

Abstract
Drug delivery to a specific site in the body typically relies on the use of targeting agents that recognize a unique biomarker. Unfortunately, it is often difficult to identify unique molecular signatures that exist only at the site of interest. An alternative strategy is to deliver energy (e.g. light) to locally trigger release from a drug carrier; however, the use of this approach is limited because energy delivery to deep tissues is often impractical or invasive. In this work, radiofrequency-responsive superparamagnetic iron oxide nanoparticles (SPIONs) are used to trigger drug release from nanoscale vesicles. Because the body is inherently non-magnetic, this approach allows for deep tissue targeting. To overcome the unfavorable meter-scale diffraction limit of SPION-compatible radiofrequency (RF) fields, a strong static gating field containing a sharp zero point is superimposed on the RF field. Only drug carriers that are at or near the zero point are susceptible to RF-triggered drug release, thereby localizing drug delivery with millimeter-scale resolution. This approach induces > 40% drug release from thermally-responsive doxorubicin-loaded liposomes within a 3.2 mm radius of the zero point with < 10% release in the surrounding area, leading to a > 2.5 therapeutic index in Huh 7 hepatocellular carcinoma cells.
Keywords: magnetic nanoparticles, targeted drug delivery, liposome, thermally-sensitive nanomaterial, magnetism
Graphical Abstract
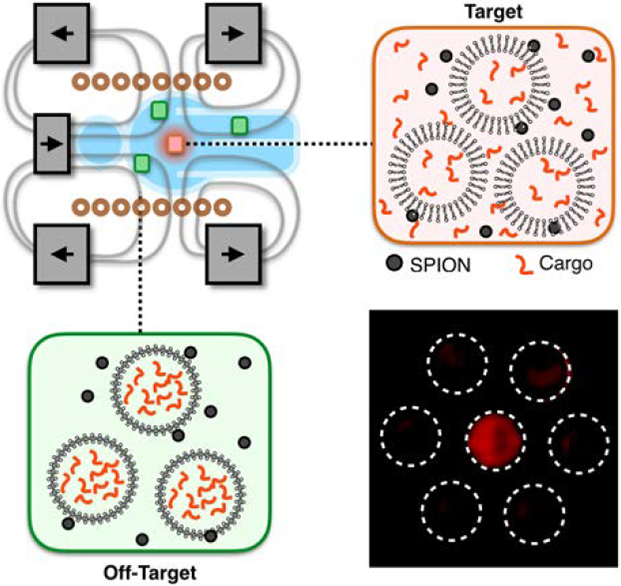
Targeted drug delivery is limited by the lack of specific biomarkers in many tissues and the difficulty of delivering energy to deep tissue. In this work, we combine radiofrequency responsive superparamagnetic iron oxide nanoparticles with a strong static gating field superimposed on an alternating field to target drug release with millimeter scale precision.
1. Introduction
When developing new therapeutics, it is generally desirable to specifically deliver drug to a particular site in the body while minimizing delivery to off-target tissues.[1,2] Drugs can be encapsulated in nanocarriers such as micelles,[3,4] polymersomes,[5,6] or liposomes[7–11] to simultaneously make the formulation more biocompatible than the free drug[2] and to allow for reduced off-target delivery.[1] Currently, spatial targeting of drug release is achieved by increasing nanocarrier accumulation at the intended site using targeting agents[12] such as antibodies[13] or by leveraging environmental factors such as low pH[14] and hypoxia[15] to induce local drug release. Unfortunately, most molecular and environmental signatures are also present at off-target sites, are heterogeneously expressed within target sites, or both.[16–18] Therefore, these methods generally suffer from poor specificity. An alternative approach involves the use of externally applied energy to trigger drug release, such as focused ultrasound[19,20] and laser-induced heating;[21] however, because tissue absorbs both ultrasound and light waves, it is difficult to specifically induce drug release without also damaging adjacent and intervening tissue or surgically exposing the target tissue.
Magnetic fields present an ideal energy source for triggering drug delivery in deep tissues while avoiding the need for surgery and damage to healthy tissue. Biological tissues are essentially transparent to magnetic fields; magnetic fields are not transformed or absorbed by biological tissues.[22] Because of this advantage, a significant body of work has been devoted to using alternating magnetic fields (AMFs) to induce drug release.[23–28] In these approaches, AMFs induce superparamagnetic iron oxide nanoparticles (SPIONs) to generate heat via Néel and/or Brownian relaxation[29–31] to drive drug release from thermally responsive nanocarriers.[23,32,33] However, AMFs, like all electromagnetic waves, are limited by diffraction. Typical frequencies for efficient energy delivery to SPIONs are on the order of hundreds of kilohertz.[34] Therefore, the diffraction limit prevents focusing of these alternating fields to resolutions of better than ~1 meter without the use of complex coil geometries.[35] As a result, most magnetically induced drug release strategies still rely on traditional targeting methods such as magnetic traps that encourage nanocarrier accumulation at superficial tissue sites[8,36] or ligand targeting[8,37] to confer specificity to a particular disease site. These targeting methods continue to be limited by off-target biological effects that diminish the advantages of using magnetism as a bio-orthogonal drug release strategy.
To address this challenge, we have developed a magnetic device that superimposes a strong static field containing a sharp zero point onto an AMF to gate SPION response to the AMF. This approach allows for spatial control of drug release from thermally sensitive liposomes. The magnetic field lines in this configuration generate an avoidance region in which the magnetic field strength |B0| goes to zero (Figure 1a). Near the zero point, SPIONs respond to the AMF and induce cargo release from thermally sensitive liposomes (Figure 1b). In the surrounding area, the static field drives the SPIONs into their saturation regime, pinning the magnetic moment of the SPIONs and suppressing their response to the AMF, thereby suppressing release of the liposomal cargo (Figure 1c). In this device, the size of the targeting region is set by the magnetic saturation of the nanoparticles and the strength of the static field (Figure 1d). A key innovation of this work is the use of three sets of static magnets (two sets of ring magnets and one set of cylindrical magnets) to create the static gating field containing the sharp zero point. By controlling the relative magnetization and position of these three magnets, it is possible to independently control the size and location of the zero point. Because the resolution of the spatial targeting region is defined by a zero in the static field rather than a peak, the resolution of the targeting region can be set arbitrarily high by using static magnets with higher field strengths (i.e. superconductors). The AMF was delivered using a solenoid wound from water-cooled hollow copper tubing to minimize convective heating. To validate this device, we demonstrated > 40% drug release from thermally responsive doxorubicin-loaded liposomes within a 3.2 mm radius targeting region with < 10% release in the surrounding area, leading to a > 2.5 therapeutic index with Huh 7 hepatocellular carcinoma cells.
Figure 1. A magnetic device containing a sharp zero point allows for magnetically driven spatially targeted drug release from thermally sensitive liposomal drug carriers.

(a) The device superimposes a strong static gating field containing a sharp zero point on an alternating magnetic field (AMF). (b) Near the zero point, the liposomes and nanoparticles are located within the targeting region. Here, the SPIONs respond to the AMF to trigger drug release from thermally-sensitive liposomes. (c) Away from the zero point, the strong static field drives the SPIONs into their saturation regime, suppressing their response to the AMF and thereby suppressing liposomal cargo release. (d) Within the targeting region, SPIONs respond to an applied AMF. However, outside of the targeting region, the SPIONs are saturated and SPION response to an applied AMF is limited. The size of the targeting region is determined by the strength of the static gating field and the magnetic saturation field of the SPIONs.
2. Results
2.1. Magnetic Device and Field Characterization
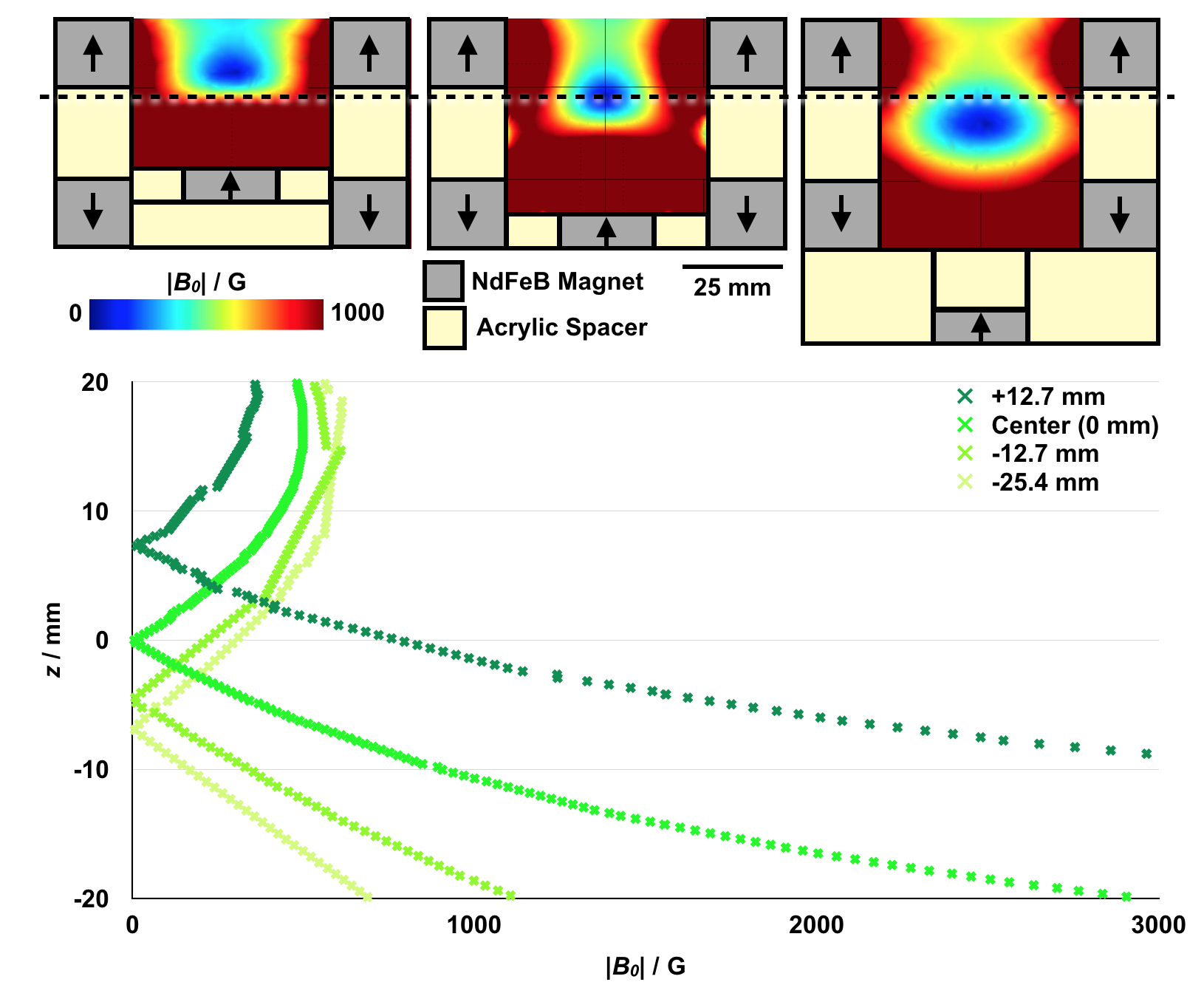
The magnetic device used to generate the zero-point-containing static gating field was assembled from NdFeB rare earth element magnets (KJ Magnetics), machined steel pieces (McMaster Carr), and laser-cut (Universal Laser) acrylic sheets (McMaster Carr) (Figure 2a–c). Two sets of ring magnets are axially aligned and assembled in an oppositely polarized configuration, z = 1.75” apart (Figure 2b) to generate an avoidance region (Figure 2d). Each set of ring magnets comprises three stacked 3” outer diameter (OD) × 1.5” inner diameter (ID) × 0.25” height (H) magnets. A cylindrical magnet is placed, axially aligned, within one set of ring magnets and is similarly comprised of three stacked 1.25” diameter (D) × 0.125” H magnets (Figure 2b). Magnetizable low-carbon steel (McMaster Carr) is used to contain the field lines (Figure 2a–c). A solenoid wound from water-cooled hollow copper tubing (Figure 2a, b) is placed within the bore of the cylindrical magnets and used to generate the AMF. To characterize the magnetic field within the device, a gaussmeter (AlphaLabs, Inc.) was used to measure the magnitude of the static field. Measurements confirm the presence of a zero-field region within the device (Figure 2d), consistent with COMSOL-simulated field lines (Figure 2d, overlay). For these simulations, the surface field of each magnet was matched to the measured surface field of the magnets. Minor differences in the magnitude of the measured field and the simulated field of ideal magnets are believed to be due to variability in the surface fields of the magnets (Figure S1). COMSOL simulations of the low-field targeting region in 3D space are shown in Figure S2.
Figure 2. The static gating field is generated using three sets of NdFeB magnets and is superimposed on an alternating magnetic field.

(a, b) The static gating field is generated using two sets of oppositely polarized NdFeB ring magnets and one set of NdFeB cylindrical magnets protected by magnetizable steel and acrylic. A water-cooled copper solenoid is used to generate the alternating magnetic field. (c) Image of device. Alternating current is passed through an amplifier and into the solenoid to generate the alternating magnetic field. The solenoid is wound from hollow copper tubing to allow for water cooling. (d) Magnetic field measurements confirm the presence of a sharp zero point at the center of the device. Overlay: magnetic field lines simulated in COMSOL.
2.2. MnZnSPION characterization
Manganese- and zinc-doped superparamagnetic iron oxide nanoparticles (MnZnSPIONs) were synthesized with a mean core size of 13.3 ± 1.6 nm (Figure 3a, b). After PEGylation, the hydrophilic particles have a hydrodynamic diameter of 20.7 nm with low polydispersity (PDI 0.177) (Figure 3c). SQUID measurements show superparamagnetic particles with a small hysteresis loop (Figure 3d).
Figure 3. MnZnSPIONs are monodisperse, hydrophilic, and magnetically responsive.

(a) MnZnSPIONs are monodisperse with a core size of 13.3 ± 1.6 nm based on transmission electron microscopy (b). (c) After PEG-ylation, the MnZnSPIONs have a hydrodynamic size of approximately 21 nm. (d) SQUID measurements show particles with a small hysteresis loop.
2.3. Gating of the MnZnSPION response to alternating magnetic fields
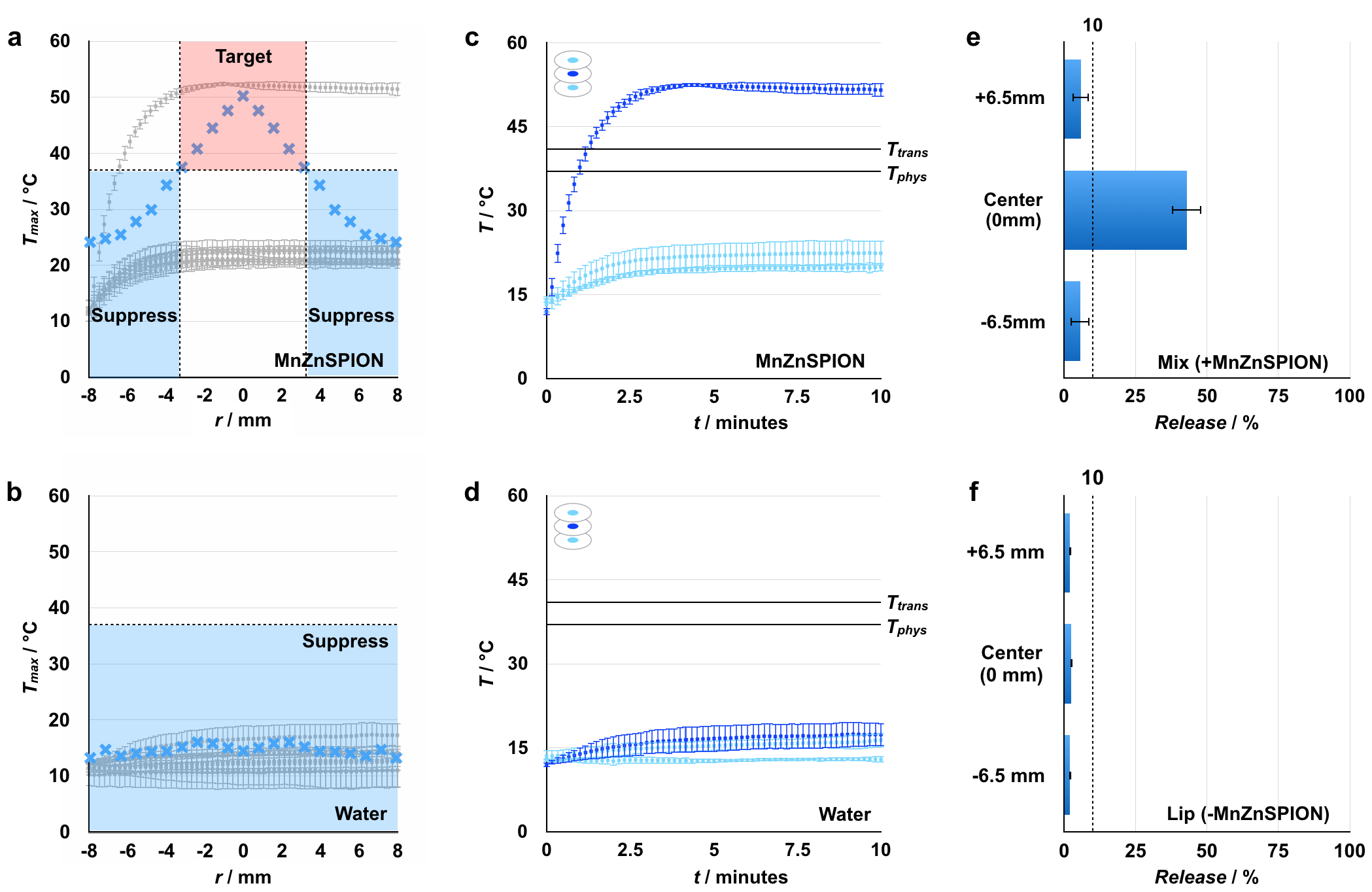
To demonstrate the principle of static field suppression, we first evaluated the capability of static fields to control MnZnSPION response to an AMF. When a 350 Oe, 500 kHz AMF is applied, 30 μL of 100 mg mL−1 MnZnSPIONs in a 2.6 mm diameter hollow Kapton tube heat approximately ΔT = 55°C compared to water over 15 minutes. However, when a 4000 G static field is superimposed on the AMF, heating of the sample is suppressed to ΔT = 2.5°C (Figure 4a). Heating is also found to be dependent on the concentration of MnZnSPIONs, validating that this effect is magnetically driven: when MnZnSPIONs are exposed to a 350 Oe, 500kHz AMF, the magnitude of heating is directly proportional to concentration, consistent with theoretical predictions.[38] When a strong 4000 G static field is superimposed on the alternating magnetic field, heating is suppressed to ΔT < 5°C at all concentrations (Figure 4b). To test the effect of static field strength on the suppression of nanoparticle heating, we measured the change in temperature of a 100 mg mL−1 sample of MnZnSPIONs exposed to an AMF with various static field strengths superimposed. We found that at low static fields (< 200 G), heating is minimally affected by the static field; however, as the amplitude of the static field increases (> 200 G), particle heating is reduced to that of the background level set by a water sample without MnZnSPIONs (Figure S3). This static field value is the minimum suppression field needed to pin the magnetic moment of the MnZnSPIONs and completely suppress their response to the AMF.
Figure 4. Spatially targeted heating is achieved using a strong space-variant static gating field.

(a) MnZnSPIONs (100 mg mL−1) heat ΔT = 55°C from baseline in the presence of a 350 Oe, 500 kHz alternating magnetic field. Heating can be suppressed to ΔT ~ 2°C with a strong (4000 G) static field. (b) In the presence of an alternating magnetic field, heat generated scales linearly with MnZnSPION concentration. At all concentrations, this effect can be suppressed with a strong (4000 G) static field. (c) At the center of the static magnetic configuration (dark blue), a sample of 100 mg mL−1 MnZnSPIONs heats to T = 50°C, while the temperature is maintained at T < 25°C at the edges (light blue), demonstrating spatially targeted induction of heating. (d) Meanwhile, the temperature of a sample of water is maintained at T < 20°C, regardless of whether it is placed at the center (dark blue) or edge (light blue). Tphys = physiological temperature, 37°C; Ttrans = liposomal transition temperature, 42°C. n = 3.
2.4. Spatially targeting AMF response using a magnetic zero
To confirm that a static field with a sharp magnetic zero can be used to spatially control MnZnSPION response to an AMF, a 30 μL sample of 100 mg mL−1 MnZnSPIONs was placed in the custom-built magnetic device and exposed to both the alternating magnetic field and the strong static gating field. Samples placed within the center targeting region of the device heat from approximately T = 12°C to approximately T = 50°C in 10 minutes. However, at the edges of the device where the SPIONs are pinned by the static field, sample temperatures do not increase past T = 25°C (Figure 4c). The temperature of a water control does not increase past T = 20°C, regardless of its location in the device (Figure 4d). As the sample is moved away from the center of the device, the amount of heating in the sample decreases because more of the sample is located outside of the targeting region. Accordingly, the heating in a sample of 100 mg mL−1 MnZnSPIONs decreases linearly with radial displacement from the zero point until it is completely suppressed (Figure S4a). Heating is suppressed below T = 37°C at any radius greater than 3.2 mm away from the center (Figure S4a), while there is no heating at any radius for a water control (Figure S4b). Moving the sample in the z-direction away from the xy-plane containing the zero point also leads to suppression of nanoparticle heating (Figure S4c), demonstrating that the zero point is a local minimum in 3D space. As before, a control sample of water is shown not to heat at any point along the z-axis (Figure S4d). Thermal imaging using an infrared camera (FLIR One) similarly shows targeted heating in the middle of the device (Figure S5). The objective in this experiment is to demonstrate a large temperature difference between the center and edge of the device, with parameters chosen accordingly. The temperature of the sample in the targeting region can be controlled by changing the amplitude of the AMF (Figure S6). When the volume of the sample is smaller than the size of the zero point, the amount of heating scales with the square of the AMF amplitude, consistent with theoretical predictions of magnetic heating in free space.[34] The baseline temperature can also be adjusted by changing the temperature of the water running through the coil. Similarly, using stronger static fields can lead to better suppression and even less heating at the edges surrounding the targeting region (Figure S3). A lower amplitude alternating field can also be used to reduce convective heating and baseline AMF heating, both of which contribute to the temperature rise at the edge of the device. This reduction in AMF amplitude would also reduce the total power delivered to improve safety.
2.5. Magnetically activated cargo release from liposomes
To demonstrate the principle of magnetically induced cargo release from thermally sensitive liposomes, we measured fluorescent dye release from liposomes in samples with MnZnSPIONs in the presence of an AMF, both with and without a strong static magnetic suppression field. First, to verify the thermal response of the liposomes, we measured dye release from liposomes at various externally set temperatures. Thermally responsive liposomes composed of a 70:30:5 molar ratio of 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) : cholesterol : 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG2000) with self-quenching concentrations of rhodamine dye encapsulated in the lumen are stable at 37°C over 15 minutes, but demonstrate 60% dye release at 45°C over the same amount of time (Figure 5a). When a sample containing both liposomes and 100 mg mL−1 MnZnSPIONs is exposed to a 350 Oe, 500 kHz alternating magnetic field for 15 minutes, 75% of the dye is released from the liposomes. However, when a strong 4000 G static suppression field is superimposed on the alternating magnetic field, liposomal dye release is suppressed to < 10%. In a sample containing liposomes but no nanoparticles, dye release is < 10% in the presence and absence of the static suppression field (Figure 5b).
Figure 5. Spatially targeted cargo release from thermally sensitive liposomes is achieved using a strong space-variant static gating field.

(a) 70:30:5 DPPC:cholesterol:DSPE-PEG2000 liposomes are thermally sensitive with a transition temperature of 42°C over 15 minutes. (b) Upon exposure to a 350 Oe, 500 kHz alternating magnetic field, thermally sensitive liposomes in a sample containing 100 mg mL−1 MnZnSPIONs release 75% of their cargo. This effect is suppressible with a strong 4000 G static field, and is not observed in the absence of MnZnSPIONs. n = 3. (c) A sample of thermally-sensitive liposomes containing 100 mg mL−1 MnZnSPIONs demonstrates spatially targeted release of 40% in the targeting region at the center of the device with background release at < 10% elsewhere. n = 3. (Fluorescence image, e). (d) However, a sample containing thermally sensitive liposomes in the absence of SPIONs shows < 10% cargo release regardless of location in the device. n = 3. (Fluorescence image, f).
2.6. Spatially targeted magnetic release from liposomes
Next, we tested whether the magnetic device could be used to spatially target cargo release from liposomes. Using dye as a stand-in for drug, we placed a 30 μL sample containing rhodamine-loaded thermally responsive liposomes and 100 mg mL−1 MnZnSPIONs in the magnetic device. The liposomes located within the targeting region of the device release approximately 40% of their cargo upon exposure to a 350 Oe, 500 kHz AMF when quantified by fluorimetry. In contrast, liposomes located outside of the targeting region release < 10% of their cargo after exposure to the same AMF (Figure 5c). When 30 μL samples of liposomes are exposed to the AMF in the absence of MnZnSPIONs, there is < 10% liposomal dye release at all locations in the device by fluorimetry (Figure 5d). The difference in dye release between the targeting region and edge of the device in the MnZnSPION-containing samples is visible by fluorescence imaging (Leica) (Figure 5e). No difference in fluorescence between the center targeting region and edge is observed by imaging in samples containing liposomes without MnZnSPIONs (Figure 5f). To demonstrate that there are also regions of high field above and below the zero point, release of dye from thermally responsive liposomes in the presence of MnZnSPIONs was also measured along the z-axis. As expected, significant dye release was observed at the zero point, but dye release was suppressed above and below the targeting region (Figure S4e). Samples lacking MnZnSPIONs demonstrate no release at any point along the z-axis (Figure S4f).
2.7. Spatially Targeted Drug Release and Inhibition of Cell Proliferation
Finally, to demonstrate that the combination of the MnZnSPIONs, the liposomes, and the magnetic device can be used to induce spatially targeted drug release for inhibition of cell proliferation, doxorubicin-loaded 70:30:5 DPPC:cholesterol:DSPE-PEG2000 liposomes were placed in the device along with 100 mg mL−1 MnZnSPIONs for 15 minutes both within the targeting region and at the edge of the device, and an AMF was applied. Because encapsulated drug has reduced toxicity compared to free drug,[39] cell proliferation should be more readily inhibited at the targeting region in center of the device. We demonstrate that at the center, the IC50 of the liposomes was 1.75 ± 0.37 μM, while the IC50 of the liposomes at the edge of the device was 4.44 ± 0.87 μM (Figure 6a). This corresponds to a therapeutic index of > 2.5 (Figure 6b).
Figure 6. Spatially targeted doxorubicin release from thermally sensitive liposomes.

(a) The cell viability curve for doxorubicin-loaded liposomes exposed to the AMF within the targeting region of the device is shifted left compared to liposomes at the edge. The concentration of doxorubicin-loaded liposomes exposed to the device at the center and edge are matched. Therefore, the total doxorubicin concentration is similarly matched between the center and edge, and any difference between cell viability at each concentration is due to differences in AMF-induced doxorubicin release between the center and the edge of the device. There is no decrease in cell viability observed when using blank liposomes loaded with PBS, or when using PBS only, either at the center or the edge. (b) Doxorubicin-loaded liposomes exposed to the AMF at the edge of the device have an IC50 of 4.44 ± 0.87 μM, while the same liposomes exposed to the AMF at the center targeting region of the device have an IC50 of 1.75 ± 0.37 μM; p = 0.0078. This corresponds to a therapeutic index of > 2.5 in Huh7 hepatocellular carcinoma cells. n = 3.
3. Discussion
In this work, we have developed a device for magnetically activated spatially targeted drug delivery. An alternating magnetic field induces magnetic nanoparticles to release cargo from thermally sensitive liposomal drug carriers. A strong static field with a sharply defined targeting region gates the effect of the alternating field to achieve spatial targeting. Previous work has used magnetic fields to trigger drug release from nanocarriers via AMF-induced heating[23,40,41] or mechanical disruption[28] of the carrier. However, because iron oxide nanoparticles are typically most responsive to frequencies in the 100–500 kHz range,[34,42–44] focusing of the AMF is limited to resolutions of approximately 1 meter. As a result, these strategies have relied on biological methods of targeting, which are limited by inherent biological variability in receptor density, ligand affinity, and off-target expression.[16] To take full advantage of the ability to use magnetic fields to trigger magnetic nanoparticles without interference from intervening tissue, it is necessary to find a means of localizing the response to the alternating magnetic field.
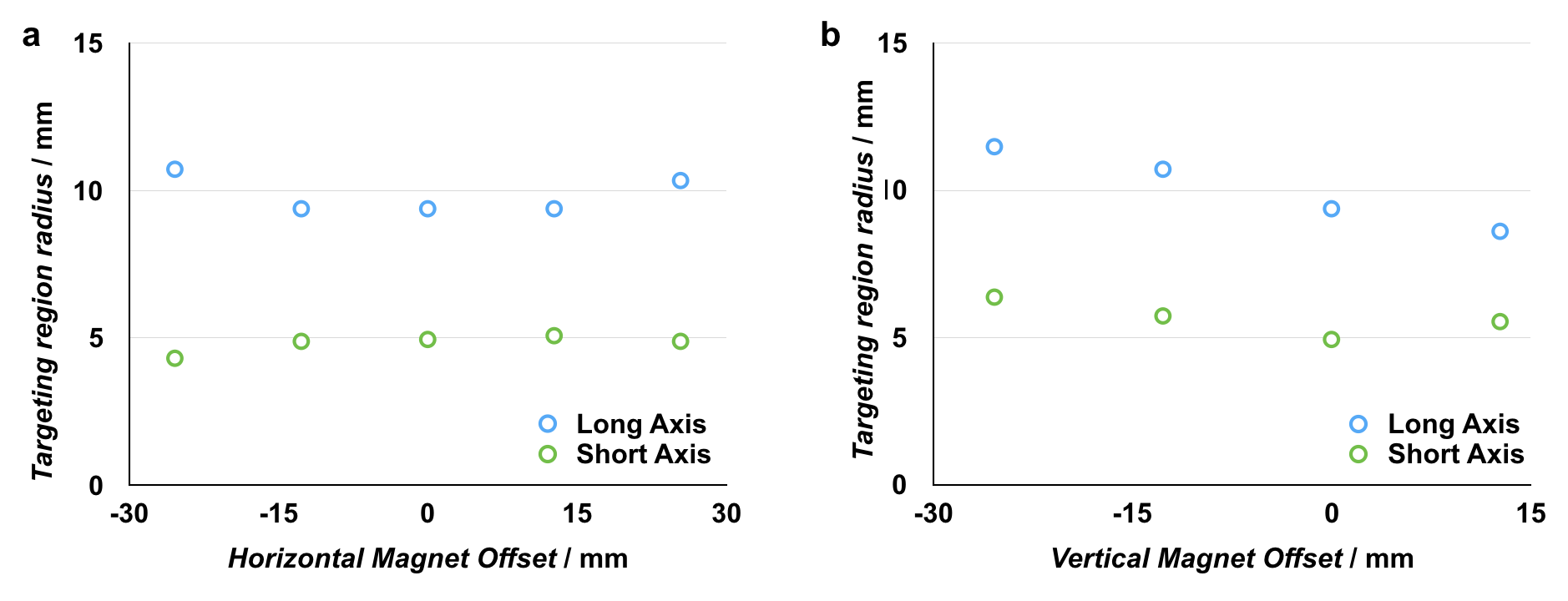
By building on previous work in the targeted hyperthermia[45–47] and high-resolution magnetic particle imaging space[47–49] in combination with the new approaches that we have reported, there are many opportunities for further development. Our prototype device is designed to have a targeting region with a 3.2 mm radius. We have demonstrated that by varying the inter-magnet distance between the ring magnets, we are able to reduce the size of the targeting region (Figure S7a). Based on empirical heating data (Figure S4a), we define the zero as the region where the static field strength is < 100 Gauss. Using this value in combination with simulation data, we show that by reducing the inter-magnet distance, we are able to reduce the radius of the targeting region to r < 1 mm (Figure S7b). At this size, we were no longer able to detect heating of the MnZnSPIONs due to the increased surface area to volume ratio of the sample (Figure S7c). Because static fields have no inherent length scale, there is no fundamental limit on the sharpness of a targeting region defined by a static zero point.[50] In addition to expanding the use of targeted magnetic heating to spatially targeted drug release, a key innovation in this work is the inclusion of a cylindrical third magnet. This magnet gives our system an additional degree of freedom that allows for independent control of the size and location of the zero point and targeting region. By moving the third magnet in the xy-plane, it is possible to move the targeting region laterally with the magnet (Figure S8); similarly, by moving the third magnet in the z-direction, it is possible to move the zero point up and down (Figure S9). Because the boundary of the zero point is determined by the strength of the static field and the saturation magnetization of the SPIONs, we show in simulation that the size of the zero point remains relatively constant as the zero is moved (Figure S10).
Further work can be done to explore methods of co-delivering SPIONs with thermally sensitive liposomes to better take advantage of nanoscale heating near the particle surface, for example, by incorporating SPIONs into the liposomal membrane.[40,51] By maintaining a close distance between SPIONs and liposomes, nanoscale heating can be used to allow for liposomal drug release without increasing the temperature of the surrounding tissue.[52,53] Next, MnZnSPIONs were used in this work because they provide approximately 4× more heating power compared to commercially available, FDA-approved SPIONs.[42] However, the presence of dopants increases the risk of nanoparticle toxicity, especially at high concentrations. Fortunately, the AMF/static field targeting system can be used with any AMF-responsive particle, and can be adapted for use with commercial SPIONs by doubling the strength of the AMF to make up for the reduction in the nanoparticles’ specific loss power. Another challenge for all magnetic targeting devices is in scaling up to larger systems, as the prototype static permanent magnets must be replaced with electromagnets. Fortunately, significant recent progress has been made in this area; for example, copper coils can be used as a resource-efficient means of generating strong fields over short timescales.[54] Finally, MnZnSPIONs can also be replaced with other particles that might be used to generate more heat at lower concentrations[55–58] to minimize the risk of nanoparticle toxicity.
4. Conclusion
Alternating-field-induced drug release from thermally sensitive nanocarriers is limited by spatial resolution. Here, we have developed a system that combines a static gating field with an alternating magnetic field to induce drug release from thermally sensitive liposomes within a 3.2 mm radius target. In contrast to current systems for targeted hyperthermia, the size and location of the targeting region in our device can be independently controlled.
5. Experimental Section
MnZnSPION synthesis and characterization:
Manganese- and zinc-doped superparamagnetic iron oxide nanoparticles (MnZnSPIONs) were synthesized as previously described.[42,43,59] Briefly, seed particles were synthesized by reacting iron (III) acetylacetonate (2 mmol, Sigma), manganese (II) chloride (0.6 mmol, Sigma), zinc (II) chloride (0.4 mmol, Fisher), 1,2-hexadecanediol (5 mmol, Sigma), oleic acid (2 mmol, Chem-Impex International), and oleylamine (6 mmol, Sigma) in benzyl ether (20 mL, Sigma) in a two-neck flask while stirring under nitrogen. The reaction was heated to 200°C for 15 minutes, then heated to reflux for 1 hour. The product was cooled to room temperature and washed with ethanol by centrifugation at 5,500 g × 15 minutes. The pellet was resuspended in toluene and large aggregates were removed by centrifugation at 3,000 g × 15 minutes. Néel size (13–15 nm) SPIONs were synthesized by reacting seed particles (80 mg) with iron (III) acetylacetonate (2 mmol, Sigma), manganese (II) chloride (0.6 mmol, Sigma), zinc (II) chloride (0.4 mmol, Fisher), 1,2-hexadecanediol (5 mmol, Sigma), oleic acid (6.3 mmol, Chem-Impex International), and oleylamine (6 mmol, Sigma) in benzyl ether (20 mL, Sigma) in a two-neck flask while stirring under nitrogen. The mixture was first heated to 140°C for 30 minutes under a flow of nitrogen to evaporate the toluene. The reaction was then heated to 200°C for 1 hour, followed by heating to reflux for 2 hours. The product was cooled to room temperature and washed with ethanol, and the pellet was resuspended in toluene as before. To transfer the hydrophobic particles to a hydrophilic phase,[60] MnZnSPIONs (66 mg) were reacted with acetic acid (18 μL, Fisher), methoxy-PEG6–9-C3-silane (1.6 mL, Gelest), and water (2 mL) in toluene (80 mL) overnight. The toluene was removed by centrifugation at 2,000 g × 3 minutes. The hydrophilic phase was then centrifuged at 12,000 g × 10 minutes to remove large aggregates and any remaining toluene. The particles in the water phase were concentrated using a centrifugal spin filter (Millipore). MnZnSPION core size was characterized by transmission electron microscopy using a Jeol-1010 electron microscope. MnZnSPION hydrodynamic diameter was quantified by dynamic light scattering (Zetasizer). MnZnSPION magnetization properties were determined using a superconducting quantum interference device (SQUID) magnetometer (Quantum Design MPMS7 Tesla with Evercool).
Liposome synthesis and measurement of cargo release:
Lipids were obtained from Avanti Polar Lipids and dissolved in chloroform at a concentration of 25 mg mL−1. Liposomes comprised of a molar ratio of 70:30:5 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) : cholesterol : 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG2000) were synthesized by thin-film hydration.[61] Briefly, DPPC (267 μL), cholesterol (60.2 μL), and DSPE-PEG2000 (72.8 μL) were mixed in a 20 mL glass sonication vial and dried under a stream of nitrogen. Residual chloroform was removed under vacuum for at least 1 hour. The lipid films were hydrated for 30 minutes at 50°C with 50 mm sulforhodamine B (1 mL, Biotium) in PBS. After hydration, the films were sonicated for 1 minute and subjected to 5 freeze-thaw cycles between liquid nitrogen and 50°C. The liposomes were then extruded through a 200 nm polycarbonate track etch filter (Sterlitech) 21 times at 70°C using a mini extruder (Avanti Polar Lipids) and purified using a Sepharose CL-4B column (Sigma) equilibrated with PBS. Cargo release (%) was calculated as (Fluorsample – Fluor0) × (Fluor100 – Fluor0)−1 × 100%, where Fluorsample is the fluorescence of the sample in question, Fluor0 is the fluorescence of the purified sample before any exposure to heat or AMF, and Fluor100 is the fluorescence of the sample when all of the cargo has been released, determined by adding 1 mm Triton X-100 (Fisher) to the sample to disrupt all liposomal membranes. Fluorescence was measured using a Jobin Yvon spectrometer (Horiba) with λex = 560 nm and λem = 590 nm.
Doxorubicin-loaded liposomes were similarly prepared as previously described.[14] The liposomes were initially prepared with 300 50 mm ammonium sulfate via thin-film hydration, extruded through a 200 nm polycarbonate track etch filter, and purified with a PD-10 column (GE Healthcare) equilibrated with 150 50 mm NaCl. After purification, 0.35 lipid equivalents of doxorubicin (LC Laboratories) dissolved in PBS were added to the ammonium sulfate-containing liposomes. The liposomes were incubated at 50°C for 24 hrs with shaking. Excess doxorubicin was removed and the liposomes were transferred to PBS using a PD-10 column, and doxorubicin concentration in the purified sample was determined by absorption at 488 nm.
In Vitro Cell Proliferation:
Cell proliferation was assessed with a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Huh 7 (pass 18) cells were seeded at 7,500 cells/well in DMEM in a 96-well plate. After 24 hours for cell attachment, the cells were incubated with doxorubicin-loaded 70:30:5 DPPC:cholesterol:DSPE-PEG2000 liposomes that were placed in the device with 100 mg mL−1 MnZnSPIONs either within or outside of the targeting region and exposed to an AMF. After 4 hours of incubation, the liposome-containing media was replaced with fresh media and the cells were allowed to proliferate for 48 hours. The cells were then incubated with MTT reagent 1 (Roche) in media for 4 hours, followed by dissolution in detergent (Roche) overnight. Cell viability was measured by absorbance at 570 nm with a background reading at 650 nm.
Statistical Analysis:
Statistical evaluation for a significant difference between the IC50 values of Dox-loaded liposomes at the center and edge of the device was performed using a Student’s t-test.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was supported by the NIH/NIBIB (R21 EB023989; DI), the NIH/NCI (R01 CA181429; AT), NIH/NINDS (T32 NS091006; JFL), the Brain Research Foundation (AT), and the Brain and Behavior Research Foundation (AT). Facility support was provided by the Singh Center for Nanotechnology, MRSEC DMR-1120901. A.T. and D.I. contributed equally to this work.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
References
- [1].Bae YH, Park K, Control J. Release 2011, 153, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Singh R, Lillard JW, Exp. Mol. Pathol 2009, 86, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Liang J, Wu W-L, Xu X-D, Zhuo R-X, Zhang X-Z, Colloids Surfaces B Biointerfaces 2014, 114, 398. [DOI] [PubMed] [Google Scholar]
- [4].Yan L, Miller J, Yuan M, Liu JF, Busch TM, Tsourkas A, Cheng Z, Biomacromolecules 2017, 18, 1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Upadhyay KK, Bhatt AN, Mishra AK, Dwarakanath BS, Jain S, Schatz C, Le Meins J-F, Farooque A, Chandraiah G, Jain AK, Misra A, Lecommandoux S, Biomaterials 2010, 31, 2882. [DOI] [PubMed] [Google Scholar]
- [6].Kartha S, Yan L, Weisshaar CL, Ita ME, Shuvaev VV, Muzykantov VR, Tsourkas A, Winkelstein BA, Cheng Z, Adv. Healthc. Mater 2017, 6, 1700500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gabizon A, Shmeeda H, Horowitz AT, Zalipsky S, Adv. Drug Deliv. Rev 2004, 56, 1177. [DOI] [PubMed] [Google Scholar]
- [8].Pradhan P, Giri J, Rieken F, Koch C, Mykhaylyk O, Döblinger M, Banerjee R, Bahadur D, Plank C, Control J. Release 2010, 142, 108. [DOI] [PubMed] [Google Scholar]
- [9].Bao Q-Y, Zhang N, Geng D-D, Xue J-W, Merritt M, Zhang C, Ding Y, Int. J. Pharm 2014, 477, 408. [DOI] [PubMed] [Google Scholar]
- [10].Kim MS, Lee D-W, Park K, Park S-J, Choi E-J, Park ES, Kim HR, Colloids Surf. B. Biointerfaces 2014, 116, 17. [DOI] [PubMed] [Google Scholar]
- [11].Pradhan L, Srivastava R, Bahadur D, Acta Biomater. 2014, 10, 2976. [DOI] [PubMed] [Google Scholar]
- [12].Sudimack J, Lee RJ, Adv. Drug Deliv. Rev 2000, 41, 147. [DOI] [PubMed] [Google Scholar]
- [13].Alley SC, Okeley NM, Senter PD, Curr. Opin. Chem. Biol 2010, 14, 529. [DOI] [PubMed] [Google Scholar]
- [14].Yan L, Crayton SH, Thawani JP, Amirshaghaghi A, Tsourkas A, Cheng Z, Small 2015, 11, 4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Thambi T, Deepagan VG, Yoon HY, Han HS, Kim S-H, Son S, Jo D-G, Ahn C-H, Suh YD, Kim K, Chan Kwon I, Lee DS, Park JH, Biomaterials 2014, 35, 1735. [DOI] [PubMed] [Google Scholar]
- [16].Muro S, Control J. Release 2012, 164, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Vaupel P, Kallinowski F, Okunieff P, Cancer Res 1989, 49, 6449. [PubMed] [Google Scholar]
- [18].Tredan O, Galmarini CM, Patel K, Tannock IF, JNCI J Natl. Cancer Inst 2007, 99, 1441. [DOI] [PubMed] [Google Scholar]
- [19].Dromi S, Frenkel V, Luk A, Traughber B, Angstadt M, Bur M, Poff J, Xie J, Libutti SK, Li KCP, Wood BJ, Clin. Cancer Res 2007, 13, 2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Grüll H, Langereis S, Control J. Release 2012, 161, 317. [DOI] [PubMed] [Google Scholar]
- [21].Sherlock SP, Tabakman SM, Xie L, Dai H, ACS Nano 2011, 5, 1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Schenck JF, Prog. Biophys. Mol. Biol 2005, 87, 185. [DOI] [PubMed] [Google Scholar]
- [23].Babincová M, Čičmanec P, Altanerová V, Altaner Č, Babinec P, Bioelectrochemistry 2002, 55, 17. [DOI] [PubMed] [Google Scholar]
- [24].Yoo D, Jeong H, Noh S-H, Lee J-H, Cheon J, Angew. Chem. Int. Ed. Engl 2013, 52, 13047. [DOI] [PubMed] [Google Scholar]
- [25].Oliveira H, Pérez-Andrés E, Thevenot J, Sandre O, Berra E, Lecommandoux S, Control J. Release 2013, 169, 165. [DOI] [PubMed] [Google Scholar]
- [26].Kumar CSSR, Mohammad F, Adv. Drug Deliv. Rev 2011, 63, 789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jain TK, Reddy MK, Morales MA, Leslie-Pelecky DL, Labhasetwar V, Mol. Pharm 2008, 5, 316. [DOI] [PubMed] [Google Scholar]
- [28].Peiris PM, Abramowski A, Mcginnity J, Doolittle E, Toy R, Gopalakrishnan R, Shah S, Bauer L, Ghaghada KB, Hoimes C, Brady-Kalnay SM, Basilion JP, Griswold MA, Karathanasis E, Cancer Res 2015, 75, 1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Rosensweig RE, J. Magn. Magn. Mater 2002, 252, 370. [Google Scholar]
- [30].Lévy M, Wilhelm C, Siaugue J-M, Horner O, Bacri J-C, Gazeau F, J. Phys. Condens. Matter 2008, 20, 204133. [DOI] [PubMed] [Google Scholar]
- [31].Suto M, Hirota Y, Mamiya H, Fujita A, Kasuya R, Tohji K, Jeyadevan B, J. Magn. Magn. Mater 2009, 321, 1493. [Google Scholar]
- [32].Satarkar NS, Hilt JZ, Control J. Release 2008, 130, 246. [DOI] [PubMed] [Google Scholar]
- [33].Shah SA, Asdi MH, Hashmi MU, Umar MF, Awan S-U, Mater. Chem. Phys 2012, 137, 365. [Google Scholar]
- [34].Laurent S, Dutz S, Häfeli UO, Mahmoudi M, Adv. Colloid Interface Sci 2011, 166, 8. [DOI] [PubMed] [Google Scholar]
- [35].Jordan A, Scholz R, Maier-Hauff K, Johannsen M, Wust P, Nadobny J, Schirra H, Schmidt H, Deger S, Loening S, Lanksch W, Felix R, J. Magn. Magn. Mater 2001, 225, 118. [Google Scholar]
- [36].Alexiou C, Arnold W, Klein RJ, Parak FG, Hulin P, Bergemann C, Erhardt W, Wagenpfeil S, Lübbe AS, Cancer Res 2000, 60, 6641. [PubMed] [Google Scholar]
- [37].Yang R, An LY, Miao QF, Li FM, Han Y, Wang HX, Liu DP, Chen R, Tang SQ, Oncotarget 2016, 7, 35894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Keblinski P, Cahill DG, Bodapati A, Sullivan CR, Taton TA, J. Appl. Phys 2006, 100, 054305. [Google Scholar]
- [39].Horowitz AT, Barenholz Y, Gabizon AA, Biochim. Biophys. Acta - Biomembr. 1992, 1109, 203. [DOI] [PubMed] [Google Scholar]
- [40].Chen Y, Bose A, Bothun GD, ACS Nano 2010, 4, 3215. [DOI] [PubMed] [Google Scholar]
- [41].Viroonchatapan E, Sato H, Ueno M, Adachi I, Tazawa K, Horikoshi I, Control J. Release 1997, 46, 263. [Google Scholar]
- [42].Jang J, Nah H, Lee J-H, Moon SHH, Kim MGG, Cheon J, Angew. Chem. Int. Ed. Engl 2009, 48, 1234. [DOI] [PubMed] [Google Scholar]
- [43].Chen R, Christiansen MG, Anikeeva P, ACS Nano 2013, 7, 8990. [DOI] [PubMed] [Google Scholar]
- [44].Mehdaoui B, Meffre A, Carrey J, Lachaize S, Lacroix L-M, Gougeon M, Chaudret B, Respaud M, Adv. Funct. Mater 2011, 21, 4573. [Google Scholar]
- [45].Hensley D, Tay ZW, Dhavalikar R, Zheng B, Goodwill P, Rinaldi C, Conolly S, Phys. Med. Biol 2017, 62, 3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ma M, Zhang Y, Shen X, Xie J, Li Y, Gu N, Nano Res. 2015, 8, 600. [Google Scholar]
- [47].Tay ZW, Chandrasekharan P, Chiu-Lam A, Hensley DW, Dhavalikar R, Zhou XY, Yu EY, Goodwill PW, Zheng B, Rinaldi C, Conolly SM, ACS Nano 2018, 12, 3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gleich B, Weizenecker J, Nature 2005, 435, 1214. [DOI] [PubMed] [Google Scholar]
- [49].Konkle JJ, Goodwill PW, Saritas EU, Zheng B, Lu K, Conolly SM, Biomed. Tech. (Berl) 2013, 58, 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Brown KA, Westervelt RM, Nano Lett. 2011, 11, 3197. [DOI] [PubMed] [Google Scholar]
- [51].Chen Y, Chen Y, Xiao D, Bose A, Deng R, Bothun GD, Colloids Surf. B. Biointerfaces 2014, 116, 452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Romero G, Christiansen MG, Stocche Barbosa L, Garcia F, Anikeeva P, Adv. Funct. Mater 2016, 26, 6471. [Google Scholar]
- [53].Chiu-Lam A, Rinaldi C, Adv. Funct. Mater 2016, 26, 3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Matter NI, Scott GC, Venook RD, Ungersma SE, Grafendorfer T, Macovski A, Conolly SM, Magn. Reson. Med 2006, 56, 1085. [DOI] [PubMed] [Google Scholar]
- [55].Peddis D, Cannas C, Musinu A, Ardu A, Orrù F, Fiorani D, Laureti S, Rinaldi D, Muscas G, Concas G, Piccaluga G, Chem. Mater 2013, 25, 2005. [Google Scholar]
- [56].Chatterjee J, Bettge M, Haik Y, Jen Chen C, J. Magn. Magn. Mater 2005, 293, 303. [Google Scholar]
- [57].Lee H, Yoon T-J, Weissleder R, Angew. Chemie Int. Ed 2009, 48, 5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Lee J-H, Jang J-T, Choi J-S, Moon SH, Noh S-H, Kim J-W, Kim J-G, Kim I-S, Park KI, Cheon J, Nat. Nanotechnol 2011, 6, 418. [DOI] [PubMed] [Google Scholar]
- [59].Sun S, Zeng H, Robinson DB, Raoux S, Rice PM, Wang SX, Li G, J. Am. Chem. Soc 2004, 126, 273. [DOI] [PubMed] [Google Scholar]
- [60].Chen Z, Xu R, Zhang Y, Gu N, Nanoscale Res. Lett 2008, 4, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Sharma A, Sharma US, Int. J. Pharm 1997, 154, 123. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.