

Abstract
The rapid emergence of antibiotic-resistant bacterial “superbugs” with concomitant treatment failure and high mortality rates presents a severe threat to global health. The superbug risk is further exacerbated by chronic infections generated from antibiotic-resistant biofilms that render them refractory to available treatments. We hypothesized that efficient antimicrobial agents could be generated through careful engineering of hydrophobic and cationic domains in a synthetic semi-rigid polymer scaffold, mirroring and amplifying attributes of antimicrobial peptides. We report the creation of polymeric nanoparticles with highly efficient antimicrobial properties. These nanoparticles eradicate biofilms with low toxicity to mammalian cells and feature unprecedented therapeutic indices against red blood cells. Most notably, bacterial resistance towards these nanoparticles was not observed after 20 serial passages, in stark contrast to clinically relevant antibiotics where significant resistance occurred after only a few passages.
Keywords: Polymeric nanoparticles, multi-drug resistance, biofilms, MRSA, therapeutic index, antimicrobials
Graphical Abstract
Introduction
Indiscriminate use of antibiotics in agricultural1 and medical fields2 has created multi-drug resistant (MDR) “superbugs” such as methicillin-resistant Staphylococcus aureus (MRSA) along with particularly refractory Gram-negative species that pose a serious threat to global health. Planktonic bacteria cause acute infections resulting in sepsis, with the threat further intensified by chronic infections from biofilms.3,4 Biofilm-associated infections frequently occur on medical implants and indwelling devices such as catheters, prosthesis and dental implants.5 Biofilm infections can also occur on or around dead tissues leading to endocarditis and chronic wound infections.6 These intractable infections are challenging due to the high resistance of these infections towards both host immune response and traditional antimicrobial therapies.7 Current biofilm treatment techniques require aggressive antibiotic therapy coupled with debridement of infected tissue.8 However, this standard regimen incurs high treatment costs and low patient compliance due to the invasive nature of the treatment.9 The therapeutic challenge is exacerbated by the increasing number of antibiotic-resistant bacterial strains, further impairing the therapeutic effectiveness of existing antibiotics.10
Antimicrobial peptides (AMPs) have emerged as an alternative to conventional antibiotic therapy, exhibiting broad spectrum activity against antibiotic-resistant bacteria.11,12 AMPs have demonstrated high therapeutic indices (TI, selectivity towards bacterial cells calculated as HC50 (Hemolytic activity)/MIC) of ~900 and ~3,30013 against planktonic bacteria, however these α-helical peptides are susceptible to proteolytic degradation, reducing their efficacy.14,15 Hostdefense peptide mimicking synthetic polymers have recently been developed, demonstrating broad spectrum activity against microbes.16–20 However, high toxicity towards mammalian cells and red blood cells, resulting in moderate therapeutic indices 16,18–20 have impaired their practical applications in clinical settings. Low toxicity to mammalian cells, in particular red blood cells is critical for effective application of antimicrobials in or on patients.29,36 Limited studies have demonstrated synthetic polymers with improved therapeutic indices (~150–550),21–24 exhibiting the ability of these polymers to kill bacteria while causing minimal hemolysis of red blood cells. However, these polymers have focused on the treatment of planktonic microbes, overlooking the more drug-resistant biofilm counterparts. To the best of our knowledge, synthetic polymers exhibiting high biofilm efficacy while maintaining low toxicity towards mammalian cells have not been reported.
We report here engineered polymers that effectively eradicate pre-formed biofilms while maintaining high therapeutic indices (>1000) against red blood cells (RBCs). In the design of these materials we hypothesized that the therapeutic window of cationic polymers could be regulated by varying hydrophobic moieties, similar to the hydrophobic residues present in the active sites of antimicrobial peptides.25 To this end we synthesized a library of quaternary ammonium poly(oxanorborneneimides) possessing different degrees of hydrophobicity (Figure 1) and screened their antimicrobial and hemolytic activities. These polymers form 10–15 nm nanoparticles in aqueous solution, increasing their overall cationic charge and molecular mass. We observed that longer hydrophobic alkyl chains that bridge the cationic head group and polymer backbone greatly enhances toxicity against planktonic bacteria while maintaining low hemolytic activity towards RBCs (TI 1250–2500). These nanoparticles readily penetrate biofilms and eradicate pre-formed biofilms while still maintaining high TI (60–165). Polymeric NPs (PNPs) demonstrated a 6-fold log reduction in bacterial colonies with no mammalian cell toxicity when tested in a biofilm-mammalian cell coculture model. Notably, we observed that bacteria did not develop any resistance against PNPs even after 20 serial passages, in stark contrast to conventional antibiotics. Overall, our engineered polymeric nanoparticle platform shows strong potential as an infectious disease therapeutic and simultaneously provides a rational approach to design novel antimicrobials for sustainably combating bacterial infections.
Figure 1.
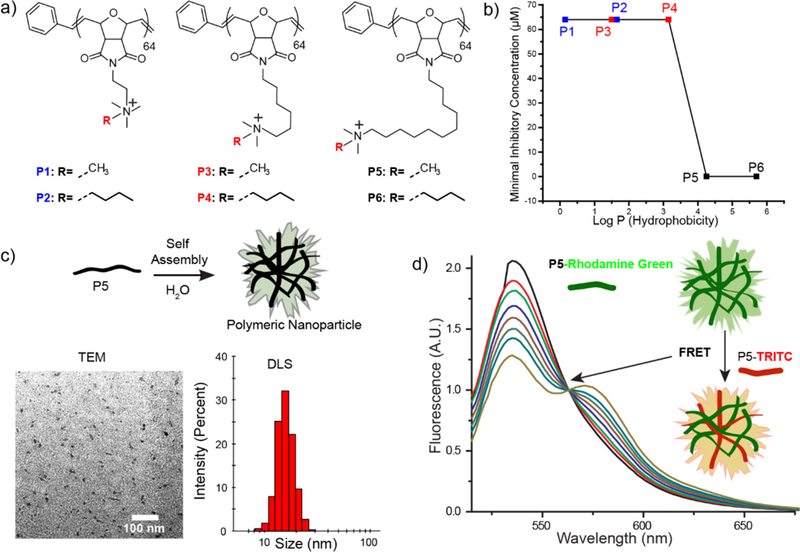
Molecular structures of a) oxanorbornene polymer derivatives. b) MIC values of polymer derivatives with different hydrophobic chain lengths. Log P represents the calculated hydrophobic values of each monomer c) Schematic representation depicting self-assembly of P5-homopolymers. Characterization of P5 PNPs using TEM imaging and DLS measurement. d) Graph for FRET experiments between P5-Rhodamine Green and P5-TRITC indicating formation of polymeric NPs.
Results and Discussions
Generation and Characterization of Polymer Nanoparticles.
Norbornene/oxanorbornene-based polymers feature conformational restrictions reminiscent of peptides, and amphiphilic cationic polymers with this backbone have shown promising antimicrobial properties.24,26 Additionally, the synthetic scalability provides a key advantage over antimicrobial peptides.27,28 The distribution of hydrophobic moieties on antimicrobial macromolecules plays a pivotal role in determining their bactericidal activity.25,29 In particular, careful consideration of “amphiphilic balance”, i.e. distribution of cationic charge and hydrophobic moieties on the polymer are critical to ensure antimicrobial selectivity towards bacteria over mammalian cells.30 We explored this design space through a library of oxanorbornene polymers (Figure 1 a, 2a) with varying unbranched alkyl chains both bridging the cationic head group and the polymer backbone itself, allowing systematic determination of structure-antimicrobial efficacy relationships. We found that polymers containing a bridged C11 alkyl chain spontaneously self-assemble into cationic PNPs (~13 nm) in aqueous solutions as confirmed by transmission electron microscopy (TEM, Figure 1c, SI 2), dynamic light scattering (DLS, Figure 1c) and Förster resonance energy transfer (FRET) experiments (Figure 1d, Structural details of dyetagged polymer is in Figure S1, Supporting Information). These micellar structures formed at low polymer concentrations: dilution experiments of encapsulated Nile Red within P5 PNPs indicated a critical micelle concentration of < 2.5 µM (Figure S6, Supporting Information).31
Figure 2.
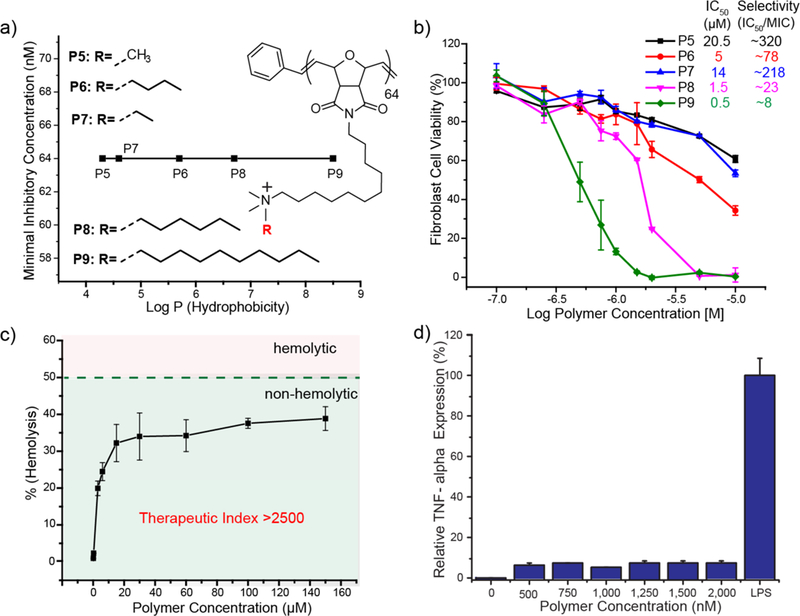
a) Graph showing minimum inhibitory concentrations (MIC) and structure details of oxanorbornene derivatives with different hydrophobicity of the cationic headgroups. Log P represents the calculated hydrophobic values of each monomer. b) Graph showing toxicity of P5-P9 polymers against 3T3 Fibroblast cells indicating increase in cytotoxicity with increased hydrophobicity of the cationic headgroup. Selectivity towards bacteria as compared to mammalian cells is calculated as (IC50/MIC). c) Hemolytic activity of PNPs at different concentrations indicates their non-hemolytic behavior at relevant therapeutic concentrations. d) TNF-α secretion of Raw 264.7 cells in the presence of PNPs. Lipopolysaccharide (LPS) was used as a positive control.
Antimicrobial Activity and Therapeutic Selectivity of PNPs Against Planktonic Bacteria.
The PNP library was screened for antimicrobial activity against an uropathogenic strain of Escherichia coli (CD-2), using broth dilution methods to evaluate their minimal inhibitory concentrations (MICs).32 We observed a 1000-fold increase in the antimicrobial activity of polymeric nanoparticles upon increasing the hydrophobicity of the alkyl chain bridging the backbone and cationic headgroup (Figure 1b). Polymers with shorter internal alkyl chains (P1-P4) displayed MICs of 64 µM, while analogs with more hydrophobic C11 chains (P5, P6) inhibited bacteria growth at 0.064 µM. We further extended the hydrophobicity on the cationic headgroup of the polymers and monitored the change in antimicrobial activity. We determined that the MICs of PNPs did not change significantly upon increasing the hydrophobicity at the cationic headgroup (Figure 2a). This result indicates that careful placement of local hydrophobic domains on polymer structure plays a crucial role in determining the antimicrobial activity of the polymer. Similar behavior has also been reported in antimicrobial peptides where the location of hydrophobic residues determines antimicrobial activity.33,34
After establishing antimicrobial efficacy, we performed cell toxicity assays on human fibroblast cell lines to determine the IC50 (half-maximal inhibitory concentration) of the most hydrophobic polymers (P5-P9) and evaluate their therapeutic selectivity.35 Therapeutic selectivity is defined as IC50/MIC that determines the ability of polymers to kill bacteria while causing minimal toxicity to mammalian cells. Polymer cytotoxicity towards fibroblasts increased with increasing hydrophobicity of the alkyl chain at cationic headgroup (Figure 2b). The least hydrophobic P5 showed an IC50 of 20.5 µM, yielding therapeutic selectivity of ~320. In contrast, the most hydrophobic counterparts (P6, P8 and P9) showed therapeutic selectivities of 78, 23 and 8 respectively. These results further indicate that careful placement of hydrophobic domains on polymer can regulate their toxicity towards mammalian cells. A related study has previously reported that colocalization of the charge and hydrophobic domains reduced the antibacterial effect, however dramatically reduced the chance of red blood cell hemolysis, thereby improving the overall selectivity of the system.30 Hence, we concluded that P5 polymer with internally hydrophobic alkyl chains demonstrated highest antimicrobial activity with least cytotoxicity.
Next, we performed hemolysis assays on human RBCs with our most potent polymer P5 and calculated their HC50 (concentration that causes 50% lysis of RBCs) to determine their biocompatibility.36,37 MIC and HC50 values were used to calculate a therapeutic index (TI = HC50/MIC) of PNPs against planktonic bacteria. PNPs P5 with undecyl-bridging alkyl chains showed minimal hemolytic character (Figure 2c). The highest antimicrobial efficiency was observed with P5 PNPs, with an MIC of 64 nM (1.8 µg.ml−1) against E. coli. P5 PNPs showed little hemolytic character (HC50 >160 µM, 4700 µg.ml−1) providing an unprecedented therapeutic index of > 2500, 5-fold higher than previous polymer-based antimicrobials. Having established P5 PNPs are non-acutely toxic, we next investigated their chronic effects in relation to inflammatory cytokine responses from macrophage RAW 264.7 cells (SI Figure 4a). P5 PNP concentrations up to 2 µM showed no significant toxicity or tumor necrosis factor alpha (TNF-α) cytokine expression (Figure 2d), suggesting in vitro immunocompatibility with mammalian immune cells.38
We next tested P5 PNPs against multiple uropathogenic clinical isolates (Table 1) to establish their broad-spectrum activity. P5 PNPs suppressed bacterial proliferation at concentrations ranging from 64–128 nM (1.8 µg.ml−1 – 3.6 µg.ml−1), once again similar or lower to previously reported antimicrobial polymers. These polymers showed similar antimicrobial activity against 5 clinical isolates of E. coli with different susceptibilities to clinical antibiotics (resistant to 1–17 drugs, SI 11), indicating their ability to evade common mechanisms of bacterial resistance. Notably, engineered polymers were effective against clinical isolates of Gram-negative P. aeruginosa and E. cloacae complex. Similarly, Gram-positive strains of S. aureus were susceptible to P5 PNPs including the highly virulent strain of methicillin-resistant S. aureus (MRSA).
Table 1.
Minimum inhibitory concentrations and therapeutic indices of P5 PNPs against multiple uropathogenic clinical isolate bacterial strains. Therapeutic indices are calculated with respect to red blood cells.
| Strain | Species | MIC (nM) | TI (HC50/MIC) |
|---|---|---|---|
| CD-23 | P, aeruginosa | 64 | ~2,500 |
| CD-1006 | P, aeruginosa | 128 | ~1,250 |
| CD-489 | S. aureus - MRSA | 64 | ~2,500 |
| CD-2 | E. coli | 64 | ~2,500 |
| CD-3 | E. coli | 64 | ~2,500 |
| CD-19 | E. coli | 64 | ~2,500 |
| CD-549 | E. coli | 128 | ~1,250 |
| CD-496 | E. coli | 128 | ~1,250 |
| CD-866 | E. cloacae complex | 128 | ~1,250 |
| CD-1412 | E. cloacae complex | 128 | ~1,250 |
| CD-1545 | E. cloacae complex | 128 | ~1,250 |
Due to the highly cationic and hydrophobic nature of our PNPs, we hypothesized their activity arose from the disruption of bacterial cell membranes.39,40 This expectation was supported through staining with membrane-impermeable propidium iodide (PI) where only cells with compromised membranes generate red fluorescence.41,42 Pathogenic E. coli (CD-2), S. aureus (CD-489) and non-pathogenic P. aeruginosa (ATCC 19660) were treated with 1 µM of P5 PNPs for 3 hours at 37 °C and subsequently stained with PI before imaging. The confocal images (Figure 3b) clearly show that the PNPs generate substantial bacterial membrane disruption in all three species, regardless of membrane composition or pathogenicity.
Figure 3.
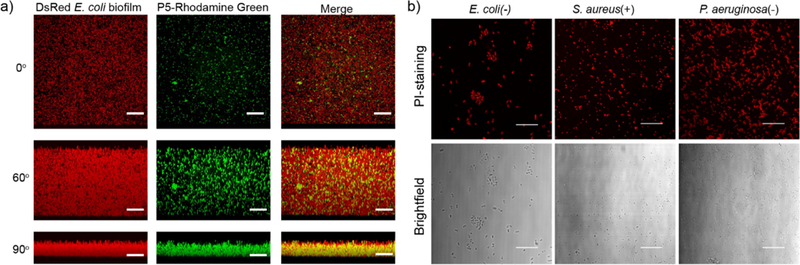
a) Representative 3D projection of confocal image stacks of E2-Crimson (Red Fluorescent Protein) expressing E. coli DH5α biofilm after 1 h treatment with P5-Rhodamine Green at 1 µM concentration. The panels are projection at 0°, 60° and 90° angle turning along X axis. Scale bars are 30 μm. b) Confocal images of E. coli (CD-2), S. aureus (MRSA, CD-489) and P. aeruginosa (ATCC 19660) stained with Propidium Iodide (PI) after treatment with PNPs. Scale bars are 30 µm.
Biofilm Penetration and Eradication by PNPs.
After establishing the efficacy of our NPs against bacterial “superbugs”, we tested their efficacy against the even more refractory bacterial biofilms. Bacteria in biofilms produce extracellular polymeric substance that provides a potent barrier against therapeutics.8 Penetration and accumulation of therapeutics inside biofilms is crucial for effective therapy of these infections,43,44 so the ability of PNPs to penetrate biofilms was determined using confocal microscopy. We treated biofilms formed by E. coli expressing E2-Crimson (a red fluorescent protein) with P5 PNPs functionalized with Rhodamine-Green fluorescent dyes. As shown in Figure 3a, fluorescently labeled nanoparticles readily penetrated and dispersed throughout the biofilms (SI Figure S3), suggesting their ability to be an effective anti-biofilm agent.
Having established biofilm penetration, the therapeutic ability of P5 PNPs against pre-formed bacterial biofilms was quantified. We chose a laboratory strain of P. aeruginosa (ATCC 19660) and 3 uropathogenic clinical isolates, P. aeruginosa (CD-1006), En. cloacae complex (CD-1412) and S. aureus (CD-489, a methicillin-resistant strain). As shown in Figure 4, P5 PNPs demonstrate minimum concentrations to eradicate 90% of biofilms (MBEC90) ranging from 1–3 µM, providing unprecedented therapeutic indices ranging from 60–165 for biofilms (TI = HC50/MBEC90, Supporting Information Figure S5). Nanoparticles could treat both Gram-negative (P. aeruginosa, and En. cloacae complex) and Gram-positive (S. aureus) bacterial strains, further highlighting their broadspectrum activity against biofilms. Notably, P5 PNPs demonstrated similar efficacy in treating MDR (CD-489, CD-1412) and non-resistant strains (CD-1006, ATCC 19660), suggesting their value as a therapeutic alternative to traditional antibiotics.
Figure 4.
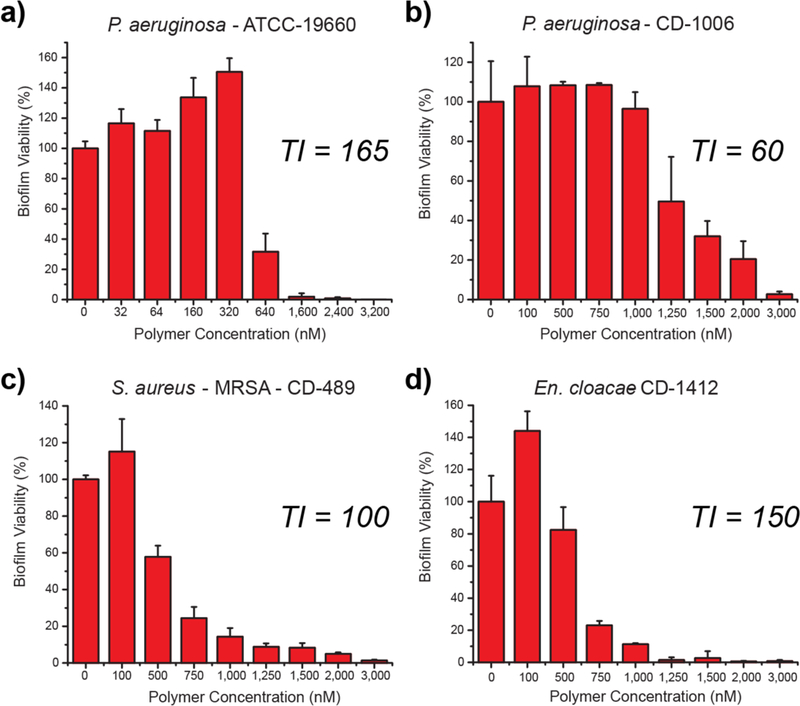
Viability of 1-day-old (a) P. aeruginosa (ATCC-19660), (b) P. aeruginosa (CD-1006), (c) S. aureus (CD-489), and (d) En. cloacae complex (CD-1412) biofilms after 3 h treatment with P5 PNPs. The data are average of triplicates, and the error bars indicate the standard deviations. TI is the therapeutic index relative to MBEC90 and hemolysis against red blood cells (HC50).
The ability to eradicate biofilms on biomedical surfaces such as medical implants and indwelling devices is a critical capability. However, treating biofilm infections on human tissues or organs is more challenging and relevant to medical settings.45 Biofilm infections on wounds significantly impair the healing process regulated by fibroblast skin cells.46
First, we investigated P5 PNPs compatibility with mammalian NIH 3T3 fibroblast cells at concentrations used to eradicate pre-formed biofilms, with no significant toxicity observed (SI Figure S4). We next used an in vitro coculture model comprised of mammalian fibroblast cells with bacterial biofilm overgrowth.47,48 In practice, P. aeruginosa bacteria were seeded on a confluent monolayer of NIH 3T3-fibroblast cells overnight to generate biofilms prior to treatment. The cocultures were treated with P5 PNPs for 3 hours, washed, and the viabilities of both bacteria and fibroblasts were determined. As shown in Figure 5a, a 4–6-fold log reduction (99.5%−99.99%) in bacterial colonies occurred at concentrations ranging from 7.5–15 µM, while no substantial loss of fibroblast viability was observed in this concentration range.
Figure 5.
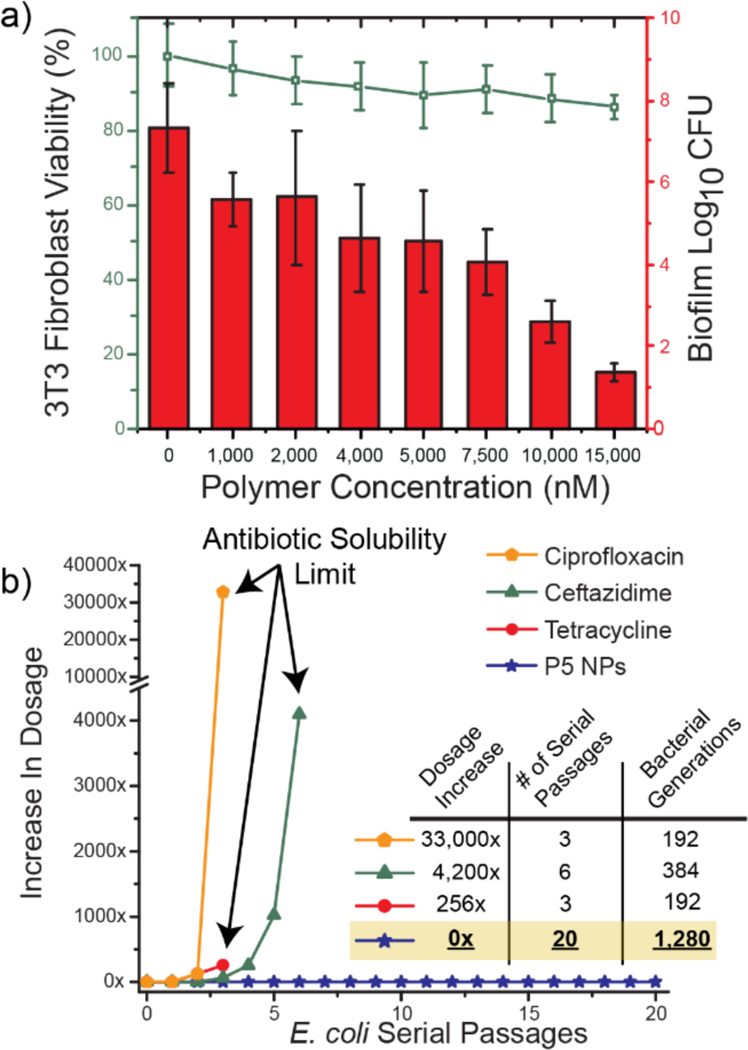
a) Viability of 3T3 fibroblast cells and E. coli biofilms in the co-culture model after 3 h treatment with P5 PNPs. Scatters and lines represent 3T3 fibroblast cell viability. Bars represent log10 of colony forming units in biofilms. The data are average of triplicates and the error bars indicate the standard deviations. b) Resistance development during serial passaging in the presence of sub-MIC levels of antimicrobials. The y axis is the highest concentration the cells grew in during passaging. The figure is representative of 3 independent experiments.
Bacterial resistance development against antibiotics vs polymer nanoparticles.
Bacteria rapidly acquire resistance towards antibiotics and other antimicrobials, limiting their long-term efficacy. Given the membrane disruption mechanism used by the PNPs, development of resistance in bacteria would require dramatic changes in the bacterial phenotype.43,45 The ability of PNPs to evade resistance was tested by subjection of uropathogenic E. coli (CD-2) to multiple serial passages of sub-MIC (66% of MIC) concentrations of P5 PNPs. The resulting bacterial population was harvested, and its MIC was evaluated. As shown in Figure 5b, even at the 20th serial passage (~1,300 bacterial generations) of CD-2, there was no change in MIC. Similar experiments were conducted on ciprofloxacin (quinolone), ceftazidime (β-lactam) and tetracycline, clinically relevant antibiotics. Respectively, there was a 33,000, 4,200 and 256-fold increase in the MICs of antibiotics against CD-2 E. coli after only a few passages. Our polymeric nanoparticles evade resistance towards bacteria longer than previously reported polymer-based nanomaterials49 (~600 generations – A. baumannii FADDI-AB156) and comparable to a recently discovered novel antibiotic, teixobactin (~1,300 generations – S. aureus ATCC 29213).50
Conclusion
We have designed and fabricated an effective polymer nanoparticle-based therapeutic platform to combat MDR bacterial and biofilm infections. Our research demonstrates the ability of these PNPs to modulate antimicrobial activity and therapeutic efficacy by structure-specific incorporation of hydrophobic and cationic moieties. These amphiphilic cationic PNPs demonstrate excellent efficiency in combating planktonic superbugs as well as their more drug-resistant biofilm counterparts. Their ability to penetrate and eradicate biofilms provides the foundation for a therapeutic strategy against biofilm infections that does not require debridement and extensive antimicrobial regimens. These PNPs function through a membrane disruption mechanism that strongly attenuates generation of tolerance or resistance. Taken together, PNP-based antimicrobial therapy has the potential to provide an effective platform to combat bacterial infections while circumventing standard antibiotic resistance pathways.
Supplementary Material
ACKNOWLEDGMENT
Clinical samples obtained from the Cooley Dickinson Hospital Microbiology Laboratory (Northampton, MA) were kindly provided by Dr. Margaret Riley. This research was supported by NIH GM077173, EB022641 and AI134770.
Footnotes
Supporting Information. Synthesis and characterization of polymer nanoparticles. Experimental procedures to determine minimum inhibitory concentrations (MICs), hemolytic activity and cell viability. Biofilm and coculture formation and treatment. Resistance development assay.
Notes
The authors declare no competing final interest.
REFERENCES
- 1.Marshall BM; Levy SB Clin. Microbiol. Rev 2011, 24 (4), 718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harris SR; Feil EJ; Holden MTG; Quail MA; Nickerson EK; Chantratita N; Gardete S; Tavares A; Day N; Lindsay JA; Edgeworth JD; De Lencastre H; Parkhill J; Peacock SJ; Bentley SD Science (80) 2010, 327 (5964), 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Amersfoort ES; Van Berkel TJC; Kuiper J Clin. Microbiol. Rev 2003, 16 (3), 379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wolcott RD; Ehrlich GD JAMA - J. Am. Med. Assoc 2008, 299 (22), 2682. [DOI] [PubMed] [Google Scholar]
- 5.Donlan RM Emerg. Infect. Dis 2001, 7 (2), 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Costerton JW; Stewart PS; Greenberg EP Science 1999, 284 (5418), 1318. [DOI] [PubMed] [Google Scholar]
- 7.Lewis K Antimicrob. Agents Chemother 2001, 45 (4), 999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fux CA; Costerton JW; Stewart PS; Stoodley P Trends Microbiol 2005, 13 (1), 34. [DOI] [PubMed] [Google Scholar]
- 9.Del Pozo JL; Patel R N. Engl. J. Med 2009, 361 (8), 787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laxminarayan R; Duse A; Wattal C; Zaidi AKM; Wertheim HFL; Sumpradit N; Vlieghe E; Hara GL; Gould IM; Goossens H; Greko C; So AD; Bigdeli M; Tomson G; Woodhouse W; Ombaka E; Peralta AQ; Qamar FN; Mir F; Kariuki S; Bhutta ZA; Coates A; Bergstrom R; Wright GD; Brown ED; Cars O Lancet Infect. Dis 2013, 13 (12), 1057. [DOI] [PubMed] [Google Scholar]
- 11.Baltzer SA; Brown MH J. Mol. Microbiol. Biotechnol 2011, 20 (4), 228. [DOI] [PubMed] [Google Scholar]
- 12.Hancock REW; Sahl HG Nat Biotech 2006, 24 (12), 1551. [DOI] [PubMed] [Google Scholar]
- 13.Jiang Z; Vasil AI; Gera L; Vasil ML; Hodges RS Chem. Biol. Drug Des 2011, 77 (4), 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marr AK; Gooderham WJ; R.E.W H Curr. Opin. Pharmacol 2006, 6 (5), 468. [DOI] [PubMed] [Google Scholar]
- 15.Sieprawska-Lupa M; Mydel P; Krawczyk K; Wójcik K; Puklo M; Lupa B; Suder P; Silberring J; Reed M; Pohl J; Shafer W; McAleese F; Foster T; Travis J; Potempa J Antimicrob. Agents Chemother 2004, 48 (12), 4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Palermo EF; Kuroda K Appl. Microbiol. Biotechnol 2010, 87 (5), 1605. [DOI] [PubMed] [Google Scholar]
- 17.Mowery BP; Lee SE; Kissounko DA; Epand RF; Epand RM; Weisblum B; Stahl SS; Gellman SH J. Am. Chem. Soc 2007, 129 (50), 15474. [DOI] [PubMed] [Google Scholar]
- 18.Patch JA; Barron AE J. Am. Chem. Soc 2003, 125 (40), 12092. [DOI] [PubMed] [Google Scholar]
- 19.Arnt L; Nüsslein K; Tew GN J. Polym. Sci. Part A Polym. Chem 2004, 42 (15), 3860. [Google Scholar]
- 20.Kuroda K; DeGrado WF J. Am. Chem. Soc 2005, 127 (12), 4128. [DOI] [PubMed] [Google Scholar]
- 21.Palermo EF; Vemparala S; Kuroda K Biomacromolecules 2012, 13 (5), 1632. [DOI] [PubMed] [Google Scholar]
- 22.Gabriel GJ; Madkour AE; Dabkowski JM; Nelson CF; Nüsslein K; Tew GN Biomacromolecules 2008, 9 (11), 2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muñoz-Bonilla A; Fernández-García M Prog. Polym. Sci 2012, 37 (2), 281. [Google Scholar]
- 24.Lienkamp K; Madkour AE; Musante A; Nelson CF; Nüsslein K; Tew GN J. Am. Chem. Soc 2008, 130 (30), 9836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yin LM; Edwards MA; Li J; Yip CM; Deber CM J. Biol. Chem 2012, 287 (10), 7738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ilker MF; Nüsslein K; Tew GN; Coughlin EB J. Am. Chem. Soc 2004, 126 (48), 15870. [DOI] [PubMed] [Google Scholar]
- 27.Lin CC; Ki CS; Shih H J. Appl. Polym. Sci 2015, 132 (8), 41563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cole JP; Lessard JJ; Lyon CK; Tuten BT; Berda EB Polym. Chem 2015, 6 (31), 5555. [Google Scholar]
- 29.Kuroda K; Caputo GA; DeGrado WF Chem. - A Eur. J 2009, 15 (5), 1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sambhy V; Peterson BR; Sen A Angew. Chemie - Int. Ed 2008, 47 (7), 1250. [DOI] [PubMed] [Google Scholar]
- 31.Chen C; Liu G; Liu X; Pang S; Zhu C; Lv L; Ji J Polym. Chem 2011, 2 (6), 1389. [Google Scholar]
- 32.Li X; Robinson SM; Gupta A; Saha K; Jiang Z; Moyano DF; Sahar A; Riley MA; Rotello VM ACS Nano 2014, 8 (10), 10682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dathe M; Wieprecht T; Nikolenko H; Handel L; Maloy WL; MacDonald DL; Beyermann M; Bienert M FEBS Lett 1997, 403 (2), 208. [DOI] [PubMed] [Google Scholar]
- 34.Huang Y; Huang J; Chen Y Protein Cell 2010, 1 (2), 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gupta A; Saleh NM; Das R; Landis RF; Bigdeli A; Motamedchaboki K; Rosa Campos A; Pomeroy K; Mahmoudi M; Rotello VM Nano Futur 2017, 1 (1), 015004. [Google Scholar]
- 36.Huo S; Jiang Y; Gupta A; Jiang Z; Landis RF; Hou S; Liang XJ; Rotello VM ACS Nano 2016, 10 (9), 8732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saha K; Moyano DF; Rotello VM Mater. Horizons 2014, 1 (1), 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parameswaran N; Patial S Crit. Rev. Eukaryot. Gene Expr 2010, 20 (2), 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nederberg F; Zhang Y; Tan JPK; Xu K; Wang H; Yang C; Gao S; Guo XD; Fukushima K; Li L; Hedrick JL; Yang YY Nat. Chem 2011, 3 (5), 409. [DOI] [PubMed] [Google Scholar]
- 40.Hayden SC; Zhao G; Saha K; Phillips RL; Li X; Miranda OR; Rotello VM; El-Sayed MA; Schmidt-Krey I; Bunz UHF J. Am. Chem. Soc 2012, 134 (16), 6920. [DOI] [PubMed] [Google Scholar]
- 41.Boulos L; Prévost M; Barbeau B; Coallier J; Desjardins R; Boulos L; Barbeau B J. Microbiol. Methods 1999, 37 (1), 77. [DOI] [PubMed] [Google Scholar]
- 42.Cox SD; Mann CM; Markham JL; Bell HC; Gustafson JE; Warmington JR; Wyllie SG J. Appl. Microbiol 2000, 88 (1), 170. [DOI] [PubMed] [Google Scholar]
- 43.Gupta A; Landis RF; Rotello VM F1000Research 2016, 5 (364), 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anderl JN; Zahller J; Roe F; Stewart PS Antimicrob. Agents Chemother 2003, 47 (4), 1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Landis RF; Li CH; Gupta A; Lee YW; Yazdani M; Ngernyuang N; Altinbasak I; Mansoor S; Khichi MAS; Sanyal A; Rotello VM J. Am. Chem. Soc 2018, 140 (19), 6176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roy S; Elgharably H; Sinha M; Ganesh K; Chaney S; Mann E; Miller C; Khanna S; Bergdall VK; Powell HM; Cook CH; Gordillo GM; Wozniak DJ; Sen CK J. Pathol 2014, 233 (4), 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anderson GG; Moreau-Marquis S; Stanton BA; O’Toole GA Infect. Immun 2008, 76 (4), 1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gupta A; Das R; Yesilbag Tonga G; Mizuhara T; Rotello VM ACS Nano 2018, 12 (1), 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lam SJ; O’Brien-Simpson NM; Pantarat N; Sulistio A; Wong EHH; Chen YY; Lenzo JC; Holden JA; Blencowe A; Reynolds EC; Qiao GG Nat. Microbiol 2016, 1, 16162. [DOI] [PubMed] [Google Scholar]
- 50.Ling L; Schneder T; Peoples A; Spoering A; Engels I; Conlon B; Mueller A; Schaberle T; Hughes D; Epstein S; Jones M; Lazarides L; Steadman V; Cohen D; Felix C; Fetterman K; Millett W; Nitti A; Zullo A; Chen C; Lewis K Nature 2015, 517 (7535), 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.