

Abstract
Congenital heart disease (CHD) is among the most common birth defect. Children with CHD frequently display long-term intellectual and behavioral disability. Emerging evidence indicates that cardiac anomalies lead to a reduction in cerebral oxygenation, which appears to profoundly impact the maturation of cerebral regions responsible for higher order cognitive functions. In this Review, we focus on the potential mechanisms by which dysregulation of cortical neuronal development during early life may lead to the significant cognitive impairments that commonly occur in children with CHD. Further understanding of the mechanisms underlying cortical dysmaturation due to CHD is required to identify strategies for neonatal neuroprotection and for mitigating developmental delays in this patient population.
Keywords: Congenital, heart, perinatal, hypoxia, cortex, interneuron
Why study disturbances in cortical development associated with congenital heart anomalies?
Human brain development is a highly dynamic process that involves proper timing and orchestration of cellular and molecular events. Unique cognitive abilities of humans likely result from a relative expansion of cortical regions responsible for intellectual and behavioral functions [1]. The frontal lobe, particularly enlarged in humans, is involved in higher-order executive functions such as planning, reasoning, decision making, attention, and personality. An appropriate balance between excitation and inhibition in the brain is critical for its proper functioning [2]. Although most human cortical neurogenesis is completed in late second or early third trimesters of gestation, recent studies indicate that substantial cortical neurogenesis continues postnatally. New neurons migrate along the lateral ventricles to the frontal lobe in the early postnatal period. These neurons differentiate into inhibitory neurons and integrate into the frontal cortex, ultimately contributing to the development of cortical regions responsible for higher-order cognitive processes [3,4].
Advances in non-invasive neuroimaging have provided a detailed picture of the dynamic human cortical expansion that occurs during late fetal and early postnatal life (from 28 to 48 weeks of gestation age [5]). The brain is broadly vulnerable throughout this key developmental period. Various insults that can cause brain damage during this period include: impairments associated with preterm birth, hypoxic-ischemic encephalopathy, and congenital heart disease (CHD) [6–8]. Widespread disturbances in brain development associated with CHD affect the maturation of many regions including the cortex, basal ganglia, thalamus and cerebral white matter [9,10].
Recent MRI studies have demonstrated decreased cortical expansion and sulcation in the fetal and neonatal brain with severe and complex CHD (Table 1). Although the hospital mortality risk of neonatal surgery for severe and complex CHD has been greatly reduced over the last 2 decades, children frequently display long-term intellectual and behavioral disabilities after complex cardiac repair [11]. Thus, there is a critical need to define mechanisms associated with cortical disturbances in order to develop therapeutic interventions to lessen the impact of these developmental insults. Although cortical neurogenesis at fetal and adult stages has been widely studied and reviewed in detail (for review, see [12,13]), the development of the frontal cortex during the perinatal period, particularly in humans, has only recently received greater attention due to methodological advances and the identification of ongoing postnatal neurogenesis in this region. It is crucial to understand the mechanisms involved in this process, since dysregulation of cortical neurogenesis likely contributes to neurodevelopmental disabilities in children with CHD (Figure 1, Key Figure). Moreover, understanding the impact of CHD on the genesis, migration, maturation, and integration of newly-migrated cortical neurons is imperative to develop rational strategies for neonatal neuroprotection.
Table 1.
Recent imaging studiesa assessing preoperative impairments in cortical maturation in CHD.
| Cohort | Gestational age | Cases (n) | Controls (n) | Technique | Findings | |
|---|---|---|---|---|---|---|
| Fetal | ||||||
| Zeng et al., 2015 [85] | Mixed | 2nd and 3rd trimester | 73 | 168 | Ultrasound | Progressive ↓ frontal lobe volume from GW28 |
| Schellen et al., 2015 [36] | TOF | 25 wks | 24 | 24 | MRI | ↓ cortical GM volume |
| Clouchoux et al., 2013 [35] | HLHS | 25–37 wks | 18 | 30 | MRI | ↓ cortical GM volume ↓ cortical surface area ↓ GI Cortical sulcation delay |
| Masoller et al., 2016 [86] | Mixed | 36–38 wks | 58 | 58 | MRI | ↓ cortical sulcation |
| Kelly et al., 2017 [37] | Mixed | 39 wks | 30 | 30 | MRI | ↓ cortical GM volume ↓ GJ |
| Postnatal preoperative | ||||||
| Ortinau et al., 2013 [38] | Mixed | Term | 15 | 12 | MRI | ↓ cortical surface area ↓ GI |
| von Rhein et al., 2015 [27] | Mixed | Term | 19 | 19 | MRI | ↓ cortical GM volume ↓ frontal, temporal, parietal and occipital lobes volume |
| De Asis-Cruz et al., 2018 [39] | Mixed | Term | 30 | 82 | rs-fc MRI | Altered functional brain connectivity |
Studies within the past 5 years
GI, gyrification index; GM, gray matter; GW, gestational week; HLHS, hypoplastic left heart syndrome; rs-fc MRI, resting state functional connectivity MRI; TOF, tetralogy of Fallot.
Figure 1, Key Figure -.
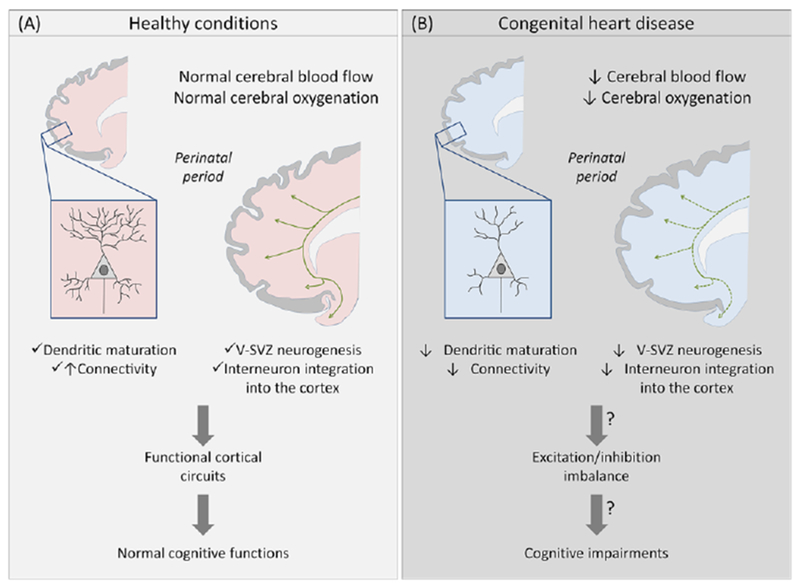
Impact of congenital heart disease on perinatal cortical maturation and subsequent cerebral impairments.
Cartoon illustrating perinatal cortical development in healthy conditions (A) and in congenital heart disease (B). Before corrective surgery, CHD reduces cerebral blood flow and oxygenation. In animal models, hypoxemia is associated with a disruption of subplate neuron maturation and connectivity prenatally, and a decrease of V-SVZ neurogenesis and interneuron populations in the frontal cortex postnatally. These alterations could result in a cortical excitation/inhibition imbalance and potentially explain the cellular basis for the spectrum of cognitive impairments observed in CHD patients. Green lines and arrows represent streams of neuroblasts in the V-SVZ migrating to the olfactory bulb and frontal lobe.
This Review focuses on cortical maturation and the deleterious effects of CHD-induced hypoxemia on brain development during the critical window of perinatal development. We first discuss aberrant brain maturation in children with CHD and the associated neurodevelopmental disabilities. We then discuss the current understanding of early postnatal neurogenesis in gyrencephalic species. We will focus on the impact of CHD on cortical maturation, particularly with respect to excitatory/inhibitory imbalance in developing cortex. We place special emphasis on the urgent need for well-designed animal studies to define disturbances in cortical neurogenesis due to perinatal brain injury. Further study of the impact of hypoxemia on brain development is of broad relevance not only for children with CHD but for other populations in which perinatal hypoxic stress may cause long-term intellectual and behavioral dysfunctions.
The CHD-neurodevelopment axis
Neurodevelopmental Disabilities Associated with CHD
CHD is among the most common birth defect and represents the leading cause of infant mortality associated with birth defects [14]. Each year, CHD affects almost 1 in every 100 infants, of whom ~25% will need catheter intervention or cardiac surgery during their first year of life [15]. Improvements in surgical techniques and therapeutic interventions allow most patients to reach adulthood. In 2010, approximately 1.4 million adults and 1 million children were estimated to be living with a CHD in the United States [16]. Although the hospital mortality risk is greatly reduced, children with CHD frequently display subsequent neurological disabilities affecting intellectual function, memory, executive function, speech and language, gross and fine motor skills, and visuo-spatial functions [17,18]. These disabilities often persist into adolescence and adulthood, and can ultimately represent long-term neurocognitive disabilities [19,20]. Apart from the impact of the neurological morbidity on the patients themselves, the toll on families and society is immense. Accordingly, the Pediatric Heart Network of the National Heart, Lung, and Blood Institute has declared that “one of the most important challenges in the 21st century for CHD is to improve neurological deficits” [21]. Elucidating the mechanisms underlying CHD-induced neurological impairments is therefore crucial for this growing population of patients.
Brain maturation and cortical development in CHD
Historically, adverse neurological outcomes observed in CHD patients were mainly attributed to neonatal cardiac interventions. It is now clear that neurological deficits in CHD involve several complex factors, often combinatorial and cumulative, including: i) intrinsic genetic factors; ii) preoperative factors related to abnormal fetal cerebral blood flow and oxygen and nutrient delivery; iii) operative factors related to cardiopulmonary bypass or deep hypothermic circulatory arrest; and iv) postoperative factors involving low cardiac output [11,17,19,22,23]. Recent advances in neonatal brain imaging revealed that CHD patients display antenatal and preoperative abnormalities in brain size, maturation, structure, metabolism and connectivity [24,25]. During the third trimester of gestation, CHD fetuses display smaller total brain volume and intracranial volume compared to healthy fetuses [26]. This decrease is also observed at birth prior to any corrective operation [27]. In neonatal care, macroscopic brain development and maturation is estimated using a ‘total maturation score’. This score is reduced pre-operatively in neonates with CHD, which supports the link between CHD and impaired brain maturation, independent of cardiac surgery [28–30]. Brain maturation can also be quantified using magnetic resonance spectroscopy through analysis of brain metabolite ratios. For instance, the ratio of N-acetyl aspartate (NAA) to choline increases over brain maturation, whereas the ratio of lactate to choline decreases. In the neonatal CHD brain, compared to control newborns, there is a 10% decrease in the NAA to choline ratio and a 28% increase in the lactate to choline ratio [9]. These findings, similar to those seen in premature infants, reflect a delay in brain development induced by CHD in utero.
In preterm birth, smaller cerebral volumes are associated with poor neurobehavioral outcomes in early childhood [31]. Specifically, cortical gray matter volume is negatively associated with motor performance and cognition at 24 months and with developmental quotient at age 3.5 years [32]. Given this established link between cortical development and neurobehavioral outcomes, we will focus here specifically on disturbances in cortical development, which are likely a key contributor to cognitive and learning disabilities in CHD. It should be noted however that CHD patients also display a large spectrum of white matter injury [33]. which likely plays a role in developmental delay associated with cardiac abnormalities, and possibly other forms of brain dysmaturation as well.
Synaptic connections and neuronal activity dramatically increase during the third trimester of pregnancy [34]. Moreover, neurons continue to migrate postnatally, particularly to the frontal lobe [3]. Deleterious factors during CHD, such as chronic hypoxemia, likely impair perinatal cortical maturation and disrupt long-term neurodevelopment (see Table 1 for recent CHD studies that assessed the role of preoperative insults on cortical maturation). For example, fetuses with hypoplastic left heart syndrome (HLHS) and tetralogy of Fallot display reduced cortical gray matter volume [35,36]. Moreover, delays in cortical gyrification, in addition to reduced cortical gray matter volume, are observed prenatally and postnatally before corrective surgery in mixed cohorts of CHD [37,38]. Notably, a recent study using resting state functional connectivity MRI (rs-fc MRI) reported alterations in connectivity in neonates with complex CHD before surgery and in the absence of brain parenchymal injury [39].
Disturbances in cerebral blood flow in CHD
In contrast to the brain, which continues to undergo profound structural changes after birth, heart development (in terms of overall organ layout) is largely completed by gestational week seven [40]. During the third trimester, the blood supply to the brain increases and represents 25% of the combined ventricular output. Hence, normal cardiovascular function is crucial for optimal brain development. Cardiac defects cause modifications in the intracardiac circulation that result in changes in cerebral blood flow characteristics [41,42]. A compensatory mechanism called “brain sparing” is established during fetal development [43]. This autoregulation reduces cerebral vascular resistance and increases cerebral blood flow in fetuses with CHD [41,44]. Despite the protective function of brain sparing, this process is insufficient to ensure optimal blood supply to the brain in CHD [41], and its effectiveness also depends on the type of cardiac anomaly. For instance, brain sparing physiology seems to be impaired in the fetus with d-transposition of the great arteries (d-TGA) in whom somatic growth is preserved relative to head growth. By contrast, HLHS fetuses display notable cerebrovascular autoregulation [45]. Interestingly, lower cerebrovascular resistance in fetuses with single ventricle anomalies are associated with impairments in early neurological development [46]. Finally, an arterial spin labeling MRI study found significant decreases in global cerebral blood flow in newborns with complex CHD, which suggests limitations in oxygen and nutrient delivery [42].
Chronic cerebral hypoxemia in CHD
Cerebral oxygenation is another critical factor in brain development in the perinatal period. During fetal life, the brain consumes half of the body’s oxygen [47]. In complex CHD (e. g., HLHS, d-TGA), the oxygen supply to the brain is limited by a decrease in blood flow (ischemia) and also by desaturated blood streamed to the brain (hypoxemia) ((Box 1 and Figure I). In addition, CHD is often associated with placental malformations such as abnormal maturation of villi, chorangiosis, inflammation, infarction and fetal thrombotic vasculopathy [48]. Recently, non-invasive placental perfusion imaging demonstrated that global placental perfusion significantly decreases with advancing gestational age in pregnancies complicated by fetal CHD [49]. Overall, impairments in placental function further contribute to the fetal cerebral hypoxemia occurring in CHD. Blood saturation can be studied in utero with the blood oxygen level–dependent magnetic resonance imaging signal, or T2*. This signal is reduced in brains of fetuses with CHD, mainly due to the desaturation of blood streamed through their cerebral vasculature [50]. Fetuses with critical forms of CHD may have 10% less cerebral oxygenation compared to normal controls, which is associated with a reduction in fetal brain volume [51]. This decrease of oxygen delivery to the brain also occurs in neonates with CHD before cardiac surgery [52,53]. Such neuroimaging studies indicate that aberrations in fetal circulation induce abnormal oxygen delivery that likely promotes developmental disturbances in CHD neonates. Although this relationship is not yet confirmed, recent advances in neonatal brain imaging have better defined brain injury mechanisms underlying CHD. It should be noted that hypoxemia also disrupts glucose delivery and metabolism, which may further contribute to aberrant cerebral development [54].
Box 1. In utero cerebral hypoxemia in complex congenital heart defects.
In the fetus, gas exchange occurs in the placenta and blood flow is distinctly different from the postnatal circulation. The stream containing the highly oxygenated blood from the placenta is preferentially directed across the foramen ovale to the left atrium, which favors the streaming of oxygen-rich blood to the brain (Figure I-A). The unique blood supply to the fetal brain is influenced by many factors including the anatomic structure of the heart [87]. In many cases of complex CHD, these beneficial systems that regulate cerebral blood flow are altered in utero [42,51].
In d-transposition of the great arteries (d-TGA), the aorta arises from the right ventricle whereas the pulmonary trunk is connected to the left ventricle (the opposite of normal circulation). The oxygen-rich blood in the left ventricle is thus directed toward the body through the pulmonary trunk and ductus arteriosus. On the other hand, the brain receives relatively deoxygenated blood that streams from the superior vena cava through the right ventricle, which causes reduced cerebral oxygenation during the fetal period (Figure I-B).
In hypoplastic left heart syndrome (HLHS), the left side of the heart is underdeveloped and dysfunctional. The oxygenated blood from the placenta and deoxygenated venous return mix in the right atrium. Blood with lower oxygen content is ejected into the pulmonary trunk and ductus arteriosus. In addition to reduced oxygenation, the blood supply to the brain is restricted by a retrograde flow into the cerebral circulation and a small diameter of the aortic arch, which significantly decreases cerebral perfusion. In this syndrome, the fetal brain is therefore underperfused with hypoxic blood (Figure I-C).
Figure I for Box 1 -.
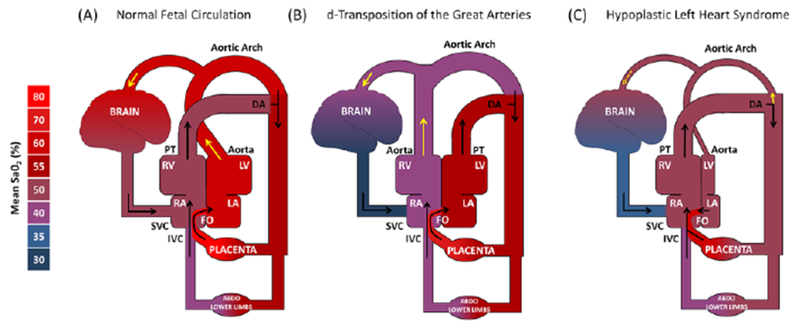
Simplified systemic hemodynamics in (A) normal fetal circulation, (B) d-transposition of the great arteries (d-TGA) and (C) hypoplastic left heart syndrome (HLHS).
Black arrows represent the blood flow. Yellow arrows represent the cerebral blood supply. The pulmonary circulation is not included because the blood flow to the lungs is highly limited due to elevated pulmonary vascular resistance and relatively low lung volume prior to birth. DA, ductus arteriosus; FO, foramen ovale; IVC, inferior vena cava; LA, left atrium; LV, left ventricle; PT, pulmonary trunk; RA, right atrium; RV, right ventricle; SVC, superior vena cava. Adapted from Sun et al. [51] with permission of the publishers.
Impact of hypoxemia on cortical development
Neonatal neurogenesis in humans
Originally believed to be complete before birth, neurogenesis and cortical maturation are now recognized to continue postnatally. In particular, early-postnatal cortical development in gyrencephalic species seems to be determined by the neurogenic activity of the ventricular-subventricular zone (V-SVZ) [3,4]. In the mammalian brain, the V-SVZ is one of the most active neurogenic niches during the postnatal period. In rodents, neural stem cells residing in the walls of the lateral ventricles give rise throughout their lifespan to neuroblasts that migrate to the olfactory bulb via the rostral migratory stream (RMS) and differentiate into interneurons. Interestingly, this stream of migrating neurons is also observed in young infants [55]. Additionally, the human infant brain displays another neuronal migration route, not reported in other vertebrates, called the medial migratory stream [55]. This route contains streams of migrating neuroblasts with elongated morphologies that divert from the proximal limb of the RMS to migrate to the frontal lobe. This supplementary stream provides interneurons to the ventromedial prefrontal cortex, an important area for social decision-making and risk processing [56,57]. Recently, numerous neuroblasts have been discovered in the frontal lobes. These cells seem to originate from various progenitor zones in the ventral forebrain, but their exact birth time and place are not yet defined (Box 2). These young neurons migrate tangentially along the lateral ventricles in the V-SVZ, and then radially to an extensive region of the anterior forebrain, including the cingulate gyrus and prefrontal cortex [3]. The migrating neuroblasts subsequently integrate within the frontal cortex. These cells differentiate into interneurons and seem to play a crucial role in postnatal plasticity (Box 3).
Box 2. Embryonic origins and diversity of interneurons.
In the mammalian developing brain, excitatory and inhibitory interneurons are generated separately from each other. Excitatory neurons originate from the dorsal telencephalon, whereas interneurons derive from specific transient germinal zones in the ventral telencephalon, namely the ganglionic eminences (GEs) and the preoptic area. The GEs are spatially subdivided into lateral, medial and caudal domains (LGE, MGE and CGE, respectively). Interneurons differentiate locally, or migrate tangentially from the subpallium to their final destination. In mice, the LGE produces inhibitory projection neurons destined for the striatum and inhibitory interneurons destined for the olfactory bulb. The MGE generates local projection neurons for the globus pallidus and the amygdala, as well as interneurons migrating to the cortex, striatum, hippocampus and amygdala. The CGE gives rise to local caudal striatal neurons, and interneurons migrating mainly to the cortex, striatum and hippocampus (for review, see [88] ).
Rodent cortical interneurons are mainly produced in the CGE and the MGE, and also in the preoptic area although to a lesser extent. The MGE is responsible for 50 to 70% of cortical interneurons, which includes two large non-overlapping subpopulations of interneurons expressing parvalbumin and somatostatin. Interestingly, the subtype of interneurons originating from the MGE depends on the temporal sequence of this generation. The CGE gives rise to late-born interneurons, which represents another population of cortical interneurons (30-40%) that mainly express the 5HT3a receptor but also other markers like calretinin, vasoactive intestinal peptide or reelin. Finally, the preoptic area produces a small fraction (5-10%) of different subtypes of cortical interneurons that frequently express neuropeptide Y [88].
Studies suggest that the neocortex contains over 20 different classes of interneurons categorized based on their morphological, molecular, electrical, and synaptic properties (for more on interneuron diversity, see [89]). As discussed above, this considerable interneuron diversity is mainly shaped by developmental spatial-temporal determinants such as spatial origins and birthdate [90]. Additionally, environmental cues play a critical role in determining subtype-specific features of cortical interneurons including settling position, morphology, synapse specificity, and afferent and efferent connectivity [89]. Modification of the environmental input due to perinatal CHD-induced brain injury could therefore interfere with interneuron diversity and influence cortical maturation.
Box 3. Roles of interneurons in the frontal cortex.
Although constituting a minority of cortical neurons (~20% in rodents) [91], GABAergic inhibitory interneurons play a critical role in fine-tuning the activity of pyramidal neurons assemblies. To date, it is not clear if the late-migrating cortical GABAergic inhibitory interneurons play a different role from those that migrate earlier, or if they simply serve as an additional mechanism of postnatal plasticity. There are more than 20 different classes of interneurons identified in the cerebral cortex [59], displaying distinct or partially overlapping functions, morphologies and properties, which make their classification highly complex (for review see [89]). Particularly, one of the criteria to classify GABAergic inhibitory interneurons is molecular marker expression for calcium binding proteins (parvalbumin, calbindin, calretinin), neuropeptides (e.g., vasoactive intestinal peptide, neuropeptide Y, reelin, somatostatin), and receptors (e. g. 5HT3R, mGluR1, CB1). Interestingly, this interneuron diversity depends on the interaction of specific genetic and environmental factors (see Box 2) [88,92]. The postnatal integration of interneurons into the frontal cortex could therefore influence this diversity and contribute to delayed plasticity during postnatal human development.
At early stages of brain development, GABA is thought to exerts a depolarizing effect on neural cells [93], and plays a critical role in circuit maturation and cortical plasticity. It allows cortical networks to generate oscillations of wider amplitude and faster frequencies that are distinctive features of circuit maturation. Additionally, GABAergic transmission can shape synaptic wiring and large-scale architecture of neuronal circuits. For instance, such transmission adjusts intracortical circuitry by inducing morphological changes at the level of dendritic spines[94]. Overall, GABAergic inhibitory interneurons play a crucial role in cortical development and maturation. They are particularly relevant in the context of perinatal brain injury, since postnatal disruption of the GABAergic system may contribute to the etiology of neurodevelopmental disorders.
Impact of hypoxemia on neurogenesis and interneuron migration
There is a growing consensus that chronic antenatal and early postnatal insults contribute to long-term neurodevelopmental disabilities in children with CHD (Table 1). Hypoxemia during CHD affects the brain from the third trimester until after birth [50–53]. It is therefore possible that CHD disrupts the recruitment of these late-migrating neurons and their connectivity.
The impact of chronic hypoxemia on cortical neurogenesis was recently studied using the neonatal piglet [4]. This species is particularly relevant because the early postnatal porcine V-SVZ shares structural features with the postnatal human V-SVZ and displays a high density of proliferative neuroblasts [58]. Moreover, similar to human, young neurons in the porcine SVZ migrate postnatally to frontal cortices, where they mainly differentiate into calretinin-positive interneurons. After chronic hypoxemia, porcine brains are smaller and exhibit a significant reduction in cortical gray matter volume and gyrification index, which is also observed in the neonatal brain with CHD. Chronic hypoxic exposure also induces a significant decrease of proliferative and non-proliferative neural stem-progenitor cells as well as neuroblasts within the V-SVZ (Figure 2). Moreover, the impaired neurogenic activity in the piglet V-SVZ after chronic hypoxemia is associated with reduced interneuron populations within frontal cortices, primarily the prefrontal cortex, which may induce imbalances between excitatory and inhibitory neurons. Interestingly, the V-SVZ of human infants that sustained abnormal fetal cerebral blood flow due to CHD, also displayed a pronounced depletion of neuroblasts [4] (Figure 2).
Figure 2 -.
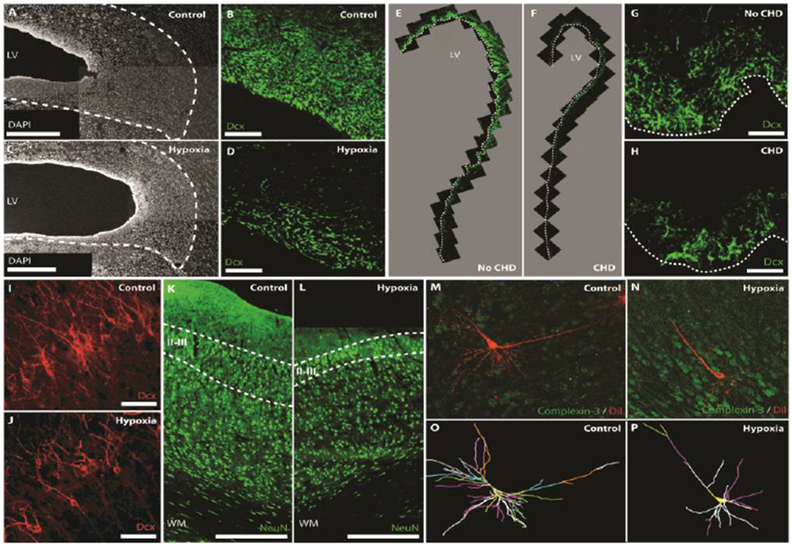
Cellular impairments in the perinatal brain associated with hypoxia and CHD.
Postnatal hypoxia in the piglet reduces the width of the SVZ (A,C; Scale bars, 500 mm) and the number of neuroblasts (DCX+) in the SVZ (B,D; Scale bars, 50 mm). Similarly, CHD reduces neuroblast numbers (DCX+) in the SVZ of human infants (E-H; Scale bars, 50 mm). Postnatal hypoxia in the piglet also reduces the number of DCX+ immature neurons (I,J; Scale bars, 50 mm) and NeuN+ mature neurons (K,L; Scale bars, 500 mm) in layers II/III of the frontal cortex. Prenatal hypoxia in the sheep results in a decrease in dendritic arborization of subplate neurons. 3D reconstructions of the soma and full basal and apical dendritic arbors (O,P) of representative sheep subplate neurons (M,N). Branch order rank is indicated by color: 1st order– bright yellow, 2nd order – white, 3rd order – purple/hot pink, 4th order – bright green, 5th order – cyan blue, 6th order – orange, 7th order – slate gray, 8th order – salmon pink, 9th order – forest green, and 10th order – bright blue. Adapted from Morton et al. [4] and McClendon et al. [75] with permission of the publishers.
It is well documented that in humans, intellectual and behavioral disabilities related to attention deficits, hyperactivity, learning and working memory are significantly associated with imbalances between excitatory and inhibitory neurons in the frontal cortex [2,59]. These types of disabilities are also commonly observed in CHD patients [60]. For instance, neuropsychological assessment of school-aged children with CHD reveals lower scores in language, attention, executive functioning, and memory, even after successful cardiac surgical correction [20,61]. Attention-deficit/hyperactivity disorder symptoms are also more prevalent in children with CHD [62]. These findings suggest that developmental disturbances in the GABAergic system contribute to the neurodevelopmental disabilities seen in CHD [63] (Figure 1).
Impact of hypoxemia on prenatal neuronal maturation and synaptic activity
Transient or chronic disturbances in fetal oxygen delivery disrupt fetal brain development [64], which may also contribute to the pathogenesis of neurodevelopmental disabilities in CHD. Instrumented fetal sheep preparations provide unique access to analyze the impact of oxygenation on the fetal brain, because of the feasibility to repeatedly sample fetal arterial blood gases and metabolites in utero in relation to a wide array of sensitive markers of disrupted brain development or injury [65]. Cerebral gray and white matter are differentially susceptible to fetal hypoxemia. White matter injury occurred infrequently in models of hypoxemia without cerebral ischemia in which a restriction in uteroplacental blood flow [66] or maternal hypoxemia [67] resulted in decreased fetal oxygen delivery and acidemia. However, moderate global cerebral ischemia in conjunction with hypoxemia appears to be a critical factor to generate a spectrum of fetal ovine white matter injury that ranges from diffuse non-necrotic lesions to severe cystic necrosis [68,69]. Late oligodendrocytes progenitors (pre-oligodendrocytes) are the primary cell type in the white matter lost in diffuse lesions arising from hypoxia-ischemia [70].
In contrast, severe global cerebral hypoxia-ischemia is required to induce pronounced fetal neuronal degeneration in cortical and subcortical gray matter structures [71]. In fact, immature fetal neurons are much less susceptible to cell death from hypoxia-ischemia than pre-oligodendrocytes [71]. This neuronal resilience at the immature stage is also observed after middle cerebral artery stroke in preterm infants, who display remarkable cortical sparing in contrast to term neonates [72]. Despite the decreased susceptibility of immature neurons to fetal hypoxia-ischemia, cortical projection neurons nevertheless sustain disturbances in maturation of both the basal dendritic arbor and spines [73]. Reduced dendritic arborization contributes significantly to a reduction in cortical volume and accounts for disturbances in cortical anisotropy defined by high field MRI measurements [73]. Neuronal dysmaturation with similar morphometric features was observed in medium spiny projection neurons of the caudate, which also displayed significant disturbances in spine density and glutamatergic synaptic activity in response to hypoxia-ischemia [74].
A central role for hypoxemia in disrupted neuronal maturation was recently defined in the subplate of preterm fetal sheep where the response to a 25-minute hypoxic-ischemic insult was compared to that of exposure to hypoxemia alone [75]. The subplate resides directly beneath the cerebral cortex and is comprised of a unique neuronal population that is transiently present during development to provide guidance cues that establish thalamocortical connectivity [76]. Four weeks after the insult, subplate neurons displayed persistent disturbances in basal dendritic arborization that were accompanied by disturbances in subplate neuron excitability and synaptic activity (Figure 2). Notably, subplate neuron dysmaturation was significantly linked to the magnitude of transient fetal hypoxemia. These findings suggest that fetal hypoxemia is sufficient to disrupt neuronal maturation. Although these findings support that transient fetal hypoxemia can disrupt subplate neuron maturation, the response to chronic hypoxemia is unclear. Future studies are also needed to determine whether hypoxemia can disrupt the maturation of cortical projections neurons or interneurons in a fashion similar to subplate neurons.
Concluding remarks and future perspectives
As mortality rates among CHD patients have gradually decreased, there has been a greater research focus on improving neurological outcomes for the growing adult population with CHD. CHD affects cerebral maturation during a broad developmental window. Hypoxemia occurs both in utero and postnatally and likely plays critical roles in cortical maturation disturbances that are frequently associated with CHD (Figure 1). In addition, multiple factors associated with cardiac surgery may injure the brain of infants with CHD, including inflammatory response, reoxygenation, reperfusion, and prolonged anesthesia exposure. Hence, in addition to chronic hypoxemia, cumulative pathological events in critical developmental time periods could contribute to the high prevalence of neurodevelopmental disturbances in the CHD population [11,19,22].
Human pathology studies are technically challenging due to limited access to autopsy tissues and the variability of postmortem tissue integrity. Hence, animal models are essential to elucidate the impact of CHD-induced insults on the developing brain. Despite the utility of rodent genetic tools, the differences between human and rodent brain development, especially regarding cortical maturation, greatly limit the study of CHD-induced disturbances. Larger mammals with gyrencephalic brains offer several advantages for elucidating mechanisms of CHD-induced injury. Fetal sheep and neonatal piglets have been the key species used to study prenatal and postnatal effects of chronic hypoxemia on brain development, respectively. Moreover, larger mammals often provide closer anatomical, physiological, and metabolic similarities to humans, which is valuable for evaluating neuroprotective strategies prior to potential clinical trials. As recent advances in genome editing, such as CRISPR-Cas9, become more available in larger mammals, it should be feasible to generate preclinical large animal models to access mechanistic questions related to the cumulative effects of hypoxemia on critical developmental events including neuronal maturation and postnatal V-SVZ neurogenesis (see Outstanding Questions).
Outstanding questions.
How can we generate new animal models to study the effects of hypoxemia during gestation and during postnatal life?
During which period is the brain more sensitive to developmental and behavioral disabilities from hypoxemia, the prenatal or the postnatal?
How does prenatal dysmaturation of subplate neurons influence postnatal integration of interneurons into the cortex?
When and where are late-migrating interneurons generated? Does hypoxemia have an impact on the generation, migration and maturation of these cells?
Does fetal neuronal dysmaturation play a role in the functional consequences of white matter injury in CHD patients?
How can we isolate the long-term neurological outcomes of CHD from the impact of corrective surgery on the brain?
To what extent can environmental enrichment play a beneficial role to improve disturbances in neuronal development arising in utero or postnatally during a highly plastic period in human brain maturation?
The development of human ‘brain organoids’ has dramatically advanced our ability to model human brain development in vitro [77]. These 3D structures are derived from human pluripotent cells (including induced pluripotent stem cells and embryonic stem cells) and recapitulate some key aspects of human brain development and function. For instance, neuronal cells organize in a multilaminar fashion in brain organoids. Additionally, these structures generate an outer SVZ, a distinctive feature of the developing human SVZ [78]. Moreover, human cortical spheroids were recently fused to human subpallial spheroids to model the migration of interneurons from the subpallium to the cortex [79]. Interneurons functionally integrated with glutamatergic neurons to form a microphysiological system [79]. This system may provide novel access to study the effects of hypoxemia on perinatal circuit assembly, and help elucidate cellular and molecular abnormalities occurring during development of specific neural circuits.
Previous clinical trials in neonates and infants who underwent cardiac surgery have led to refinements in surgical methods that may improve neurodevelopmental outcomes of CHD patients [80]. During this manuscript’s preparation, an interventional clinical trial is underway to test the effect of maternal hyperoxygenation in CHD (https://clinicaltrials.gov/ct2/show/NCT03136835). This approach likely results in a diffuse increase in fetal oxygenation which may influence fetal blood flow and heart dimensions [81,82]. Maternal hyperoxygenation may thus reduce the deleterious effects of hypoxemia on fetal systemic and brain development [81]. However, it should be noted that the response to maternal hyperoxygenation is highly cardiac-lesion dependent. For instance, the beneficial effect of hyperoxygenation is prevented by the presence a restrictive/intact atrial septum or ventricular septal defects [83,84].
Improving neurodevelopmental outcomes in CHD is one of the main challenges for pediatric cardiology. Recent advances in our understanding of fetal neuronal susceptibility to hypoxemia may have relevance both to infants with CHD and those with other causes of intermittent or chronic postnatal hypoxemia, such as apnea of prematurity or chronic lung disease. The identification of persistent human perinatal neurogenesis that targets the frontal cortex has further opened new opportunities that may lead to regeneration and repair of the dysmature cortex in children with CHD. The abundance of available tools to study the complexity of cellular and molecular mechanisms of cortical development and dysmaturation will likely help identify novel strategies to treat and improve outcomes in children suffering from intellectual and behavioral disabilities.
Highlights.
Patients with congenital heart disease (CHD) frequently exhibit a broad spectrum of neurological outcomes. Currently, the neurological deficits exhibited by CHD patients are irreversible.
Fetal cerebral hypoxemia is correlated with impaired cortical development in infants with CHD.
The perinatal period encompasses several critical windows for cortical maturation, including neurogenesis, neuronal migration, dendritic maturation and circuit formation.
Recent animal studies show that both transient fetal hypoxemia and chronic perinatal hypoxemia have deleterious effects on neuronal development.
Studies with large animal models are essential to unravel the complex molecular and cellular mechanisms underlying cortical dysmaturation associated with CHD.
Acknowledgments
The authors would like to kindly thank Drs. Evan Goldstein and Joshua Corbin for critically reading the manuscript. This work was supported by R01HL139712 from NIH/NHLBI (N. I.), 1RO1NS054044 and R37NS045737 from NIH/NINDS (S.A.B), 1R01AG031892-01 from NIH/NIA (S.A.B), 17GRNT33370058 from the American Heart Association (S.A.B), District of Columbia Intellectual and Developmental Disabilities Research Center (DC-IDDRC) (V.G.), U54HD090257 from NIH/NICHD (V.G.), and R37NS109478 (Javits Award) from NIH/NINDS (V.G.). The authors have no conflicts of interest relating to this work. The content is solely the responsibility of the authors and does not necessarily represent the official views of the DC-IDDRC or the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Passingham RE and Smaers JB (2014) Is the prefrontal cortex especially enlarged in the human brain allometric relations and remapping factors. Brain. Behav. Evol. 84, 156–66 [DOI] [PubMed] [Google Scholar]
- 2.Southwell DG et al. (2014) Interneurons from Embryonic Development to Cell-Based Therapy. Science (80- ). 344, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paredes MF et al. (2016) Extensive migration of young neurons into the infant human frontal lobe. Science 354, aaf7073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morton PD et al. (2017) Abnormal neurogenesis and cortical growth in congenital heart disease. Sci. Transl. Med. 9, eaah7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dubois J and Dehaene-Lambertz G (2015) Fetal and Postnatal Development of the Cortex: MRI and Genetics. Brain Mapp. An Encycl. Ref. 2, 11–19 [Google Scholar]
- 6.Salmaso N et al. (2014) Neurobiology of premature brain injury. Nat. Neurosci. 17, 341–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fatemi A et at. (2009) Flypoxic-ischemic encephalopathy in the term infant. Clin. Perinatol. 36, 835–58, vii [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mebius MJ et al. (2017) Brain Injury and Neurodevelopmental Outcome in Congenital Heart Disease: A Systematic Review. Pediatrics 140, e20164055. [DOI] [PubMed] [Google Scholar]
- 9.Miller SP et al. (2007) Abnormal Brain Development in Newborns with Congenital Heart Disease. N. Engl. J. Med. 357, 1928–1938 [DOI] [PubMed] [Google Scholar]
- 10.Morton PD et al. (2015) Congenital cardiac anomalies and white matter injury. Trends Neurosci. 38, 353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wernovsky G and Licht DJ (2016) Neurodevelopmental Outcomes in Children With Congenital Heart Disease-What Can We Impact? Pediatr. Crit. Care Med. 17, S232–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bergmann O et al. (2015) Adult Neurogenesis in Humans. Cold Spring Harb. Perspect. Biol. 7, a018994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stiles J and Jernigan TL (2010) The basics of brain development. Neuropsychol. Rev. 20, 327–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang Q et al. (2006) Racial differences in infant mortality attributable to birth defects in the United States, 1989–2002. Birth Defects Res. Part A Clin. Mol. Teratol. 76, 706–713 [DOI] [PubMed] [Google Scholar]
- 15.Marelli AJ et al. (2014) Lifetime Prevalence of Congenital Heart Disease in the General Population From 2000 to 2010. Circulation 130, 749–756 [DOI] [PubMed] [Google Scholar]
- 16.Gilboa SM et al. (2016) Congenital Heart Defects in the United States. Circulation 134, 101–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Newburger JW et al. (2012) Early developmental outcome in children with hypoplastic left heart syndrome and related anomalies: The single ventricle reconstruction trial. Circulation 125, 2081–2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Naef N et al. (2017) Neurodevelopmental Profiles of Children with Congenital Heart Disease at School Age. J. Pediatr. 188, 75–81 [DOI] [PubMed] [Google Scholar]
- 19.Marelli A et al. (2016) Brain in congenital heart disease across the lifespan: The cumulative burden of injury. Circulation 133, 1951–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bellinger DC et al. (2011) Adolescents with d-transposition of the great arteries corrected with the arterial switch procedure: neuropsychological assessment and structural brain imaging. Circulation 124, 1361–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaltman JR et al. (2010) Report of the Pediatric Heart Network and National Heart, Lung, and Blood Institute Working Group on the Perioperative Management of Congenital Heart Disease. Circulation 121, 2766–2772 [DOI] [PubMed] [Google Scholar]
- 22.Morton PD et al. (2017) Neurodevelopmental Abnormalities and Congenital Heart Disease: Insights Into Altered Brain Maturation. Circ. Res. 120, 960–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Homsy J et al. (2015) De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 350, 1262–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Y et al. (2015) Neurodevelopmental delay with critical congenital heart disease is mainly from prenatal injury not infant cardiac surgery: current evidence based on a meta-analysis of functional magnetic resonance imaging. Ultrasound Obstet. Gynecol. 45, 639–48 [DOI] [PubMed] [Google Scholar]
- 25.Peyvandi S et al. (2018) The neonatal brain in critical congenital heart disease: Insights and future directions. Neuroimage DOI: 10.1016/J.NEUROIMAGE.2018.05.045 [DOI] [PubMed] [Google Scholar]
- 26.Limperopoulos C et al. (2010) Brain volume and metabolism in fetuses with congenital heart disease: evaluation with quantitative magnetic resonance imaging and spectroscopy. Circulation 121, 26–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.von Rhein M et al. (2015) Severe Congenital Heart Defects Are Associated with Global Reduction of Neonatal Brain Volumes. J. Pediatr. 167, 1259–1263.e1 [DOI] [PubMed] [Google Scholar]
- 28.Licht DJ et al. (2009) Brain maturation is delayed in infants with complex congenital heart defects. J. Thorac. Cardiovasc. Surg. 137, 529–36-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andropoulos DB et al. (2010) Brain immaturity is associated with brain injury before and after neonatal cardiac surgery with high-flow bypass and cerebral oxygenation monitoring. J. Thorac. Cardiovasc. Surg. 139, 543–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dimitropoulos A et al. (2013) Brain injury and development in newborns with critical congenital heart disease. Neurology 81, 241–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monson BB et al. (2016) Examination of the Pattern of Growth of Cerebral Tissue Volumes From Hospital Discharge to Early Childhood in Very Preterm Infants. JAMA Pediatr. 170, 772. [DOI] [PubMed] [Google Scholar]
- 32.Keunen K et al. (2016) Brain Volumes at Term-Equivalent Age in Preterm Infants: Imaging Biomarkers for Neurodevelopmental Outcome through Early School Age. J. Pediatr. 172, 88–95 [DOI] [PubMed] [Google Scholar]
- 33.Guo T et al. (2018) White matter injury in term neonates with congenital heart diseases: Topology & comparison with preterm newborns. Neuroimage DOI: 10.1016/J.NEUROIMAGE.2018.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McQuillen PS et al. (2010) Effects of congenital heart disease on brain development. Prog. Pediatr. Cardiol. 29, 79–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clouchoux C et al. (2013) Delayed Cortical Development in Fetuses with Complex Congenital Heart Disease. Cereb. Cortex 23, 2932–2943 [DOI] [PubMed] [Google Scholar]
- 36.Schellen C et al. (2015) Fetal MRI detects early alterations of brain development in Tetralogy of Fallot. Am. J. Obstet. Gynecol. 213, 392.e1–392.e7 [DOI] [PubMed] [Google Scholar]
- 37.Kelly CJ et al. (2017) Impaired development of the cerebral cortex in infants with congenital heart disease is correlated to reduced cerebral oxygen delivery. Sci. Rep. 7, 15088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ortinau C et al. (2013) Cortical Folding Is Altered before Surgery in Infants with Congenital Heart Disease. J. Pediatr. 163, 1507–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Asis-Cruz J et al. (2018) Aberrant brain functional connectivity in newborns with congenital heart disease before cardiac surgery. Neuroimage. Clin 17, 31–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Srivastava D (2006) Making or Breaking the Heart: From Lineage Determination to Morphogenesis. Cell 126, 1037–1048 [DOI] [PubMed] [Google Scholar]
- 41.Donofrio MT et al. (2003) Autoregulation of Cerebral Blood Flow in Fetuses with Congenital Heart Disease: The Brain Sparing Effect. Pediatr. Cardiol. 24, 436–443 [DOI] [PubMed] [Google Scholar]
- 42.Nagaraj UD et al. (2015) Impaired Global and Regional Cerebral Perfusion in Newborns with Complex Congenital Heart Disease. J. Pediatr. 167, 1018–1024 [DOI] [PubMed] [Google Scholar]
- 43.Giussani DA (2016) The fetal brain sparing response to hypoxia: physiological mechanisms. J. Physiol. 594, 1215–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Masoller N et al. (2014) Evidence of second-trimester changes in head biometry and brain perfusion in fetuses with congenital heart disease. Ultrasound Obstet. Gynecol. 44, 182–187 [DOI] [PubMed] [Google Scholar]
- 45.Berg C et al. (2009) Doppler indices of the middle cerebral artery in fetuses with cardiac defects theoretically associated with impaired cerebral oxygen delivery in utero: is there a brain-sparing effect? Ultrasound Obstet. Gynecol. 34, 666–672 [DOI] [PubMed] [Google Scholar]
- 46.Williams IA et al. (2013) The association of fetal cerebrovascular resistance with early neurodevelopment in single ventricle congenital heart disease. Am. Heart J. 165, 544–550.e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prsa M et al. (2014) Reference Ranges of Blood Flow in the Major Vessels of the Normal Human Fetal Circulation at Term by Phase-Contrast Magnetic Resonance Imaging. Circ. Cardiovasc. Imaging 7, 663–670 [DOI] [PubMed] [Google Scholar]
- 48.Rychik J et al. (2018) Characterization of the Placenta in the Newborn with Congenital Heart Disease: Distinctions Based on Type of Cardiac Malformation. Pediatr. Cardiol. 39, 1165–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zun Z et al. (2017) Non-lnvasive Placental Perfusion Imaging in Pregnancies Complicated by Fetal Heart Disease Using Velocity-Selective Arterial Spin Labeled MRI. Sci. Rep. 7, 16126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lauridsen MH et al. (2017) Cerebral Oxygenation Measurements by Magnetic Resonance Imaging in Fetuses With and Without Heart Defects. Circ. Cardiovasc. Imaging 10, e006459. [DOI] [PubMed] [Google Scholar]
- 51.Sun L et al. (2015) Reduced fetal cerebral oxygen consumption is associated with smaller brain size in fetuses with congenital heart disease. Circulation 131, 1313–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jain V et al. (2014) Cerebral oxygen metabolism in neonates with congenital heart disease quantified by MRI and optics. J. Cereb. Blood Flow Metab. 34, 380–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lim JM et al. (2016) Cerebral oxygen delivery is reduced in newborns with congenital heart disease. J. Thorac. Cardiovasc. Surg. 152, 1095–1103 [DOI] [PubMed] [Google Scholar]
- 54.Rudolph AM (2016) Impaired cerebral development in fetuses with congenital cardiovascular malformations: Is it the result of inadequate glucose supply? Pediatr. Res. 80, 172–177 [DOI] [PubMed] [Google Scholar]
- 55.Sanai N et al. (2011) Corridors of migrating neurons in the human brain and their decline during infancy. Nature 478, 382–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van den Bos W and GOroglu B (2009) The role of the ventral medial prefrontal cortex in social decision making. J. Neurosci. 29, 7631–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schonberg T et al. (2012) Decreasing Ventromedial Prefrontal Cortex Activity During Sequential Risk-Taking: An fMRI Investigation of the Balloon Analog Risk Task. Front. Neurosci 6, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Costine BA et al. (2015) The Subventricular Zone in the Immature Piglet Brain: Anatomy and Exodus of Neuroblasts into White Matter after Traumatic Brain Injury. Dev. Neurosci 37, 115–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marín O (2012) Interneuron dysfunction in psychiatric disorders. Nat. Rev. Neurosci. DOI: 10.1038/nrn3155 [DOI] [PubMed] [Google Scholar]
- 60.Marino BS et al. (2012) Neurodevelopmental Outcomes in Children With Congenital Heart Disease: Evaluation and Management: A Scientific Statement From the American Heart Association. Circulation 126, 1143–1172 [DOI] [PubMed] [Google Scholar]
- 61.Miatton M et al. (2007) Neuropsychological Performance in School-Aged Children with Surgically Corrected Congenital Heart Disease. J. Pediatr 151, 73–78.e1 [DOI] [PubMed] [Google Scholar]
- 62.Hansen E et al. (2012) Prevalence of ADHD symptoms in patients with congenital heart disease. Pediatr. Int. 54, 838–843 [DOI] [PubMed] [Google Scholar]
- 63.Edden RAE et al. (2012) Reduced GABA concentration in attention-deficit/hyperactivity disorder. Arch. Gen. Psychiatry 69, 750–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rees S and Inder T (2005) Fetal and neonatal origins of altered brain development. Early Hum. Dev. 81, 753–761 [DOI] [PubMed] [Google Scholar]
- 65.Back SA et al. (2012) The instrumented fetal sheep as a model of cerebral white matter injury in the premature infant. Neurotherapeutics 9, 359–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rees S et al. (1997) The vulnerability of the fetal sheep brain to hypoxemia at mid-gestation. Dev. Brain Res. 103, 103–118 [DOI] [PubMed] [Google Scholar]
- 67.Penning DH et al. (1994) Neuropathology of the near-term and midgestation ovine fetal brain after sustained in utero hypoxemia. Am. J. Obstet. Gynecol. 170, 1425–32 [DOI] [PubMed] [Google Scholar]
- 68.Riddle A et al. (2011) Histopathological correlates of magnetic resonance imaging-defined chronic perinatal white matter injury. Ann. Neurol 70, 493–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Riddle A et al. (2012) Differential susceptibility to axonopathy in necrotic and non-necrotic perinatal white matter injury. Stroke 43, 178–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Back SA et al. (2002) Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J. Neurosci 22, 455–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Riddle A et al. (2006) Spatial heterogeneity in oligodendrocyte lineage maturation and not cerebral blood flow predicts fetal ovine periventricular white matter injury. J. Neurosci 26, 3045–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.van der Aa NE et al. (2016) Cortical Sparing in Preterm Ischemic Arterial Stroke. Stroke 47, 869–871 [DOI] [PubMed] [Google Scholar]
- 73.Dean JM et al. (2013) Prenatal cerebral ischemia disrupts MRI-defined cortical microstructure through disturbances in neuronal arborization. Sci. Transl. Med. 5, 168ra7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McClendon E et al. (2014) Prenatal cerebral ischemia triggers dysmaturation of caudate projection neurons. Ann. Neurol. 75, 508–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McClendon E et al. (2017) Transient Hypoxemia Chronically Disrupts Maturation of Preterm Fetal Ovine Subplate Neuron Arborization and Activity. J. Neurosci. 37, 11912–11929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kanold PO and Luhmann HJ (2010) The Subplate and Early Cortical Circuits. Annu. Rev. Neurosci. 33, 23–48 [DOI] [PubMed] [Google Scholar]
- 77.Arlotta P (2018) Organoids required! A new path to understanding human brain development and disease. Nat. Methods 15, 27–29 [DOI] [PubMed] [Google Scholar]
- 78.Qian X et al. (2016) Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 165, 1238–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Birey F et al. (2017) Assembly of functionally integrated human forebrain spheroids. Nature 545, 54–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jonas RA et al. (2003) The influence of hemodilution on outcome after hypothermic cardiopulmonary bypass: results of a randomized trial in infants. J. Thorac. Cardiovasc. Surg. 126, 1765–1774 [DOI] [PubMed] [Google Scholar]
- 81.Porayette P et al. (2016) MRI reveals hemodynamic changes with acute maternal hyperoxygenation in human fetuses with and without congenital heart disease. Prenat. Diagn 36, 274–281 [DOI] [PubMed] [Google Scholar]
- 82.Co-Vu J et al. (2017) Maternal hyperoxygenation: A potential therapy for congenital heart disease in the fetuses? A systematic review of the current literature. Echocardiography 34, 1822–1833 [DOI] [PubMed] [Google Scholar]
- 83.Szwast A et al. (2010) Vasoreactive Response to Maternal Hyperoxygenation in the Fetus With Hypoplastic Left Heart Syndrome. Circ. Cardiovasc. Imaging 3, 172–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kohl T (2010) Chronic intermittent materno-fetal hyperoxygenation in late gestation may improve on hypoplastic cardiovascular structures associated with cardiac malformations in human fetuses. Pediatr. Cardiol. 31, 250–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zeng S et al. (2015) Volume of intracranial structures on three-dimensional ultrasound in fetuses with congenital heart disease. Ultrasound Obstet. Gynecol. 46, 174–181 [DOI] [PubMed] [Google Scholar]
- 86.Masoller N et al. (2016) Mid-gestation brain Doppler and head biometry in fetuses with congenital heart disease predict abnormal brain development at birth. Ultrasound Obstet. Gynecol. 47, 65–73 [DOI] [PubMed] [Google Scholar]
- 87.McQuillen PS and Miller SP (2010) Congenital heart disease and brain development. Ann. N. Y. Acad. Sci 1184, 68–86 [DOI] [PubMed] [Google Scholar]
- 88.Bandler RC et al. (2017) Cortical interneuron specification: the juncture of genes, time and geometry. Curr. Opin. Neurobiol 42, 17–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tremblay R et al. (2016) GABAergic Interneurons in the Neocortex: From Cellular Properties to Circuits. Neuron 91, 260–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Butt SJB et al. (2005) The temporal and spatial origins of cortical interneurons predict their physiological subtype. Neuron 48, 591–604 [DOI] [PubMed] [Google Scholar]
- 91.Tamamaki N et al. (2003) Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J. Comp. Neurol 467, 60–79 [DOI] [PubMed] [Google Scholar]
- 92.Wamsley B and Fishell G (2017) Genetic and activity-dependent mechanisms underlying interneuron diversity. Nat. Publ. Gr 18, [DOI] [PubMed] [Google Scholar]
- 93.Represa A and Ben-Ari Y (2005) Trophic actions of GABA on neuronal development. Trends Neurosci. 28, 278–283 [DOI] [PubMed] [Google Scholar]
- 94.Le Magueresse C and Monyer H (2013) GABAergic Interneurons Shape the Functional Maturation of the Cortex. Neuron 77, 388–405 [DOI] [PubMed] [Google Scholar]