPO 35,445 nmol·120 min·l−1 (SD 17,432), RE 36,716 nmol·120 min·l−1 (SD 24,915); P = 0.642].
Cross-Sectional Muscle Area
Cross-sectional muscle area as a proxy for muscle cell volume reduced significantly from baseline after HYPO [Δ −2.9% (SD 2.7); P = 0.003] but not after RE [Δ 0.0% (SD 2.1) P = 0.936; Table 3).
Muscle Water Content
Pre-OGTT muscle biopsies showed that muscle water content was lower during HYPO [760.5 g/kg (SD 13.2)] compared with RE [771.6 g/kg (SD 17.8)], but this difference dissipated after OGTT [HYPO 778.6 g/kg (SD 15.1), RE 780.2 g/kg (SD 20.0); trial F = 3.183, P = 0.135; time F = 15.36, P = 0.011; trial × time F = 6.265, P = 0.055; Fig. 4).
Fig. 4.
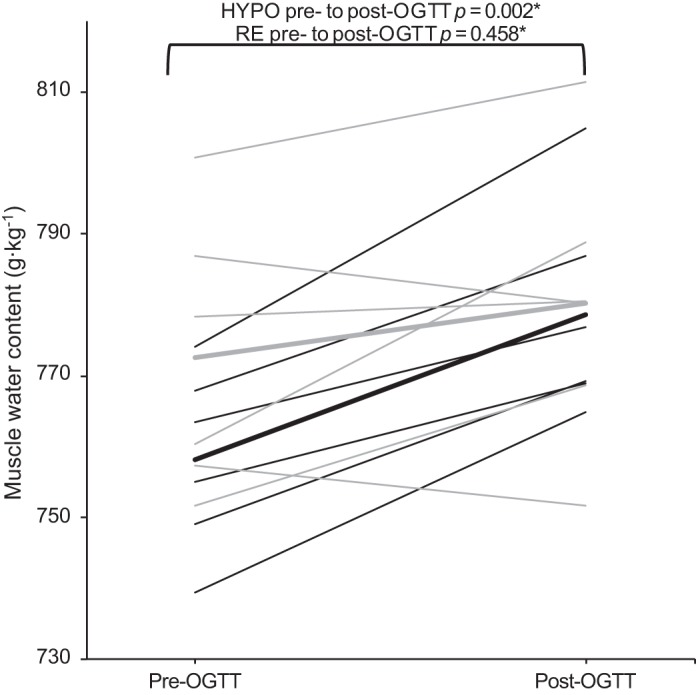
Average (thick lines) and individual (thin lines) muscle water content before and after oral glucose tolerance test (OGTT) during hypohydrated trial arm (HYPO; black) and rehydrated trial arm (RE; gray) (n = 6; trial F = 3.183, P = 0.135; time F = 15.36, P = 0.011; trial × time F = 6.265, P = 0.055). *P value Bonferroni adjusted for multiple comparisons.
Resting Energy Expenditure
Resting metabolic rate was similar between trial arms in the fasted [HYPO 96.32 kJ·kg−1·day−1 (SD 11.94), RE 95.11 kJ·kg−1·day−1 (SD 13.09), P = 0.400] and postprandial (trial F = 0.015, P = 0.904; time F = 10.130, P = 0.001; trial × time F = 0.140, P = 0.798) states. Participants had similar fasting respiratory exchange ratios [HYPO 0.84 (SD 0.05), RE 0.84 (SD 0.06); P = 0.900], although carbohydrate oxidation after glucose ingestion had a tendency to increase to a greater extent in the RE trial at 60 min [HYPO 0.88 (SD 0.04), RE 0.93 (SD 0.09)] and 120 min [HYPO 0.87 (SD 0.07), RE 0.91 (SD 0.07); trial F = 3.650, P = 0.078; time F = 14.693, P < 0.001; trial × time F = 3.754, P = 0.039].
Plasma Volume
Because of an error in postural control during the baseline (euhydrated) venipuncture, the plasma volume data were invalid. We have included details of how plasma volume was measured and the data obtained in the data set available at https://researchdata.bath.ac.uk/id/eprint/547.
Sensitivity Analysis
Those who had muscle biopsies demonstrated a distinct stress response in plasma copeptin (Fig. 3B), ACTH, and cortisol concentrations (shown below). Removing those who had any biopsies did not alter the findings.
Copeptin.
Removing those who had any biopsies did not meaningfully affect the trend in plasma copeptin concentration throughout the OGTT (n = 7; trial F = 13.517, P = 0.010; time F = 4.081, P = 0.040; trial × time F = 1.489, P = 0.262), although the overall AUC was lower [n = 7; HYPO 11,916 pmol·120 min·l−1 (SD 1,661), RE 390 pmol·120 min·l−1 (SD 177); P = 0.028].
ACTH.
Removing those who had opted in for muscle biopsies eliminated the time effect and highlighted a small though nonsignificant trend toward higher ACTH concentrations during RE compared with HYPO (n = 7; trial F = 4.203, P = 0.086; time F = 0.989, P = 0.361; trial × time F = 1.729, P = 0.219). When biopsy participants were removed the AUCs were lower [n = 7; HYPO 292 pmol·120 min·l−1 (SD 139), RE 355 pmol·120 min·l−1 (SD 86); P = 0.176], but there was still no difference between HYPO and RE.
Cortisol.
In accordance with the plasma copeptin and ACTH responses, plasma cortisol concentration also increased after biopsy. Removing these participants did not meaningfully alter the results, although the time effect was no longer evident (n = 7; trial F = 0.278, P = 0.617; time F = 5.172, P = 0.055; trial × time F = 0.260, P = 0.686). Despite a postbiopsy peak in plasma cortisol concentration, the AUC was higher when the biopsy participants were removed [n = 7; HYPO 37,920 nmol·120 min·l−1 (SD 25,008), RE 40,989 nmol·120 min·l−1 (SD 37,507); P = 0.735], with no difference between HYPO and RE.
Resting metabolic rate.
In the subgroup who had the additional measurement at 30 min after glucose ingestion (n = 9), no difference in resting metabolic rate (trial F = 0.346, P = 0.573; time F = 6.087, P = 0.009; trial × time F = 0.586, P = 0.508) or respiratory exchange ratio (trial F = 0.433, P = 0.529; time F = 17.330, P < 0.001; trial × time F = 0.467, P = 0.607) was apparent according to hydration status.
DISCUSSION
This randomized crossover trial is the first to show that neither fasted nor postprandial glycemia and insulinemia are influenced by hydration status in healthy adults, contrary to our hypothesis. The key implication of this work is that clinicians and researchers may not have to control for hydration status when investigating glycemic regulation in healthy adults. Participants replicated their diet and activity 4 days before trial, reducing known confounding influences. Average body mass loss during HYPO was 1.9%, which is within the typical range to induce thirst and is not uncommon in the general population (2), increasing the external validity of these findings. In the hypohydrated state, copeptin (a surrogate marker of AVP) concentrations increased from levels seen in healthy adults to levels reported in those at highest risk of metabolic syndrome (10), with an accompanying reduction in muscle cell volume of ~3%, hypothesized to be detrimental to glycemic regulation (16). Therefore the level of hypohydration achieved was sufficient to induce physiological changes that theoretically have meaningful health implications; such changes did not occur when participants were rehydrated.
Despite these physiological changes hypothesized to cause higher glycemia, fasted and postprandial glycemia were similar between HYPO and RE. These results are in contrast to similar work in those with diabetes (5, 18). In both previous studies, participants were required to withdraw from diabetes medication; accordingly, Burge et al. (5) found higher glucosuria when participants were euhydrated compared with hypohydrated, potentially explaining the lower glycemic response. As this effect of glucosuria has previously been alluded to (31), it is a possible (at least partial) explanation for the findings of Johnson et al. (18). This hypothesis could be tested by comparing glycemic regulation in those with diabetes during medication withdrawal versus prescription. Glucosuria should not occur in healthy adults, potentially explaining why we did not find a lower glycemia during RE compared with HYPO.
Similarly, our findings conflict with research in healthy adults. Keller et al. (20) administered intravenous saline and/or desmopressin to induce changes specifically in extracellular osmolality, finding higher fasted glucose concentrations during hyperosmolality. This method of dehydration, however, is not representative of whole body water losses, potentially explaining the discordance between our findings. The present study also contradicts our own pilot work (8), most likely because of the lack of rigorous pretrial standardization of diet (verbal 24-h recall), physical activity (self-reported replication), and hydration status (no preintervention measures) in the pilot.
A key mechanism by which elevations in AVP induce poor glycemic control is through the hypothalamic-pituitary-adrenal axis, stimulating ACTH and cortisol secretion. Despite the increase we observed in plasma copeptin concentrations, plasma ACTH and cortisol concentrations were not different between HYPO and RE, suggesting that in healthy adults short-term hypohydration is not a sufficient stimulus to induce a stress response along the hypothalamic-pituitary-adrenal axis. This theory is in accordance with earlier work highlighting that the role of AVP in ACTH secretion is predominantly a response to physical stress (15). Nevertheless, we did not find a difference in ACTH or cortisol by hydration status in those who had the muscle biopsies, despite higher copeptin responses during HYPO. In previous work in those with type 2 diabetes, cortisol concentrations did increase 45 min after glucose ingestion when hypohydrated (18), perhaps suggesting an interaction between hydration status and feeding in those with metabolic impairments. However, as copeptin did not increase postprandially in our study, the mechanism for this cortisol response is unlikely to be AVP mediated.
Another mechanism proposed to link hypohydration to higher glycemia is changes to cell volume influencing insulin and glucagon secretion, although a limitation of our study is that glucagon was not measured. Muscle cell volume was reduced by ~3% from baseline, compared with no change when participants were rehydrated—a reduction that may deteriorate glucose tolerance (16, 20). The pQCT data were confirmed by the muscle biopsy samples, which showed lower pre-OGTT total muscle water content in HYPO vs. RE. Interestingly, after OGTT the muscle water content difference diminished, suggesting that the introduction of glucose to the cell created a strong enough stimulus to move water from other compartments into skeletal muscle. Conversely, muscle water content did not change from before to after OGTT during RE, perhaps showing greater overall water balance as no extra water was needed intracellularly for glycogen storage (25).
Hydration status could influence metabolic health over longer time periods by altering thermogenesis. Although it has been shown, albeit inconsistently, that water ingestion induces greater thermogenesis (27), our study did not support these findings, supporting evidence that the thermogenic effect is from consuming (cold) water rather than altering hydration status (27). There was a tendency for higher carbohydrate utilization during RE compared with HYPO, perhaps indicating greater hepatic glucose output during RE to account for the higher carbohydrate utilization; however, these findings should be interpreted cautiously, as the study was not powered for these outcomes.
There are several other speculative theories that could explain our null findings that we did not test. First, it could be that healthy adults have a greater capacity to handle metabolic challenges such as acute bouts of hypohydration, and although physiological changes occur to handle this flux, such deviations ensure maintenance of homeostasis and therefore minimize metabolic disruption. Accordingly, both serum osmolality and plasma copeptin concentrations increased during HYPO vs. RE, suggesting higher AVP secretion to help maintain homeostasis. Considering that the level of HYPO we induced is not uncommon in the population (2) and caused an increase in copeptin concentrations associated with poor cardiometabolic health (10, 11), it may be that such commonly achieved levels of hypohydration have detrimental longer-term health effects not captured by our acute study design.
A further hypothesis is that the conflicting roles of vasopressin receptors (VRs) in adipose tissue insulin sensitivity led to a null effect on glucose tolerance during HYPO. Specifically, V1aR−/− mice have reduced adipose tissue glucose tolerance, whereas V1bR−/− mice have increased adipose tissue insulin sensitivity (1, 14, 28). If these findings translate into humans (and assuming a similar propensity for VR binding), this could mean that HYPO-induced AVP secretion, such as that achieved in our study, leads to AVP binding on both V1aR and V1bR, resulting in net zero effect on glycemic regulation. However, the present study was not equipped to examine this speculative theory.
Overall, despite physiologically and clinically meaningful increases in plasma copeptin and serum osmolality, and a notable reduction in cell volume supporting a systemic difference in hydration status between trial arms, we demonstrated for the first time that acute hypohydration did not alter the glycemic response. Our findings suggest that when OGTTs are conducted in healthy adults hydration status may not necessarily need to be strictly controlled for. Although longer-term research is needed to understand the glucoregulatory effects of chronic hypohydration, these data suggest that acute manipulations of hydration status in healthy adults do not impact fasted or postprandial glycemic regulation.
GRANTS
This work was supported by the Economic and Social Research Council (Grant ES/J50015X/1) and the European Hydration Institute Graduate Research Grant.
DISCLAIMERS
Neither funding source had any role in the study design, data analysis, or writing or publishing of this article.
DISCLOSURES
H. A. Carroll has accepted conference fees from Danone. O. Melander has received consultancy honoraria from Danone Research. L. J. James has previously received funding for hydration-related research from PepsiCo Inc., the European Hydration Institute, and Volac International Ltd. and has performed consultancy work for Lucozade Ribena Suntory. L. Johnson has received funding from Kellogg Europe and Danone Baby Nutrition. J. A. Betts has received funding from Lucozade Ribena Suntory, PepsiCo Inc., and Kenniscentrum Suiker. None of the other authors has any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
H.A.C. conceived and designed research; H.A.C., I.T., Y.-C.C., R.M.E., E.K.B., J.T.J., G.P., T.D.R., W.L.D., and J.A.B. performed experiments; H.A.C., I.T., Y.-C.C., R.J., K.T., W.G., O.M., and L.J.J. analyzed data; H.A.C., I.T., Y.-C.C., R.J., K.T., W.G., O.M., L.J.J., and J.A.B. interpreted results of experiments; H.A.C. prepared figures; H.A.C. drafted manuscript; H.A.C. edited and revised manuscript; H.A.C., I.T., Y.-C.C., R.M.E., E.K.B., J.T.J., G.P., T.D.R., W.L.D., R.J., K.T., W.G., O.M., D.T., L.J.J., L.J., and J.A.B. approved final version of manuscript.
ENDNOTE
At the request of the authors, readers are herein alerted to the fact that additional materials related to this manuscript may be found at the institutional Web site of the authors, which at the time of publication they indicate is: https://researchdata.bath.ac.uk/id/eprint/547. These materials are not a part of this manuscript and have not undergone peer review by the American Physiological Society (APS). APS and the journal editors take no responsibility for these materials, for the Web site address, or for any links to or from it.
ACKNOWLEDGMENTS
We thank Dr. Oliver Perkin for invaluable technical help with the pQCT scanner and interpreting these results.
REFERENCES
- 1.Aoyagi T, Birumachi J, Hiroyama M, Fujiwara Y, Sanbe A, Yamauchi J, Tanoue A. Alteration of glucose homeostasis in V1a vasopressin receptor-deficient mice. Endocrinology 148: 2075–2084, 2007. doi: 10.1210/en.2006-1315. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong LE. Hydration biomarkers during daily life: recent advances and future potential. Nutr Today 47: S3–S6, 2012. doi: 10.1097/NT.0b013e31826266cf. [DOI] [Google Scholar]
- 3.Bergström J. Percutaneous needle biopsy of skeletal muscle in physiological and clinical research. Scand J Clin Lab Invest 35: 609–616, 1975. doi: 10.3109/00365517509095787. [DOI] [PubMed] [Google Scholar]
- 4.Betts JA, Thompson D. Thinking outside the bag (not necessarily outside the lab). Med Sci Sports Exerc 44: 2040, 2012. doi: 10.1249/MSS.0b013e318264526f. [DOI] [PubMed] [Google Scholar]
- 5.Burge MR, Garcia N, Qualls CR, Schade DS. Differential effects of fasting and dehydration in the pathogenesis of diabetic ketoacidosis. Metabolism 50: 171–177, 2001. doi: 10.1053/meta.2001.20194. [DOI] [PubMed] [Google Scholar]
- 6.Carroll HA, Betts JA, Johnson L. An investigation into the relationship between plain water intake and glycated Hb (HbA1c): a sex-stratified, cross-sectional analysis of the UK National Diet and Nutrition Survey (2008–2012). Br J Nutr 116: 1–11, 2016. doi: 10.1017/S0007114516003688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carroll HA, Davis MG, Papadaki A. Higher plain water intake is associated with lower type 2 diabetes risk: a cross-sectional study in humans. Nutr Res 35: 865–872, 2015. doi: 10.1016/j.nutres.2015.06.015. [DOI] [PubMed] [Google Scholar]
- 8.Carroll HA, Johnson L, Betts JA. Effect of hydration status on glycemic control: a pilot study. Med Sci Sports Exerc 48: 745, 2016. doi: 10.1249/01.mss.0000487236.11983.e6 [DOI] [Google Scholar]
- 9.Corney RA, Horina A, Sunderland C, James LJ. Effect of hydration status and fluid availability on ad-libitum energy intake of a semi-solid breakfast. Appetite 91: 399–404, 2015. doi: 10.1016/j.appet.2015.04.075. [DOI] [PubMed] [Google Scholar]
- 10.Enhörning S, Struck J, Wirfält E, Hedblad B, Morgenthaler NG, Melander O. Plasma copeptin, a unifying factor behind the metabolic syndrome. J Clin Endocrinol Metab 96: E1065–E1072, 2011. doi: 10.1210/jc.2010-2981. [DOI] [PubMed] [Google Scholar]
- 11.Enhörning S, Wang TJ, Nilsson PM, Almgren P, Hedblad B, Berglund G, Struck J, Morgenthaler NG, Bergmann A, Lindholm E, Groop L, Lyssenko V, Orho-Melander M, Newton-Cheh C, Melander O. Plasma copeptin and the risk of diabetes mellitus. Circulation 121: 2102–2108, 2010. doi: 10.1161/CIRCULATIONAHA.109.909663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evans GH, James LJ, Shirreffs SM, Maughan RJ. Optimizing the restoration and maintenance of fluid balance after exercise-induced dehydration. J Appl Physiol (1985) 122: 945–951, 2017. doi: 10.1152/japplphysiol.00745.2016. [DOI] [PubMed] [Google Scholar]
- 13.Frayn KN. Calculation of substrate oxidation rates in vivo from gaseous exchange. J Appl Physiol Respir Environ Exerc Physiol 55: 628–634, 1983. doi: 10.1152/jappl.1983.55.2.628. [DOI] [PubMed] [Google Scholar]
- 14.Fujiwara Y, Hiroyama M, Sanbe A, Aoyagi T, Birumachi J, Yamauchi J, Tsujimoto G, Tanoue A. Insulin hypersensitivity in mice lacking the V1b vasopressin receptor. J Physiol 584: 235–244, 2007. doi: 10.1113/jphysiol.2007.136481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gibbs DM. Vasopressin and oxytocin: hypothalamic modulators of the stress response: a review. Psychoneuroendocrinology 11: 131–139, 1986. doi: 10.1016/0306-4530(86)90048-X. [DOI] [PubMed] [Google Scholar]
- 16.Häussinger D, Lang F, Gerok W. Regulation of cell function by the cellular hydration state. Am J Physiol Endocrinol Metab 267: E343–E355, 1994. doi: 10.1152/ajpendo.1994.267.3.E343. [DOI] [PubMed] [Google Scholar]
- 17.James LJ, Shirreffs SM. Fluid and electrolyte balance during 24-hour fluid and/or energy restriction. Int J Sport Nutr Exerc Metab 23: 545–553, 2013. doi: 10.1123/ijsnem.23.6.545. [DOI] [PubMed] [Google Scholar]
- 18.Johnson EC, Bardis CN, Jansen LT, Adams JD, Kirkland TW, Kavouras SA. Reduced water intake deteriorates glucose regulation in patients with type 2 diabetes. Nutr Res 43: 25–32, 2017. doi: 10.1016/j.nutres.2017.05.004. [DOI] [PubMed] [Google Scholar]
- 19.Kant AK, Graubard BI, Atchison EA. Intakes of plain water, moisture in foods and beverages, and total water in the adult US population—nutritional, meal pattern, and body weight correlates: National Health and Nutrition Examination Surveys 1999–2006. Am J Clin Nutr 90: 655–663, 2009. doi: 10.3945/ajcn.2009.27749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keller U, Szinnai G, Bilz S, Berneis K. Effects of changes in hydration on protein, glucose and lipid metabolism in man: impact on health. Eur J Clin Nutr 57, Suppl 2: S69–S74, 2003. doi: 10.1038/sj.ejcn.1601904. [DOI] [PubMed] [Google Scholar]
- 21.Keppens S, de Wulf H. The nature of the hepatic receptors involved in vasopressin-induced glycogenolysis. Biochim Biophys Acta 588: 63–69, 1979. doi: 10.1016/0304-4165(79)90371-4. [DOI] [PubMed] [Google Scholar]
- 22.Minshull C, James L. The effects of hypohydration and fatigue on neuromuscular activation performance. Appl Physiol Nutr Metab 38: 21–26, 2013. doi: 10.1139/apnm-2012-0189. [DOI] [PubMed] [Google Scholar]
- 23.Mora-Rodríguez R, Sanchez-Roncero A, Fernández-Elías VE, Guadalupe-Grau A, Ortega JF, Dela F, Helge JW. Aerobic exercise training increases muscle water content in obese middle-age men. Med Sci Sports Exerc 48: 822–828, 2016. doi: 10.1249/MSS.0000000000000848. [DOI] [PubMed] [Google Scholar]
- 24.Morgenthaler NG, Struck J, Jochberger S, Dünser MW. Copeptin: clinical use of a new biomarker. Trends Endocrinol Metab 19: 43–49, 2008. doi: 10.1016/j.tem.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 25.Olsson KE, Saltin B. Variation in total body water with muscle glycogen changes in man. Acta Physiol Scand 80: 11–18, 1970. doi: 10.1111/j.1748-1716.1970.tb04764.x. [DOI] [PubMed] [Google Scholar]
- 26.Roussel R, Fezeu L, Bouby N, Balkau B, Lantieri O, Alhenc-Gelas F, Marre M, Bankir L; D.E.S.I.R. Study Group . Low water intake and risk for new-onset hyperglycemia. Diabetes Care 34: 2551–2554, 2011. doi: 10.2337/dc11-0652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stookey JJ. Negative, null and beneficial effects of drinking water on energy intake, energy expenditure, fat oxidation and weight change in randomized trials: a qualitative review. Nutrients 8: E19, 2016. doi: 10.3390/nu8010019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanoue A. New topics in vasopressin receptors and approach to novel drugs: effects of vasopressin receptor on regulations of hormone secretion and metabolisms of glucose, fat, and protein. J Pharmacol Sci 109: 50–52, 2009. doi: 10.1254/jphs.08R15FM. [DOI] [PubMed] [Google Scholar]
- 29.Whitton PD, Rodrigues LM, Hems DA. Stimulation by vasopressin, angiotensin and oxytocin of gluconeogenesis in hepatocyte suspensions. Biochem J 176: 893–898, 1978. doi: 10.1042/bj1760893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolever TM. Effect of blood sampling schedule and method of calculating the area under the curve on validity and precision of glycaemic index values. Br J Nutr 91: 295–301, 2004. doi: 10.1079/BJN20031054. [DOI] [PubMed] [Google Scholar]
- 31.Zerbe RL, Vinicor F, Robertson GL. Plasma vasopressin in uncontrolled diabetes mellitus. Diabetes 28: 503–508, 1979. doi: 10.2337/diab.28.5.503. [DOI] [PubMed] [Google Scholar]