

Abstract
Obscurins, expressed from the single OBSCN gene, are a family of giant, modular, cytoskeletal proteins that play key structural and regulatory roles in striated muscles. They were first implicated in the development of heart disease in 2007 when two missense mutations were found in a patient diagnosed with hypertrophic cardiomyopathy (HCM). Since then, the discovery of over a dozen missense, frameshift, and splicing mutations that are linked to various forms of cardiomyopathy, including HCM, dilated cardiomyopathy (DCM), and left ventricular non-compaction (LVNC), has highlighted OBSCN as a potential disease-causing gene. At this time, the functional consequences of the identified mutations remain largely elusive, and much work has yet to be done to characterize the disease mechanisms of pathological OBSCN variants. Herein, we describe the OBSCN mutations known to date, discuss their potential impact on disease development, and provide future directions in order to better understand the involvement of obscurins in heart disease.
Keywords: Obscurin, Sarcomeric mutations, Cardiomyopathy, Heart failure
Introduction
Cardiomyopathies comprise a heterogeneous group of myocardial disorders that are characterized by abnormal pumping of blood and/or the inability to maintain normal electrical rhythm [13]. They are routinely grouped into six major subtypes according to the clinical presentation of patients and the structural and functional maladaptations that manifest in the diseased myocardium [12, 16, 45, 75]. The most common subtype, hypertrophic cardiomyopathy (HCM), occurs with an incidence of 1 in 500 and is the leading cause of sudden cardiac death in young athletes. Dilated cardiomyopathy (DCM) affects 1 in 2500 individuals and is the third leading cause of heart failure in the USA after coronary artery disease and hypertension. The remaining subtypes are more rare forms of heart disease and include restrictive cardiomyopathy (RCM), arrhythmogenic right ventricular cardiomyopathy (ARVC), left ventricular non-compaction (LVNC), and inflammatory cardiomyopathy (IC) [12, 16, 45, 75].
The causes of cardiomyopathy are variable and complex, often driven by environmental factors (e.g., industrialism, pollution), lifestyle choices (e.g., alcohol or drug abuse, obesity), other diseases (e.g., viral or bacterial infections, diabetes, thyroid, or autoimmune disease), and idiosyncratic factors (e.g., age, gender, and race), as well as genetic etiologies (e.g., inherited mutations and/or genetic predisposition). Over the last three decades, particular emphasis has been given on the familial transmission of mutations in genes encoding sarcomeric, Ca2+ sensitive and Ca2+ cycling, signaling, storage, and metabolic proteins highlighting the importance of genetics in heart disease [13, 18, 37, 48, 68]. As such, remarkable progress has been made towards the identification of genetically based cardiomyopathies that had been previously classified as idiopathic.
The discovery of hundreds of pathogenic mutations in genes encoding sarcomeric proteins has established “sarcomeric cardiomyopathies” as an entity. Depending on the severity of the mutation, sarcomeric cardiomyopathies can develop either early in life or later in adulthood. Early onset cases represent a common cause of pediatric cardiomyopathy often associated with sudden cardiac death, whereas adult onset cases develop between 20 and 50 years of age [71]. The clinical presentation of affected patients is highly diverse, exhibiting hypertrophic (the most commonly observed pathology), dilated, non-compaction, or restrictive cardiac phenotypes [13, 69, 71].
OBSCN, encoding obscurins, has been recently added to the long list of affected sarcomeric genes due to the identification of missense, frameshift, and splicing mutations associated with different forms of cardiomyopathy [46, 74]. Obscurins comprise a multifaceted family of modular proteins that range in size between 50 and 870 kDa [3]. Although the molecular architecture of the giant isoforms has been well characterized, the structural composition of the intermediate and small obscurins is still unclear [3]. Nevertheless, giant obscurins, referred to as obscurin-A (~ 720 kDa) and obscurin-B (~ 870 kDa), consist of a series of immunoglobulin (Ig) and fibronectin-III (Fn-III) domains followed by a tandem array of signaling motifs, including two Ser/Thr kinase domains, referred to as Kin1 and Kin2, that play key structural and regulatory roles (Fig. 1; for a detailed description of the structure, localization, and proposed roles of obscurins, we refer the interested reader to the following recent reviews: [39, 43, 73]).
Fig. 1.
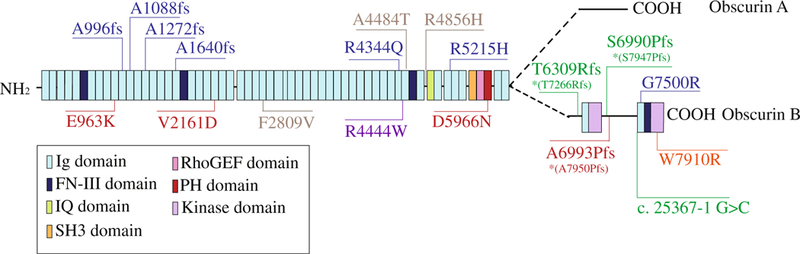
Domain schematic of giant obscurins and localization of known OBSCN mutations linked to myopathy (based on accession numbers NP- 001092093 or NP-001258152 when mutations are marked with an asterisk, *). To date, there are seven known mutations associated with HCM (shown in blue), four mutations associated with DCM (shown in red), three mutations associated with LVNC (shown in green), one mutation associated with a case of systolic heart failure with hereditary ataxia (shown in orange), and one mutation linked to distal myopathy (shown in purple). Three polymorphisms that have been classified as non-pathogenic are shown in gray
Herein, we present a synopsis of the currently known mutations identified in the OBSCN gene linked to heart disease, and how we envision the involvement of mutant or truncated obscurins in the development of cardiomyopathy given their roles as structural and signaling mediators in the sarcomeric cytoskeleton.
Obscurins and cardiomyopathy
Despite the major technological advancements in highthroughput DNA sequencing methodologies, the involvement of OBSCN in the development of sarcomeric cardiomyopathies has only begun to be interrogated. There are likely several reasons for this prior oversight in including OBSCN in large-scale genetic screens for familial heart disease. First, the relatively recent discovery of OBSCN in 2001 [78], especially compared to other heavily mutated sarcomeric genes such as MYBPC3, TTN, and MYH7 that have been known and investigated for several decades, has obviously contributed to OBSCN being understudied [73]. Second, its large size (> 170 kb) and molecular complexity arising from the presence of at least two promoters, three possible translation initiation sites, and multiple transcript variants [3, 64] may have also deterred cardiovascular geneticists from examining whether mutations in OBSCN are causatively linked to familial cardiomyopathy. Third, although many reports over the past 15 years have demonstrated that obscurins may act as structural and signaling mediators in muscle cells [1, 2, 6, 11, 17, 28, 34–36, 38, 54, 56, 58, 59, 78], their exact roles in muscle pathophysiology are still unclear. Neglecting to include OBSCN in the genetic evaluation of heart failure patients was slowly reversed when obscurin transcript levels were found increased in a mouse HCM model [9] and a canine tachycardia-induced DCM model [76], and isoform switching was reported in the left ventricles of severely diseased DCM patients [41]. Following these early observations, a number of studies have identified the presence of mutations in OBSCN that are associated with different forms of cardiomyopathy.
HCM-linked OBSCN mutations
HCM is characterized by left ventricular hypertrophy, reduced chamber size, fibrosis, impaired relaxation, and increased susceptibility to arrhythmia [13, 18]. In the absence of pathologies such as hypertension or aortic stenosis that may underlie or contribute to disease development, HCM is considered a monogenic disorder caused by dominant mutations in genes encoding sarcomeric proteins [69, 71]. It is therefore regarded as the most common cardiac manifestation of sarcomeric cardiomyopathy.
The first indication that obscurins may be causatively involved in the development of familial sarcomeric cardiomyopathy came in 2007 with the discovery of two OBSCN mutations in a 19-year-old patient with HCM from Japan [5]. Genomic linkage analysis identified two heterozygous missense mutations, R4344Q (c.13031 G>A in exon 51) and A4484T (c.13450 G>A in exon 52) in obscurin Ig58 and Ig59 domains, respectively, that were most likely inherited from the mother of the proband who was also diagnosed with HCM (Fig. 1 and Table 1) [5]. Neither of these mutations was present in a cohort of 288 normal subjects of Japanese origin [5].
Table 1.
OBSCN mutations linked to myopathy
| OBSCN (NP-001092093) | OBSCN (NP-001258152) | |||||
|---|---|---|---|---|---|---|
| Mutation | Domain | Mutation | Domain | Disease | Genotype | Reference |
| Missense mutations | ||||||
| E963K | Ig10 | E1055K | Ig11 | DCM | Het | Marston et al. 2015 |
| V2161D | Ig23 | V2536D | Ig27 | DCM | Compound Het | Marston et al. 2015 |
| F2809V | Ig29 | F3238 V | Ig34 | DCM | Compound Het | Marston et al. 2015 |
| R4344Q | Ig47 | R5304Q | Ig58 | HCM | Compound Het | Arimura et al. 2007 |
| R4444W | Ig48 | R5401W | Ig59 | Distal muscular dystrophy | Compound Het | Rossi et al. 2017 |
| A4484T | Ig48 | A5441T | Ig59 | HCM | Compound Het | Arimura et al. 2007 |
| R4856H | Between Ig50 and IQ | R5813H | Between Ig61 and Ig62 | DCM | Het | Marston et al. 2015 |
| R5215H | Ig52 | R6172H | Ig63 | HCM | Het | Xu et al. 2015 |
| G7500R | Ig58 | G8457R | Ig69 | HCM | Het | Xu et al. 2015 |
| D5966N | PH | D6923N | PH | DCM | Het | Marston et al. 2015 |
| W7910R | Kin2 | W8865R | Kin2 | Systolic HF/ hereditary ataxia | Het | Catalano et al. 2018 |
| Frameshift mutations | ||||||
| A996fs | Ig11 | A1088fs | Ig12 | HCM | Het | Xu et al. 2015 |
| A1088fs | Ig12 | A1180fs | Ig13 | HCM | Het | Xu et al. 2015 |
| A1272fs | Ig14 | A1364fs | Ig15 | HCM | Het | Xu et al. 2015 |
| A1640fs | Ig18 | A2015fs | Ig22 | HCM | Het | Xu et al. 2015 |
| T6309Rfs*53 | Between Ig56 and Ig57 | T7266Rfs*53 | Between Ig67 and Ig68 | LVNC | Het | Rowland et al. 2015 |
| S6990Pfs*82 | Between Kin1 and Ig58 | S7947Pfs*82 | Between Kin1 and Ig69 | LVNC | Het | Rowland et al. 2015 |
| A6993Pfs*79 | Between Kin1 and Ig58 | A7950Pfs*79 | Between Kin1 and Ig69 | DCM | Het | Rowland et al. 2015 |
| Splicing mutations | ||||||
| c. 25367–1 G>C | Ig58 | c. 25367–1 G>C | Ig58 | LVNC | Het | Rowland et al. 2015 |
Ig immunoglobulin, IQ isoleucine/glutamine, PH pleckstrin homology, Kin1 obscurin kinase 1, Kin2 obscurin kinase 2, HCM hypertrophic cardiomyopathy, DCM dilated cardiomyopathy, HF heart failure, LVNC left ventricular non-compaction
The Ig58/Ig59 module of obscurin, where the R4344Q and A4484T mutations reside, has been shown to support binding to the Ig9/Ig10 domains of titin [78]. Titin is a modular 3–4- MDa protein that extends longitudinally across a half-sarcomere, and functions as a molecular blueprint, regulator of passive stiffness, and mechanosensor [19, 21–23, 40]. In vitro studies indicated that the presence of R4344Q, but not A4484T, modestly diminishes binding of the obscurin Ig58/59 module to the titin Ig9/10 domains [5]. Furthermore, immunofluorescence experiments and modeling analyses indicated that R4344Q also affects the Z-disk incorporation of mutant Ig58/Ig59 and interferes with the structure of Ig58 [5]. Due to the lack of a functional effect observed from the presence of A4484T alone, Arimura and colleagues suggested that the A4484T variation might not be pathogenic. This is consistent with its presence in the genome of wild-type C57Bl6/J mice, further lending credence to the classification of the A4484T variation as a polymorphism rather than a driving mutation [27].
Further evaluation of the frequency of the R4344Q variant in larger and more diverse databases resulted in its reclassification from “hypertrophic cardiomyopathy-causing” to “hypertrophic cardiomyopathy-causing” and later to “benign” due to its high prevalence (~ 15%) among African Americans (Human Gene Mutation Database 2017.4) [42]. Notably though, its prevalence remained relatively low among white Americans (~ 0.3%). Although minor allele frequency is a key determinant of classifying a variant as pathogenic or non-pathogenic, one could argue that given that the African American population exhibits considerably higher rates of heart disease compared to white Americans (44.4 versus 36.6% for males and 48.9 versus 32.4% for females, respectively) [20], the R4344Q variant may contribute or predispose carriers to cardiomyopathy along with other influences, such as idiosyncratic factors, lifestyle choices, and socio-economical factors.
Consistent with this notion, homozygous knock-in mice carrying the R4344Q mutation develop arrhythmia by 1 year of age under sedentary conditions, consisting of significant tachycardia and frequent episodes of premature ventricular contractions [27]. Isolated cardiomyocytes exhibit enhanced Ca2+ transients and accelerated contractility kinetics due to increased expression and activity of SERCA2. Detailed structural and biochemical work demonstrated that enhanced SERCA2 activity might result from sequestration of phospholamban, the major SERCA2 regulator in cardiac cells, via enhanced binding to mutant obscurin [27]. Of note, the expression and localization of titin were indistinguishable between wild-type and homozygous knock-in mice. Moreover, young adult homozygous knock-in mice subjected to sustained pathological mechanical stress via transaortic constriction develop a DCM-like phenotype characterized by left ventricular dilation and fibrosis [27]. Taken together, these findings indicate that the presence of the R4344Q mutation leads to pathogenicity and disease development by interfering with binding interactions of the Ig58 domain that contribute to the regulation of Ca2+ cycling.
Almost a decade after the identification of the R4344Q and A4484T variants, Xu and colleagues reported the presence of six novel, rare, pathogenic OBSCN variants linked to HCM [77]. These variants, identified by whole exome sequencing (WES) analysis in a cohort of 74 Chinese patients diagnosed with sporadic HCM, were absent from 2000 control subjects [77]. Remarkably, OBSCN was identified in the top 10 putative HCM-associated genes out of 92 candidate genes interrogated [77]. All six variants were present in a dominant fashion and included four frameshift and two missense mutations (Fig. 1 and Table 1). Notably, the four frameshift mutations, Ala996fs, Ala1088fs, Ala1272fs, Ala1640fs, cluster in the NH2-terminal Ig12, Ig13, Ig15, and Ig22 domains that are shared by both obscurin-A and obscurin-B. These mutations likely result in truncated proteins and thus “loss of function.” The two missense mutations that were identified, R5215H and G7500R, localize to the Ig63 and Ig69 domains, respectively. The Ig63 domain is present in both giant isoforms, whereas the Ig69 domain precedes the most COOH-terminal Ser/Thr kinase domain and is restricted to obscurin-B and the smaller obscurin-kinase isoforms. At this time, the molecular, biochemical, and functional implications of these mutations are largely unknown, which appears to be the case for almost all of the OBSCN mutations known to date (with the exception of R4344Q discussed above).
DCM-linked OBSCN mutations
Mutations in OBSCN have also been associated with the development of DCM. Left ventricular dilation accompanied by reduced wall thickness and systolic dysfunction is the hallmark of DCM [13, 18]. Approximately 40% of all DCM cases have been attributed to monogenic causes, involving ~ 50 genes mainly encoding sarcomeric proteins [26, 44]. Notably, truncating mutations in TTN account for ~ 25% of all inherited DCM cases identified to date [25, 61].
OBSCN was recently included in the list of sarcomeric genes linked to DCM when Marston and colleagues identified five novel heterozygous missense mutations, E963K, V2161D, F2809V, R4856H, and D5966N, via WES in 4 out of 30 patients diagnosed with end-stage heart failure and familial DCM (Fig. 1 and Table 1) [47]. Of these five mutations, V2161D and F2809V were found in a compound heterozygous state. Moreover, F2809Vand R4856H were classified as non-pathogenic polymorphisms due to high prevalence in reference databases and lack of conservation among species, respectively [47]. The three potentially pathogenic mutations E963K, V2161D, and D5966N are spread throughout the length of giant obscurins in domains that are shared by both giant isoforms, localizing in Ig11, Ig27, and the PH domain, respectively. Interestingly, DCM biopsies carrying the three pathogenic mutations exhibited reduced expression levels of mutant obscurins compared to DCM samples without OBSCN mutations, HCM samples, and healthy controls, suggesting that these mutations may function via haploinsufficiency [47].
In addition to the DCM associated OBSCN mutations identified by Marston et al., Rowland and colleagues also reported a dominant frameshift mutation, A7950Pfs*ter79, residing between Kin1 and Ig69, which is restricted to kinase-bearing obscurins (Fig. 1 and Table 1) [63]. No biochemical work followed the identification of this mutation, and it is therefore unknown whether it may also act through haploinsufficiency.
LVNC-linked OBSCN mutations
In addition to HCM and DCM, mutations in OBSCN have also been linked to LVNC [63]. LVNC is a relatively rare form of cardiomyopathy (0.014–1.3% occurrence in the general population). Although it can be sporadic, 40–50% of cases exhibit a familial cause [15]. LVNC is characterized by the presence of prominent left ventricular trabeculation and deep intertrabecular recesses, most frequently in the apex that are filled with blood with no evidence of communication with the epicardial coronary artery system [4, 70]. Mutations in ~ 10 sarcomeric genes, including MYH7, MYBPC3, ACTC, TNNT2, and TPM1, have been associated with the development of LVNC [15, 29, 50]. Using the TruSight One-Sequence panel, Rowland and coworkers interrogated over 4800 genes associated with clinical phenotypes in a population of 335 cardiomyopathy patients, 325 of which were diagnosed with DCM and the remaining 10 with LVNC [63]. Three novel, dominant, variants in OBSCN were identified in the 10 LVNC patients that all cluster in the unique COOH-terminus of obscurin-B (Fig. 1 and Table 1). These variants include two frameshift mutations, T7266Rfs*ter53 and S7947Pfs*ter82, and one splicing mutation, c. 25367–1 G>C, which localize between Ig67 and Ig68, between Kin1 and Ig69, and Ig69, respectively. Given the prevalence of DCM samples in the cohort, it is tempting to speculate that OBSCN mutations affecting the expression, regulation, and/or enzymatic activity of the two obscurin Ser/Thr kinase domains may be more commonly associated with the pathogenesis of LVNC rather than DCM or HCM.
Additional myopathy-linked OBSCN mutations
OBSCN mutations associated with other muscle diseases have also been reported, although the details regarding the clinical presentation of the carriers are currently missing. A novel dominant missense mutation, W7910R, was identified in Kin2 via next-generation sequencing in a patient with chronic systolic heart failure, unspecified hereditary ataxia, and a family history of heart failure (Fig. 1 and Table 1) [14]. In another study, WES and gene-based association analysis revealed nominal significant association of OBSCN variants (along with MYLK, DYNC2H1, and RNF213 variants) with the development of multifocal fibromuscular dysplasia (FMD) [30]. FMD comprises a group of non-atherosclerotic and non-inflammatory vascular diseases leading to stenosis, aneurysm, and dissection primarily of renal arteries and carotids, which manifests as hypertension, dizziness, transient ischemic attack, or stroke [49]. Lastly, a missense mutation identified in Ig59, c.13330C>T (p.R4444W) in combination with the FLNC c.5161delG frameshift mutation, may underlie the development and/or increase the penetrance of distal muscular dystrophy (Fig. 1 and Table 1) [62]. Contrary to the A4484T polymorphism that also resides in Ig59, the R4444W mutation significantly reduces binding of the obscurin Ig58/Ig59 module to titin Ig9/Ig10 region by ~ 15-fold [62].
Future directions: what we know and what we need to learn
The roles of obscurins in healthy muscle have been heavily investigated since their discovery in 2001, though their involvement in heart disease has only begun to be interrogated. Through their diverse binding partners and different subcellular locations, obscurins have been implicated in several cellular processes [33, 60, 73]. In particular, it has been postulated that obscurins serve as structural linkers between internal membranes and the sarcomeric cytoskeleton as well as the sarcolemma and superficial myofibrils, maintain the subsarcolemmal microtubule network, contribute to the targeting of dystrophin to costameres, play key roles in the assembly and stabilization of sarcomeric M- and A-bands and the SR membranes, participate in the regulation of Ca2+ homeostasis, mediate RhoA and PI3K signaling cascades, and are involved in cell adhesion and mechanotransduction pathways [2, 6, 8, 10, 17, 28, 34, 36, 38, 52, 53, 55, 58, 59, 65]. Given the many processes that obscurins are implicated within a healthy cell, it is reasonable to speculate that mutations in OBSCN may lead to structural and/or signaling alterations that result in disease development.
To date, a total of 10 missense, 1 splicing, and 7 frameshift mutations have been identified in OBSCN that are linked to various forms of cardiomyopathy (Fig. 1 and Table 1). Similar to the majority of sarcomeric mutations, mutations in OBSCN are inherited in an autosomal dominant fashion. Mutations that result in frameshift and premature chain terminations are more obviously pathogenic, as they result in truncated proteins and thus “loss of function” or “dominant negative” effects. On the other hand, it is less clear how missense mutations lead to disease development. Similar to missense mutations in TTN, encoding titin, and NEB, encoding nebulin, it is simultaneously astonishing and puzzling how a single amino acid substitution in a protein that contains from ~ 8000 (obscurin and nebulin) up to ~ 34,000 (titin) residues leads to pathogenicity.
There are several possible explanations for the detrimental effects of missense mutations, including alterations in the targeting and incorporation of the mutant protein into sarcomeres, altered stability at the mRNA or protein level, or changes in the binding interactions of the specific domain containing the mutation. In agreement with these hypotheses, Marston and colleagues showed that although the targeting of mutant missense obscurins at sarcomeric M-bands and Z-disks was unaffected, their expression levels were significantly diminished in cardiac biopsies from DCM patients, suggesting haploinsufficiency as a possible pathogenic mechanism [47]. Although it is currently unknown how obscurin transcripts and proteins are targeted for degradation in normal cardiac cells, work from breast cancer epithelial cells, in which obscurins are heavily mutated and expressed in low levels, demonstrated that they undergo lysosomal degradation [51]. Conversely, missense mutations may stabilize mutant obscurins either at the mRNA or protein level resulting in allelic imbalance and therefore affecting disease progression and severity.
The presence of pathogenic substitutions could also affect the folding and/or surface charge distribution of the specific domain that carries the mutation. This could render strong binding interactions into weak and unfavorable, or transient and dynamic binding interactions into stable ones. In support of this mechanism, recent work utilizing a knock-in mouse model carrying the HCM-linked R4344Q substitution demonstrated that the presence of R4344Q alters the topography of the electrostatic charges on the surface of Ig58 leading to enhanced binding between mutant Ig58 and PLN [27]. The sequestration of PLN by mutant obscurin Ig58 leads to deregulation of Ca2+ cycling via enhanced SERCA activity and thus the development of arrhythmia [27].
Moreover, an area that has remained unexplored concerns the potential correlation of disease development with direct or indirect alterations in post-translational modifications of obscurin. As such, obscurins appear to be regulated through phosphorylation [32, 78], and their phosphorylation status changes drastically in response to endurance exercise [24] or following electrically evoked maximal intensity contractions [53]. Future work should investigate the effect of mutations on the post-translational modification profile of obscurins as a possible mechanism of pathogenicity, in addition to those discussed above.
Although no hot spots have been identified along the length of obscurins, it is notable that 6 out of the 18 currently known mutations associated with cardiomyopa-thy are restricted to obscurin-B, suggesting disease mechanisms that are driven by alterations in kinase-mediated cascades. Earlier work has shown that both kinases are enzymatically active and undergo autophosphorylation, which might regulate their activity [28]. While Kin1 binds and phosphorylates N-cadherin, Kin2 binds to Na/K ATPase subunit 1; at this time, it is unknown whether Na/K ATPase subunit 1 is also a substrate of Kin2. Given the presence of kinase-bearing obscurins at the intercalated disc [28], alterations in the regulation and/or enzymatic activity of Kin1 and Kin2 could perhaps affect the mechanical and electrical coupling of neighboring cardiomyocytes resulting in cardiac remodeling, contractile impairment, and the development of arrhythmias ultimately leading to cardiomyopathy and heart failure.
As a testament to the diversity of human diseases causatively linked to OBSCN mutations, OBSCN single nucleotide polymorphisms (SNP) may underlie the pathogenesis of aspirin exacerbated respiratory disease (AERD) by contributing to aspirin sensitivity in asthmatics [31]. Kim and colleagues suggested that altered sarcoplasmic reticulum (SR) structure in airway smooth muscle cells bearing variant OBSCN alleles may result in defective Ca2+ signaling and bronchoconstriction upon aspirin ingestion. Moreover, a number of solid tumors have been shown to possess mutant obscurins [7, 57, 67]. The first indication that mutant obscurins may be associated with tumor formation and progression came with the observation that the OBSCN gene is bisected within the first intron by a chromosomal translocation associated with the childhood kidney disease, Wilms’ tumor [72]. Recent data further suggests that OBSCN mutations may contribute to the formation of several solid cancers. During large-scale sequencing efforts aiming to identify commonly mutated genes in breast and colorectal cancers, OBSCN was identified as one of only two genes (the other one being TP53) which, when mutated, could drive the formation of both tumor types [67]. In a follow-up study, OBSCN mutations were also detected in glioblastoma and melanoma [7]. This accumulated evidence led our group to investigate the role of obscurins in cancer. We found that the expression of giant obscurins is dramatically reduced in breast, skin, and colon cancer cells relative to their nonmalignant counterparts [51], resulting in increased tumorigenicity and metastasis via deregulation of the RhoA and PI3K signaling cascades [52, 65]. Thus, similar to the haploinsufficiency observed in DCM samples by Marston and colleagues [47], epithelial cancer cell lines and advanced stage human biopsies containing mutant obscurins express considerably low levels, and this phenotype correlates with disease progression [51, 66].
Though there are increasing reports of OBSCN mutations linked to disease, a major caveat of the current research is the lack of evidence for co-segregation with disease development among blood-related family members. This is obviously due to the types of analyses that have been performed, having mainly used biobank samples obtained during transplantation surgery following end-stage heart failure. Moreover, cardiac biopsies are often unavailable, and thus, the genetic characterization originates from analysis of blood samples. Inclusion of OBSCN as a potential pathological gene in large-scale genetic screenings could potential remedy the fact that it was previously somewhat neglected.
Furthermore, with the exception of the HCM-linked R4344Q mutation, which our lab has recently started to characterize after generating the respective knock-in mouse model (please see above) [27], there are no preclinical animal models at this time carrying known OBSCN mutations. Generating animal models that express the identified OBSCN mutations, or lack specific domains that are potential hot spots, will be crucial for understanding the pathogenic mechanisms underlying OBSCN mutations linked to cardiomyopathy.
There is still much to learn about the potential involvement of obscurins in the development of heart disease. It is currently unknown whether OBSCN variants may be monogenic causes of cardiomyopathy or whether they contribute to disease development via compound heterozygosity with variations in other genes [46]. It is therefore our anticipation and prediction that more research will focus on OBSCN and its causative association with disease development in the near future. Inclusion of OBSCN in the list of clinically pathogenic genes, co-segregation analysis, biochemical and biophysical studies, the generation of preclinical animal models, and the use of human biopsies (Table 2) are therefore essential in order to interrogate the functional consequences of known OBSCN mutations and further identify novel OBSCN mutations and their potentially causative association to heart disease.
Table 2.
Future directions for deciphering the involvement of OBSCN in cardiomyopathy
| Future directions |
|---|
| • Inclusion of OBSCN in large-scale genetic screens to identify novel disease-causing variants |
| • Co-segregation analyses of OBSCN variants in blood-related family members |
| • Biochemical, biophysical, and functional studies of known OBSCN variants |
| • Investigation of post-translational modification status as a potential disease mechanism |
| • Generation of preclinical animal models carrying known OBSCN mutations |
Acknowledgments
Funding information This work was supported by grants from the American Heart Association (16GRNT31290010 to AKK), Muscular Dystrophy Association (313579 to AKK), and NIH/MIAMS (T32AR7592 to AG).
Footnotes
Compliance with ethical standards
Conflict of interest The authors declare that they have no conflict of interest.
This article is part of the special issue on Sarcomeric Mutations in Pflügers Archiv – European Journal of Physiology
References
- 1.Ackermann MA, Hu LY, Bowman AL, Bloch RJ, Kontrogianni-Konstantopoulos A (2009) Obscurin interacts with a novel isoform of MyBP-C slow at the periphery of the sarcomeric M-band and regulates thick filament assembly. Mol Biol Cell 20:2963–2978. 10.1091/mbc.E08-12-1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ackermann MA, King B, Lieberman NAP, Bobbili PJ, Rudloff M, Berndsen CE, Wright NT, Hecker PA, Kontrogianni-Konstantopoulos A (2017) Novel obscurins mediate cardiomyocyte adhesion and size via the PI3K/AKT/mTOR signaling pathway. J Mol Cell Cardiol 111:27–39. 10.1016/j.yjmcc.2017.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ackermann MA, Shriver M, Perry NA, Hu LY, Kontrogianni-Konstantopoulos A (2014) Obscurins: Goliaths and Davids take over non-muscle tissues. PLoS One 9:e88162 10.1371/journal.pone.0088162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arbustini E, Favalli V, Narula N, Serio A, Grasso M (2016) Left ventricular noncompaction: a distinct genetic cardiomyopathy? J Am Coll Cardiol 68:949–966. 10.1016/j.jacc.2016.05.096 [DOI] [PubMed] [Google Scholar]
- 5.Arimura T, Matsumoto Y, Okazaki O, Hayashi T, Takahashi M, Inagaki N, Hinohara K, Ashizawa N, Yano K, Kimura A (2007) Structural analysis of obscurin gene in hypertrophic cardiomyopathy. Biochem Biophys Res Commun 362:281–287. 10.1016/j.bbrc.2007.07.183 [DOI] [PubMed] [Google Scholar]
- 6.Bagnato P, Barone V, Giacomello E, Rossi D, Sorrentino V (2003) Binding of an ankyrin-1 isoform to obscurin suggests a molecular link between the sarcoplasmic reticulum and myofibrils in striated muscles. J Cell Biol 160:245–253. 10.1083/jcb.200208109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balakrishnan A, Bleeker FE, Lamba S, Rodolfo M, Daniotti M, Scarpa A, van Tilborg AA, Leenstra S, Zanon C, Bardelli A (2007) Novel somatic and germline mutations in cancer candidate genes in glioblastoma, melanoma, and pancreatic carcinoma. Cancer Res 67:3545–3550. 10.1158/0008-5472.CAN-07-0065 [DOI] [PubMed] [Google Scholar]
- 8.Borisov AB, Kontrogianni-Konstantopoulos A, Bloch RJ, Westfall MV, Russell MW (2004) Dynamics of obscurin localization during differentiation and remodeling of cardiac myocytes: obscurin as an integrator of myofibrillar structure. J Histochem Cytochem 52: 1117–1127. 10.1369/jhc.3A6183.2004 [DOI] [PubMed] [Google Scholar]
- 9.Borisov AB, Raeker MO, Kontrogianni-Konstantopoulos A, Yang K, Kurnit DM, Bloch RJ, Russell MW (2003) Rapid response of cardiac obscurin gene cluster to aortic stenosis: differential activation of rho-GEF and MLCK and involvement in hypertrophic growth. Biochem Biophys Res Commun 310:910–918 [DOI] [PubMed] [Google Scholar]
- 10.Borisov AB, Sutter SB, Kontrogianni-Konstantopoulos A, Bloch RJ, Westfall MV, Russell MW (2006) Essential role of obscurin in cardiac myofibrillogenesis and hypertrophic response: evidence from small interfering RNA-mediated gene silencing. Histochem Cell Biol 125:227–238. 10.1007/s00418-005-0069-x [DOI] [PubMed] [Google Scholar]
- 11.Bowman A, Catino D, Strong J, Randall W, Kontrogianni-Konstantopoulos A, Bloch R (2008) The rho-guanine nucleotide exchange factor domain of obscurin regulates assembly of titin at the Z-disk through interactions with Ran binding protein 9. Mol Biol Cell 19:3782–3792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Braunwald E (2017) Cardiomyopathies: an overview. Circ Res 121: 711–721. 10.1161/CIRCRESAHA.117.311812 [DOI] [PubMed] [Google Scholar]
- 13.Burke MA, Cook SA, Seidman JG, Seidman CE (2016) Clinical and mechanistic insights into the genetics of cardiomyopathy. J Am Coll Cardiol 68:2871–2886. 10.1016/j.jacc.2016.08.079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Catalano J, Paynton B, Kaniper S, Gerhard G, Alvarez R (2018) Identification of a novel obscurin protein variant in nonischemic cardiomyopathy. J Am Coll Cardiol 71:A743 [Google Scholar]
- 15.Connolly H, Attenhofer-Jost C Isolated left ventricular noncompaction UpToDate. https://www.uptodate.com/contents/isolated-left-ventricular-noncompaction. Accessed May 2018
- 16.Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, Kühl U, Maisch B, McKenna WJ, Monserrat L, Pankuweit S, Rapezzi C, Seferovic P, Tavazzi L, Keren A (2008) Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 29:270–276. 10.1093/eurheartj/ehm342 [DOI] [PubMed] [Google Scholar]
- 17.Ford-Speelman DL, Roche JA, Bowman AL, Bloch RJ (2009) The rho-guanine nucleotide exchange factor domain of obscurin activates rhoA signaling in skeletal muscle. Mol Biol Cell 20:3905–3917. 10.1091/mbc.E08-10-1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garfinkel AC, Seidman JG, Seidman CE (2018) Genetic pathogenesis of hypertrophic and dilated cardiomyopathy. Heart Fail Clin 14: 139–146. 10.1016/j.hfc.2017.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gautel M (2011) Cytoskeletal protein kinases: titin and its relations in mechanosensing. Pflugers Arch 462:119–134. 10.1007/s00424-011-0946-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB, Subcommittee AHASCaSS (2013) Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation 127:e6–e245. 10.1161/CIR.0b013e31828124ad [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Granzier H, Labeit S (2002) Cardiac titin: an adjustable multifunctional spring. J Physiol 541:335–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Granzier HL, Labeit S (2004) The giant protein titin: a major player in myocardial mechanics, signaling, and disease. Circ Res 94:284–295. 10.1161/01.RES.0000117769.88862.F8 [DOI] [PubMed] [Google Scholar]
- 23.Granzier HL, Labeit S (2006) The giant muscle protein titin is an adjustable molecular spring. Exerc Sport Sci Rev 34:50–53 [DOI] [PubMed] [Google Scholar]
- 24.Guo H, Isserlin R, Emili A, Burniston JG (2017) Exercise-responsive phosphoproteins in the heart. J Mol Cell Cardiol 111: 61–68. 10.1016/j.yjmcc.2017.08.001 [DOI] [PubMed] [Google Scholar]
- 25.Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, Teodorescu DL, Cirino AL, Banner NR, Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM, Ackerman MJ, Mitchell RN, Murry CE, Lakdawala NK, Ho CY, Barton PJ, Cook SA, Mestroni L, Seidman JG, Seidman CE (2012) Truncations of titin causing dilated cardiomyopathy. N Engl J Med 366:619–628. 10.1056/NEJMoa1110186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hershberger RE, Hedges DJ, Morales A (2013) Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol 10:531–547. 10.1038/nrcardio.2013.105 [DOI] [PubMed] [Google Scholar]
- 27.Hu L-YR, Ackermann MA, Hecker PA, Prosser BL, King B, O’Connell KA, Grogan A, Meyer LC, Berndsen CE, Wright NT, Lederer WJ, Kontrogianni-Konstantopoulos A (2017) Deregulated Ca2+ cycling underlies the development of arrhythmia due to mutant obscurin. Sci Adv 3:e1603081 10.1126/sciadv.1603081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu LY, Kontrogianni-Konstantopoulos A (2013) The kinase domains of obscurin interact with intercellular adhesion proteins. Faseb J 27:2001–2012. 10.1096/fj.12-221317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ichida F, Tsubata S, Bowles KR, Haneda N, Uese K, Miyawaki T, Dreyer WJ, Messina J, Li H, Bowles NE, Towbin JA (2001) Novel gene mutations in patients with left ventricular noncompaction or Barth syndrome. Circulation 103:1256–1263 [DOI] [PubMed] [Google Scholar]
- 30.Kiando SR, Barlassina C, Cusi D, Galan P, Lathrop M, Plouin PF, Jeunemaitre X, Bouatia-Naji N (2015) Exome sequencing in seven families and gene-based association studies indicate genetic heterogeneity and suggest possible candidates for fibromuscular dysplasia. J Hypertens 33:1802–1810; discussion 1810. 10.1097/HJH.0000000000000625 [DOI] [PubMed] [Google Scholar]
- 31.Kim JH, Park BL, Pasaje CF, Kim Y, Bae JS, Park JS, Uh ST, Kim YH, Kim MK, Choi IS, Cho SH, Choi BW, Koh I, Park CS, Shin HD (2010) Contribution of the OBSCN nonsynonymous variants to aspirin exacerbated respiratory disease susceptibility in Korean population. DNA Cell Biol 31:1001–1009. 10.1089/dna.2011.1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kirk JA, Holewinski RJ, Kooij V, Agnetti G, Tunin RS, Witayavanitkul N, de Tombe PP, Gao WD, Van Eyk J, Kass DA (2014) Cardiac resynchronization sensitizes the sarcomere to calcium by reactivating GSK-3β. J Clin Invest 124:129–138. 10.1172/JCI69253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kontrogianni-Konstantopoulos A, Ackermann MA, Bowman AL, Yap SV, Bloch RJ (2009) Muscle giants: molecular scaffolds in sarcomerogenesis. Physiol Rev 89:1217–1267. 10.1152/physrev.00017.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kontrogianni-Konstantopoulos A, Catino DH, Strong JC, Randall WR, Bloch RJ (2004) Obscurin regulates the organization of myosin into A bands. American journal of physiology Cell physiology 287:C209–C217. 10.1152/ajpcell.00497.2003 [DOI] [PubMed] [Google Scholar]
- 35.Kontrogianni-Konstantopoulos A, Catino DH, Strong JC, Sutter S, Borisov AB, Pumplin DW, Russell MW, Bloch RJ (2006) Obscurin modulates the assembly and organization of sarcomeres and the sarcoplasmic reticulum. FASEB J 20:2102–2111. 10.1096/fj.06-5761com [DOI] [PubMed] [Google Scholar]
- 36.Kontrogianni-Konstantopoulos A, Jones EM, Van Rossum DB, Bloch RJ (2003) Obscurin is a ligand for small ankyrin 1 in skeletal muscle. Mol Biol Cell 14:1138–1148. 10.1091/mbc.E02-07-0411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Landstrom AP, Ackerman MJ (2012) Beyond the cardiac myofilament: hypertrophic cardiomyopathy-associated mutations in genes that encode calcium-handling proteins. Curr Mol Med 12:507–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lange S, Ouyang K, Meyer G, Cui L, Cheng H, Lieber RL, Chen J (2009) Obscurin determines the architecture of the longitudinal sarcoplasmic reticulum. J Cell Sci 122:2640–2650. 10.1242/jcs.046193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin BL, Song T, Sadayappan S (2017) Myofilaments: movers and rulers of the sarcomere. Compr Physiol 7:675–692. 10.1002/cphy.c160026 [DOI] [PubMed] [Google Scholar]
- 40.Linke W (2017) Titin gene and protein functions in passive and active muscle. Annu Rev Physiol 80:389–411. 10.1146/annurev-physiol-021317-121234 [DOI] [PubMed] [Google Scholar]
- 41.Makarenko I, Opitz CA, Leake MC, Neagoe C, Kulke M, Gwathmey JK, del Monte F, Hajjar RJ, Linke WA (2004) Passive stiffness changes caused by upregulation of compliant titin isoforms in human dilated cardiomyopathy hearts. Circ Res 95:708–716. 10.1161/01.RES.0000143901.37063.2f [DOI] [PubMed] [Google Scholar]
- 42.Manrai AK, Funke BH, Rehm HL, Olesen MS, Maron BA, Szolovits P, Margulies DM, Loscalzo J, Kohane IS (2016) Genetic misdiagnoses and the potential for health disparities. N Engl J Med 375:655–665. 10.1056/NEJMsa1507092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Manring HR, Dorn LE, Ex-Willey A, Accornero F, Ackermann MA (2018) At the heart of inter- and intracellular signaling: the intercalated disc. Biophys Rev 10:961–971. 10.1007/s12551-018-0430-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marian AJ, van Rooij E, Roberts R (2016) Genetics and genomics of single-gene cardiovascular diseases: common hereditary cardiomyopathies as prototypes of single-gene disorders. J Am Coll Cardiol 68:2831–2849. 10.1016/j.jacc.2016.09.968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB, Association AH, Council on Clinical Cardiology HaFaTC, Groups QoCaORaFGaTBIW, Prevention CoEa (2006) Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 113:1807–1816. 10.1161/CIRCULATIONAHA.106.174287 [DOI] [PubMed] [Google Scholar]
- 46.Marston S (2017) Obscurin variants and inherited cardiomyopathies. Biophys Rev 9:239–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marston S, Montgiraud C, Munster AB, Copeland O, Choi O, Dos Remedios C, Messer AE, Ehler E, Knöll R (2015) OBSCN mutations a ssoc iated w ith dilated cardiomyopathy a nd haploinsufficiency. PLoS One 10:e0138568 10.1371/journal.pone.0138568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Masarone D, Kaski JP, Pacileo G, Elliott PM, Bossone E, Day SM, Limongelli G (2018) Epidemiology and clinical aspects of genetic cardiomyopathies. Heart Fail Clin 14:119–128. 10.1016/j.hfc.2017.12.007 [DOI] [PubMed] [Google Scholar]
- 49.Olin JW, Froehlich J, Gu X, Bacharach JM, Eagle K, Gray BH, Jaff MR, Kim ES, Mace P, Matsumoto AH, McBane RD, Kline-Rogers E, White CJ, Gornik HL (2012) The United States registry for fibromuscular dysplasia: results in the first 447 patients. Circulation 125:3182–3190. 10.1161/CIRCULATIONAHA.112.091223 [DOI] [PubMed] [Google Scholar]
- 50.Online Mendelian Inheritance in Man, OMIM. McKusick-Nathans Institute of Genetic Medicine Johns Hopkins University, Baltimore [Google Scholar]
- 51.Perry NA, Shriver M, Mameza MG, Grabias B, Balzer E, Kontrogianni-Konstantopoulos A (2012) Loss of giant obscurins promotes breast epithelial cell survival through apoptotic resistance. FASEB J 26:2764–2775. 10.1096/fj.12-205419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Perry NA, Vitolo MI, Martin SS, Kontrogianni-Konstantopoulos A (2014) Loss of the obscurin-RhoGEF downregulates RhoA signaling and increases microtentacle formation and attachment of breast epithelial cells. Oncotarget 5:8558–8568. 10.18632/oncotarget.2338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Potts GK, McNally RM, Blanco R, You JS, Hebert AS, Westphall MS, Coon JJ, Hornberger TA (2017) A map of the phosphoproteomic alterations that occur after a bout of maximal-intensity contractions. J Physiol 595:5209–5226. 10.1113/JP273904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Raeker M, Russell MW (2011) Obscurin depletion impairs organization of skeletal muscle in developing zebrafish embryos. J Biomed Biotechnol 2011:479135–479115. 10.1155/2011/479135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Raeker MO, Bieniek AN, Ryan AS, Tsai HJ, Zahn KM, Russell MW (2010) Targeted deletion of the zebrafish obscurin a RhoGEF domain affects heart, skeletal muscle and brain development. Dev Biol 337:432–443. 10.1016/j.ydbio.2009.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Raeker MO, Su F, Geisler SB, Borisov AB, Kontrogianni-Konstantopoulos A, Lyons SE, Russell MW (2006) Obscurin is required for the lateral alignment of striated myofibrils in zebrafish. Dev Dyn 235:2018–2029. 10.1002/dvdy.20812 [DOI] [PubMed] [Google Scholar]
- 57.Rajendran BK, Deng CX (2017) Characterization of potential driver mutations involved in human breast cancer by computational approaches. Oncotarget 8:50252–50272. 10.18632/oncotarget.17225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Randazzo D, Blaauw B, Paolini C, Pierantozzi E, Spinozzi S, Lange S, Chen J, Protasi F, Reggiani C, Sorrentino V (2017) Exercise-induced alterations and loss of sarcomeric M-line organization in the diaphragm muscle of obscurin knockout mice. Am J Physiol Cell Physiol 312:C16–C28. 10.1152/ajpcell.00098.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Randazzo D, Giacomello E, Lorenzini S, Rossi D, Pierantozzi E, Blaauw B, Reggiani C, Lange S, Peter AK, Chen J, Sorrentino V (2013) Obscurin is required for ankyrinB-dependent dystrophin localization and sarcolemma integrity. J Cell Biol 200:523–536. 10.1083/jcb.201205118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Randazzo D, Pierantozzi E, Rossi D, Sorrentino V (2017) The potential of obscurin as a therapeutic target in muscle disorders. Expert Opin Ther Targets 21:897–910 [DOI] [PubMed] [Google Scholar]
- 61.Roberts AM, Ware JS, Herman DS, Schafer S, Baksi J, Bick AG, Buchan RJ, Walsh R, John S, Wilkinson S, Mazzarotto F, Felkin LE, Gong S, MacArthur JA, Cunningham F, Flannick J, Gabriel SB, Altshuler DM, Macdonald PS, Heinig M, Keogh AM, Hayward CS, Banner NR, Pennell DJ, O’Regan DP, San TR, de Marvao A, Dawes TJ, Gulati A, Birks EJ, Yacoub MH, Radke M, Gotthardt M, Wilson JG, O’Donnell CJ, Prasad SK, Barton PJ, Fatkin D, Hubner N, Seidman JG, Seidman CE, Cook SA (2015) Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med 7:270ra276. 10.1126/scitranslmed.3010134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rossi D, Palmio J, Evilä A, Galli L, Barone V, Caldwell TA, Policke RA, Aldkheil E, Berndsen CE, Wright NT, Malfatti E, Brochier G, Pierantozzi E, Jordanova A, Guergueltcheva V, Romero NB, Hackman P, Eymard B, Udd B, Sorrentino V (2017) A novel FLNC frameshift and an OBSCN variant in a family with distal muscular dystrophy. PLoS One 12:e0186642 10.1371/journal.pone.0186642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rowland T, Graw S, Sweet M, Gigli M, Taylor M, Mestroni L (2016) Obscurin variants in patients with left ventricular noncompaction. J Am Coll Cardiol 68:2237–2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Russell MW, Raeker MO, Korytkowski KA, Sonneman KJ (2002) Identification, tissue expression and chromosomal localization of human Obscurin-MLCK, a member of the titin and Dbl families of myosin light chain kinases. Gene 282:237–246 [DOI] [PubMed] [Google Scholar]
- 65.Shriver M, Marimuthu S, Paul C, Geist J, Seale T, Konstantopoulos K, Kontrogianni-Konstantopoulos A (2016) Giant obscurins regulate the PI3K cascade in breast epithelial cells via direct binding to the PI3K/p85 regulatory subunit. Oncotarget 7:45414–45428. 10.18632/oncotarget.9985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shriver M, Stroka KM, Vitolo MI, Martin S, Huso DL, Konstantopoulos K, Kontrogianni-Konstantopoulos A (2015) Loss of giant obscurins from breast epithelium promotes epithelial-to-mesenchymal transition, tumorigenicity and metastasis. Oncogene 34:4248–4259. 10.1038/onc.2014.358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sjöblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, Dawson D, Willson JK, Gazdar AF, Hartigan J, Wu L, Liu C, Parmigiani G, Park BH, Bachman KE, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE (2006) The consensus coding sequences of human breast and colorectal cancers. Science 314:268–274. 10.1126/science.1133427 [DOI] [PubMed] [Google Scholar]
- 68.Tardiff JC (2011) Thin filament mutations: developing an integrative approach to a complex disorder. Circ Res 108:765–782. 10.1161/CIRCRESAHA.110.224170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tardiff JC, Carrier L, Bers DM, Poggesi C, Ferrantini C, Coppini R, Maier LS, Ashrafian H, Huke S, van der Velden J (2015) Targets for therapy in sarcomeric cardiomyopathies. Cardiovasc Res 105:457–470. 10.1093/cvr/cvv023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Towbin JA, Lorts A, Jefferies JL (2015) Left ventricular non-compaction cardiomyopathy. Lancet 386:813–825. 10.1016/S0140-6736(14)61282-4 [DOI] [PubMed] [Google Scholar]
- 71.van der Velden J, Ho CY, Tardiff JC, Olivotto I, Knollmann BC, Carrier L (2015) Research priorities in sarcomeric cardiomyopathies. Cardiovasc Res 105:449–456. 10.1093/cvr/cvv019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vernon EG, Malik K, Reynolds P, Powlesland R, Dallosso AR, Jackson S, Henthorn K, Green ED, Brown KW (2003) The parathyroid hormone-responsive B1 gene is interrupted by a t(1;7)(q42;p15) breakpoint associated with Wilms’ tumour. Oncogene 22:1371–1380. 10.1038/sj.onc.1206332 [DOI] [PubMed] [Google Scholar]
- 73.Wang L, Geist J, Grogan A, Hu LR, Kontrogianni-Konstantopoulos A (2018) Thick filament protein network, functions, and disease association. Compr Physiol 8(2):631–709. 10.1002/cphy.c170023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang L, Geist J, Grogan A, Hu LR, Kontrogianni-Konstantopoulos A (2018) Thick filament protein network, functions, and disease association. Compr Physiol 8:631–709. 10.1002/cphy.c170023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wexler RK, Elton T, Pleister A, Feldman D ( 2009) Cardiomyopathy: an overview. Am Fam Physician 79:778–784 [PMC free article] [PubMed] [Google Scholar]
- 76.Wu Y, Bell SP, Trombitas K, Witt CC, Labeit S, LeWinter MM, Granzier H (2002) Changes in titin isoform expression in pacing-induced cardiac failure give rise to increased passive muscle stiffness. Circulation 106:1384–1389 [DOI] [PubMed] [Google Scholar]
- 77.Xu J, Li Z, Ren X, Dong M, Li J, Shi X, Zhang Y, Xie W, Sun Z, Liu X, Dai Q (2015) Investigation of pathogenic genes in Chinese sporadic hypertrophic cardiomyopathy patients by whole exome sequencing. Sci Rep 5:16609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Young P, Ehler E, Gautel M (2001) Obscurin, a giant sarcomeric rho guanine nucleotide exchange factor protein involved in sarcomere assembly. J Cell Biol 154:123–136 [DOI] [PMC free article] [PubMed] [Google Scholar]