

Abstract
Research into post-transcriptional processing and modification of RNA continues to speed forward, as their ever-emerging role in the regulation of gene expression in biological systems continues to unravel. Liquid chromatography tandem mass spectrometry (LC-MS/MS) has proven for over two decades to be a powerful ally in the elucidation of RNA modification identity and location, but the technique has not proceeded without its own unique technical challenges. The throughput of LC-MS/MS modification mapping experiments continues to be impeded by tedious and time-consuming spectral interpretation, particularly during for the analysis of complex RNA samples. RNAModMapper was recently developed as a tool to improve the interpretation and annotation of LC-MS/MS data sets from samples containing post-transcriptionally modified RNAs. Here, we delve deeper into the methodology and practice of RNAModMapper to provide greater insight into its utility, and remaining hurdles, in current RNA modification mapping experiments.
Keywords: Transfer RNA, Modified nucleosides, Bioinformatics, Sequence Analysis, Modification placement
1. Introduction
1.1. Mapping of Modifications in RNA
Although interest in the enzymatic modifications of deoxynucleic acid (DNA) came to prominence first with the discovery of 5-methyldeoxycytidine (m5C) [1], the detection of the first ribonucleic acid (RNA) modification, pseudouridine (Ψ)[2], followed only a few years later. Research focused on DNA modifications has a rich history dating back to 1948, but the enzymatic and xenobiotic modifications to RNA have recently grabbed the attention of researchers as we continue to understand more about their chemical diversity, dynamic behavior, and ultimate effect on the biological system. Post-transcriptional modifications of RNA cannot only directly impact gene expression[3] and protein translation accuracy[4], but also have more subtle effects on RNA stability, structure, and function[5, 6]. Modifications have been elucidated in all types of RNA, including messenger RNA (mRNA), ribosomal RNA (rRNA), and microRNA (miRNA), but they are found to be most abundant in transfer RNA (tRNA) [7]. In tRNA, these modifications not only play a critical role in the formation of the structure necessary for codon:anticodon recognition[4], but can also be a determinant for aminoacyl synthetases[8]. The presence of modifications, or lack thereof, in RNA may also correlate to important human diseases[9].
The unambiguous assignment of modification location in RNA has proven to be a non-trivial challenge to researchers in the field. Even the analysis of tRNA, where modifications are the most abundant, can be a difficult due to the wide range of chemical diversity, number of unique sequences to assign those modifications, and interference from other co-isolated RNA species. Two main modern approaches to RNA modification mapping have emerged, which identify and locate modifications through either mass spectrometry (MS) or next-generation sequencing (NGS). The NGS technique, commonly referred to as RNA-seq, is a high throughput approach[10] capable of transcriptome wide detection of modifications [11, 12] with low sample amounts but requires modification specific strategies[13]. The MS technique, coupled to liquid chromatography with MS performed in tandem (LC-MS/MS), allows direct detection of all RNA modifications that result in an increase in the mass of the canonical nucleoside, but requires more sample and is more time-consuming. As both LC-MS/MS and RNA-seq techniques take different, complimentary approaches to RNA modification detection, no obvious choice has emerged. In fact, future studies using both techniques on similar systems may offer validation of RNA modifications identified from the other.
The most common LC-MS/MS approach to the modification mapping of RNA is through the adaptation of ribonuclease (RNase) mapping, pioneered by early work from the McCloskey[14, 15] and McLuckey laboratories[16, 17]. Once a modified nucleoside profile is obtained, typically through LC-MS/MS nucleoside analysis[18], these modifications are placed back onto their originating sequences through the generation of oligonucleotides by RNase digestion and their subsequent analysis by LC-MS/MS. While the liquid chromatographic separation reduces the number of the oligonucleotides being detected by the MS at any given point in the analysis, the first of two tandem stages of MS allows direct detection of precursor ions with mass shifts corresponding to the presence of modifications in the oligonucleotide. A second tandem, or MS/MS, event then generates a sequence of fragment (product) ions through collision-induced dissociation (CID) of the precursor ion with inert gases (typically He or Ar). Using the nomenclature developed by McLuckey[16], the product ions observed for oligonucleotides can be identified by type (c-, y-, w-, a-B), where the mass differences in a particular ion series allows for the determination of the exact oligonucleotide sequence – revealing the precise location of the modification within the oligonucleotide. While a powerful and effective approach to RNA modification mapping, the primary limiting factor for throughput is the interpretation of data. The interpretation of LC-MS/MS spectra involves not only the assignment of individual precursor m/z values to each oligonucleotide detected, but also the assignment of each (and many) product ions in the MS/MS spectra. Only then can an RNase digestion product containing a modified nucleoside be used to map the modification back onto its location in the corresponding sequence.
1.2. Moving Beyond Manual Interpretation of Spectra
The manual interpretation of MS/MS spectra - when performed by an MS expert – is the most accurate way of validating precursor and product ion spectra from theoretically determined ions when synthetic and well-characterized standards are not available[19]. However, the complexity and sheer number of spectra in a typical oligonucleotide mapping experiment presents a significant through-put hurdle. Several computational tools have been previously developed to address this issue, providing aids to help automate the spectral interpretation step of the analysis. The first was the Simple Oligonucleotide Sequencer (SOS), developed by Rozenski and McCloskey[20], which allowed ab initio oligonucleotide sequencing from MS/MS data in an interactive software environment. Another important development from Nyakas et al.[21] was OMA and OPA, which can analyze MS and MS/MS spectra of oligonucleotides, their derivatives, and adducts with metal ions or drugs. While each represented important developments, neither tool is capable of large-scale RNA modification mapping due to the lack of batch-processing of large LC-MS/MS data files.
Ariadne, developed by Nakayama et al.[22], was the first computational platform capable of determining the location of RNA modifications at scale. Using a web-based sequence database search engine, Ariadne uses MS/MS data from RNA oligonucleotides to identify particular RNAs in biological samples. It also allows the user to select the types of chemical modifications known to be present in the sample, though a limited number of organisms are present in the database. RMM, another database search program[23], is capable of whole prokaryotic genome or RNA FASTA sequence databases. More recently RoboOligo[24] was created to handle both the manual and automated de novo analysis of modified oligonucleotide MS/MS spectra specifically for modification dense tRNAs but is limited to single sequences.
Continuing to build off the efforts and milestones of these previously developed computation tools, a new stand-alone program capable of improving the throughput of LC-MS/MS data analysis was recently reported by our group[25]. RNAModmapper, or RAMM, was developed to both interpret the large number of spectra obtained from typical LC-MS/MS modification mapping experiments and map the modified (and unmodified) oligonucleotides back onto the RNA sequence(s). RAMM can perform data analysis in two independent modes, fixed and variable position mapping, depending on what is already known about the modification profile of the target sequence(s). The user can select from 120 chemical modifications/motifs included in the program or define custom (synthetic or derivatized) modifications. To improve the accuracy of the spectral interpretations, a two-component scoring function, with user-defined scoring thresholds, was implemented. The utility of RAMM for the mapping of modification in complex RNA samples has previously been shown in the cases of Escherichia coli total tRNA and Streptomyces griseus rRNA[25].
Here we delve deeper into the operation and outcomes provided by RAMM. Saccharomyces cerevisiae tRNAPhe was used as a model compound for these investigations. Two LC-MS platforms and four different acquisition modes (ion-trap CID, orbitrap CID, orbitrap-higher energy collision dissociation (HCD), and time-of-flight beam-type CID) were utilized to identify any platform or CID acquisition mode characteristics of automated sample analysis by RAMM. The impact of each acquisition mode on fixed and variable mapping outcomes was evaluated along with the performance of the scoring functions, and their impact of the number of correct and incorrect interpretations. Taken together, improved and – in some cases – optimized approaches have been identified that improve the utility of RAMM for interpreting and annotating LC-MS/MS data during RNA modification mapping analyses.
2. Materials and Methods
2.1. Materials and Reagents
RNase T1 and bacterial alkaline phosphatase (BAP) were purchased from Worthington Biochemical (Lakewood, NJ). S. cerevisiae tRNAPhe and ammonium acetate (LC-MS grade) were purchased from Sigma Aldrich. Acetic acid (LC-MS grade) and methanol (LC-MS grade) were purchased from Fisher Scientific. High purity (18 MOhm) water was generated in-house with a Barnstead Nanopure System (Thermo Scientific).
2.2. Preparation of RNA Digests
S. cerevisiae tRNAPhe was digested with RNase T1 (28 units/µg) and BAP (0.01 units/µg) in 110 mM ammonium acetate at 37°C for 2 h. Digests were then evaporated to dryness and reconstituted in the HPLC mobile phase prior to analysis.
2.3. LC-MS/MS analysis of RNA oligonucleotides
Liquid chromatographic separation of oligonucleotides was performed in HILIC mode on a 2.0 × 150mm Shodex HILICpak VN-50 column[26]. Mobile phases were prepared by pre-mixed aqueous ammonium acetate (15 mM, pH 5.5) and acetonitrile. An aqueous gradient consisting of 1.5 min hold at 30%, ramping to 56% at 30 min with 20 min re-equilibration at initial conditions was delivered at 220 µL/min. The column was thermostatted at 50°C.
Two different LC-MS/MS platforms were employed for oligonucleotide analysis. One consisted of an ultra-high performance liquid chromatography (UHPLC) system (Vanquish Flex Quaternary, Thermo Fisher Scientific, San Jose, CA) interfaced to an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific) by a heated electrospray ionization (H-ESI) source. The other consisted of an Ultimate 3000 UPLC (Thermo Scientific) interfaced to a Waters Synapt G2-S with electrospray source.
For the Orbitrap Fusion Lumos, the analyses were carried out in negative polarity (static spray, 2.8 kV), ion funnel radiofrequency (RF) level at 50%, sheath gas (36 a.u.), auxiliary gas (16 a.u.), sweep gas (0 a.u.), ion transfer tube temperature of 327 °C, vaporizer temperature of 283 °C, mass range 220–2000 m/z, automatic gain control (AGC) 5.0×105, injection time (IT) 100 ms, intensity threshold 1.0×103, charge state selection 1–4, and each full scan spectrum consisted of accumulation of 1 microscan. Data dependent MS/MS (top 5) were acquired with a 2 m/z quadrupole isolation mode, AGC 1.0×105, IT 200 ms, and each MS/MS spectrum consisted of accumulation of 1 microscan. Full scan and MS/MS data were acquired employing either the ion trap or the orbitrap as mass analyzer. When the ion trap was employed, both full scan and MS/MS data were acquired by the same mass analyzer with rapid scan rate and normal mass range. When the orbitrap was used, both full scan and MS/MS data were also acquired by the same mass analyzer, with normal mass range and 15k resolution. MS/MS data were acquired employing either CID or HCD as activation type. For the CID experiments, collision energy (CE) was set at 21% and activation Q at 0.25. HCD studies were carried out with CE 20 a.u. and first mass m/z set as 50. Aiming to cover different sets of oligonucleotide data generated using trap instruments, the Orbitrap Fusion Lumos data herein presented consisted of three different combinations of mass analyzer and MS/MS activation type: (i) ion trap and CID (for low resolution and low mass accuracy data); (ii) orbitrap and CID (for high resolution and high mass accuracy data); and (iii) orbitrap and HCD (for high resolution and high mass accuracy data in which an alternative activation mode is employed).
MS data acquisition on the Waters Synapt G2-S was performed in negative polarity and sensitivity (V-mode) with resolving power of at least 15 000 FWHM (full width half maximum). Electrospray source conditions were capillary voltage of 2.5 kV, sampling cone 30 V, source temperature 120 °C, desolvation temperature 400 °C, and cone and desolvation gas flow rates of 20 and 800 L/h, respectively. MS/MS data acquisition was performed in Fast DDA mode with MS and MS/MS scan times of 0.3 s and 1.0 s, respectively, and MS scan range of 200–2000 m/z and MS/MS scan range of 50–2000 m/z. A mass-to-charge dependent collision energy ramp (12–15 at m/z 200, 45–55 at m/z 2000) was applied in the “trap” region of the Triwave. Lockspray correction was performed using leucine enkephalin (200 pg/µL), m/z = 554.2615, infused at 2 µL/min and collected for 1 s every 30 s.
2.4. RNA Modification Mapping Workflow
The RNA modification mapping and data analysis workflow is shown in Figure 1. Briefly, the RNA sample is subjected to enzymatic digestion to yield oligonucleotides analyzed by LC-MS/MS. The LC-MS/MS experiment will generate a data file that contains the precursor and product ion information in a vendor specific format that is then converted to a non-vendor specific file format. Using the MS/MS data file, user generated sequences, and modifications RAMM interprets the spectra, scores and ranks the interpretations, then performs sequence alignment and mapping results for those that meet scoring function threshold. Additional details on data analysis will be described in the following sections.
Figure 1.
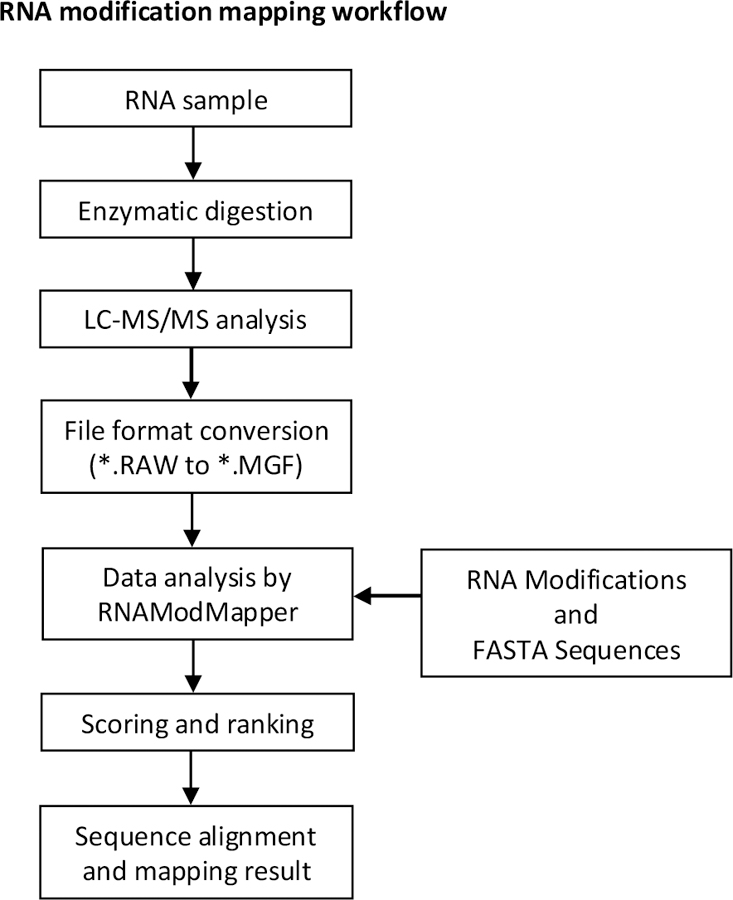
RNA modification mapping and data analysis workflow.
2.5. Pre-Processing of Raw Data
To analyze LC-MS/MS data with RAMM, data must first be converted from its original, vendor specific RAW format to a non-vendor specific format. In this manuscript, RAW refers to a data file format and should be distinguished from actual raw data. RAMM uses the MGF file format as the universal file input format. The MGF format contains the information (precursor m/z, retention time, MS/MS spectral data) needed to perform the MS/MS interpretation. If the MS/MS data is acquired in profile mode, it must also be centroided for use with RAMM. No additional pre-processing of the data (i.e. noise reduction, deisotoping, etc.) is required, but can be performed prior to analysis by RAMM.
To pre-process data generated on the Fusion Lumos, MSConvertGUI (64-bit, proteowizard.sourceforge.net) was used to convert the RAW file to MGF format file. The data acquired in this study was profile data, and MSConvertGUI was also used to convert the profile data to centroid data. To pre-process the data generated on the Waters Synapt G2-S, PLGS (ProteinLynx Global Server, Waters Corp) was used for Lockspray correction of the RAW file, which was exported in mzML format. MSConvertGUI was then used to convert the mzML to MGF format.
2.6. Data Analysis
2.6.1. Software and Computer Details
RAMM was used to perform data analysis. The software is available for free download from http://bearcatms.uc.edu/new/limbachgroup_publication/rna-modmapper-software.html. RAMM runs on Window 7 or higher version (32 or 64 bit). Minimal recommended hardware includes an Intel i3 processor and 8GB RAM, while better performance can be expected with more advanced configurations.
2.6.2. Input Files
There are two approaches for RNA modification mapping in RAMM: fixed sequence position and variable sequence position. Both functions take an MGF format MS/MS data file and a FASTA format sequence file as input files. For the fixed sequence approach, the FASTA format sequence file will include the expected modification (Figure 2A). For the variable sequence position approach, the FASTA format sequence file can be RNA or DNA gene sequences (Figure 2B).
Figure 2.
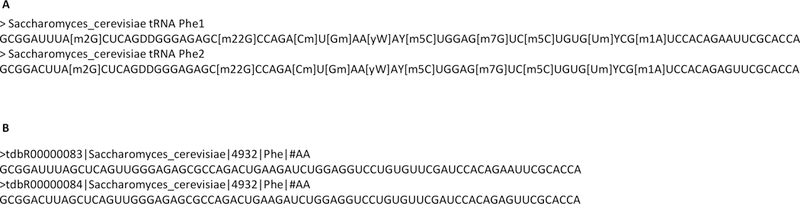
(A) S. cerevisiae modified tRNAPhe sequences used in fixed position modification mapping analysis. (B) Unmodified (genomic) S. cerevisiae tRNAPhe sequences used in variable position modification mapping analysis.
2.6.3. Graphical User Interface and User Inputs
The user interfaces for fixed sequence position and variable sequence position are very similar as shown in Figure 3A. For variable sequence position, the modified nucleosides can be selected by the user as shown in Figure 3B. RAMM provides a variety of user-selected conditions including the enzyme, the 3’-end identity (e.g., phosphate, cyclic phosphate, no phosphate), number of allowable missed cleavages, the mass types and the mass tolerances for precursor and product ions, which are set according to the instrument used for the analysis. RAMM uses two product ions channels, c/y- and a-B/w-type ions, for evaluating the quality of the data via the scoring function. The default c/y-type ions weighting factor is set to 0.7 based on our previous study[25] and this value can be changed by the user based on the relative contribution of the c/y dissociation channels in the experiment. RAMM also supports custom modifications and user-input oligonucleotide sequences.
Figure 3.

(A) Screenshot of Fixed position modification mapping window from GUI where data file and sequences are selected. Data processing parameters are also defined here. (B) Screenshot of Modifications window from GUI where modifications are identified during Variable position modification mapping.
To process low-resolution MS and MS/MS data, precursor and product mass tolerances were both kept at 1.0 m/z consistent with the expected mass accuracy of this acquisition approach. For high-resolution MS and MS/MS data collected on the Waters G2-S, the precursor and product mass tolerances were kept at 0.06 and 0.1 m/z (30 ppm and 100 ppm at m/z 2000, respectively). A higher product ion mass tolerance was used because the Lockspray correction was only performed on MS data. For high-resolution MS and MS/MS data collected on the Thermo Orbitrap Fusion Lumos, the precursor and product mass tolerances initially were both set to 0.06 m/z (30 ppm at m/z 2000). However, it was realized that oligonucleotides containing an isotopic distribution where the 13C isotope peak is the most abundant require larger precursor ion tolerances (e.g., ≥ 0.33 for a −3 charge state) for accurate MS/MS data interpretation. The exact oligonucleotide length where the 13C peak becomes more abundant than the 12C peak will depend on the sequence composition and extent of modification, but generally occurs for unmodified RNA oligonucleotides 8–9 nucleotides in length (~ 2700 Da).
2.6.4. In-silico Digestion
During data analysis, RAMM generates a temporary, local database of in-silico digestion products to determine theoretical m/z values for precursor ions based on user defined parameters. RAMM supports five different ribonucleases for in silico digestion: RNase T1, RNase U2, RNase A, RNase MC1 and Cusativin. Within RAMM, the following RNase cleavage rules are implemented recognizing that actual enzyme cleavage patterns may have different selectivity’s towards modified nucleosides [27–29]: RNase T1 cleaves at the 3'-end of guanosine and N2-methylguanosine (m2G); RNase U2 cleaves at the 3'-end of guanosine and adenosine; RNase A cleaves at the 3'-end of unmodified pyrimidines; Cusativin cleaves at the 3'-end of cytidine; and RNase MC1 cleaves at the 5'-end of uridine. To add additional flexibility based on enzyme behavior, the program supports up to five missed cleavages for any ribonuclease.
2.6.5. Fixed-Sequence and Variable-Sequence Modification Mapping
RAMM allows for RNA modification mapping in two different modes, fixed-sequence and variable-sequence mapping. This is a user selected option within the software that allows flexibility of data analysis depending on the goal of the mapping experiment and/or data analysis. In fixed-sequence mode, in addition to the pre-processed LC-MS/MS data, the user must supply a FASTA file containing RNA sequences with pre-annotated modified nucleosides. This option allows the user to determine if known modified RNA digestion products were detected during LC-MS/MS analysis. In addition to the 120 chemical modifications included within RAMM, fixed-sequence mode allows additional, user-defined, modifications not already programmed into the software. This allows the detection of, for example, pseudouridine through commonly used derivatizing agents[27, 30], or other synthetic modifications. Since positional isomers of modified nucleobases (e.g., 1-methyladenosine (m1A) vs N6-methyladenosine (m6A) cannot be distinguished during routine MS/MS analysis of oligonucleotides, selection of only a single one of these motifs (if more than one is present in the sample) can reduce computation time and improve throughput of post-processing review of interpretations without compromising sequence coverage.
In variable-sequence mapping mode, the user provides the unmodified (genomic) RNA sequences in addition to the pre-processed LC-MS/MS data. The user must then select all the modified nucleosides to be evaluated by the program during MS/MS data interpretation. Variable-sequencing mapping is more computationally expensive than the fixed position approach, with important variables including the computer hardware, number of FASTA sequences, number of modified nucleosides to be evaluated, and number of MS/MS scans in the MGF file. Using a standard desktop computer configuration, a typical tRNA modification mapping experiment (e.g. 22 genomic sequences, 32 modifications) usually takes less than 12 h on a conventional hard drive (HDD), which can be reduced further (~33% faster in our experience) by using a solid-state drive.
2.6.6. MS/MS Spectral Interpretation
To interpret MS/MS spectra, a local database of theoretical digestion products is generated in-silico based on the enzyme, FASTA sequences, and modifications defined by the user prior to data analysis. The molecular weight is then calculated for each potential digestion product to serve as an initial comparison with a precursor mass in the MS/MS data in the MGF file. If the calculated mass matches the precursor mass within the precursor mass tolerance, the set of predicted product ions are generated and then compared against the actual peaks in the MS/MS spectrum. Like precursor ion matching, product ions must fall within the product ion mass tolerance to be considered a match. The MS/MS data is then scored and ranked. An interpretation of an MS/MS spectrum is only returned by RAMM if it meets the pre-defined user criteria, including precursor and product ion tolerances, and p-score and dot product thresholds.
All RAMM results presented in this manuscript were manually reviewed for accuracy. The automated interpretation is considered correct if manual review matches the top ranked sequence interpretation returned by RAMM. Additionally, the retention time of the MS/MS spectrum must also have a chromatographic retention time consistent with that previously determined through manual review of the data (Qual Browser for Thermo data, MassLynx for Waters data). To not bias the results between different vendor platforms and MS/MS sampling rates, only one correct (or incorrect) interpretation was reported for a chromatographic peak. As is typical in untargeted modification mapping experiments, unmodified monomers and dimers were excluded from data analysis due to their ambiguity. Those digestion products detected during variable mapping and appearing in more than one high-resolution acquisition mode, but not previously reported digestion products of S. cerevisiae tRNAPhe, were excluded from the analysis. These digestion products are believed to be present due to minor contaminating RNAs co-isolated in the purchased commercial S. cerevisiae tRNAPhe.
2.6.7. Scoring Function
RAMM uses a combination of a normalized binomial distribution probability[31] and dot product [32] scoring to assess the quality of the MS/MS spectral interpretation. These approaches have been adopted in RAMM for the fragmentation of oligonucleotides which generate c-, y-, w-, and a-B type ions during CID. Because the binomial distribution probability (P-value) will be very small for a true positive (Supplemental Equation 1) and will depend on the length of the oligonucleotide and number of matched ions, it is more useful to convert the P-value to a P-score that is normalized based on the length of the oligonucleotide. Therefore, if all theoretical ions are found in the MS/MS spectrum, the P-score (S(P), Supplemental Equation 2) will be 100 independent of oligonucleotide length. It has been shown that that the relative abundance of c- and y- type ions are greater than w- and a-B ions in RNA[33]. RAMM allows the P-score to be weighted by the expected (or observed) differences in relative abundance (Supplemental Equation 3). The default weighting factors for c- and y-type ions and w- and a-B ions are 70% and 30%, respectively. However, these values can be adjusted by the user to match experimental data. A P-score threshold can be defined by the user to eliminate any interpretations below the set value. A significant P-score threshold should be set based on the goals of the experiment, knowledge of the sample, and experience of the analyst. In this work, a P-score of 55 and above is considered to be significant, meaning the MS/MS interpretation contains the minimum number of assigned product ions to be accepted with high confidence.
The main limitation of using the P-score alone is that it only takes into account whether the theoretical product ion was present but ignores ion abundance in the spectrum. To improve the ability of RAMM to distinguish between true and false positives, the dot product score (Supplemental Equation 4) is also incorporated in the scoring function. The dot product provides a quantitative measure of the similarity between the observed and reconstructed spectra. To determine the dot product score, a reconstructed spectrum is generated that contains only experimental m/z values that match theoretical m/z values calculated for each digestion product during in-silico digestion that fall within the user-defined product m/z tolerances. An experimental spectrum where all of the most abundant ions correspond to only those generated in-silico would have a dot product of 1. In addition to the most common (c/y) product ions, RAMM also takes nucleobase loss observed during CID into account. For certain modifications (e.g., 7-methyl-guanosine (m7G), queuosine (Q), N6-threonylcarbamoyladenosine (t6A), lysidine (k2C)), nucleobase loss from the precursor ion can predominate an MS/MS spectrum for a digestion product containing these modifications[34]. The RAMM scoring function accounts for these losses from the precursor ion during the dot product calculation. It should be noted that these losses are not accounted for when a product ion contains these modifications. This can impact RAMM’s ability to interpret MS/MS spectra containing these modifications. However, if these are known to be present in the RNA sample, a lower dot product threshold can be defined as a work-around.
2.5.8. Sequence Alignment
After interpretation of MS/MS spectra is complete, the oligonucleotides that meet the user criteria are then mapped back onto the RNA sequence. Common to most RNA modification mapping experiments, when interpreted digestion products (i.e., MS/MS data) could map onto more than one region of a single RNA or onto more than one single RNA sequence, the software cannot identify the “correct” RNA sequence (or region) for mapping. In these cases, RAMM will match the interpreted MS/MS data to all possible RNA sequences. Future updates to the software will compile mapping results from experiments where multiple RNases digests generated from more than one enzyme are used to improved confidence in modification mapping results through overlapping sequence coverage[28].
2.5.9. Output
An output file containing the results of the interpreted and scored MS/MS spectra can be exported as a .CSV file. The file contains information for each interpreted MS/MS spectrum, including retention time (RT), measured m/z of precursor, exact m/z of precursor, calculated charge state, rank of each interpretation (when more than one interpretation per MS/MS event), score, sequence, and number of a-B, c, w, and y-type ions found.
3. Results and Discussion
To further refine and improve RAMM as a tool to enable higher-throughput analysis of LC-MS/MS data from RNA modification mapping experiments, a comparative study using a well-defined tRNA was conducted. The motivation for this work was to better understand the experimental conditions and software settings that would improve the accuracy of RNA modification mapping analyses. In addition, we sought to define more clearly when manual review of RAMM data is required as well as when the user can have increased confidence that the MS/MS data interpretations returned by RAMM were correct. Taken together, our goal was to continue to improve the rate-limiting step in LC-MS/MS based RNA modification mapping experiments.
LC-MS/MS data for an RNase T1 digestion of S. cerevisiae tRNAPhe was acquired on two different MS platforms (Waters Synapt G2-S and Thermo Orbitrap Fusion Lumos) under four different acquisition modes (TOF-CID, ion-trap CID, orbitrap-CID and orbitrap-CD). The number of incorrect MS/MS interpretations returned by RAMM using standard processing parameters in both fixed and variable modes was also evaluated. A summary of the parameters used is shown in Table 1.
Table 1.
Summary of standard data processing parameters used for MS platforms and acquisition types.
| Processing Parameter | Low-Resolution Orbitrap Fusion Lumos |
High-Resolution Orbitrap Fusion Lumos |
High-Resolution Synapt G2-S |
|---|---|---|---|
| Precursor Tolerance (Da) | 1.0 | 0.06 | 0.06 |
| Product Ion Tolerance (Da) | 1.0 | 0.061 | 0.10 |
| P-score Threshold, Standard | 55 | 55 | 55 |
| P-score Threshold, Unique CID | 25 | 25 | 25 |
| Dot Product Threshold, Standard | 0.65 | 0.65 | 0.65 |
May require increase if isotope peak other than monoisotopic peak is most abundant in MS
3.1. Fixed Position Mapping
Fixed position modification mapping requires a FASTA sequence file that includes fully (or partially) modified sequences. The FASTA sequences used for fixed position mapping of S. cerevisiae tRNAPhe are provided in Figure 2A and a summary of the detected digestion products are provided in Table 2. The number of correct and incorrect interpretations found for the RNase T1 digest in all four acquisition modes are given in Figure 4A. In fixed mapping mode, RAMM was capable of correctly interpreting 10 of the 12 expected digestion products using the processing parameters in Table 1 for trap CID in both low (ion-trap) and high (Orbitrap) resolution acquisition modes. The sequence alignment is shown in Supplemental Figure S1. The same digestion products were interpreted for Orbitrap HCD and TOF CID; however each of these acquisition modes did not return one digestion product due to poor MS/MS signal quality and low MS abundance, respectively. Examples of correctly interpreted MS/MS spectra are shown in Figure 5.
Table 2.
Summary of detected digestion products for S. cerevisiae tRNAPhe for fixed and variable mapping for all MS acquisition types. Monomer and dimer digestion products were excluded from the data analysis.
| Digestion Product | IT-CID Fixed |
IT-CID Variable |
Orbi-CID Fixed |
Orbi-CID Variable |
Orbi- HCD Fixed |
Orbi- HCD Variable |
TOF-CID Fixed |
TOF-CID Variable |
|---|---|---|---|---|---|---|---|---|
| AUUUA[m2G] | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| ACUUA[m2G] | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| CUCAG | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| DDG | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| DD[Gm]G | N/D1 | ✓ | N/D1 | ✓ | N/D1 | ✓ | N/D1 | ✓ |
| C[m22G]CCAG | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | N/D2 | ✓ |
| A[Cm]U[Gm]AA[yW]AU[m5C]UG | N/D3 | N/D4 | N/D3, 5 | N/D4 | N/D3, 5 | N/D4 | N/D3, 5 | N/D4 |
| [m7G]UC[m5C]UG | N/D3 | N/D3 | N/D3 | N/D3 | N/D3 | N/D3 | N/D3 | N/D3 |
| [m5U]ѰCG | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| [m1A]UCCACAG | ✓ | ✓ | ✓ | ✓ | N/D6 | ✓ | ✓ | ✓ |
| AAUUCG | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| UUCG | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| CACCA | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Total | 10 | 11 | 10 | 11 | 9 | 10 | 9 | 10 |
N/D – Not Detected
Digestion product not included in fixed sequence analysis
Low MS response did not result in MS/MS triggering
Lower P-score threshold needed due to neutral base loss of yW or m7G
Multiple modified Gs that result in missed cleavage not detected in current version
A higher precursor ion tolerance (0.33) is needed due to selection of more abundant 13C isotope
Low MS/MS abundance
Figure 4.

(A) Number of correct and incorrect interpretations for fixed and variable mapping for all four acquisition types using standard processing conditions (p-score = 55, dot product = 0.65). (B) Number of interpretations for variable mapping with p-score = 55 at various dot product scores.
Figure 5.

Examples of a correctly interpreted MS/MS spectra for digestion product AUUUA[m2G] from (A) beam-type CID on Synapt (B) HCD on Fusion Lumos and (C)ion-trap CID on Fusion Lumos. Additional information on fragment ion assignments and mass errors can be found in Supplemental Figure S6.
The two digestion products not returned by RAMM using standard processing parameters under all acquisition conditions were the m7G and yW containing oligonucleotides. Due to the labile nature of these modified RNA nucleobases under CID conditions[34], the predominant ions in the MS/MS spectra of these oligonucleotides are neutral base losses. Although RAMM does account for neutral base loses from the precursor m/z value (M-BH), these neutral base losses will also be detected for any product ions that contain these modifications (e.g. all c-type ions in [m7G]UC[m5C]UG). These fragmentation channels result in detected m/z values shifted by the characteristic loss (m7G =165.065, yW=374.133) from the calculated m/z value, which will not be detected by RAMM. An example is provided in Figure 6A, where all m7G containing product ions (i.e. all c-type ions) were not assigned. Because these product ions were not identified by RAMM, what would be manually verified as a correct MS/MS spectrum gives a P-score value below the standard threshold of 55. For RAMM to successfully return the m7G and yW containing oligonucleotides, a lower P-score threshold of 25 was required.
Figure 6.

Interpreted MS/MS spectrum obtained on the Synapt G2-S for (A) [m7G]UC[m5C]UG and (B) A[Cm]U[Gm]AA[yW]AU[m5C]UG digestion products from S. cerevisiae tRNAPhe. Due to the significant neutral base losses of [m7G] of [yW] during CID, [M-BH] and product ions containing these modified bases are not assigned.
In addition to the two missed digestion products containing unique fragmentation channels, one additional digestion product was not returned with the Orbitrap HCD and TOF CID ([m1A]UCCACCAG and C[m22G]CCAG, respectively). For the missed digestion product during HCD acquisition, the correct precursor ion was detected during the MS scan at sufficient abundance to trigger MS/MS, but too few correct product ions were found due to over-fragmentation during CID. In the case of TOF CID, the corresponding precursor ion was not of sufficient abundance to trigger an MS/MS event. In both cases poor ion abundance leads to the lack of an interpretation for a known digestion product present in the sample. This result underscores the importance of manual review of the raw data where gaps in sequence coverage are reported by RAMM.
The number of incorrect interpretations for fixed position mapping using the four acquisition modes also is presented in Figure 4A. The number of incorrect interpretation in fixed position modification mapping was low, which is expected as data analysis is very targeted. Examples of false positive spectra are shown in Supplemental Figures S2 and S3. Although a combination of two separate scoring functions were included to improve MS/MS spectral interpretation performance and limit the number of incorrect interpretations, false positives are still a possibility. Incorrect interpretations were those that met the P-score and dot product thresholds but could not be manually validated. Three incorrect interpretations were returned for the low-resolution data acquisition, whereas only a single incorrect interpretation was returned in all three high-resolution modes. The higher number of incorrect interpretations for low-resolution data is expected due to the wider precursor and product ion tolerances used for lower mass accuracy acquisition. The wider mass tolerances will lead to more sequence arrangements that meet the scoring criteria, which lead to more ambiguous interpretations that can only be eliminated manually. The most commonly observed false positives in high-resolution acquisitions are due to both the assignment of lower abundant product ions with simultaneous incorrect assignment of high abundance product ions. This can be common when the precursor mass falls within the defined tolerance, but is the incorrect charge state. To reduce the number of incorrect interpretations, future versions of RAMM will include direct determination of precursor charge states through analysis of isotopic spacing.
Another issue that can arise when working with high resolution MS and MS/MS data is the mis-assignment of the all 12C peak. For example, the interpretation of the yW containing digestion product using high-resolution acquisition on the Fusion Lumos was not possible using the standard precursor ion tolerance of 0.06. Because the 13C isotope peak was the most abundant ion in the MS spectrum, MS/MS isolation was centered around this isotope, which is 0.33 m/z (charge state = −3) higher than the monoisotopic peak (see Supplemental Figure S4). While the mass spectral behavior was found to be similar between the Fusion Lumos and the Synapt G2-S, the Waters software (MassLynx) stored the monoisotopic peak in the raw data while the Thermo software (XCalibur) stored the 13C peak in the raw data. In such cases, the analyst should increase the precursor mass tolerance in RAMM for successful interpretation of higher molecular weight RNase digestion products (e.g., > 2700 Da). Alternatively, adjusting the mass spectrometer acquisition conditions (e.g., performing the acquisition in MIPS (monoisotopic precursor selection) on the Fusion Lumos), or incorporating isotopic and charge state calculations for precursor ions may eliminate the need for increased precursor ion tolerances when using RAMM.
3.2. Variable Position Mode
Unlike fixed position mode, variable position sequencing uses unmodified RNA sequences and requires selection of the individual modified nucleosides suspected or known to be present in the sample. This mapping mode is used for experiments where little is known about the location of individual modifications. To demonstrate performance of RAMM for variable position modification mapping, the FASTA sequences for S. cerevisiae tRNAPhe shown in Figure 2B were used. The modifications were selected in the “Modifications” menu in the GUI (Figure 3B). The same processing parameters as those in the fixed position mode, including identical P-score and dot product thresholds (55 and 0.65), were used.
The digestion products correctly interpreted in fixed mode were also returned in variable mapping mode using the standard scoring threshold, with one exception. An additional digestion product, DD[Gm]G, not detected in fixed mode was found in variable mode. This is a known, lower abundance modification present in S. cerevisiae tRNAPhe[35], but it is not reported in common RNA sequence databases[36, 37]. The removal of the constraint for each nucleotide to have a single identity in variable position mapping mode allowed for the interpretation of this MS/MS spectrum. The same two digestion products missed in fixed mapping mode, the m7G and yW containing oligonucleotides, were also missed in variable mapping mode. However, the yW containing oligonucleotide could not be detected even when using a lower P-score threshold. This is due to a limitation in RAMM that does not allow for the interpretation of digestion products containing more than one modified G that results in no enzymatic cleavage (e.g., m7G, m22G, Gm, yW). While this outcome in our experience is quite uncommon, it serves as an important reminder to the user that the actual behavior of an enzyme may be different than those currently identified by the software. In this case here, because the yW containing digestion product contains both Gm and yW, RAMM was not capable of interpreting this digestion product. However, this specific limitation will be addressed in a future update to RAMM.
The number of incorrect interpretations in variable mapping mode are shown in Figure 4A. More incorrect interpretations were given in variable mode than fixed mode for all four acquisition modes. This behavior was also noticed in the receiver operating characteristic (ROC) curves originally reported for RAMM[25]. Because variable mapping is less restrictive from a data analysis perspective, this behavior could be expected. Incorrect interpretations in variable mode included those found in fixed mode for the previously stated reasons, but also include modifications placed at incorrect positions. This outcome not only decreases overall data processing throughput due to additional manual review of interpretations, but may also increase the number of incorrect or ambiguous modification location assignments for samples that are not well-characterized. In our experience, the performance of the software is similar for a sample containing either a single sequence or multiple sequences. The increase in the number of incorrect interpretations is most notable for the low-resolution MS/MS data, where an approximately equal number of correct and incorrect interpretations were returned by RAMM. The use of a high-resolution, high mass accuracy mass analyzer minimizes the number of incorrect interpretations returned by RAMM, improving throughput and leading to higher confidence in RNA sequence annotation.
3.3. Selection of Appropriate Scoring Thresholds
The P-score and dot product thresholds have already been shown to affect the number of correct and incorrect interpretations returned by the software. In general, raising these values will reduce the number of returned MS/MS interpretations, while decreasing these values can increase the number of returned MS/MS interpretations. As noted in the previously reported ROC[25], there is always a trade-off between having RAMM return a correct interpretation and the number of incorrect interpretations that would also be returned for the same MS/MS data file. Thus, the user must find the appropriate balance based on experience and MS/MS data quality.
Because the P-score represents the minimum number of product ions that must be found during data analysis, it should be set at a value that represents established lab and experiment criteria and the intended experimental goal and/or future use of the data. It is our experience that a P-score of 55 consistently gives the minimum number of product ions needed to have a high confidence in the interpretation. As the P-score is independent of length, a P-score threshold of 55 corresponds to approximately 70% of predicted product ions detected. Because this includes ALL product ions (c-, y-, a-B, and W), a lower value may still yield a confident interpretation for modification mapping purposes if at least one complimentary c/a-B or y/w ion provides complete coverage of the sequence. The dot product threshold, however, can be adjusted based on the desired accuracy and throughput of the analysis. To demonstrate the impact of the number of incorrect and correct MS/MS interpretations given by RAMM as the dot product threshold is changed, a range of dot product scoring thresholds (0.30 to 0.90) was evaluated for all four acquisition types. The P-score was maintained at the standard value of 55.
The number of correct and incorrect interpretations under variable position modification mapping for low-resolution and high-resolution ion-trap CID is shown in Figure 4B. It should be noted that similar results were obtained for all high-resolution modes (see Supplemental Figure S5). Two major observations can be made from these results. First, the number of correct interpretations increases until all possible oligonucleotides are detected (dot product = 0.65), with a decreasing dot product threshold. This behavior should be expected, since an arbitrarily high dot product threshold may be too restrictive and exclude correct interpretations (false negatives). The second significant observation is that continuing to lower the dot product threshold only increases the number of incorrect interpretations. Similar to results shown in Figure 4A, variable mapping returned more incorrect interpretations than fixed mapping mode in all cases. The results in Figure 4B illustrate the importance of appropriate dot product threshold selection. Raising it may improve data analysis throughput by reducing the number of interpretations that must be manually reviewed and rejected. However, raising it too high will also increase the chance to miss correct interpretations. Our experience has found that a dot product of 0.65 provides an acceptable trade-off between throughput and RNA modification mapping accuracy.
4. Conclusion
RNAModmapper, or RAMM, was previously developed as a program capable of improving the throughput of LC-MS/MS data acquired on commonly used MS platforms for RNA modification mapping experiments. Here, a fixed and variable modification mapping study was performed using a commercially available modified tRNA sequence to underscore the importance and impact of appropriate dot product and P-score data processing thresholds. Adjusting these values allows the user direct control over the number of correct and incorrect interpretations returned by the software, which can have a significant effect on both sequence coverage and overall data processing throughput. Additionally, the compromises of performing modification mapping experiments using low-resolution MS, and the similarity in data processing outcomes for commonly implemented CID platforms was illustrated. Perhaps most importantly, although this and previous studies have illustrated RAMM’s utility in improving the tedious task of MS/MS spectral interpretation for the user, it cannot completely eliminate the manual review of modification mapping data.
Supplementary Material
Highlights.
RNAModMapper Software evaluated
Examined impact of instrumental conditions on RNA mapping
Identified optimal software settings and improved approach for reducing data analysis time and effort
Acknowledgements
Financial support for this work is provided by the National Institutes of Health (NIH GM058843). The generous support of the Rieveschl Eminent Scholar Endowment and the University of Cincinnati for these studies is also appreciated.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Hotchkiss RD, The quantitative separation of purines, pyrimidines, and nucleosides by paper chromatography, The Journal of biological chemistry, 175 (1948) 315–332. [PubMed] [Google Scholar]
- [2].Cohn WEV, E., Nucleoside-5’-phosphates from ribonucleic acid, Nature, 167 (1951) 483–484. [Google Scholar]
- [3].Roundtree IA, Evans ME, Pan T, He C, Dynamic RNA Modifications in Gene Expression Regulation, Cell, 169 (2017) 1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Agris PF, Narendran A, Sarachan K, Vare VYP, Eruysal E, The Importance of Being Modified: The Role of RNA Modifications in Translational Fidelity, The Enzymes, 41 (2017) 1–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Helm M, Post-transcriptional nucleotide modification and alternative folding of RNA, Nucleic Acids Res, 34 (2006) 721–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lewis CJ, Pan T, Kalsotra A, RNA modifications and structures cooperate to guide RNA-protein interactions, Nature reviews. Molecular cell biology, 18 (2017) 202–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bjork GR, Ericson JU, Gustafsson CE, Hagervall TG, Jonsson YH, Wikstrom PM, Transfer RNA modification, Annu Rev Biochem, 56 (1987) 263–287. [DOI] [PubMed] [Google Scholar]
- [8].Senger B, Auxilien S, Englisch U, Cramer F, Fasiolo F, The modified wobble base inosine in yeast tRNAIle is a positive determinant for aminoacylation by isoleucyl-tRNA synthetase, Biochemistry, 36 (1997) 8269–8275. [DOI] [PubMed] [Google Scholar]
- [9].Bohnsack MT, Sloan KE, The mitochondrial epitranscriptome: the roles of RNA modifications in mitochondrial translation and human disease, Cellular and molecular life sciences : CMLS, 75 (2018) 241–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tserovski L, Marchand V, Hauenschild R, Blanloeil-Oillo F, Helm M, Motorin Y, High-throughput sequencing for 1-methyladenosine (m(1)A) mapping in RNA, Methods, 107 (2016) 110–121. [DOI] [PubMed] [Google Scholar]
- [11].Schwartz S, Bernstein DA, Mumbach MR, Jovanovic M, Herbst RH, Leon-Ricardo BX, Engreitz JM, Guttman M, Satija R, Lander ES, Fink G, Regev A, Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA, Cell, 159 (2014) 148–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Li X, Xiong X, Wang K, Wang L, Shu X, Ma S, Yi C, Transcriptome-wide mapping reveals reversible and dynamic N(1)-methyladenosine methylome, Nat Chem Biol, 12 (2016) 311–316. [DOI] [PubMed] [Google Scholar]
- [13].Helm M, Motorin Y, Detecting RNA modifications in the epitranscriptome: predict and validate, Nature reviews. Genetics, 18 (2017) 275–291. [DOI] [PubMed] [Google Scholar]
- [14].Kowalak JA, Pomerantz SC, Crain PF, McCloskey JA, A novel method for the determination of post-transcriptional modification in RNA by mass spectrometry, Nucleic Acids Research, 21 (1993) 4577–4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ni J, Pomerantz C, Rozenski J, Zhang Y, McCloskey JA, Interpretation of oligonucleotide mass spectra for determination of sequence using electrospray ionization and tandem mass spectrometry, Anal Chem, 68 (1996) 1989–1999. [DOI] [PubMed] [Google Scholar]
- [16].McLuckey SA, Van Berkel GJ, Glish GL, Tandem mass spectrometry of small, multiply charged oligonucleotides, J Am Soc Mass Spectrom, 3 (1992) 60–70. [DOI] [PubMed] [Google Scholar]
- [17].McLuckey SA, Habibi-Goudarzi S, Ion trap tandem mass spectrometry applied to small multiply charged oligonucleotides with a modified base, J Am Soc Mass Spectrom, 5 (1994) 740–747. [DOI] [PubMed] [Google Scholar]
- [18].Cai WM, Chionh YH, Hia F, Gu C, Kellner S, McBee ME, Ng CS, Pang YLJ, Prestwich EG, Lim KS, Ramesh Babu I, Begley TJ, Dedon PC, A Platform for Discovery and Quantification of Modified Ribonucleosides in RNA: Application to Stress-Induced Reprogramming of tRNA Modifications, Methods in enzymology, 2015, pp. 29–71. [DOI] [PMC free article] [PubMed]
- [19].Nichols AM, White FM, Manual validation of peptide sequence and sites of tyrosine phosphorylation from MS/MS spectra, Methods in molecular biology (Clifton, N.J.), 492 (2009) 143–160. [DOI] [PubMed] [Google Scholar]
- [20].Rozenski J, McCloskey JA, SOS: a simple interactive program for ab initio oligonucleotide sequencing by mass spectrometry, J Am Soc Mass Spectrom, 13 (2002) 200–203. [DOI] [PubMed] [Google Scholar]
- [21].Nyakas A, Blum LC, Stucki SR, Reymond JL, Schurch S, OMA and OPA--software-supported mass spectra analysis of native and modified nucleic acids, J Am Soc Mass Spectrom, 24 (2013) 249–256. [DOI] [PubMed] [Google Scholar]
- [22].Nakayama H, Akiyama M, Taoka M, Yamauchi Y, Nobe Y, Ishikawa H, Takahashi N, Isobe T, Ariadne: a database search engine for identification and chemical analysis of RNA using tandem mass spectrometry data, Nucleic Acids Res, 37 (2009) e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Matthiesen R, Kirpekar F, Identification of RNA molecules by specific enzyme digestion and mass spectrometry: software for and implementation of RNA mass mapping, Nucleic Acids Res, 37 (2009) e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sample PJ, Gaston KW, Alfonzo JD, Limbach PA, RoboOligo: software for mass spectrometry data to support manual and de novo sequencing of post-transcriptionally modified ribonucleic acids, Nucleic Acids Res, 43 (2015) e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yu N, Lobue PA, Cao X, Limbach PA, RNAModMapper: RNA Modification Mapping Software for Analysis of Liquid Chromatography Tandem Mass Spectrometry Data, Anal Chem, 89 (2017) 10744–10752. [DOI] [PubMed] [Google Scholar]
- [26].Lobue PA, Jora M, Addepalli B, Limbach PA, Oligonucleotide Analysis by Hydrophilic Interaction Liquid Chromatography-Mass Spectrometry in the Absence of Ion-Pair Reagents, submitted for publication (2018). [DOI] [PMC free article] [PubMed]
- [27].Mengel-Jorgensen J, Kirpekar F, Detection of pseudouridine and other modifications in tRNA by cyanoethylation and MALDI mass spectrometry, Nucleic Acids Res, 30 (2002) e135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Addepalli B, Lesner NP, Limbach PA, Detection of RNA nucleoside modifications with the uridine-specific ribonuclease MC1 from Momordica charantia, RNA, 21 (2015) 1746–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Addepalli B, Venus S, Thakur P, Limbach PA, Novel ribonuclease activity of cusativin from Cucumis sativus for mapping nucleoside modifications in RNA, Anal Bioanal Chem, 409 (2017) 5645–5654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Patteson KG, Rodicio LP, Limbach PA, Identification of the mass-silent post-transcriptionally modified nucleoside pseudouridine in RNA by matrix-assisted laser desorption/ionization mass spectrometry, Nucleic Acids Res, 29 (2001) E49–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP, A probability-based approach for high-throughput protein phosphorylation analysis and site localization, Nat Biotechnol, 24 (2006) 1285–1292. [DOI] [PubMed] [Google Scholar]
- [32].Yen CY, Houel S, Ahn NG, Old WM, Spectrum-to-spectrum searching using a proteome-wide spectral library, Mol Cell Proteomics, 10 (2011) M111 007666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Huang TY, Kharlamova A, Liu J, McLuckey SA, Ion trap collision-induced dissociation of multiply deprotonated RNA: c/y-ions versus (a-B)/w-ions, J Am Soc Mass Spectrom, 19 (2008) 1832–1840. [DOI] [PubMed] [Google Scholar]
- [34].Ross R, Cao X, Yu N, Limbach PA, Sequence mapping of transfer RNA chemical modifications by liquid chromatography tandem mass spectrometry, Methods, 107 (2016) 73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hori H, Suzuki T, Sugawara K, Inoue Y, Shibata T, Kuramitsu S, Yokoyama S, Oshima T, Watanabe K, Identification and characterization of tRNA (Gm18) methyltransferase from Thermus thermophilus HB8: domain structure and conserved amino acid sequence motifs, Genes Cells, 7 (2002) 259–272. [DOI] [PubMed] [Google Scholar]
- [36].Boccaletto P, Machnicka MA, Purta E, Piątkowski P, Bagiński B, Wirecki TK, de Crécy-Lagard V, Ross R, Limbach PA, Kotter A, Helm M, Bujnicki JM, MODOMICS: a database of RNA modification pathways. 2017 update, Nucleic Acids Research, 46 (2018) D303–D307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sprinzl M, Horn C, Brown M, Ioudovitch A, Steinberg S, Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res 26 (1998) 148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.